

Abstract
Breast cancer was the second leading cause of cancer related mortality for females in 2014. Recent studies suggest histone H1 phosphorylation may be useful as a clinical biomarker of breast and other cancers because of its ability to recognize proliferative cell populations. Although monitoring a single phosphorylated H1 residue is adequate to stratify high-grade breast tumors, expanding our knowledge of how H1 is phosphorylated through the cell cycle is paramount to understanding its role in carcinogenesis. H1 analysis by bottom-up MS is challenging because of the presence of highly homologous sequence variants expressed by most cells. These highly basic proteins are difficult to analyze by LC-MS/MS because of the small, hydrophilic nature of peptides produced by tryptic digestion. Although bottom-up methods permit identification of several H1 phosphorylation events, these peptides are not useful for observing the combinatorial post-translational modification (PTM) patterns on the protein of interest. To complement the information provided by bottom-up MS, we utilized a top-down MS/MS workflow to permit identification and quantitation of H1 proteoforms related to the progression of breast cells through the cell cycle. Histones H1.2 and H1.4 were observed in MDA-MB-231 metastatic breast cells, whereas an additional histone variant, histone H1.3, was identified only in nonneoplastic MCF-10A cells. Progressive phosphorylation of histone H1.4 was identified in both cell lines at mitosis (M phase). Phosphorylation occurred first at S172 followed successively by S187, T18, T146, and T154. Notably, phosphorylation at S173 of histone H1.2 and S172, S187, T18, T146, and T154 of H1.4 significantly increases during M phase relative to S phase, suggesting that these events are cell cycle-dependent and may serve as markers for proliferation. Finally, we report the observation of the H1.2 SNP variant A18V in MCF-10A cells.
Histones are the proteins around which DNA is wrapped. Two copies each of the four core histones H2A, H2B, H3, and H4 constitute the histone octamer, which is bound by ∼147 base pairs of nucleosomal DNA. Linker histone H1 is involved in chromatin compaction and binds in the linker region where the DNA enters and exits the nucleosome. Because of their proximity to and inherent interaction with the genetic material, histone post-translational modifications, including those to H1, have an effect on all chromatin-templated processes (1). The genomic instability underlying carcinogenesis may be initiated by a combination of aberrant epigenetic modifications (to both DNA and histones) and genetic mutations in oncogenes and tumor suppressors (2). Modifications to the core histones have been reported as potential biomarkers in bladder (3, 4), prostate (5), lung (6), renal (7), pancreatic (8), and breast (9) cancers. Despite analytical challenges associated with the study of H1, novel and specific roles of this protein and its sequence variants have been reported in the literature in recent years (10).
Breast cancer is the second leading cause of cancer related mortality of females in the United States and was estimated to account for 232,650 new diagnoses and 40,000 deaths in 2014 (11). Novel biomarkers of breast cancer are desirable, particularly to combat false-positive mammograms occurring in ∼10% of cases that may result in invasive treatment (12). The activity of modifying enzymes with specificity for histones has been implicated in breast cancer (13, 14). There is evidence to support the existence of crosstalk networks between well-characterized DNA methylation and histone methylation events occurring in breast cancer (15, 16). Furthermore, this crosstalk is modulated by histone H1 in mouse ES cells (16). Moreover, H1 variants may have specific roles in transcriptional regulation (17, 18). A key factor in the ability of all histone H1 variants to play a regulatory role is the abundance of reversible post-translational modifications (PTMs)1 in their N- and C-terminal domains. These regulatory mechanisms are known to be combinatorial in nature. For instance, H1 phosphorylation may preclude methylation or acetylation of neighboring lysine residues (19). Like core histone acetylation, H1 acetylation has been linked to transcriptional activation and cell cycle progression (19, 20). Phosphorylation of H1 results in dynamic changes to the chromatin that may include condensation or relaxation, but the functional and contextual contributions of individual proteoforms remain an ongoing area of research interest. A proteoform here refers to a single permutation of PTMs present on a histone protein (21). Nonallelic H1 sequences (i.e. H1.2, H1.3, H1.4) are referred to as variants independently of modifications to the protein sequences.
Because H1 phosphorylation is maximal during M phase (22–26), it has been used to distinguish mitotic cell populations in proliferative cancers, including head and neck (27–30), cervical (31), bladder (32), and breast (33) cancers and holds great potential as a clinical biomarker of other hyperproliferative cancers. H1 phosphorylation occurs at specific sites as a function of the cell cycle (34–40). However, a lack of commercially available antibodies restricts the study of many of these modifications beyond identification. We previously used a polyclonal antibody against phosphorylated H1 residue threonine 146 (T146ph) to stratify high- and low-grade bladder and breast tumors by the use of immunohistochemistry (32, 33). Others have shown that the same phosphorylation event cooperates with acetylation of the p53 C-terminal domain to enable p53-dependent transcription upon chemically induced DNA damage (41, 42). These observations suggest multifaceted functions of T146ph. Based on the sequence similarity in the proximity of the T146 residue in H1.2, H1.3, and H1.4, antibody cross-reactivity is highly likely and problematic to the thorough study of this important event. Because of the lack of highly specific antibodies, the study of H1 PTMs has been mostly based on bottom-up MS. Analysis of H1 variants by this approach is complicated by H1's highly basic sequences containing numerous lysine-rich regions. In fact, there are few tryptic peptides greater than four amino acids in length in the C-terminal domains of H1.2, H1.3, or H1.4. Sequence homology within the more MS-conducive globular domains renders the variants indistinguishable by bottom-up methods (10).
A significant limitation of PTM identification by bottom-up MS is its inability to associate concomitant PTMs within the intact protein. Information about co-occurring modifications in a proteoform is inherently lost during the process of enzymatic digestion unless they are present on the same peptide. Middle-down digestion poses an alternative strategy for mapping combinatorial PTMs, but the H1 sequences and the location of common PTM sites are such that digestion by those proteases do not result in a dramatically simplified peptide containing most of the combinatorial complexity and with variant specificity (10). For example, the use of GluC for H1 analysis results in approximately a third of the sequence being indistinguishable as to the variant of origin because of identical peptides. The use of AspN produces only two peptides and adds little value over a direct top-down experiment. In both cases, all information about the overall modified state (or proteoform) is lost because common PTMs are found throughout the entire sequence and not clustered in the N-terminal domain, as is the case with H3 and H4.
Top-down MS/MS has been applied to the study of linker histone PTMs in multiple systems, including H. sapiens (35), T. thermophila (43), and D. melanogaster (44) and has also been used to localize core histone PTMs (45–47). Untargeted bottom-up MS is not quantitative for PTM identification because of sampling bias associated with an under-abundance of the modified peptide. Similar oversampling may occur if phosphorylation is enriched at the protein or peptide level. Thus, the degree of modification remains unknown without additional experiments such as stable isotope labeling, multiple reaction monitoring (MRM), or Western blotting. To overcome the limitations of bottom-up MS and a lack of antibodies against modified H1 sites, we have implemented a top-down MS/MS-based strategy to identify breast cancer-relevant H1 modified sites with proteoform level specificity. By fragmenting intact histone proteins, we can capture quantitative information about the global modification status of the protein. Complete characterization by use of complementary MS techniques is integral to our understanding of the functionality of individual proteoforms, particularly for studying cancer and other diseases governed, at least in part, by epigenetics (47, 48).
In the present study, we examine H1 PTMs in the metastatic cell line MDA-MB-231 and the nonneoplastic cell line MCF-10A. Histones were extracted from cells grown asynchronously or synchronized chemically with nocodazole or thymidine. Proteoforms associated with M phase (mitosis; nocodazole blocked) likely play roles in the processes associated with nuclear division, whereas those specific to S phase (synthesis; thymidine blocked) may be players in DNA replication. Electron capture dissociation (ECD) was applied to intact histones to generate fragment ions. ECD spectra of variants in different modification states were subjected to database search by novel in-house software and ProSight PTM software for fragmentation mapping (49–52). The relative abundance of each proteoform at different phases of the cell cycle was determined as validation of our previously reported phospho-H1 profiling of breast cancer cells (33). We also report the identification of an amino acid substitution A18V on H1.2, which has been previously observed twice by hydrophilic interaction liquid chromatographic (HILIC) methods (53, 54). However, it has never been reported by top-down mass spectrometry at the intact protein level and never in MCF-10A cells. Furthermore, we report the PTMs in the A18V SNP variant for the first time. The implementation of a top-down MS/MS-based approach provides information that is complementary to our previous study, but provides identification of multiply occurring modifications at the proteoform level and distinguishes analogous PTM sites in different variants. We are thus able to determine the occupancy of the phosphorylation sites across multiple variants.
EXPERIMENTAL PROCEDURES
Cell Growth
MDA-MB-231 and MCF-10A cell lines were obtained from and passaged as prescribed by the American Type Culture Collection. Cells were incubated at 37 °C with 5% CO2. Growth medium for MDA-MB-231 cells was adapted from Fogh (55) and contained high-glucose DMEM with l-glutamine (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 50 U/mL penicillin and 50 μg/mL streptomycin (Life Technologies). Growth medium for MCF-10A cells was adapted from Soule et al. (56) and contained DMEM supplemented with 5% horse serum (Life Technologies), 20 ng/mL recombinant human epidermal growth factor (PeproTech, Rocky Hill, NJ), 0.5 mg/mL hydrocortisone (Sigma-Aldrich), 10 μg/mL recombinant human insulin (Sigma-Aldrich), 10 ng/mL cholera toxin (Sigma-Aldrich), penicillin, and streptomycin.
Cell Synchronization
Asynchronously grown cells were harvested at ∼70% confluency. An S phase block was performed with two rounds of thymidine administration as performed previously by our lab (33). Briefly, a thymidine (Sigma-Aldrich) solution in PBS (Life Technologies) was added at a final concentration of 2 mm to cells grown to ∼30% confluency. Cells were incubated for 18 h then supplemented with normal growth medium. Cells were grown an additional 9 h then blocked again in thymidine for 18 h and collected. M phase block was performed as performed previously by our lab (33). Briefly, nocodazole (Sigma-Aldrich) was added at a final concentration of 100 nm to cells grown to ∼70% confluency. Cells were treated for ∼22 h and collected. Cells were stored at −80 °C prior to histone extraction.
Nuclei Isolation
Frozen cell pellets containing 50 m cells were thawed on ice. Nuclei isolation buffer (15 mm Tris-HCl, pH 7.5, 60 mm KCl, 15 mm NaCl, mL 5mm MgCl2, 1 mm CaCl2, 10% Nonidet P-40, and 250 mm sucrose) was added at a ratio of 10:1 to the cell pellets. The cells were gently mixed and incubated for 5 min at 4 °C. Pellets were centrifuged at 600 × g for 5 min at 4 °C and gently washed with nuclei isolation buffer (without Nonidet P-40) at a ratio of 10:1 multiple times. Supernatant was discarded and the remaining isolated nuclei were used promptly for acid extraction.
Acid Extraction
0.2 m H2SO4 was added to the isolated nuclei at a ratio of 5:1 (v:v), and any clumps of nuclei were disrupted by vortexing. The pellets were held on a rotator in a cold room for ∼2 h for histone extraction by H2SO4. The mixture was pelleted at 3400 × g for 5 min and the supernatant containing the bulk histones was transferred to several 1.5 mL conical tubes. 25% of the supernatant volume of 100% trichloroacetic acid was added to the conical tubes and kept on ice for ∼60 min to precipitate histones. The mixture of histones was centrifuged at 10,000 rpm for 5 min, and the supernatant was discarded. The precipitated histones were washed with 1.0 mL of acetone with 0.1% HCl once, followed by a wash with 100% acetone. The mixture of histones was centrifuged at 10,000 rpm for 5 min and was air-dried overnight for HPLC separation.
HPLC Separation
Extracted histones were subjected to purification and separation by reversed-phase high-performance liquid chromatography (HPLC) with a Dionex UltiMate 3000 × 2 Dual Analytical system (Thermo Scientific, Waltham, MA). Extracted histones were dissolved in 350 μl mobile phase A (95% H2O, 5% CH3CN, 0.2% TFA) and loaded onto a 4.6 × 250 mm, 5 μm, 300 Å pore size C18 column (218TP52, GRACE/Vydac, Columbia, MD) as reported elsewhere (57–59). Mobile phase B consisted of 5% H2O, 95% CH3CN, and 0.188% TFA, increasing linearly from 5% to 30% in 5 min, and then increasing 0.3% B/min to 60% at 105 min. Fractions were collected with an UltiMate 3000 Autosampler (Thermo Scientific) and dried by vacuum centrifugation (model SPD121P; Thermo Scientific). The histone H1 fraction was dissolved in H2O and further diluted to ∼1.0 μm in H2O/CH3CN (50/50) with 1.0% formic acid solution for MS analysis.
9.4 T FT-ICR Mass Spectrometry
Intact protein mass measurement and electron capture dissociation (ECD) were performed with a custom-built Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer equipped with a 220 mm horizontal room-temperature bore 9.4 T magnet (60). Positive ions generated by ESI were introduced into vacuum via a heated capillary, and focused by a dual ion funnel (61–63). Ions emanating from the dual ion funnel were transferred through a differential pressure chamber by a quadrupole ion guide operated at 2.0 MHz and ∼300 Vp-p rf amplitude. Ions of interest were mass-selected by a quadrupole mass filter before accumulation in an external octopole ion trap (64), then transferred to an open-ended cylindrical ICR trap (65).
An electron beam was generated for ECD experiments by an on-axis 3 mm diameter dispenser cathode-based electron gun mounted at the back end of the instrument. Electrons were injected into the ICR cell for 50 ms, followed by a 100 ms electron cleanup event. −2.5 V and +10 V dc potentials were applied to the cathode and accelerating grid during electron injection. Otherwise, the cathode and accelerating grid were kept at +10 V and −200 V. To optimize the overlap of the ion cloud and electron beam inside the ICR trap for maximized fragmentation efficiency, a ∼50 ms delay was added prior to ECD analysis to compensate for ion magnetron motion in the ICR trap. Instrument control, data acquisition, and data analysis were carried out with a modular ICR data station (66). Ions were excited by frequency-sweep (50 Hz/μs) and the ∼950 ms time-domain ICR transient (resolving power at m/z 400 is ∼170 K) was Hanning apodized, zero-filled once, and Fourier transformed to produce a magnitude-mode spectrum that was converted to a mass-to-charge ratio spectrum by a two term calibration equation (67,68).
Proteoform Identification
Novel in-house software (Young Algorithm, Internal Release June 2014) was used to localize all PTMs (lysine acetylation; serine, threonine and tyrosine phosphorylation) in a variant-specific manner to determine specific proteoforms. Magnitude mode MS/MS spectra were converted to absorption-mode spectra by phase correction before PTM localization (69). Briefly, a custom database of six known histone H1 variants derived from the UniProt database was used: H1.0 (accession number P07305), H1.1 (accession number Q02539), H1.2 (accession number P16403), H1.3 (accession number P16402), H1.4 (accession number P10412), and H1.5 (accession number P16401). As described in the text below, several other sequences and PTMs (formylation, dimethylation, and single nucleotide polymorphisms) were later considered. The isotopic distributions of all potential fragment ions of all proteoforms (produced by combining allowed PTMs) were enumerated. An objective function based on the sum of peak heights of all valid peaks (with appropriate flanking isotopes and within a mass tolerance of 3, 5, or 10 ppm) at the m/z of the theoretically most abundant fragment ion was optimized against all possible proteoforms. PTM localizations were also conducted by use of ProSight PTM 1.0, based on a 10 ppm error tolerance for comparison with the in-house software. In this case, absorption-mode ECD spectra were processed by use of a THRASH algorithm (70) to generate fragment monoisotopic masses followed by ProSight PTM analyses for product ion assignments. In all cases, both software packages agreed on the identities of all PTM localizations/proteoforms. To verify important findings, key peaks for proteoform identification and PTM localization were manually validated. The mass errors for most peaks were substantially less than the search tolerance and those outside the expected distribution were further manually scrutinized.
Relative Abundance of PTMs
The absolute abundance of each proteoform was determined from the highest magnitude isotopic peak in the broadband mass spectrum. The peak heights of proteoforms were averaged based on two biological replicates, each of which has three technical replicates, and standard deviations of relative abundance were calculated based on the total of six replicates. The absolute abundance of each proteoform was normalized to the total abundance (summation of absolute abundance of all identified proteoforms in the same broadband spectrum) to determine the relative abundance of each proteoform. During total phosphorylation determination, the relative abundances of the phosphorylated proteoforms were normalized to the total (phosphorylated plus nonphosphorylated) relative abundance of a specific sequence variant, H1.2, H1.3, or H1.4. ANOVA tests were performed to analyze relative abundance of different proteoforms, histone H1 variants, or phosphorylation sites among asynchronous, S phase, and M phase cells. The relative abundances were further examined by t-tests between S phase and M phase, following the ANOVA tests. Relative abundances of different proteoforms, histone H1 variants, or phosphorylation sites between MDA-MB-231 and MCF-10A cell lines were analyzed by t-tests. All statistical analysis was based on six replicates.
RESULTS
MS1 Analysis of Histone H1 PTMs in Breast Cell Lines
A top-down MS/MS approach was employed to analyze histone H1 variants in metastatic and nonneoplastic cells. As shown in Fig. 1, histone variants H1.2, H1.2 SNP (A18V), H1.3, H1.4, and numerous post-translationally modified forms were identified. Fig. 2 depicts representative mass spectra of histone H1 variants, H1.2 (35+), H1.3 (37+), and H1.4 (36+) from MDA-MB-231 and MCF-10A cells grown asynchronously and chemically blocked at S and M phases of the cell cycle. A complete list of identified proteoforms and their relative abundances is provided in supplemental Table S1. Broadband mass spectra of histone H1 at different stages of the cell cycle with charge states ranging from 34+ to 41+ are shown in supplemental Fig. S1. Histones H1.2, H1.3, and H1.4 were observed in MCF-10A cells. As reported previously, the H1.3 variant was absent in the MDA-MB-231 cells (33). For both cell lines, histone H1.2 was invariably observed with an N-terminal acetylation, and all further PTMs coexist with this constitutive PTM (71, 72). Monophosphorylation of H1.2 was also identified in both cell lines. Note that the H1.2 SNP variant A18V was observed in MCF-10A cells but is absent from MDA-MB-231 cells. Supplemental Table S2 shows the experimental mass of the H1.2 SNP variant A18V, determined from two biological replicates, each repeated three times (three technical replicates). The averaged experimental intact mass difference for the H1.2 SNP variant A18V in the present study is 28.037 Da with a standard deviation of 0.012 Da. Given the calculated mass difference for H1.2 SNP variant A18V is 28.031 Da, the experimental error is 0.0061 Da or 0.52σ. With 11 degrees of freedom (Supplemental Table S2 includes 12 measurements), the 95% confidence interval is ±0.026 Da. The calculated mass difference for the A18V SNP is actually closer to the mean experimental mass than over 50% of the experimental distribution, placing it well within the range of experimental error. We considered the possibility of dimethylation, which has an identical mass difference as SNP variant A18V. After MS/MS analysis, the possibility of dimethylation is low or ruled out based on observed fragments, as discussed in later sections. The complete absence of mono and trimethylated MS1 peaks further controverts the possibility of a dimethylation event. We also considered the possibility of formylation, which has a calculated mass difference of 27.995 Da. In that case, the experimental error is 0.043 Da or 3.61σ. With 11 degrees of freedom, the true formylation mass falls outside of the 99.5% confidence interval for the distribution. Because of low relative abundance of H1.2ac at M phase, isotopic peaks for this proteoform were buried in noise. Therefore, the mass information for H1.2ac at M phase was not considered during peak assignment shown in supplemental Table S2. In addition, monophosphorylation of histone H1.2 SNP variant A18V was observed for asynchronous and S phase MCF-10A cells, whereas diphosphorylation of the H1.2 SNP variant A18V was observed in M phase MCF-10A cells, as shown in Fig. 2. Acetylation and monophosphorylation of histone H1.3 were identified only for MCF-10A cells. For both cell lines, acetylated and nonacetylated histone H1.4 were observed. The nonacetylated histone H1.4 exists at low abundance and always exhibits monophosphorylation, a connection on which we expand below. Multiple phosphorylations were observed for acetylated histone H1.4 from both cell lines. S phase synchronization of both cell lines resulted in the identification of mono and diphosphorylation of histone H1.4. At M phase, H1.4 proteoforms containing tri-, tetra-, and pentaphosphorylation were also observed. These results are in accordance with similar work by Zheng et al. in the HeLa S3 cell line (35).
Fig. 1.

Top-down MS/MS-identified post-translational modifications of histone variants H1.2, H1.2SNP (A18V), H1.3, and H1.4.
Fig. 2.

Positive ESI 9.4 T FT-ICR mass spectra of identified proteoforms from cell lines MDA-MB-231 and MCF-10A.
MS/MS ECD of Histone H1 Variants and PTM Localizations
Database searches of ECD MS/MS data against UniProt human histone H1 sequences result in localization of PTMs of histone H1.2, H1.3, and H1.4. Fig. 3 shows a representative annotated ECD fragmentation spectrum and ion map of histone H1.4 exhibiting three phosphorylations from MDA-MB-231 cells. Other ECD fragmentation maps of histone variants with modifications are shown in supplemental Figs. S2 and S3. Annotated spectra for all proteoforms are also available in the Supplemental Information. For both cell lines, acetylation of histone H1.2 occurs at the N terminus (supplemental Fig. S2). In both cell lines, a single phosphorylation of acetylated histone H1.2 was observed at serine 173 (S173). A previous MS/MS analysis reported the identification of threonine 146 (T146) phosphorylation on histone H1.2 (33). Therefore, in order to determine if T146 and S173 phosphorylation coexist on H1.2, experimental ECD fragmentation spectra were searched against the histone H1.2 sequence with T146 phosphorylation. However, no matched fragments distinguishing T146 from S173 were identified as shown in red in supplemental Fig. S2. This result suggests that phosphorylation on T146 of histone H1.2 does not exist at a levels sufficient to contribute unique ECD fragments to the spectrum. This observation is in line with the requirement of phosphopeptide enrichment to detect phosphorylated T146 of H1.2 in our previous report (33). Histone H1.4 does exhibit phosphorylation at T146 in the presence of S172 phosphorylation (a site directly analogous to H1.2 S173 phosphorylation). The ability of Zheng et al. (35) to detect T146 phosphorylation of H1.2 by top-down MS/MS indicates that this modification may be present at significantly different levels across cell types.
Fig. 3.

Fig. 3A: ECD product ion FT-ICR mass spectrum of histone H1.4 with N-terminus acetylation and triphosphorylations from cell line MDA-MB-231. Fig. 3B (Next Page) ECD fragmentation mapping by custom software.
Fig. 3.

B ECD fragmentation map of the spectrum.
ECD fragmentation analysis identifies H1.2 SNP variant A18V from cell line MCF-10A. Fragmentation ions were numbered without considering the N-terminal methionine, which does not exist in the histones of the present study. Several segments of ECD product ion mass spectra for asynchronous and S phase histone H1.2 SNP variant A18V with N-terminal acetylation clearly show identification of c162+, c182+, c192+, c202+, c212+, and c213+ ions and localization of the SNP variant A18V, as shown in Fig. 4. Lysine 21 (K21) dimethylation of histone H1.2 was also considered because of its identical precursor mass compared with H1.2 SNP variant A18V. Observation of c162+ and c202+ ions suggests that the 28.0374 Da mass shift occurs between K17 and K21. Moreover, observation of c182+ and c192+ ions, which is 28.037 Da higher than calculated mass of c182+ and c192+ ions from unmodified H1.2, further pinpoints the mass shift at either A18 or proline 19 (P19). Because neither A18 nor P19 is a suitable dimethylation substrate, the possibility of a dimethylation of histone H1.2 is ruled out. As a result, the proteoform observed here is histone H1.2 SNP variant A18V.
Fig. 4.
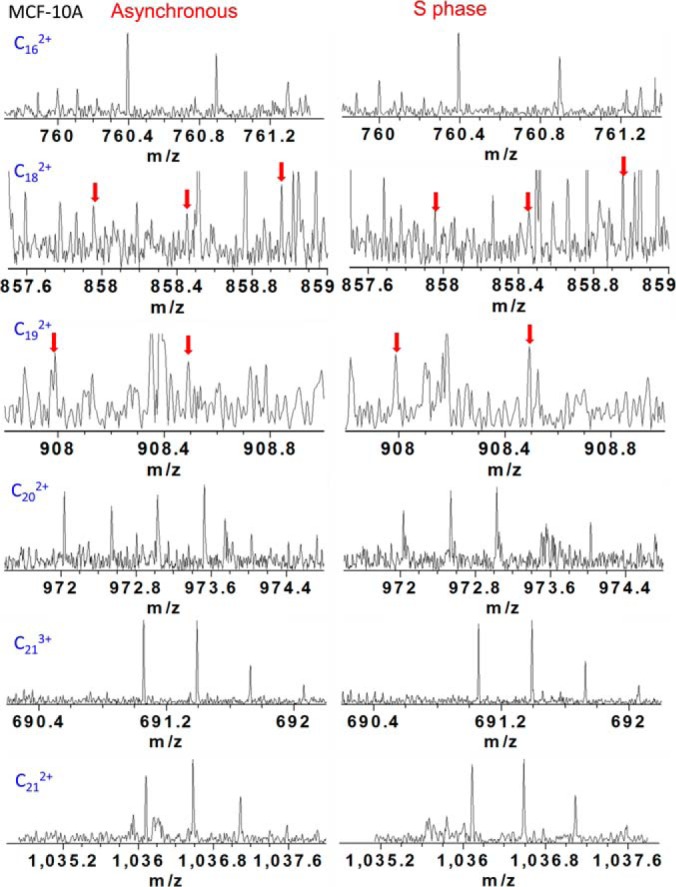
Mass scale-expanded segment of ECD product ion FT-ICR mass spectra of histone H1.2 SNP A18V from asynchronous and s phase cells of cell line MCF-10A, showing identification of c162+, c182+, c192+, c202+, c212+, and c213+, ions.
A mass consistent with H1.2 SNP variant A18V of M phase histone H1.2 was also observed in the broadband spectrum from cell line MCF-10A; however, precursor ion abundance was insufficient for ECD fragmentation. Therefore, ECD product ion mass spectra for histone H1.2 SNP variant A18V in M phase were not included. Moreover, c162+, c182+, c192+, c202+, c212+, c213+, c222+, and c223+ ions were also observed for a phosphorylated H1.2 SNP variant, confirming H1.2 SNP A18V, as shown in supplemental Fig. S4. Note that because of N-terminal methionine excision and the convention within the field of histone H1 studies to include the N-terminal methionine in the amino acid numbering scheme, numbering of c type ions is shifted by −1 relative to the numbering of the amino acids. Mass errors for relevant c type ions for determination of variant A18V are listed in Tables I–III below ∼2.0 ppm. Peak annotations are shown in supplemental Figs. S5–S7. Corresponding ECD fragmentation maps are shown in supplemental Fig. S3.
Table I. Selected ECD fragmentation ions identified in asynchronous H1.2SNP A18V Nα-ac.
| Ion Type | Charge State | MS m/z | Theoretical m/z | Mass Error (ppm) |
|---|---|---|---|---|
| c16 | 1+ | 1519.7726 | 1519.7738 | 0.80 |
| c16 | 2+ | 760.3913 | 760.3903 | 1.35 |
| c18 | 2+ | 858.4491 | 858.4516 | 2.89 |
| c19 | 2+ | 907.9872 | 907.9851 | 2.33 |
| c20 | 2+ | 972.5354 | 972.5343 | 1.17 |
| c21 | 2+ | 1036.5820 | 1036.5818 | 0.19 |
| c21 | 3+ | 691.3911 | 691.3901 | 1.43 |
| c22 | 2+ | 1100.6301 | 1100.6294 | 0.68 |
| c22 | 3+ | 734.0892 | 734.0888 | 0.54 |
| c23 | 2+ | 1136.1484 | 1136.1475 | 0.78 |
| c23 | 3+ | 757.7680 | 757.7673 | 0.98 |
| c24 | 3+ | 781.4468 | 781.4467 | 0.15 |
| c25 | 2+ | 1235.7156 | 1235.7142 | 1.13 |
| c25 | 3+ | 824.1454 | 824.1444 | 1.22 |
| c26 | 3+ | 866.8439 | 866.8431 | 0.94 |
Table II. Selected ECD fragmentation ions identified in S Phase H1.2SNP A18V Nα-ac.
| Ion Type | Charge State | MS m/z | Theoretical m/z | Mass Error (ppm) |
|---|---|---|---|---|
| c16 | 1+ | 1519.7741 | 1519.7738 | 0.18 |
| c16 | 2+ | 760.3909 | 760.3903 | 0.82 |
| c18 | 2+ | 858.4520 | 858.4516 | 0.47 |
| c19 | 2+ | 907.9858 | 907.9851 | 0.77 |
| c20 | 2+ | 972.5338 | 972.5343 | 0.47 |
| c21 | 2+ | 1036.5813 | 1036.5818 | 0.49 |
| c21 | 3+ | 691.3907 | 691.3901 | 0.85 |
| c22 | 2+ | 1100.6281 | 1100.6294 | 1.14 |
| c22 | 3+ | 734.0890 | 734.0888 | 0.27 |
| c23 | 2+ | 1136.1467 | 1136.1475 | 0.72 |
| c23 | 3+ | 757.7677 | 757.7673 | 0.59 |
| c24 | 3+ | 781.4465 | 781.4467 | 0.23 |
| c25 | 2+ | 1235.7129 | 1235.7142 | 1.06 |
| c25 | 3+ | 824.1451 | 824.1444 | 0.86 |
Table III. Selected ECD fragmentation ions identified in asynchronous H1.2SNP A18V Nα-ac S173ph.
| Ion Type | Charge State | MS m/z | Theoretical m/z | Mass Error (ppm) |
|---|---|---|---|---|
| c16 | 1+ | 1519.7735 | 1519.7738 | 0.21 |
| c16 | 2+ | 760.3914 | 760.3903 | 1.48 |
| c18 | 2+ | 858.4522 | 858.4516 | 0.70 |
| c19 | 2+ | 907.9835 | 907.9851 | 1.76 |
| c20 | 2+ | 972.5359 | 972.5343 | 1.69 |
| c21 | 2+ | 1036.5829 | 1036.5818 | 1.05 |
| c21 | 3+ | 691.3909 | 691.3901 | 1.14 |
| c22 | 2+ | 1100.6297 | 1100.6294 | 0.31 |
| c22 | 3+ | 734.0893 | 734.0888 | 0.67 |
| c23 | 3+ | 757.7681 | 757.7673 | 1.12 |
| c24 | 3+ | 781.4460 | 781.4467 | 0.87 |
| c25 | 2+ | 1235.7137 | 1235.7142 | 0.41 |
| c25 | 3+ | 824.1459 | 824.1444 | 1.83 |
| c26 | 3+ | 866.8442 | 866.8431 | 1.29 |
Phosphorylation was identified at S173 of histone H1.2 SNP A18V from asynchronous cells as shown in the ECD fragmentation map, supplemental Fig. S3. The abundance of the phosphorylated histone H1.2 SNP A18V ions from S and M phases was not sufficiently high for ECD MS/MS analysis; therefore, ECD experiments were performed only for asynchronous cells. A diphosphorylated form of H1.2 SNP A18V is observed in M phase of MCF-10A cells but was not localized. In all cases, H1.2 SNP A18V was N-terminally acetylated. This result is the first proteoform level analysis of the PTMs present in H1.2 SNP A18V.
Acetylation at the N terminus of histone H1.3 in the MCF-10A cell line was identified, as shown in supplemental Fig. S3. Although monophosphorylation was also observed on histone H1.3, ECD fragmentation was not performed for phosphorylation localization because of low ion abundance. Although H1.3 has a serine at position 174 that aligns with the abundant H1.4-S172 and H1.2-S173 phosphorylation sites, it lacks the trailing proline that is known to be important in kinase targeting (73), making it incorrect to assume that S174 is the likely phosphorylation localization. Furthermore, the observed H1.3 phosphorylation does not exhibit the cell cycle dependence observed at sites in the other variants analogous to S174. Given that the H1.2/H1.4 phosphorylation at S173/S172 appears to be required for further phosphorylation of the other variants observed, this difference in sequence may play an important role in conferring to H1.3 a function distinct from these other variants.
Monophosphorylation of the nonacetylated histone H1.4 for both cell lines was localized on serine 2 (S2) through different phases of the cell cycle as shown in supplemental Fig. S2. Sequence coverage of the ECD fragmentation map of histone H1.4 monophosphorylation (in the absence of N-terminal acetylation) is not sufficient to distinguish it from threonine 4 phosphorylation because of the low precursor ion abundance. However, S2 phosphorylation on histone H1.4 was previously reported by mass spectrometry (10), consistent with our results. All other proteoforms of histone H1.4 were identified with an acetylation at the N terminus. These observations suggest a possible antagonistic mechanism by which S2 phosphorylation (located on the side chain of the N-terminal residue) and Nα-acetylation of the N terminus inhibit each other. For S phase and asynchronous histone H1.4, monophosphorylation (S172ph) and diphosphorylation (S172ph and S187ph) were identified. Although triphosphorylation of asynchronous and S phase histone H1.4 was also observed, its relative abundance was not sufficient for ECD experiments to localize the third phosphorylation site. However, histone H1.4 was identified with five M phase phosphorylations for both cell lines. As shown in supplemental Figs. S1–S3, phosphorylation of histone H1.4 occurs sequentially: first at S172, second at S187, third at T18, fourth at T146, and fifth at T154. This type of hierarchical relationship of PTMs at the single molecule level we describe here can be revealed only by top-down MS/MS. Furthermore, our top-down analysis gives variant specificity, played out at least in these cell lines primarily on H1.4. Note that H1.3 lacks an analogous site to H1.4S172 and H1.2 lacks a site analogous to H1.4S187, the first and second sites for the sequential phosphorylation of H1.4. Histone H1.2 phosphorylation at S173 and histone H1.4 phosphorylation at T18, T146, T154, S172, and S187 have all been characterized as consensus cyclin-dependent kinase (CDK) sites (10).
Characterization of Histone H1 PTMs Across the Cell Cycle
Previous studies have suggested accumulated phosphorylation of histone H1 throughout the cell cycle reaches a maximum during M phase (22–26). Here, we monitored all phosphorylation sites at different phases of the cell cycle with proteoform specificity to discover changes in phosphorylation as cells advance through the cell cycle. Quantitative changes of all modifications to histone H1 at S and M phases of the cell cycle, as well as in asynchronous cells, are shown in Fig. 5, and p values from ANOVA and t-tests are listed in supplemental Table S3. S173 phosphorylation occurs on N-terminally acetylated histone H1.2 (H12ac), leading to a decrease of the H12ac proteoform and an increase in phosphorylation for both cell lines during M phase. Consistent with that observation, mono and diphosphorylated histone H1.2 SNP A18V (H12SNPacph and H12SNPacph2) also increase at M phase compared with S phase. Acetylated and phosphorylated histone H1.3 in MCF-10A cells maintain stable levels as the cells enter M phase. S2 phosphorylation (S2ph) of histone H1.4 maintains stable levels across the entire cell cycle in the MDA-MB-231 cell line, whereas it decreases by about twofold as MCF-10A cells enter M phase. Note that these cell cycle behaviors of H1.4-S2ph and H1.3ph are markedly different than the other H1 phosphorylation events, all of which dramatically increase in M phase. For both cell lines, exclusively acetylated histone H1.4 dramatically decreases as a result of progressive phosphorylation at S172, S187, T18, T146, and T154 as the cells enter M phase, as shown in Fig. 5. S2ph is not observed in these hyperphosphorylated species despite being present as a monophosphorylated species, and thus appears to be part of an unrelated pathway. At M phase, H1.4 diphosphorylation increases by 66.4% and 94.2% for MDA-MB-231 and MCF-10A cells relative to S phase; triphosphorylation increases by more than 14-fold in MDA-MB-231 cells and more than fivefold in MCF-10A cells. Notably, H1.4 tetra- and pentaphosphorylation were not detected at S phase in either cell line; however, those respective proteoforms are characterized as 7.0% and 2.5% of total histone H1 observed in MDA-MB-231 and 5.4% and 1.3% of total histone H1 observed in MCF-10A cells at M phase. Our results suggest that H1 phosphorylation occurs predominantly at M phase in these cell lines, a finding that is consistent with previous studies (32–39). Our results also reveal that some less abundant H1 phosphorylations, H1.4-S2ph and H1.3ph, do not follow this prevailing cell cycle dependent behavior.
Fig. 5.

Relative abundances of the proteoforms observed from cell lines MDA-MB-231 and MCF-10A at different cell cycle stages. Asterisk denotes p value less than 0.05.
Global phosphorylation of histone H1.2 and H1.4 normalized to the total population of each variant is shown in Fig. 6. p values from the associated ANOVA and t-tests are listed in supplemental Table S4. Histone H1.2 phosphorylation in the MDA-MB-231 cell line occurs solely at S173 as represented by the proteoform H12acph. In contrast, the H1.2 phosphorylation profile of the MCF-10A cell line includes multiple phosphorylated proteoforms: H12acph, H12SNPacph, and H12SNPacph2. Sites of histone H1.4 phosphorylation include S2, S172, S187, T18, T146, and T154. Overall phosphorylation of histone H1.2 increases by 58.3% and 111.2% for MDA-MB-231 and MCF-10A cells blocked in M phase compared with S phase. The decrease of histone H1.3 phosphorylation from 35.9% at S phase to 32.2% at M phase is within experimental error. Global phosphorylation of histone H1.4 increases by 62.1% and 64.2% for the MDA-MB-231 and MCF-10A cell lines in M phase relative to S phase. Our results suggest that phosphorylation of histone H1.2 and H1.4 occurs mainly during M phase for both cell lines examined.
Fig. 6.
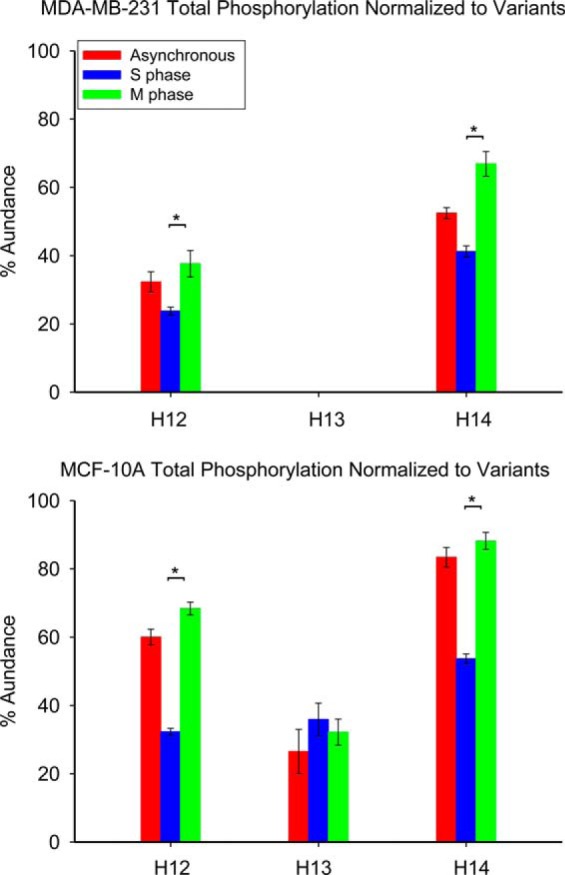
Normalized total phosphorylations of histone variants from cell lines MDA-MB-231 and MCF-10A at different cell cycle stages. Asterisk denotes p value less than 0.05.
Fig. 7 depicts the drastic variation in phosphorylation at individual sites through the cell cycle for the present cell lines. p values from ANOVA and t-tests are listed in supplemental Table S5. Phosphorylation of histone H1.3 was not included because of low ion abundance of an unidentified phosphorylation site. Phosphorylation of histone H1.2 on S173 increases by 58.4% and 111.8% for MDA-MB-231 and MCF-10A cells at M phase compared with S phase. Phosphorylation of histone H1.4 on S2 varies only slightly through the cell cycle in both cell lines. In contrast, the phosphorylation of histone H1.4 at S172, S187, and T18 increases by 26.3%, 2.5-fold and 25-fold in MDA-MB-231 cells at M phase compared with S phase; phosphorylation at these sites increases by 75.9%, 1.9-fold and 6.6-fold in MCF-10A cells. Phosphorylations at T146 and T154 occur only at M phase, with relative abundances of 11.5% and 2.5% in MDA-MB-231 cells, and 9.1% and 1.5% for MCF-10A cells.
Fig. 7.
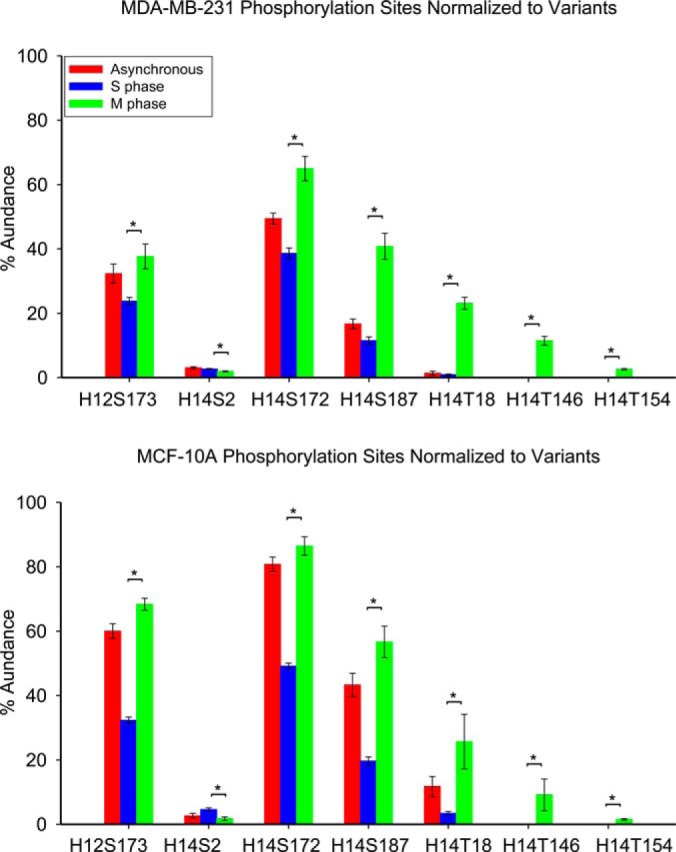
Normalized phosphorylations at each identified modification site of histone variants from cell lines MDA-MB-231 and MCF-10A at different cell cycle stages. Asterisk denotes p value less than 0.05.
DISCUSSION
Histone H1.2 SNP Variant A18V
In the present study, we identified the H1.2 SNP variant A18V and its PTMs for the first time by use of top-down MS/MS. This SNP, but not its PTMs, has been previously identified by Lindner and coworkers by use of HILIC chromatography in K562 erythroleukemic cells (53). A homozygous GCC to GTC shift was found at codon 18, confirming the A18V substitution at the genome level. That SNP has been assigned SNP ID rs2230653. The N-terminal region of histone H1.2 exhibits random coil composition in solution (74). However, it acquires a substantial amount of α-helical structure on binding to DNA (75). According to these authors' NMR results, the helical region of histone H1.0 encompasses residues K11 to D23. The N-terminal domains of other histone H1 variants may function similarly. For example, the N-terminal tail of histone H1 from breast cancer cells may adopt its native conformation when it binds to chromatin. Because valine has less stabilizing effects on α-helical structure, the replacement of alanine by valine may affect the α-helical structure at the N-terminal region of histone H1. Note that H1.2 SNP variant A18V exhibits a detectable level of diphosphorylation whereas H1.2 does not. This SNP-specific second phosphorylation is observed only in M phase whereas the other H1.2/H1.2 SNP phosphorylation events are observed throughout the cell cycle.
Phosphorylation Occurs at M Phase for both Cell Lines
Previous studies have shown that histone H1 phosphorylation is associated with chromatin relaxation in interphase and condensation in M phase (22, 24, 29). In the present study, we applied a top-down MS/MS workflow to study phosphorylation profiles of a metastatic breast cancer cell line (MDA-MB-231) and a nonneoplastic breast epithelial cell line (MCF-10A). We observe phosphorylation changes for H1.2 and H1.4 throughout the cell cycle. Phosphorylation of H1.2 occurs at S2 and S173. S2ph appears to occur independent of the cell cycle, whereas S173ph increases in M phase enriched cell populations relative to the S phase cell population. In contrast to H1.2, up to five concurrent phosphorylation events are observed for H1.4. We are particularly interested in H1.4, based on observations by the Jordan lab that shRNA-mediated knockdown of H1.4 results in severe growth deficiencies in MCF-10A cells and cell death in T47D breast cancer cells (17). We surmise that phosphorylation of this variant may be involved in the regulation of processes necessary for normal cell proliferation. In the present study, H1.4 phosphorylation during interphase occurs mainly at S172 and S187, with a smaller fraction at T18. Upon block in M phase, phosphorylation levels at these residues increases, and T146ph and T154ph become detectable. Based upon the identification of the proteoforms corresponding to H1.4ph, H1.4ph2, H1.4ph3, H1.4ph4, and H1.4ph5, these phosphorylations occur sequentially; i.e., S187ph was not detected unless S172ph was also present; T18ph was not detected unless both S172ph and S187ph were present; and so on through T146ph and T154ph. The sequential phosphorylation of histone H1 has previously been reported in numerous systems (43, 76, 77), including human cells by the Lindner lab (34). Interphase phosphorylation of serine residues contained within the SP(A/K)K motif is in line with a previous report (34). We can further specify the motif observed here as SP(A/K)KAK. Additional motif analyses may reveal variant-specific sequences that attract context-dependent phosphorylation events.
Comparison between Cell Lines MDA-MB-231 and MCF-10A
Fig. 8 provides a comparison of the relative abundances of proteoforms observed in the two cell lines examined, with p values from the t-tests available in supplemental Table S6. Note that acetylated H1.4 is particularly enriched in S phase in both cell lines. Figs. 9 and 10 show the extent of phosphorylation of H1.2 and H1.4 in this study. p values are listed in supplemental Tables S7–S8. The present data show a clear increase in global phosphorylation of H1.2 and H1.4 as cells progress through the cell cycle and reach maximum phosphorylation during mitosis. The phosphorylation at T146 and T154 is detectable exclusively in M phase blocked cells and supports other reports that phosphorylation at these sites can mark mitotic cells. Our findings support the notion that H1 phosphorylation increases because the cells are proliferating and that this phosphorylation increases irrespective of whether or not the cells have been transformed into cancer cells. This finding explains our previous observation of greater T146ph stain intensity in highly proliferative tumor cells relative to the more slowly dividing cells in adjacent normal tissue.
Fig. 8.

Relative abundance of proteoforms from cell lines MDA-MB-231 and MCF-10A: (a) asynchronous cells; (b) S phase cells; (c) M phase cells. Asterisk denotes p value less than 0.05.
Fig. 9.
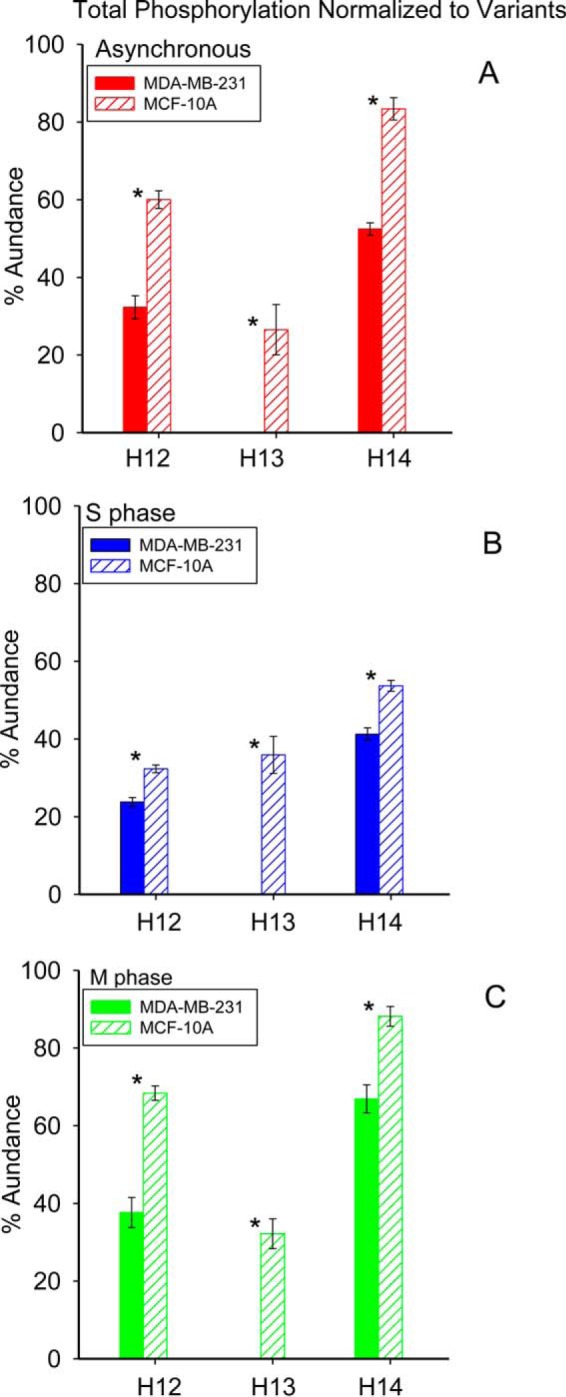
Normalized phosphorylation from cell lines MDA-MB-231 and MCF-10A: (a) asynchronous cells; (b) S phase cells; (c) M phase cells. Asterisk denotes p value less than 0.05.
Fig. 10.

Normalized phosphorylation at different modification sites from cell lines MDA-MB-231 and MCF-10A: (a) asynchronous cells; (b) S phase cells; (c) M phase cells. Asterisk denotes p value less than 0.05.
Translational Impact
Commercial H1 antibodies are cross-reactive with multiple histone H1 sequences. Limitations in antibody availability and specificity restrict the study of variant-specific PTM patterns. The present top-down MS/MS analysis clearly shows differences in H1 phosphorylation between major variants H1.2 and H1.4 through the cell cycle. These data are intriguing in that most studies performed on linker histones are limited to the H1.2 variant because of an available commercial antibody. The observed sequential phosphorylation specific to H1.4 suggests the involvement of multiple H1 kinases, as has been reported previously (36). Our localization of T146ph exclusively to H1.4 is of interest because of a previously reported functional role played by DNA-PK mediated phosphorylation of T146 on H1.2 in regulating p53-dependent transcription (41, 42). Our data suggest that cell cycle mediated T146 phosphorylation is variant-specific. Kinase inhibitors blocking the phosphorylation of H1 residues may also prove to be useful therapeutics (78). The specificity of such inhibitors is crucial. For example, the promiscuous kinase inhibitor staurosporine blocks progression of cells from G2 to M phase because of inhibition of H1- and H3-modifying kinases (79). This effect may be due in part to substrate similarity of serine and threonine residues. In addition to the structural effects H1 phosphorylation imparts on the chromatin during M phase, there are likely other outcomes of phosphorylation, such as recruitment of histone readers with phospho-recognition domains. The present results pave the road for studies involving individual and combinatorial amino acid mutations to understand the importance of individual phosphorylation events as they occur in breast cancer. An assay capable of monitoring M phase-specific phosphorylation events (i.e., T146 and T154 and, to a lesser degree, T18) would provide a more accurate snapshot of H1 phosphorylation associated with cellular proliferation in vivo, as opposed to global H1 phosphorylation or phosphorylation of an individual mitotic residue. More important, the results of this study may prioritize development of novel antibodies recognizing H1 modifications and increase awareness of the need to distinguish analogous modification sites on different H1 variants, particularly for the often-ignored H1.4.
Supplementary Material
Footnotes
Author contributions: M.A.F. and N.L.Y. designed research; Y.C., M.E.H., X.D., A.A.S., X.G., and N.L.Y. performed research; Y.C., M.E.H., M.A.F., and N.L.Y. analyzed data; Y.C., M.E.H., A.G.M., M.A.F., and N.L.Y. wrote the paper.
* This work was supported by the National Science Foundation, NSF Division of Materials Research through DMR-11–57490, NSF CHE-1019193, and the State of Florida. MAF acknowledges support from The Ohio State University, the Leukemia and Lymphoma Society and the NIH CA107106.
 This article contains supplemental Figs S1 to S7 and Tables S1 to S8.
This article contains supplemental Figs S1 to S7 and Tables S1 to S8.
Supplemental Information Available. Figures showing broadband spectra of histone H1 from asynchronous cells and cells blocked at S and M phases in cell line MDA-MB-231 and MCF-10A. ECD fragmentation maps of all proteoforms observed in histone H1 from cell lines MDA-MB-231 and MCF-10A. Segments of ECD product ion mass spectra of phosphorylated histone H1.2 SNP variant A18V from asynchronous cells of cell line MCF-10A. Peak annotations in ECD product ion mass spectra of asynchronous and S phase histone H1.2 SNP variant A18V, and asynchronous phosphorylated histone H1.2 SNP variant A18V. In addition, we provide annotated ECD product ion mass spectra for all identified proteoforms as validation of correct identification. This information is available free of charge via the internet at http://pubs.acs.org/.
1 The abbreviations used are:
- PTM
- post-translational modification
- DMEM
- Dulbecco's modified Eagle medium
- ECD
- electron capture dissociation
- ESI
- electrospray ionization
- FT-ICR
- Fourier transform ion cyclotron resonance
- HILIC
- hydrophilic interaction liquid chromatography
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- MRM
- multiple reaction monitoring
- MS/MS
- tandem mass spectrometry
- NP-40 Alternative
- (Nonylphenol nonylglycol ether)
- PBS
- phosphate-buffered saline
- SNP
- single nucleotide polymorphism
- TFA
- trifluoroacetic acid.
REFERENCES
- 1. Strahl B. D., and Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 3. Schneider A.-C., Heukamp L. C., Rogenhofer S., Fechner G., Bastian P. J., von Ruecker A., Müller S. C., and Ellinger J. (2011) Global histone H4K20 trimethylation predicts cancer-specific survival in patients with muscle-invasive bladder cancer. BJU Int. 108, E290–E296 [DOI] [PubMed] [Google Scholar]
- 4. Cheung W. L., Albadine R., Chan T., Sharma R., and Netto G. J. (2009) Phosphorylated H2AX in noninvasive low grade urothelial carcinoma of the bladder: correlation with tumor recurrence. J. Urol. 181, 1387–1392 [DOI] [PubMed] [Google Scholar]
- 5. Seligson D. B., Horvath S., Shi T., Yu H., Tze S., Grunstein M., and Kurdistani S. K. (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262–1266 [DOI] [PubMed] [Google Scholar]
- 6. Barlési F., Giaccone G., Gallegos-Ruiz M. I., Loundou A., Span S. W., Lefesvre P., Kruyt F. A. E., and Rodriguez J. A. (2007) Global histone modifications predict prognosis of resected nonsmall-cell lung cancer. J. Clin. Oncol. 25, 4358–4364 [DOI] [PubMed] [Google Scholar]
- 7. Seligson D. B., Horvath S., McBrian M. A., Mah V., Yu H., Tze S., Wang Q., Chia D., Goodglick L., and Kurdistani S. K. (2009) Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 174, 1619–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Manuyakorn A., Paulus R., Farrell J., Dawson N. A., Tze S., Cheung-Lau G., Hines O. J., Reber H., Seligson D. B., Horvath S., Kurdistani S. K., Guha C., and Dawson D. W. (2010) Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J. Clin. Oncol. 28, 1358–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elsheikh S. E., Green A. R., Rakha E. A., Powe D. G., Ahmed R. A., Collins H. M., Soria D., Garibaldi J. M., Paish C. E., Ammar A. A., Grainge M. J., Ball G. R., Abdelghany M. K., Martinez-Pomares L., Heery D. M., and Ellis I. O. (2009) Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 69, 3802–3809 [DOI] [PubMed] [Google Scholar]
- 10. Harshman S. W., Young N. L., Parthun M. R., and Freitas M. A. (2013) H1 histones: current perspectives and challenges. Nucleic Acids Res. 41, 9593–9609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. American Cancer Society. Cancer Facts & Figures 2014 Atlanta: American Cancer Society; 2014 [Google Scholar]
- 12. Fletcher S. W., and Elmore J. G. (2003) Mammographic screening for breast cancer. N. Engl. J. Med. 348, 1672–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamane K., Tateishi K., Klose R. J., Fang J., Fabrizio L. A., Erdjument-Bromage H., Taylor-Papadimitriou J., Tempst P., and Zhang Y. (2007) PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 25, 801–812 [DOI] [PubMed] [Google Scholar]
- 14. Shi L., Sun L., Li Q., Liang J., Yu W., Yi X., Yang X., Li Y., Han X., Zhang Y., Xuan C., Yao Z., and Shang Y. (2011) Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 7541–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okitsu C. Y., and Hsieh C. L. (2007) DNA methylation dictates histone H3K4 methylation. Mol. Cell Biol. 27, 2746–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang S. M., Kim B. J., Toro L. N., and Skoultchi A. I. (2013) H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc. Natl. Acad. Sci. U.S.A. 110, 1708–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sancho M., Diani E., Beato M., and Jordan A. (2008) Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet. 4, e1000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Millan-Arino L., Islam A. B., Izquierdo-Bouldstridge A., Mayor R., Terme J. M., Luque N., Sancho M., Lopez-Bigas N., and Jordan A. (2014) Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 42, 4474–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Terme J. M., Millan-Arino L., Mayor R., Luque N., Izquierdo-Bouldstridge A., Bustillos A., Sampaio C., Canes J., Font I., Sima N., Sancho M., Torrente L., Forcales S., Roque A., Suau P., and Jordan A. (2014) Dynamics and dispensability of variant-specific histone H1 Lys-26/Ser-27 and Thr-165 post-translational modifications. FEBS Lett. 588, 2353–2362 [DOI] [PubMed] [Google Scholar]
- 20. Kamieniarz K., Izzo A., Dundr M., Tropberger P., Ozretic L., Kirfel J., Scheer E., Tropel P., Wisniewski J. R., Tora L., Viville S., Buettner R., and Schneider R. (2012) A dual role of linker histone H1.4 Lys 34 acetylation in transcriptional activation. Genes Dev. 26, 797–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith L. M., Kelleher N. L., and Consortium for Top Down, P. (2013) Proteoform: a single term describing protein complexity. Nat. Methods 10, 186–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bradbury E. M., Inglis R. J., Matthews H. R., and Sarner N. (1973) Phosphorylation of very-lysine-rich histone in Physarum polycephalum. Eur. J. Biochem. 33, 131–139 [DOI] [PubMed] [Google Scholar]
- 23. Gurley L. R., D'Anna J. A., Barham S. S., Deaven L. L., and Tobey R. A. (1978) Histone phosphorylation and chromatin structure during mitosis in chinese hamster cells. Eur. J. Biochem. 84, 1–15 [DOI] [PubMed] [Google Scholar]
- 24. Matsumoto Y.-i., Yasuda H., Mita S., Marunouchi T., and Yamada M.-a. (1980) Evidence for the involvement of H1 histone phosphorylation in chromosome condensation. Nature 284, 181–183 [DOI] [PubMed] [Google Scholar]
- 25. D'Anna J. A., Gurley L. R., and Deaven L. L. (1978) Dephosphorylation of histones H1 and H3 during the isolation of metaphase chromosomes. Nucleic Acids Res. 5, 3195–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ajiro K., Borun T. W., and Cohen L. H. (1981) Phosphorylation states of different histone 1 subtypes and their relationship to chromatin functions during the HeLa S-3 cell cycle. Biochemistry 20, 1445–1454 [DOI] [PubMed] [Google Scholar]
- 27. Burstein D. E., Nagi C., Kohtz D. S., Lumerman H., and Wang B. Y. (2006) Immunohistochemical detection of GLUT1, p63 and phosphorylated histone H1 in head and neck squamous intraepithelial neoplasia: evidence for aberrations in hypoxia-related, cell cycle- and stem-cell-regulatory pathways. Histopathology 48, 708–716 [DOI] [PubMed] [Google Scholar]
- 28. Burstein D. E., Nagi C., Kohtz D. S., Lee L., and Wang B. (2006) Immunodetection of GLUT1, p63, and phospho-histone H1 in invasive head and neck squamous carcinoma: correlation of immunohistochemical staining patterns with keratinization. Histopathology 48, 717–722 [DOI] [PubMed] [Google Scholar]
- 29. Burstein D. E., Oami S., Dembitzer F., Chu C., Cernaianu G., Leytin A., Misilim E., Jammula S. R., Strauchen J., and Kohtz D. S. (2002) Monoclonal antibody specific for histone H1 phosphorylated by cyclin-dependent kinases: a novel immunohistochemical probe of proliferation and neoplasia. Mod. Pathol. 15, 705–711 [DOI] [PubMed] [Google Scholar]
- 30. Wu M., Kafanas A., Gan L., Kohtz D. S., Zhang L., Genden E., and Burstein D. E. (2008) Feasibility of immunocytochemical detection of tumor markers (XIAP, phosphohistone H1, and p63) in FNA cellblock samples from head and neck squamous cell carcinoma. Diagn. Cytopathol. 36, 797–800 [DOI] [PubMed] [Google Scholar]
- 31. Kafanas A., Wang B. Y., Kalir T., Gan L., Bodian C., Fish H., Kohtz D. S., and Burstein D. E. (2003) Immunohistochemical visualization of histone H1 phosphorylation in squamous intraepithelial lesions of the gynecologic tract. Hum. Pathol. 34, 166–173 [DOI] [PubMed] [Google Scholar]
- 32. Telu K. H., Abbaoui B., Thomas-Ahner J. M., Zynger D. L., Clinton S. K., Freitas M. A., and Mortazavi A. (2013) Alterations of histone H1 phosphorylation during bladder carcinogenesis. J. Proteome Res. 12, 3317–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harshman S. W., Hoover M. E., Huang C., Branson O. E., Chaney S. B., Cheney C. M., Rosol T. J., Shapiro C. L., Wysocki V. H., Huebner K., and Freitas M. A. (2014) Histone H1 phosphorylation in breast cancer. J. Proteome Res. 13, 2453–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sarg B., Helliger W., Talasz H., Forg B., and Lindner H. H. (2006) Histone H1 phosphorylation occurs site-specifically during interphase and mitosis: identification of a novel phosphorylation site on histone H1. J. Biol. Chem. 281, 6573–6580 [DOI] [PubMed] [Google Scholar]
- 35. Zheng Y., John S., Pesavento J. J., Schultz-Norton J. R., Schiltz R. L., Baek S., Nardulli A. M., Hager G. L., Kelleher N. L., and Mizzen C. A. (2010) Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell Biol. 189, 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hergeth S. P., Dundr M., Tropberger P., Zee B. M., Garcia B. A., Daujat S., and Schneider R. (2011) Isoform-specific phosphorylation of human linker histone H1.4 in mitosis by the kinase Aurora B. J. Cell Sci. 124, 1623–1628 [DOI] [PubMed] [Google Scholar]
- 37. Chu C. S., Hsu P. H., Lo P. W., Scheer E., Tora L., Tsai H. J., Tsai M. D., and Juan L. J. (2011) Protein kinase A-mediated serine 35 phosphorylation dissociates histone H1.4 from mitotic chromosome. J. Biol. Chem. 286, 35843–35851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Talasz H., Sarg B., and Lindner H. H. (2009) Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma 118, 693–709 [DOI] [PubMed] [Google Scholar]
- 39. Valdez J. G. (1995) Characterization of the mitotic specific phosphorylation site of histone H1. J. Biol. Chem. 270, 27653–27660 [DOI] [PubMed] [Google Scholar]
- 40. Green A., Sarg B., Green H., Lonn A., Lindner H. H., and Rundquist I. (2011) Histone H1 interphase phosphorylation becomes largely established in G1 or early S phase and differs in G1 between T-lymphoblastoid cells and normal T cells. Epigenet. Chromatin 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim K., Choi J., Heo K., Kim H., Levens D., Kohno K., Johnson E. M., Brock H. W., and An W. (2008) Isolation and characterization of a novel H1.2 complex that acts as a repressor of p53-mediated transcription. J. Biol. Chem. 283, 9113–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim K., Jeong K. W., Kim H., Choi J., Lu W., Stallcup M. R., and An W. (2012) Functional interplay between p53 acetylation and H1.2 phosphorylation in p53-regulated transcription. Oncogene 31, 4290–4301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia B. A., Joshi S., Thomas C. E., Chitta R. K., Diaz R. L., Busby S. A., Andrews P. C., Ogorzalek Loo R. R., Shabanowitz J., Kelleher N. L., Mizzen C. A., Allis C. D., and Hunt D. F. (2006) Comprehensive phosphoprotein analysis of linker histone H1 from Tetrahymena thermophila. Mol. Cell Proteomics 5, 1593–1609 [DOI] [PubMed] [Google Scholar]
- 44. Bonet-Costa C., Vilaseca M., Diema C., Vujatovic O., Vaquero A., Omenaca N., Castejon L., Bernues J., Giralt E., and Azorin F. (2012) Combined bottom-up and top-down mass spectrometry analyses of the pattern of post-translational modifications of Drosophila melanogaster linker histone H1. J. Proteomics 75, 4124–4138 [DOI] [PubMed] [Google Scholar]
- 45. Tian Z., Tolic N., Zhao R., Moore R. J., Hengel S. M., Robinson E. W., Stenoien D. L., Wu S., Smith R. D., and Pasa-Tolic L. (2012) Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 13, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dang X., Scotcher J., Wu S., Chu R. K., Tolic N., Ntai I., Thomas P. M., Fellers R. T., Early B. P., Zheng Y., Durbin K. R., Leduc R. D., Wolff J. J., Thompson C. J., Pan J., Han J., Shaw J. B., Salisbury J. P., Easterling M., Borchers C. H., Brodbelt J. S., Agar J. N., Pasa-Tolic L., Kelleher N. L., and Young N. L. (2014) The first pilot project of the consortium for top-down proteomics: a status report. Proteomics 14, 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moradian A., Kalli A., Sweredoski M. J., and Hess S. (2014) The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications. Proteomics 14, 489–497 [DOI] [PubMed] [Google Scholar]
- 48. Savaryn J. P., Catherman A. D., Thomas P. M., Abecassis M. M., and Kelleher N. L. (2013) The emergence of top-down proteomics in clinical research. Genome Med. 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. LeDuc R. D., Taylor G. K., Kim Y.-B., Januszyk T. E., Bynum L. H., Sola J. V., Garavelli J. S., and Kelleher N. L. (2004) ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 32, W340–W345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taylor G. K., Kim Y. B., Forbes A. J., Meng F. Y., McCarthy R., and Kelleher N. L. (2003) Web and database software for identification of intact proteins using “top-down” mass spectrometry. Anal. Chem. 75, 4081–4086 [DOI] [PubMed] [Google Scholar]
- 51. Zamdborg L., LeDuc R. D., Glowacz K. J., Kim Y. B., Viswanathan V., Spaulding I. T., Early B. P., Bluhm E. J., Babai S., and Kelleher N. L. (2007) ProSight PTM 2.0: improved protein identification and characterization for top-down mass spectrometry. Nucleic Acids Res. 35, W701–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. D. LeDuc R., and L. Kelleher N. (2002) Using Prosight PTM and related tools for targeted protein identification and characterization with high mass accuracy tandem MS data. Curr. Protoc. Bioinformatics, John Wiley & Sons, Inc. [DOI] [PubMed] [Google Scholar]
- 53. Sarg B., Green A., Soderkvist P., Helliger W., Rundquist I., and Lindner H. H. (2005) Characterization of sequence variations in human histone H1.2 and H1.4 subtypes. FEBS J. 272, 3673–3683 [DOI] [PubMed] [Google Scholar]
- 54. Ohe Y., Hayashi H., and Iwai K. (1989) Human spleen histone H1. Isolation and amino acid sequences of three minor variants, H1a, H1c, and H1d. J. Biochem. 106, 844–857 [DOI] [PubMed] [Google Scholar]
- 55. Fogh J. (1975) Human Tumor Cells in Vitro, Plenum Press, New York [Google Scholar]
- 56. Soule H. D., Maloney T. M., Wolman S. R., Peterson W. D. Jr., Brenz R., McGrath C. M., Russo J., Pauley R. J., Jones R. F., and Brooks S. C. (1990) Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50, 6075–6086 [PubMed] [Google Scholar]
- 57. Plazas-Mayorca M. D., Zee B. M., Young N. L., Fingerman I. M., LeRoy G., Briggs S. D., and Garcia B. A. (2009) One-pot shotgun quantitative mass spectrometry characterization of histones. J. Proteome Res. 8, 5367–5374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Harshman S. W., Chen M. M., Branson O. E., Jacob N. K., Johnson A. J., Byrd J. C., and Freitas M. A. (2013) Isolation and analysis of linker histones across cellular compartments. J. Proteomics 91, 595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Molden R. C., and Garcia B. A. (2014) Middle-down and top-down mass spectrometric analysis of co-occurring histone modifications. Curr. Protoc. Protein Sci. 77, 21–23, 27, 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaiser N., Quinn J., Blakney G., Hendrickson C., and Marshall A. (2011) A novel 9.4 Tesla FTICR mass spectrometer with improved sensitivity, mass resolution, and mass range. J. Am. Soc. Mass. Spectrom. 22, 1343–1351 [DOI] [PubMed] [Google Scholar]
- 61. Shaffer S. A., Prior D. C., Anderson G. A., Udseth H. R., and Smith R. D. (1998) An ion funnel interface for improved ion focusing and sensitivity using electrospray ionization mass spectrometry. Anal. Chem. 70, 4111–4119 [DOI] [PubMed] [Google Scholar]
- 62. Kelly R. T., Tolmachev A. V., Page J. S., Tang K., and Smith R. D. (2010) The ion funnel: theory, implementations, and applications. Mass Spectrom. Rev. 29, 294–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ibrahim Y., Tang K., Tolmachev A., Shvartsburg A., and Smith R. (2006) Improving mass spectrometer sensitivity using a high-pressure electrodynamic ion funnel interface. J. Am. Soc. Mass. Spectrom. 17, 1299–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wilcox B., Hendrickson C., and Marshall A. (2002) Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. J. Am. Soc. Mass. Spectrom. 13, 1304–1312 [DOI] [PubMed] [Google Scholar]
- 65. Kaiser N. K., Savory J. J., McKenna A. M., Quinn J. P., Hendrickson C. L., and Marshall A. G. (2011) Electrically compensated Fourier transform ion cyclotron resonance cell for complex mixture mass analysis. Anal. Chem. 83, 6907–6910 [DOI] [PubMed] [Google Scholar]
- 66. Blakney G. T., Hendrickson C. L., and Marshall A. G. (2011) Predator data station: a fast data acquisition system for advanced FT-ICR MS experiments. Int. J. Mass spectrom. 306, 246–252 [Google Scholar]
- 67. Ledford E. B., Rempel D. L., and Gross M. L. (1984) Space charge effects in Fourier transform mass spectrometry. II. Mass calibration. Anal. Chem. 56, 2744–2748 [DOI] [PubMed] [Google Scholar]
- 68. Shi S. D. H., Drader J. J., Freitas M. A., Hendrickson C. L., and Marshall A. G. (2000) Comparison and interconversion of the two most common frequency-to-mass calibration functions for Fourier transform ion cyclotron resonance mass spectrometry. Int. J. Mass spectrom. 195, 591–598 [Google Scholar]
- 69. Xian F., Hendrickson C. L., Blakney G. T., Beu S. C., and Marshall A. G. (2010) Automated broadband phase correction of Fourier transform ion cyclotron resonance mass spectra. Anal. Chem. 82, 8807–8812 [DOI] [PubMed] [Google Scholar]
- 70. Horn D. M., Zubarev R. A., and McLafferty F. W. (2000) Automated reduction and interpretation of high-resolution electrospray mass spectra of large molecules. J. Am. Soc. Mass. Spectrom. 11, 320–332 [DOI] [PubMed] [Google Scholar]
- 71. Wisniewski J. R., Zougman A., Kruger S., and Mann M. (2007) Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell. Proteomics 6, 72–87 [DOI] [PubMed] [Google Scholar]
- 72. Garcia B. A., Busby S. A., Barber C. M., Shabanowitz J., Allis C. D., and Hunt D. F. (2004) Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry. J. Proteome Res. 3, 1219–1227 [DOI] [PubMed] [Google Scholar]
- 73. Lu K. P., Liou Y. C., and Zhou X. Z. (2002) Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 12, 164–172 [DOI] [PubMed] [Google Scholar]
- 74. Bradbury E. M., Cary P. D., Chapman G. E., Crane-Robinson C., Danby S. E., Rattle H. W. E., Boublik M., Palau J., and Aviles F. J. (1975) Studies on the role and mode of operation of the very-lysine-rich histone H1 (F1) in eukaryote chromatin. Eur. J. Biochem. 52, 605–613 [DOI] [PubMed] [Google Scholar]
- 75. Vila R., Ponte I., Collado M., Arrondo J. L. R., Jiménez M. A., Rico M., and Suau P. (2001) DNA-induced α-helical structure in the NH2-terminal domain of histone H1. J. Biol. Chem. 276, 46429–46435 [DOI] [PubMed] [Google Scholar]
- 76. Gurley L. R., Walters R. A., and Tobey R. A. (1975) Sequential phosphorylation of histone subfractions in the Chinese hamster cell cycle. J. Biol. Chem. 250, 3936–3944 [PubMed] [Google Scholar]
- 77. Mizzen C. A., Dou Y., Liu Y., Cook R. G., Gorovsky M. A., and Allis C. D. (1999) Identification and mutation of phosphorylation sites in a linker histone: phosphorylation of macronuclear H1 is not essential for viability in tetrahymena. J. Biol. Chem. 274, 14533–14536 [DOI] [PubMed] [Google Scholar]
- 78. Wang L., Harshman S. W., Liu S., Ren C., Xu H., Sallans L., Grever M., Byrd J. C., Marcucci G., and Freitas M. A. (2010) Assaying pharmacodynamic endpoints with targeted therapy: flavopiridol and 17AAG induced dephosphorylation of histone H1.5 in acute myeloid leukemia. Proteomics 10, 4281–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Th'ng J. P., Guo X. W., Swank R. A., Crissman H. A., and Bradbury E. M. (1994) Inhibition of histone phosphorylation by staurosporine leads to chromosome decondensation. J. Biol. Chem. 269, 9568–9573 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.