

Abstract
Rituximab is a mouse/human chimeric monoclonal antibody targeted toward CD20. It is efficient as first-line therapy of CD20-positive B-cell malignancies. However, a large fraction of treated patients relapse with rituximab-resistant disease. So far, only modest progress has been made in treatment options for rituximab refractory patients. One of the mechanisms for rituximab resistance involves the upregulation of CD46, which is a key cell surface protein that blocks the activation of complement. We have recently developed a technology that depletes CD46 from the cell surface and thereby sensitizes tumor cells to complement-dependent cytotoxicity. This technology is based on a small recombinant protein, Ad35K++ that binds with high affinity to CD46. In preliminary studies using a 6 × histidinyl tagged protein, we had demonstrated that intravenous Ad35K++ injection in combination with rituximab was safe and increased rituximab-mediated killing of CD20-positive target cells in mice and nonhuman primates (NHPs). The presence of the tag, while allowing for easy purification by Ni-NTA chromatography, has the potential to increase the immunogenicity of the recombinant protein. For clinical application, we therefore developed an Ad35K++ protein without His-tag. In the present study, we performed preclinical studies in two animal species (mice and NHPs) with this protein demonstrating its safety and efficacy. These studies estimated the Ad35K++ dose range and treatment regimen to be used in patients. Furthermore, we showed that intravenous Ad35K++ injection triggers the shedding of the CD46 extracellular domain in xenograft mouse tumor models and in macaques. Shed serum CD46 can be measured in the serum and can potentially be used as a pharmacodynamic marker for monitoring Ad35K++ activity in patient undergoing treatment with this agent. These studies create the basis for an investigational new drug application for the use of Ad35K++ in combination with rituximab in the treatment of patients with B-cell malignancies.
Introduction
B-cell non-Hodgkin’s lymphoma (NHL) is the seventh most common cancer in the United States with approximately 72,000 new diagnoses each year.1 The 1-, 5-, and 10-year survival rates for NHL are 79, 63, and 51%, respectively. Rituximab is a mouse/human chimeric monoclonal antibody targeting CD20 and induces cell killing through complement-dependent cytotoxicity (CDC), antibody-dependent cell cytotoxicity, antibody-dependent cell-mediated phagocytosis, and direct cell apoptosis.2–6 It is effective as a single agent and in combination with chemotherapy in the treatment of CD20-postive B-cell malignancies. While efficient as first-line therapy, rituximab is less effective in patients with relapsed lymphoma.7–9 For example, only 40% of the patients with low-grade NHL responded again to rituximab when they had received prior rituximab treatment.10–12 Therefore, these patients often will require alternative treatments to control the lymphoma.13–15
CD46 is a membrane-linked glycoprotein that is expressed in humans on all cells except red blood cells. One of its functions is to block complement-mediated killing of autologous cells. CD46 attaches to complement fragments C3b and C4b that are bound to host cells and then serves as a cofactor for their targeted destruction by the plasma serine protease Factor I. Many cancer cells upregulate CD46.16 For example, we and others have shown that CD46 levels on blood cell malignancies were at least one order of magnitude higher than on normal peripheral blood mononuclear cells (PBMCs).17,18 Considering that rituximab kills lymphoma cells in part through CDC, increased levels of CD46 may contribute to the ineffectiveness of rituximab in treating recurring NHL.19
CD46 is also a receptor for a number of pathogens, including measles virus, Neisseria gonorrhoeae and meningitides, group A streptococcus, and human Herpes virus 6. We and others reported that CD46 is a high-affinity receptor for a series of human adenovirus (Ad) serotypes.20,21 Among the adenovirus serotypes that use CD46 is serotype 35 (Ad35). Ad35 engages CD46 via residues in its C-terminal trimeric fiber knob domain. We have shown that a recombinant fiber knob produced in Escherichia coli binds to recombinant CD46 cross-linking several CD46 molecules. Subsequently, a PCR mutation strategy was used to enhance the avidity of the trimer and a series of increasingly tight binding mutant knob proteins were identified. The highest affinity (KD of 0.63; 23.2-fold higher than that of the wild-type Ad35 fiber knob) was that of a knob mutant with a double Asp207Gly, Thr245Ala substitution. The protein was named Ad35K++. For purification purposes, Ad35K++ contained an N-terminal 6-His tag (Ad35K++His). Binding of Ad35K++His to cells results in transient removal of CD46 from cells.17 In a previous study, we had demonstrated that Ad35K++His increased the efficacy of lymphoma cell killing by rituximab both in vitro in primary and established human CD20-positive lymphoma/leukemia cells, and in vivo in tumor xenograft models.17 To extend these studies to nonhuman primates (NHPs), we first defined a rituximab dose that did not deplete CD20-positive peripheral blood cells in macaques.17 Using this dose of rituximab, we then demonstrated that pretreatment with Ad35K++His reconstituted near complete elimination of B cells. Ad35K++ enhancement of rituximab was more pronounced on CD20+CD46high cells, i.e., a subset of CD20-positive cells that resembles lymphoma cells.22 Further studies demonstrated that the treatment was well-tolerated and safe.22
All published safety and efficacy studies in mice and NHPs previously had been conducted with His-tagged Ad35K++His.17,22 There are numerous reasons to pursue the clinical development of a non-His tagged protein: (i) The presence of the tag itself can lead to undesirable protein properties including increased immunogenicity or decreased function as exemplified in a study with recombinant TRAIL.23 (ii) protein purification via immobilized nickel affinity chelate resins is more expensive than standard chromatography, and (iii) due to the risk of leachable nickel the product might be unsuitable for patients with nickel hypersensitivities. We therefore developed an Ad35K++ protein that did not contain a His-tag (Ad35K++). This report describes the safety, pharmacokinetics, pharmacodynamics, and tissue biodistribution studies in mouse models and in macaques. Based on these studies, we have defined an Ad35K++ dose range for upcoming clinical studies. We have also identified shed serum CD46 as a potential biomarker for Ad35K++ activity.
Results
Characterization of Ad35K++
Ad35K++ is composed of three self-trimerizing monomers consisting of an affinity-enhanced fiber knob and a fiber shaft motif. To enhance the affinity to CD46 Asp207Gly and Thr245Ala mutations were introduced into the Ad35 fiber knob.24 Only the trimerized fiber knob efficiently binds to CD46. As shown in Figure 1a, when boiled under reducing conditions, the trimeric fiber knob dissociated into monomers migrating on the gel in close agreement with the predicted molecular mass of 22.7 kDa for Ad35K++. Without boiling, Ad35K++ appears as a trimer with a predicted mass around 68.0 kD. The activity of Ad35K++ was assessed using a CDC assay with Raji cells (Figure 1b). Raji cells are CD20-positive, human Burkitt’s lymphoma cells. When incubated with the humanized anti-CD20 monoclonal antibody rituximab in the presence of normal human serum as a source for complement factors, rituximab kills about 50% of Raji cells by CDC under the given assay conditions. CDC activity increased as expected when Raji cells were preincubated for 10 hours with Ad35K++.
Figure 1.
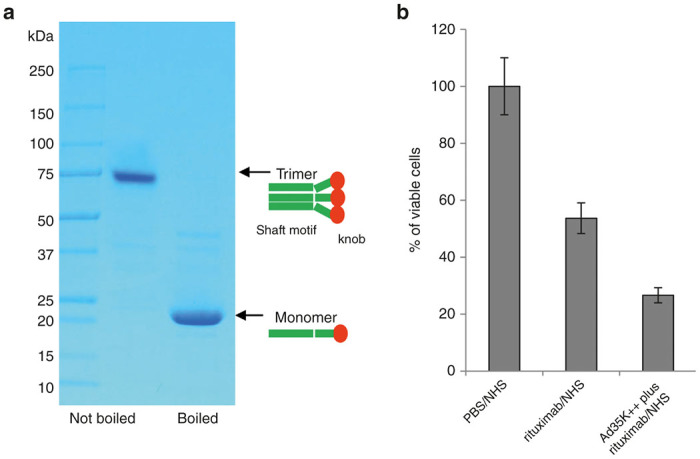
Characterization of Ad35K++. (a) Polyacrylamide sodium dodecyl sulfate (SDS) gel analysis of Ad35K++. Five micrograms of protein samples were loaded in Laemmli buffer containing β-mercaptoethanol with or without boiling for 5 minutes. The right graphic shows a schematic of Ad35K++ in the various states: Native Ad35K++ contains three monomers comprising the C-terminal, affinity-enhanced fiber knob, which interacts with CD46 and a shaft motif required for trimerization. During production, the monomer spontaneously trimerizes. When boiled under denaturing and reducing conditions, the trimer dissociates into monomers. (b) Ad35K++ activity in the rituximab complement-dependent cytotoxicity assay using Raji cells. Raji cells were incubated with Ad35K++ (25 µg/ml) for 8 hours followed by rituximab (15 µg/ml). After 30 minutes, normal human serum (NHS) was added and viable cells were counted 3 hours later based on trypan blue exclusion. Controls included incubation with phosphate-buffered saline (PBS)/NHS or rituximab/NHS. The viability of PBS/NHS-treated cells was taken as 100%. N = 3 independent experiments. Rituximab/NHS versus Ad35K++/rituximab/NHS: P < 0.001.
Studies in human CD46 transgenic mice
In contrast to humans where CD46 is expressed on all nucleated cells, in mice, CD46 is expressed only in the testes. For PK and BD studies, we therefore used human CD46 (hCD46) transgenic mice that express CD46 in a human-like pattern.25 Mice were injected intravenously with Ad35K++ at a dose of 2 mg/kg. Ad35K++ concentrations in serum samples were measured by enzyme-linked immunosorbent assay (ELISA). The ELISA detects only Ad35K++ capable of binding to CD46. The serum clearance curve shows a rapid decline during the first 24 hours with some residual Ad35K++ present at 72 hours (Figure 2a). Ad35K++ had a low clearance (CL = 0.23 l/hour/kg) and low volume of distribution (Vss = 0.07 l/kg). The value of AUC0-72 hour was 144,293 ng*hour/ml and the value of AUC0-∞ was 148,491 ng*hour/ml. AUC or “area under the curve” is the time-averaged concentration of a drug circulating in blood. The average half-life is about 5.1 hours. For BD studies, hCD46 transgenic mice were injected with Ad35K++ at a dose of 2 mg/kg. Six hours later blood was flushed from the circulation and Ad35K++ concentrations in tissue lysates were measured by ELISA (Figure 2b). Compared to Ad35K++ concentrations per ml of serum, concentrations per gram tissue were at least two orders of magnitude lower. Relatively high levels in the lungs could be due to residual blood or uptake by the lung reticulo-endothelial system. Presence of Ad35K++ in the kidneys could indicate a potential clearance mechanism.
Figure 2.

Pharmacokinetics (PK) and biodistribution studies in CD46tg mice. (a) Ad35K++ serum clearance. Ad35K++ serum concentrations were measured by enzyme-linked immunosorbent assay. Left panel: Summary of data from six animals. Right panel: Semi-log plots of average serum concentrations versus time profile (used for calculating the PK values). (b) Ad35K++ concentration in tissues at 6 hours after intravenous injection into hCD46 transgenic mice. Shown are Ad35K++ concentrations normalized to total protein as measured using the Bradford assay. N = 3.
Blood analysis.
Blood samples collected at 15 minutes, 1 hour, 6 hours, 24 hours, 48 hours, and 72 hours were analyzed for blood cell counts. Blood samples, collected at 72 hours, were analyzed for blood chemistry parameters including AST, Alk Phos, and ALT by the UW Medicine Clinical Laboratory. There were no abnormalities observed in the hematology panel and serum chemistry when comparing the mice treated with Ad35K++ to untreated animals (data not shown).
In vivo efficacy
To establish a xenograft lymphoma model, we intravenously injected Raji cells into immunodeficient CB17-SCID/beige mice. In previous studies, we found that in this model, 100% of mice that did not receive treatment developed hind-leg paralysis between day 17 and 19 after Raji cell injection.17 The paralysis is due to tumor growth in the bone marrow, which affects the spinal cord. Raji tumors also form in the spleen as well as in axillary and mesenteric lymph nodes.17 This model therefore reflects tumor cell growth in patients with B-cell malignancies.
Mice received three cycles of treatment at day 8, 11, and 14 after Raji cell injection (Figure 3a). Five groups were compared: (i) phosphate-buffered saline (PBS); (ii) Ad35K++ (2 mg/kg per injection), (iii) rituximab (2 mg/kg per injection, i.v.); (iv) Ad35K++ followed 10 hours later by rituximab (“Ad35K++→ rituximab”, this regimen was used in previous studies based on the kinetics of CD46 removal from the cell surface17); and (v) Ad35K++ in combination with rituximab (“Ad35K++ plus rituximab”). Figure 3a shows the Kaplan-Meier survival data. Endpoint timing for this study was the day of onset of hind-leg paralysis. The study had to be stopped at day 37 because of skin infections in surviving mice due to multiple treatment rounds. Median survival for the PBS-treated group was 18 days. Treatment with Ad35K++ increased the median survival by 2.5 days (from 18 to 20.5 days) (P < 0.05). The Rituximab alone group showed an increase in survival by 4 days (from 18 to 22 days) (P < 0.05), with one animal surviving until day 32. Treatment by “Ad35K++→rituximab” resulted in 60% survival at day 37. Combined injection of Ad35K++ and rituximab resulted in survival of 20%. The differences between the survival curves for “Ad35K++→rituximab” versus “Ad35K++ plus rituximab” were not significant (P = 0.150).
Figure 3.
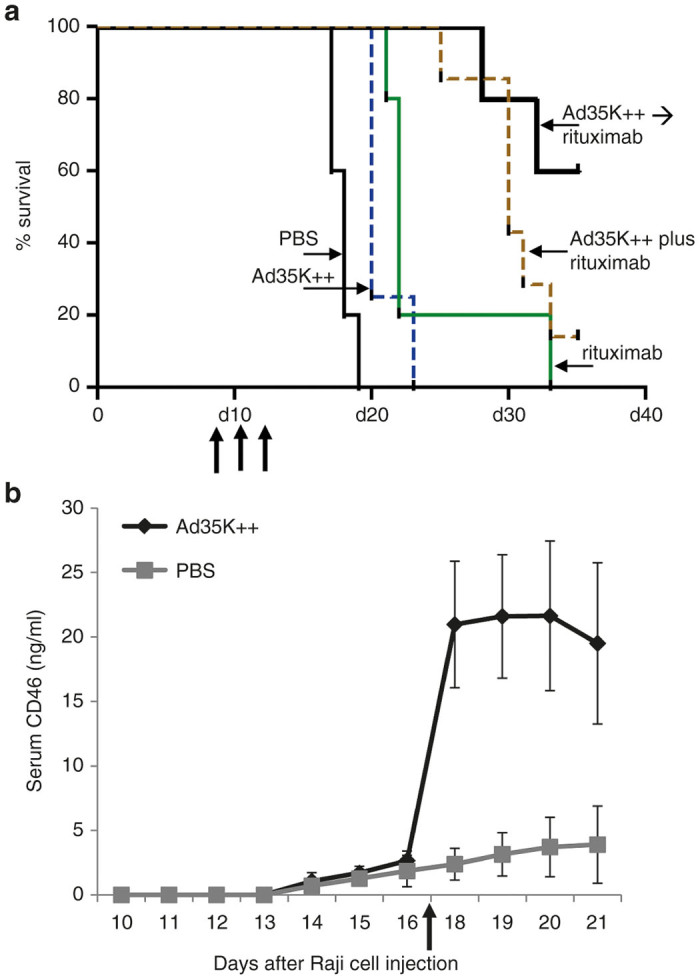
In vivo activity of Ad35K++ in the Raji tumor xenograft mouse model. Immunodeficient CB17-SCID/beige mice received human lymphoma Raji cells via tail vein injection. (a) Three cycles of treatment were given at days 8, 11, and 14 after Raji cell injection (see arrows). Treatment groups were: (i) phosphate-buffered saline (PBS), (ii) Ad35K++ (2 mg/kg), (iii) rituximab (2 mg/kg), (iv) Ad35K++ followed 10 hours later by rituximab (“Ad35K++ → rituximab”), and (v) Ad35K++ given together with rituximab (“Ad35K++ plus rituximab”). Kaplan-Meier survival analysis with days of onset of hind leg paralysis as endpoints. N = 5. (b) CD46 shedding after Ad35K++ injection. Mice with established Raji tumors were intravenously injected with Ad35K++ at 2 mg/kg on day 17 after Raji cell injection (see arrow). The control group received PBS. Serum samples before and after Ad35K++/PBS injection were collected and CD46 was measured by enzyme-linked immunosorbent assay using antibodies that recognize the extracellular domain of CD46. N = 5.
CD46 shedding
Our studies on human tumor cell lines suggested that Ad35K++His triggers the removal of CD46 from the cell surface through internalization and subsequent degradation.17 Considering reports that CD46 at the surface of human T cells is shed by matrix metalloproteases upon ligation with anti-CD46 antibodies,26 we found that Ad35K++ also triggers matrix metalloprotease-mediated CD46 removal from the cell surface of human CD4+ T-cells.27 We speculate that both processes, CD46 internalization and shedding, are connected. Therefore, we measured the concentration of shed CD46 in serum of mice with Raji tumor following injection of Ad35K++ (Figure 3b). In control mice that did not receive Ad35K++, a slow steady increase in serum sCD46 levels was observed. This indicates that CD46 is shed from Raji cells at low levels. The mechanism for this process remains to be studied. Importantly, upon intravenous Ad35K++ injection, serum sCD46 levels increased ~5-fold reaching a peak two days after Ad35K++ injection. This reflects an interaction of Ad35K++ with CD46 on Raji cells in vivo likely resulting in CD46 shedding. Serum CD46 levels can therefore serve a potential pharmacodynamics marker for Ad35K++ in vivo activity.
In summary, our PK and BD studies in hCD46tg mice demonstrated low uptake of intravenously injected Ad35K++ by normal tissues with a serum half-life of 5.1 hours. Efficacy studies in the Raji tumor xenograft model showed that Ad35K++ significantly increased the therapeutic effect of rituximab. Potentially, CD46 serum levels can be used as a pharmacodynamic marker for the monitoring in vivo activity of Ad35K++.
Studies in nonhuman primates
Justification for using Macaca fascicularis
Unlike most animals, in humans, CD46 is expressed on all nucleated cells. Of all mammals, only NHPs express CD46 in a pattern similar to humans.28 Our recent studies have shown that Ad35K++His increases rituximab-mediated CDC of primary M. fascicularis CD20+ cells.22 Rituximab cross-reacts with macaque CD20 and depletes B-cells after intravenous injection29 similar to humans. Also, this species has been used before in safety studies for other CD46-targeting therapeutics such as oncolytic measles viruses.30 M. fascicularis is therefore an adequate animal model to test the safety and efficacy of Ad35K++ in preparation for an investigational new drug submission.
NHP Ad35K++ dose range finding study
In our previous studies in M. fascicularis, intravenous injection of 4 mg/kg Ad35K++His (alone and in combination with rituximab) was well tolerated.22 To determine the lowest effective dose as well as the highest nonseverely toxic dose and correlative pharmacokinetic attributes for non His-tag Ad35K++, four M. fascicularis were intravenously injected with 0.05, 0.2, 2, and 20 mg/kg of Ad35K++. Blood samples were taken daily. A full necropsy was performed at day 7 after Ad35K++ injection.
Toxicology
The animals were monitored by an independent group of veterinarians and pathologists at the Washington State Northwest Primate Research Center at the University of Washington. Physical examinations were performed daily. No changes in health, behavior, and weight were observed. Food intake and body temperature were normal.
Hematology and blood chemistry parameters did not show treatment-related changes. Caging and handling of animals before infusion of Ad35K++ caused stress-related reactions, including an elevation in neutrophils and decrease in lymphocytes shortly after infusion (4-hour time point) in all animals regardless of the Ad35K++ dose (Figure 4a). Cell counts returned to normal by 12 hours. One out of four animals (0.2 mg/kg dose) had an elevation in AST levels. However, ALT levels did not increase, but rather trended to decrease in a dose-related fashion without toxicological significance and all values remained within the laboratory reference range. Because of daily blood sampling, erythrocyte counts and hemoglobin concentrations decreased over time (Figure 4b). In three out of four animals C3 complement levels declined slightly. The RBC, Hb, and C3 values were however all in the normal range for this species. No changes in partial prothrombin time were observed at day 7 indicating normal coagulation.
Figure 4.

Blood analysis of macaques that received intravenous injection of Ad35K++. Ad35K++ doses were 0.05, 0.2, 2, and 20 mg/kg. (a) Changes in neutrophil and lymphocyte counts as well as transaminase levels that can be attributed to stress-related reactions during infusion. ALT, alanine transaminase (aka SGPT), AST, aspartate transaminase (aka SGOT). (b) Changes in erythrocyte counts and hemoglobin levels that can be attributed to blood loss due to sampling. The bottom panels show concentrations of complement factor C3 and partial thromboplastin times, an indicator for coagulation activity.
Serum levels of the proinflammatory cytokines IFNγ, IL-6, IL-17a, and TNFα as well as levels indicative for Tregs (IL-10) were analyzed by a Luminex-based assay (Figure 5). TFNα was not detected in samples with the assay used. IFNγ levels were increased in animals that received 2 and 20 mg/kg of Ad35K++ starting 10 minutes after injection. For the 2 mg/kg dose, IFNγ levels returned to pretreatment levels by 8 hours, for 20 mg/kg by day 1. This indicates a dose-dependent response without defined toxicologic relevance. IL-6 levels increased between 30 minutes and day 1 without any correlation to the Ad35K++ dose suggesting that IL-6 elevation could be related to the infusion process. In animals injected with 2 and 20 mg/kg Ad35K++, IL-10 levels were elevated between 1 and 4 hours postinjection, with a peak at 2 hours. The 2mg/kg dose animal showed stronger IL-10 responses than the animal that received 20mg/kg. In the same animal, IL-17A levels declined about 50% during the first hour after injection. This observation needs to be further evaluated in additional animals. Notably, variations in baseline levels and responses in individual animals are not uncommon in NHP studies.
Figure 5.

Serum cytokine concentrations in M. fascicularis. Interferon-γ (IFNγ), interleukin 6 (IL-6), and IL-17a are proinflammatory cytokines. IL-10 is an anti-inflammatory cytokine considered to be a marker for regulatory T cells.
At necropsy there were no remarkable changes in gross examination in any tissue or apparent treatment-related changes in histological analysis of sections for 30 tissues from each animal.
Pharmacokinetics
Blood samples were collected at 10 and 30 min, 1, 2, 4, 8, 12, 24 hours and day 2, 3, 5, 7 postdosing. Ad35K++ serum concentrations were measured by ELISA (Figure 6). The linear plots for serum concentration versus time profiles that were used to calculate the PK parameters are shown in Figure 6 and the semi-log plots are presented in Supplementary Figure S1. In animals that received 0.05 mg/kg, Ad35K++ was only detectable during the first 2 hours after injection. Data for this group were therefore not included into the PK analyses. The total body clearance (CL) of Ad35K++ was 0.049 l/hour/kg at the 0.2 mg/kg dose and decreased to 0.002 l/hour/kg at 2 and 20 mg/kg indicating nonlinear clearance of Ad35K++ at lower doses (Table 1). This higher clearance of Ad35K++ at lower doses (0.2 mg/kg) resulted in a shorter half-life of Ad35K++ (2.2 hours) compared to a longer half-life of 20 and 49 hours for the 2 and 20 mg/kg doses, respectively. The volume of distribution at steady-state (Vss) of Ad35K++ was also low (mean value = 0.11 l/kg) suggesting that Ad35K++ distributed mainly in the systemic circulation.
Figure 6.

Ad35K++ serum concentrations versus time profiles following intravenous administration of Ad35K++ at 0.05, 0.2, 2, or 20 mg/kg in M. fascicularis. Serum dilutions ranging from 1:10 to 1:100,000 were used to achieve the optical density (OD) readings that were in the range of the standard curve. The dilution factor is shown in the lower right corner of each panel.
Table 1. Ad35K++ PK parameters in M. fascicularis.
| Species | Body weight (kg) | Dose (mg/kg) | CL (l/hour/kg) | Vss (l/kg) | t1/2 (hour) | AUC0-last (ng*hour/ml) | AUC0-inf (ng*hour/ml) |
|---|---|---|---|---|---|---|---|
| A14172 | 6.23 | 0.2 | 0.049 | 0.14 | 2.2 | 3,957 | 4,046 |
| A14396 | 5.17 | 2 | 0.002 | 0.05 | 20 | 916,165 | 999,851 |
| A14391 | 3.65 | 20 | 0.002 | 0.13 | 49 | 68,25,169 | 102,78,766 |
Serum anti-Ad35K++ antibody responses
Ad35K++-specific IgG, IgM, and IgA antibodies in serum samples were measured by ELISA (Supplementary Figure S2). Compared to pretreatment levels, no detectable increase in immune responses was observed over the 7-day observation period for all time points and all doses. The animal that received the highest Ad35K++ dose had higher baseline anti-Ad35K++ antibody levels which could be the result of a previous exposure to Ad35 virus.
Pharmacodynamics
Soluble serum CD46
We measured the concentration of shed CD46 in serum using antibodies that are specific to the extracellular domain of CD46 and cross-react with macaque CD46 (Figure 7). At the lower doses of Ad35K++, i.e., 0.05 and 0.2 mg/kg no increase in serum CD46 levels was evident (Figure 7a). However, at the higher doses of Ad35K++, a clear effect was observed. At a dose of 2 mg/kg CD46 levels started to rise at 12 hours postinjection, peaked at day 3 and declined to pretreatment levels by day 7. For the 20 mg/kg doses the decline was slower, consistent with the longer half-life of Ad35K++ in the animal injected with this dose. The serum CD46 concentrations in animals used for the efficacy and biodistribution studies are shown in Figure 7b,c.
Figure 7.

Soluble CD46 serum levels in M. fascicularis. CD46 was measured by enzyme-linked immunosorbent assay with human CD46 extracellular domain (ECD)-specific antibodies that cross-react with macaque CD46. (a) Results from animals that were in the dose finding study which went for 7 days. (b,c) Results from animals that were in the 14-day efficacy study (b) and 6-hour biodistribution study (c). Ad35K++ doses are indicated in each graph.
CD46 levels on PBMC subsets
Flow cytometry was performed on freshly isolated PBMCs. CD46 is expressed on all cells in nonhuman primates and binding of Ad35K++ should trigger a decline in CD46 levels on these cells. In our study we focused on CD20+ B-cells as well as CD4+, CD8+, and PD-1+ T-cells. PD1, or Programmed cell Death protein 1, is a coinhibitor receptor expressed on T-cells that negatively regulates their activation. At a dose of 0.05 mg/kg, Ad35K++ had no effect on CD46 levels on these subsets (Figure 8a). At 0.2 mg/kg, CD46 levels decreased rapidly but returned to pretreatment levels at day 3 after Ad35K++ injection. Considering a serum half-life of ~2 hours for this dose, it is unlikely that the observed decline is due to CD46 masking. Both higher doses—2 and 20 mg/kg—resulted in decreased CD46 levels on CD20+, CD4+, CD8+, and PD-1+ cells over the time period of analysis with a trend to rise again at day 7, which is consistent with our previous study with Ad35K++His.22 None of the Ad35K++ doses changed the percentage of CD20+, CD4+, CD8+, and PD-1+ cells (Figure 8b). This was expected because no CDC inducing antibody like rituximab was included in this study. This finding also supports the fact that Ad35K++ injection up to a dose of 20 mg/kg is well tolerated and does not negatively affect PBMCs.
Figure 8.

Flow cytometry on M. fascicularis PBMC. (a) CD46 levels were assessed based on the mean fluorescence intensity (MFI) after staining with a goat polyclonal anti-CD46 antibody followed by an anti-goat Alexa Fluor 488 antibody. The relative percentage of CD46 MFI compared to pretreatment controls is shown. (b) Percentage relative to pretreated controls of CD20-, CD4-, CD8-, and PD-1-positive PBMC subsets. Differences in the baseline levels of subsets between individual animals are common in NHPs.
Ex vivo CDC activity in CD20-positive cells from treated animals
To test whether Ad35K++-mediated removal of CD46 from the surface of CD20+ cells increases sensitivity to rituximab, we collected PBMCs from animals at 4 hours and day 1, 2, 3, 5, and 7 after Ad35K++ injection, isolated CD20+ cells by magnetic-activated cell sorting, and exposed them to rituximab (15 and 3 µg/ml) in the presence of normal M. fascicularis serum to trigger CDC. Figure 9 shows the relative viability of CD20+ cells compared to cells that were not treated with rituximab. Overall, the higher rituximab concentration (15 µg/ml) caused stronger CDC than the lower 3 µg/ml dose. CD20+ cells collected from the animal that received 0.05 mg/kg of Ad35K++ showed the same relative viability at all time points, indicating that this dose is not effective. For the 0.2 mg/kg Ad35K++ dose, reduced viability was observed at 4 hours and day 1 in the setting with 3 µg/ml rituximab which appears to be more sensitive to Ad35K++ effects. A clear effect of Ad35K++ on CDC sensitivity was however observed with CD20+ cells from animals injected with 2 and 20 mg/kg Ad35K++ starting at 4 hours postinjection. Similar to the kinetics of serum CD46 levels, an effect of Ad35K++ was seen until day 5. The return in cell viability percentages to pretreatment levels at day 7 indicates a loss of Ad35K++ effectiveness after this time frame.
Figure 9.

Rituximab complement-dependent cytotoxicity (CDC) assay with CD20+ cells from Ad35K++-injected M. fascicularis. PBMCs were collected at the indicated time points and CD20+ cells were isolated. CD20+ cells were subjected to treatment with rituximab (15 and 3 µg/ml) and fresh serum from an animal that did not receive Ad35K++ (as a source of complement) and viable cells were counted. The viability of untreated cells (no rituximab/complement) was taken as 100%. “Pre”: Relative viability of CD20+ cells taken from animals before Ad35K++ injection. These levels reflect the sensitivity of CD20+ cells to rituximab CDC. The bars show the relative viability of CD20+ cells compared to untreated cells (no rituximab/complement).
Biodistribution
One animal was injected with Ad35K++ at a dose of 2 mg/kg and euthanized 6 hours later. Blood was flushed from the circulation and samples from 28 tissues were collected. No treatment-related changes were recognized during gross and histological examinations upon necropsy. Tissue samples were also collected from the 4 animals used in the dose-finding study. These animals were sacrificed at day 7 after Ad35K++ injection. Tissues samples were lysed, clarified by centrifugation and analyzed for Ad35K++ by ELISA. Figure 10 shows Ad35K++ in tissues normalized to total protein concentration. At 6 hours, the highest Ad35K++ levels were found in the spleen. Importantly, these levels were more than 200-fold lower than Ad35K++ serum levels suggesting that Ad35K++ stayed in the systemic circulation which coincides with extremely low Vss of Ad35K++. Other tissues with notable Ad35K++ levels included the kidneys, lymph nodes, liver, gall bladder, lung, and pancreas. These measurements could be due to residual blood in these tissues and/or reflect potential Ad35K++ clearance mechanisms. A comparison of Ad35K++ levels in tissues of animals sacrificed at 6 hour and 7 days after injection of 2 mg/kg Ad35K++ indicates that most Ad35K++ is cleared by day 7. A comparison of Ad35K++ levels at day 7 in animals injected with 0.05, 0.2, 2, and 20 mg/kg revealed dose dependence of Ad35K++ presence in tissues (Figure 10a; Supplementary Figure S3). As expected, the highest tissue levels were found in animals injected with 20 mg/kg. Specifically, residual levels (comparable to those found at 6 hours after injection of 2 mg/kg) were found in the liver, pancreas, and thyroids (Figure 10b). Overall, the Ad35K++ levels found in tissues were considered trivial compared to the Ad35K++ levels in the circulation. PBMCs from this animal were also used to measure CD46 levels on PBMC subfractions and sensitivity to rituximab CDC in vitro (Supplementary Figure S4) and the data support our findings described for Figures 8 and 9.
Figure 10.

Ad35K++ concentrations in tissues 6 hours after injection into M. fascicularis. A total of 100 mg of tissue in phosphate-buffered saline (PBS)/0.1% Tween 20 were lysed by freeze/thawing and sonication. Lysates were cleared by centrifugation and subjected to an Ad35K++ ELISA. Corresponding lysates of each tissue from an untreated animal were spiked with Ad35K++ protein for standard curves. Ad35K++ concentrations were normalized to total protein concentrations in each sample. (a) Comparison of Ad35K++ tissue concentrations at 6 hours (animal A14401, see Supplementary Table S1) and at day 7 (animal A14369) after injection of 2 mg/kg of Ad35K++. (b) Comparison of Ad35K++ tissue concentrations at 6 hours after injection of 2 mg/kg Ad35K++ (animal A14401) and at day 7 after injection of 20 mg/kg Ad35K++ (animal A14361).
Efficacy study with Ad35K++
Rituximab cross-reacts with macaque CD20 and depletes B-cells after intravenous injection similar to what is observed in humans. Previously, we established that rituximab at a dose of 0.01 mg/kg had no effect on CD20+ cell levels.22 We then showed that injection of Ad35K++His two or three days before injection of 0.01 mg/kg of rituximab resulted in a sharp decrease in the percentage of CD20+ cells suggesting that Ad35K++-mediated downregulation of CD46-sensitized CD20 cells to rituximab CDC when given at dose of 0.01 mg/kg. CD20 depletion lasted for ~5 days. To assess the potency of our new Ad35K++ preparation, we used the same treatment regimen as in the previous study. One animal received Ad35K++ at a dose of 4 mg/kg followed 2 days later by rituximab (0.01 mg/kg) (Figure 11). CD46, CD20, and CD4 on PBMCs were monitored for 14 days. In agreement with our dose-finding study (see Figure 8), after Ad35K++ injection, CD46 levels declined on CD20+ cells as well as other PBMC subsets, e.g., CD4 cells and returned to pretreatment levels by day 14. Injection of rituximab resulted in a >30% decline in the percentage of peripheral blood CD20 cells. CD20 levels returned to preinjection levels by day 14. In contrast, rituximab had no effect on the level of cells that do not express the CD20 target, e.g., CD4 cells. These data are in agreement with our previous study performed with Ad35K++His and indicate that the two Ad35K++ preparations have comparable potencies with regards to enhancing rituximab CDC in vivo.
Figure 11.

Effect of Ad35K++ on rituximab killing of CD20 cells in M. fascicularis. Ad35K++ (4 mg/kg) was injected 10 minutes after preinjection samples (pre) were collected. Rituximab (0.01 mg/kg) was injected 10 minutes after day 2 blood samples were collected. The upper graph shows CD46 levels on CD20 cells (red lines) and the percentage of CD20+ cells compared to baseline (blue line). The lower graphs show a similar analysis of CD4+ cells.
Summary of NHP studies
In summary, the dose range finding study in M. fascicularis did not show dose-limiting toxicities, which potentially warrants the analysis of higher doses of Ad35K++ to assess dose-limiting toxicity in this species. Therapeutic effects in NHP of Ad35K++ doses lower than 0.2 mg/ml are not expected. The therapeutic effect on PBMCs at 2 and 20 mg/kg did not differ greatly. However, this does not necessarily apply to lymphoma cells in vivo where CD46 levels are higher than on PBMCs. The PK and PD data indicate that the period of Ad35K++ activity in vivo is 5 days with a half-life in blood of at least 20 hours. The study also corroborated the value of serum CD46 levels as a PD marker.
Discussion
To date, only modest progress has been made in treatment options for patients with rituximab-refractory B-NHL with the most effective agents demonstrating progression-free survival of less than 1 year. Furthermore, outcomes for patients with more aggressive histologies and rituximab-resistance are even more dismal.7–9 We developed Ad35K++ as a cotherapeutic to enhance rituximab therapy and, potentially, overcome treatment resistance. Ad35K++ transiently removes CD46 from cells and thereby sensitizes them to CDC triggered by rituximab. CD46 shedding and CD46 flow cytometry data indicate that the period of decreased surface CD46 expression lasts about 3 days in the Raji xenogaft models and about 5 days in NHPs. Several mechanisms for CD46 removal by Ad35K++ can be considered: Our previous in vitro data using tumor cells indicate internalization of CD46 and subsequent degradation.17 Internalization of CD46 through macropinocytosis in lymphoid and nonlymphoid cells was also reported upon CD46 crosslinking by multivalent antibodies or by measles virus.31 In this study, incubation with monoclonal antibodies against CD46 did not trigger long-term CD46 downregulation. The latter is consistent with our observation that anti-CD46 mAbs did not significantly enhance rituximab CDC in vitro.17 Another potential pathway of Ad35K++ mediated CD46 removal from the cell surface is cleavage and shedding of the extracellular domain. In epithelial cells32 and T cells,26 upon CD46-crosslinking, the CD46 ectodomain is shed after cleavage by matrix metalloproteinases, and its two intracytoplasmic tails are cleaved by the presenilin-gamma secretase enzymatic complex. Previous studies in primary human T cells also showed that Ad35K++ triggered CD46 shedding.27 It was therefore not surprising that we observed shed CD46 in the serum of macaques upon Ad35K++ injection. Our report here in the Raji xenograft model demonstrated Ad35K++-mediated shedding from tumor cells in vivo. This study also demonstrated that low levels of CD46 shedding even without Ad35K++ appears to be an ongoing process from the tumor cells. In this context, it is notable that shed CD46 was documented in the sera of cancer patients.33 These findings on in vivo CD46 shedding have implications for our upcoming clinical trial in patients with B cell malignancies and indicate that serum CD46 can serve as a valuable marker for the in vivo function of Ad35K++.
Efficacy studies in a Raji tumor xenograft model are relevant for the clinical translation of our approach. While rituximab treatment in this aggressive xenograft lymphoma model had only a marginal effect on survival, this was increased significantly with the addition of Ad35K++ more than doubling survival times in comparison to rituximab alone. Our previous studies—which used the research grade Ad35K++(His) in combination with rituximab—were performed with an interval of 6 to 12 hours between Ad35K++(His) dosing and rituximab dosing. Most rituximab refractory patients that would be candidates to be treated with Ad35K++ in upcoming trials will have residual rituximab in the blood due to the long half-life of the antibody. Considering this, the kinetics of CD46 downregulation in vitro,17,22 and the convenience of concomitant administration in the clinic, in this report we tested simultaneous injection of Ad35K++ and rituximab. Differences between the simultaneous and sequential treatment groups were not statistically significant although there appeared to be a trend of the sequential groups to be slightly superior in terms of survival. It has to be considered that the 5.1 hour half-life of Ad35K++ in hCD46tg mice is much shorter than the 20 hour one we found for macaques and most likely humans, which limits the predictive value of the murine dosing schedule studies.
Ad35K++ injection alone in mice had a small but significant therapeutic effect. This is in agreement with our previous in vitro CDC studies in which Ad35K++His triggered cell death in the absence of rituximab and complement.17 The function of CD46 is not only restricted to blocking the complement cascade. A series of studies show that CD46 ligation modulates the phenotype of T- and B-cells.34,35 Binding of pathogenic bacteria to CD46 on epithelial cells is associated with apoptotic cell death.36 Signaling upon Ad35K++ binding to lymphoma cells has not been studied yet. However, our data suggest that it might affect tumor cell viability to some degree.
The safety of Ad35K++ infusion was assessed in the hCD46-transgenic mouse model with 2 mg/kg of Ad35K++ and in M. fascicularis with a maximum dose of 20 mg/kg. In both species, no remarkable Ad35K++ treatment-related changes in hematological parameters were found. Tissue histology upon necropsy was normal. In hCD46tg mice and macaques, we found only minimal amounts of Ad35K++ taken up by normal tissues. CD46 levels on normal tissues are low and CD46 in epithelial tissues is not readily accessible to intravenously injected ligands.17,18,37,38 In contrast, CD46 is overexpressed on circulating malignant cells.18 We therefore speculate that this confers some preference of Ad35K++ interaction with lymphoma cells. In this context, we have previously shown that Ad35K++ strongly increased rituximab efficacy in a double transgenic (human CD46/human CD20) mouse model with syngeneic hCD20-positive tumors that expressed hCD46 at levels seen in human lymphoma cells.17
The efficacy of Ad35K++ in NHPs was evaluated based on four parameters: (i) concentrations of shed CD46 in the serum, (ii) removal of CD46 from the surface of PBMC subsets by flow cytometry, (iii) enhancement of rituximab-triggered cell killing in vitro, and (iv) enhancement of rituximab-triggered CDC of CD20+ cells in vivo. These studies suggest that a dose of 2 mg/kg may be effective in humans while doses below 0.2 mg/kg have no effect. Analysis of Ad35K++ pharmacodynamics indicated an interval of demonstrable effectiveness of approximately 5 days and would be well suited to pairing with the standard weekly rituximab cycle. Based on this, weekly treatment with Ad35K++/rituximab will be incorporated into the clinical trial design. In our first-in-human clinical trial, we plan to combine Ad35K++ with rituximab given at the standard therapeutic dose (375 mg/m2). Lower rituximab doses and alternate schedules could be considered for later stage trials. As our studies in NHPs demonstrated, Ad35K++ can convert a subtherapeutic dose of rituximab (0.01 mg/kg or about 0.12 mg/m2) into a B-cell depleting dose. Notably, monkey B-cells appear to be more sensitive to rituximab than human B-cells.39
Ad35K++ is a protein derived from an adenovirus with low sero-prevalence of pre-existing anti-Ad35 antibodies in humans.17 However, our previous study in NHPs showed that antibodies were generated after Ad35K++ infusion.22 The effect of multiple cycles of Ad35K++/rituximab infusion on the development of anti-Ad35K++ antibodies and their influence on safety and Ad35K++ efficacy remains to be assessed in future studies. Ad35K++ immunogenicity is unlikely to be a significant issue in NHL patients who have a suppressed immune system, both related to their disease and due to rituximab treatment which leads to depletion of B-cells.6,40–42 In fact, rituximab is used clinically to suppress antibody production in order to prevent humoral-mediated transplant rejection.40 Further, with the non-His-tag-engineered version of Ad35K++, the risk of generating an immune response may be further reduced. Our studies in NHPs have, so far, not assessed the effect of multiple cycles of Ad35K++/rituximab injection on the development of anti-Ad35K++ antibodies and their influence on safety and efficacy. The design of our clinical trial involves four treatment cycles with an interval of 1 week and a potential maintenance phase with treatment every 2 months for up to 2 years. Before investigational new drug submission, we will perform an additional NHP study with four cycles of Ad35K++/rituximab treatment and evaluate anti-Ad35K++ antibody levels and safety after each treatment cycle. In parallel, to assess Ad35K++ efficacy, sCD46 serum levels and CD46 levels on PBMCs will be measured before and 3 days after each treatment cycle. If there is evidence of anti-Ad35K++ antibody associated toxicity or reduced effectiveness after multiple cycles, we will pursue approaches to suppress antibody development. This will include approaches that have been tested clinically (e.g., pentostatin, cyclophosphamide, prednisolone,43,44 or others that are currently under development.45,46
Overall, the preclinical studies reported here have created the basis for selection of key trial parameters for human clinical testing and provide new insights in the mechanism of action of our cotherapeutic Ad35K++.
Materials and Methods
Ad35K++
Ad35K++ was produced by EirGenix (Taipei, Taiwan) (Lot H15003) in a 2-l fermentor based on a non-His tagged construct in E. coli. Purification of Ad35K++ involved three chromatography steps. Endotoxin levels were 1.7 EU/mg. The protein was stored at −80 °C in 25 mmol/l Tris pH 7.0/15% glycerol.
Cell lines
Human lymphoma Raji cells (ATCC CCL-86) were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and with L-glutamine and Pen/Strep in a humidified CO2 incubator at 37 °C as previously described.8
Antibodies
Rituximab (NDC 50242-051-21) was obtained from Genentech (South San Francisco, CA). Anti-Ad35K++His was generated in rabbits immunized with Ad35K++His using services of Custom-Hybridoma.com (A division of PickCell Laboratories, Amsterdam, the Netherlands). The mouse anti-human CD46 monoclonal antibody (clone 344519) and the goat anti-human CD46 polyclonal antibody were from R&D Systems (Minneapolis, MN). Another mouse anti-human CD46 monoclonal antibody (clone J4-48) was from Chemicon (Raleigh, NC). The goat anti-mouse IgG-horseradish peroxidase (HRP), PE mouse anti-human CD20 (clone 2H7), PE mouse anti-human CD4 (clone M-T477), PE mouse anti-human CD8 (clone RPA-T8), PE mouse anti-human CD14 (clone M5E2), and PE-Cy7 mouse anti-human CD25 (clone M-A251) were from BD Biosciences (San Jose, CA). PE mouse anti-human CD279 (PD-1) (clone EH12.2E7) was from BioLegend (San Diego, CA). Alexa Fluor 488 donkey anti-goat was from Life Technologies (Carlsbad, CA). FcR blocking reagents human was from Miltenyi Biotec (San Diego, CA). Goat-anti-monkey IgG+IgA+IgM-HRP was from MyBioSource (San Diego, CA).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Samples were either boiled at 95 °C for 5 minutes in a water bath to evaluate Ad35K++ monomer or not boiled to evaluate the Ad35K++ homotrimer which unlike many other proteins does not dissociate in sodium dodecyl sulfate without heat treatment. Laemmli sample buffer and β-mercaptoethanol were added to all samples. A total of 5 μg of protein sample were loaded per lane of 4–15% Mini-PROTEAN TGX gels. Gels were stained with Coomassie Blue.
Complement-dependent cell killing assay (CDC)
Raji cells were incubated with PBS or 25 μg/ml Ad35K++. Eight hours later, 15 µg/ml of rituximab was added to cells and incubated at room temperature for 30 minutes. Normal human serum was then added to a final dilution of 1:5 and cells were incubated at 37 °C for another 3 hours. Viable cells in each well were counted after trypan blue staining. Each sample was tested in triplicate and each well was counted four times.
sCD46 ELISA
The ELISA was performed using the goat polyclonal anti-CD46 antibody and the mouse monoclonal antibody J4-48 directed against human CD46. Recombinant CD46 extracellular domain protein (sCD46, Sino Biological, Beijing, China) was used for the standard curve. The detection limit of the sCD46 ELISA was 0.5 ng/ml. The antibodies cross-react with macaque CD46.
Ad35K++ ELISA
The ELISA consisted of a polyclonal rabbit antibody directed against the Ad35 fiber knob as capture antibody. Binding of Ad35K++ was detected with sCD46 followed by anti-CD46 mAb and goat anti-mouse IgG-HRP. Mouse serum dilutions of 1:500 with blocking buffer resulted in optical density values that were in the linear range of the standard curve. For the standard curve, a 1:500 dilution of normal hCD46tg mouse serum was spiked with purified Ad35K++, respectively. For the measurement of Ad35K++ in monkey serum, the dilutions indicated in the figure legends were used. Corresponding dilution of serum from a nontreated macaque were spiked with purified Ad35K++. The detection limit of the ELISA is 0.015 μg/ml.
Animal studies—mice
All experiments involving animals were conducted in accordance with the institutional guidelines set forth by the University of Washington. Mice were housed in specific-pathogen-free facilities.
hCD46-transgenic mice
C57Bl/6-based transgenic mice that contained the human CD46 genomic locus and provide CD46 expression at a level and in a pattern similar to humans were described earlier.25 These mice are also interferon receptor knock-out. Ad35K++ was injected retroorbitally. Blood samples were taken by submandibular bleeding.
Preparation of tissues for Ad35K++ ELISA
Six hours after Ad35K++ injection, animals were sedated and blood was flushed from the circulation with 5 ml of PBS using the heart as a pump. Hundred milligrams of tissues in PBS-0.05%Tween20 were homogenized using the TissueRuptor system (Qiagen, Valencia, CA), sonicated for 20 seconds, and subjected to three freeze/thaw cycles. Cell debris was spun down and supernatants from lysed tissues were used in the Ad35K++ ELISA at 1:10 dilutions. For standard curves, 1:10 dilutions of lysates from corresponding tissues from a nontreated mouse were spiked with purified Ad35K++.
Animal studies—NHPs
All NHP studies were performed by the Washington National Primate Research Center at the University of Washington under IACUC protocol 3108-03. Ages and weights of all animals are shown in Supplementary Table S1. All blood cell counts and chemistry analyses were done by UW Laboratory Medicine. The day before infusion the required amount of Ad35K++ was dialyzed against 2 l of PBS overnight at 4 °C. The material was then filtered through a 0.2-µm filter and injected into the arm vein of sedated animals over a time period of 8 minutes.
Pharmacokinetics analysis
PK analysis of Ad35K++ serum concentration versus time data was performed using Phoenix for WinNonlin Program, version 6.3 (Phoenix WinNonlin Copyright © 2012, Pharsight Corporation, Mountain View, CA). PK parameters were estimated using a noncompartmental model method. Area under the curve (AUC) was calculated using the linear trapezoidal rule. Last three concentration time points were used to determine the half-life.
PBMC flow cytometry
Blood samples were drawn into ethylene-diamine-tetra-acetic acid tubes. Per 0.3 ml of whole blood, 3 ml of 1× Pharm Lyse Buffer (BD Biosciences, San Jose, CA) were added and incubated at room temperature for 15 minutes. Cells were spun down, supernatant was removed, and the pellet was washed with PBS supplemented with 1% heat-inactivated FBS (fluorescence-activated cell sorting buffer). After a second spin, cells were resuspended in 100 μl fluorescence-activated cell sorting buffer supplemented with 1 μl human Fc blocking reagent and incubated on ice for 10 minutes. Hundred microliters of antibody staining solution were added with the following antibodies and allowed to incubate on ice for 30 minutes: PE mouse anti-human CD20 (clone 2H7), PE mouse anti-human CD4 (clone M-T477), PE mouse anti-human CD8 (clone RPA-T8), PE mouse anti-human CD14 (clone M5E2), PE-Cy7 mouse anti-human CD25 (clone M-A251), PE anti-human CD279 (clone EH12.2E7), and goat polyclonal anti-human CD46. In addition, corresponding isotype-matched controls were used: PE mouse IgG1, PE mouse IgG2a, PE mouse IgG2a, and PE-Cy7 mouse IgG1. Cells were washed once and resuspended in 100 µl fluorescence-activated cell sorting buffer supplemented with 1 μl of Alexa Fluor 488 donkey anti-goat and incubated on ice for 30 minutes. Samples were washed once more and analyzed on a BS LSRII flow cytometer (BD Biosciences). PBMCs were gated for size first, then, doublets were excluded. Single cells were gated for the respective cell surface markers.
Magnetic-activated cell sorting for CD20+ cells
PBMCs of 1 ml of whole blood were isolated via RBC lysis and CD20+ cells were purified using nonhuman primate CD20 microbeads (Miltenyi Biotec) according to the manufacturer’s instructions.
Ex vivo analysis of complement-dependent cell killing using PBMCs from Ad35K++-treated animals
Purified CD20+ cells from the sorting described above were divided into wells of a 96-well plate and taken up in 100 μl of RPMI 1640 medium supplemented with 10% heat-inactivated FCS and 1× penicillin/streptomycin. Rituximab was added at 15 and 3 μg/ml, respectively and incubated at 37 °C for 30 minutes. Next, 7.5 μl normal nonhuman primate serum were added and cells were incubated at 37 °C for 3 hours. Cells were transferred back to microcentrifuge tubes, spun down, and the supernatant removed leaving 10 μl medium on the cells. Samples were mixed with 10 μl Trypan blue staining solution and enumerated using a hemocytometer.
Cytokine detection
Serum samples were analyzed using the Milliplex nonhuman primate cytokine premixed immunoassay kit from Millipore (Billerica, MA).
Anti-Ad35K++ antibody ELISA
Polyclonal Ad35 fiber antibody was absorbed on ELISA plates followed by Ad35K++ protein at a saturating concentration. Dilutions of monkey serum (1:10, 1:100, 1,000) were added and binding was detected with goat-anti monkey IgG+IgA+IgM-HRP.
Acknowledgments
This study was supported by NIH grant R44CA162582 and a gift from BRIM Biotechnology, Inc. The NHP studies were supported by the National Primate Research Center at the University of Washington, NIH grant RR00166 and the National Center for Research Resources and the Office of Research Infrastructure Programs of the National Institutes of Health through Grant Number OD 010425. We thank Uma Prabhakar for editing the manuscript.
A.L. and D.C. are co-owners of Compliment Corp., a start-up company that is involved in the clinical development of the Ad35K++ technology.
References
- Siegel, RL, Miller, KD and Jemal, A (2015). Cancer statistics, 2015. CA Cancer J Clin 65: 5–29. [DOI] [PubMed] [Google Scholar]
- Di Gaetano, N, Cittera, E, Nota, R, Vecchi, A, Grieco, V, Scanziani, E et al. (2003). Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol 171: 1581–1587. [DOI] [PubMed] [Google Scholar]
- Golay, J, Cittera, E, Di Gaetano, N, Manganini, M, Mosca, M, Nebuloni, M et al. (2006). The role of complement in the therapeutic activity of rituximab in a murine B lymphoma model homing in lymph nodes. Haematologica 91: 176–183. [PubMed] [Google Scholar]
- Reff, ME, Carner, K, Chambers, KS, Chinn, PC, Leonard, JE, Raab, R et al. (1994). Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 83: 435–445. [PubMed] [Google Scholar]
- Bellosillo, B, Villamor, N, López-Guillermo, A, Marcé, S, Esteve, J, Campo, E et al. (2001). Complement-mediated cell death induced by rituximab in B-cell lymphoproliferative disorders is mediated in vitro by a caspase-independent mechanism involving the generation of reactive oxygen species. Blood 98: 2771–2777. [DOI] [PubMed] [Google Scholar]
- van der Kolk, LE, Grillo-López, AJ, Baars, JW, Hack, CE and van Oers, MH (2001). Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol 115: 807–811. [DOI] [PubMed] [Google Scholar]
- Coiffier, B, Radford, J, Bosly, A, Martinelli, G, Verhoef, G, Barca, G et al. 415 study investigators. (2013). A multicentre, phase II trial of ofatumumab monotherapy in relapsed/progressive diffuse large B-cell lymphoma. Br J Haematol 163: 334–342. [DOI] [PubMed] [Google Scholar]
- Gisselbrecht, C, Glass, B, Mounier, N, Singh Gill, D, Linch, DC, Trneny, M et al. (2010). Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol 28: 4184–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morschhauser, FA, Cartron, G, Thieblemont, C, Solal-Céligny, P, Haioun, C, Bouabdallah, R et al. (2013). Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J Clin Oncol 31: 2912–2919. [DOI] [PubMed] [Google Scholar]
- Chao, MP (2013). Treatment challenges in the management of relapsed or refractory non-Hodgkin’s lymphoma - novel and emerging therapies. Cancer Manag Res 5: 251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, TA, Grillo-López, AJ, White, CA, McLaughlin, P, Czuczman, MS, Link, BK et al. (2000). Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin’s lymphoma: safety and efficacy of re-treatment. J Clin Oncol 18: 3135–3143. [DOI] [PubMed] [Google Scholar]
- Martín, A, Conde, E, Arnan, M, Canales, MA, Deben, G, Sancho, JM et al. Grupo Español de Linfomas/Trasplante Autólogo de Médula Osea (GEL/TAMO Cooperative Group). (2008). R-ESHAP as salvage therapy for patients with relapsed or refractory diffuse large B-cell lymphoma: the influence of prior exposure to rituximab on outcome. A GEL/TAMO study. Haematologica 93: 1829–1836. [DOI] [PubMed] [Google Scholar]
- Coiffier, B, Lepage, E, Briere, J, Herbrecht, R, Tilly, H, Bouabdallah, R et al. (2002). CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 346: 235–242. [DOI] [PubMed] [Google Scholar]
- Hiddemann, W, Kneba, M, Dreyling, M, Schmitz, N, Lengfelder, E, Schmits, R et al. (2005). Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 106: 3725–3732. [DOI] [PubMed] [Google Scholar]
- Gopal, AK, Kahl, BS, de Vos, S, Wagner-Johnston, ND, Schuster, SJ, Jurczak, WJ et al. (2014). PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med 370: 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, D and Lieber, A (2014). Protein engineering to target complement evasion in cancer. FEBS Lett 588: 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H, Liu, Y, Li, ZY, Fan, X, Hemminki, A and Lieber, A (2010). A recombinant adenovirus type 35 fiber knob protein sensitizes lymphoma cells to rituximab therapy. Blood 115: 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong, HT, Timm, MM, Greipp, PR, Witzig, TE, Dispenzieri, A, Russell, SJ et al. (2006). Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol 34: 713–720. [DOI] [PubMed] [Google Scholar]
- Zell, S, Geis, N, Rutz, R, Schultz, S, Giese, T and Kirschfink, M (2007). Down-regulation of CD55 and CD46 expression by anti-sense phosphorothioate oligonucleotides (S-ODNs) sensitizes tumour cells to complement attack. Clin Exp Immunol 150: 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggar, A, Shayakhmetov, DM and Lieber, A (2003). CD46 is a cellular receptor for group B adenoviruses. Nat Med 9: 1408–1412. [DOI] [PubMed] [Google Scholar]
- Arnberg, N (2012). Adenovirus receptors: implications for targeting of viral vectors. Trends Pharmacol Sci 33: 442–448. [DOI] [PubMed] [Google Scholar]
- Beyer, I, Cao, H, Persson, J, Wang, H, Liu, Y, Yumul, R et al. (2013). Transient removal of CD46 is safe and increases B-cell depletion by rituximab in CD46 transgenic mice and macaques. Mol Ther 21: 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K, Wang, Y, Wang, X, Wang, Y and Ma, Y (2012). Tagged and untagged TRAIL show different activity against tumor cells. Oncol Lett 4: 1301–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H, Liu, Y, Li, Z, Tuve, S, Stone, D, Kalyushniy, O et al. (2008). In vitro and in vivo properties of adenovirus vectors with increased affinity to CD46. J Virol 82: 10567–10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper, C, Leung, M, Stephensen, CB, Pinkert, CA, Liszewski, MK, Cattaneo, R et al. (2001). Membrane cofactor protein (MCP; CD46) expression in transgenic mice. Clin Exp Immunol 124: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Choileain, S, Weyand, NJ, Neumann, C, Thomas, J, So, M and Astier, AL (2011). The dynamic processing of CD46 intracellular domains provides a molecular rheostat for T cell activation. PLoS One 6: e16287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay, J, Carter, D, Lieber, A and Astier, AL (2014). Recombinant Ad35 adenoviral proteins as potent modulators of human T cell activation. Immunology 144: 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, EC, Dörig, RE, Sarangi, F, Marcil, A, Iorio, C and Richardson, CD (1997). Artificial mutations and natural variations in the CD46 molecules from human and monkey cells define regions important for measles virus binding. J Virol 71: 6144–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaufin, T, Pattison, M, Gautam, R, Stoulig, C, Dufour, J, MacFarland, J et al. (2009). Effect of B-cell depletion on viral replication and clinical outcome of simian immunodeficiency virus infection in a natural host. J Virol 83: 10347–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ and Peng, KW (2009). Measles virus for cancer therapy. Curr Top Microbiol Immunol 330: 213–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimeen-Irwin, B, Ellis, S, Christiansen, D, Ludford-Menting, MJ, Milland, J, Lanteri, M et al. (2003). Ligand binding determines whether CD46 is internalized by clathrin-coated pits or macropinocytosis. J Biol Chem 278: 46927–46937. [DOI] [PubMed] [Google Scholar]
- Weyand, NJ, Calton, CM, Higashi, DL, Kanack, KJ and So, M (2010). Presenilin/gamma-secretase cleaves CD46 in response to Neisseria infection. J Immunol 184: 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seya, T, Hara, T, Iwata, K, Kuriyama, S, Hasegawa, T, Nagase, Y et al. (1995). Purification and functional properties of soluble forms of membrane cofactor protein (CD46) of complement: identification of forms increased in cancer patients’ sera. Int Immunol 7: 727–736. [DOI] [PubMed] [Google Scholar]
- Ni Choileain, S and Astier, AL (2012). CD46 processing: a means of expression. Immunobiology 217: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabara, HH, Angelini, F, Brodeur, SR and Geha, RS (2011). Ligation of CD46 to CD40 inhibits CD40 signaling in B cells. Int Immunol 23: 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lövkvist, L, Sjölinder, H, Wehelie, R, Aro, H, Norrby-Teglund, A, Plant, L et al. (2008). CD46 Contributes to the severity of group A streptococcal infection. Infect Immun 76: 3951–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo N, et al. (2006). Evaluation of adenovirus vectors containing seroytpe 35 fibers for tumor targeting. Cancer Gene Ther 13: 1072–1081. [DOI] [PubMed] [Google Scholar]
- Wang, H, Li, ZY, Liu, Y, Persson, J, Beyer, I, Möller, T et al. (2011). Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med 17: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vugmeyster, Y, Howell, K, Bakshl, A, Flores, C and Canova-Davis, E (2003). Effect of anti-CD20 monoclonal antibody, Rituxan, on cynomolgus monkey and human B cells in a whole blood matrix. Cytometry A 52: 101–109. [DOI] [PubMed] [Google Scholar]
- Jordan, SC, Kahwaji, J, Toyoda, M and Vo, A (2011). B-cell immunotherapeutics: emerging roles in solid organ transplantation. Curr Opin Organ Transplant 16: 416–424. [DOI] [PubMed] [Google Scholar]
- Lanini, S, Molloy, AC, Prentice, AG, Ippolito, G and Kibbler, CC (2013). Infections in patients taking Rituximab for hematologic malignancies: two-year cohort study. BMC Infect Dis 13: 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, CP, Brovarney, MR, Dabbagh, K, Birnböck, HF, Richter, WF and Del Nagro, CJ (2013). Subcutaneous versus intravenous administration of rituximab: pharmacokinetics, CD20 target coverage and B-cell depletion in cynomolgus monkeys. PLoS One 8: e80533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan, R, Miller, AC, Sharon, E, Thomas, A, Reynolds, JC, Ling, A et al. (2013). Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med 5: 208ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastan, I and Hassan, R (2014). Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res 74: 2907–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hareendran, S, Balakrishnan, B, Sen, D, Kumar, S, Srivastava, A and Jayandharan, GR (2013). Adeno-associated virus (AAV) vectors in gene therapy: immune challenges and strategies to circumvent them. Rev Med Virol 23: 399–413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.