

Abstract
Repurposing of drugs to novel disease indications has a promise of faster clinical translation. However, identifying best drugs for a given pathological context is not trivial. We developed an integrated random walk-based network framework that combines functional biomolecular relationships and known drug-target interactions as a platform for contextual prioritization of drugs, genes and pathways. We show that the use of gene-centric or drug-centric data, such as gene expression data or a phenotypic drug screen, respectively, within this network platform can effectively prioritize drugs and pathways, respectively, to the studied biological context. We demonstrate that various genomic data can be used as contextual cues to effectively prioritize drugs to the studied context, while similarly, phenotypic drug screen data can be used to effectively prioritize genes and pathways to the studied phenotypic context. As a proof-of-principle, we showcase the use of our platform to identify known and novel drug indications against different subsets of breast cancers through contextual prioritization based on genome-wide gene expression, shRNA and drug screen and clinical survival data. The integrated network and associated methods are incorporated into the NetWalker suite for functional genomics analysis (http://netwalkersuite.org).
Introduction
Small molecule drugs used in the clinic usually possess an inherent promiscuity, which, while a potential source of off-target effects and adverse reactions in patients, can also prove beneficial in some pathological contexts other than their primary indications. In addition to such repurposing of drugs to novel protein targets (target repositioning), drugs may also be repurposed to a novel indication based on their known targets (disease repositioning). Biological systems are characterized by remarkable modularity, where molecular machineries can perform different functions in different biological contexts. Therefore, a drug developed against a target gene in one disease may prove beneficial in another due to its unappreciated role in that disease.
Significant amount of work in the drug-repositioning field has been dedicated to the discovery of novel drug-target pairings (target repositioning) using drug-to-drug chemical and functional similarity approaches. One of the most notable resources for such analyses is the connectivity map (cmap) dataset, where gene expression responses of cells to some ~1,400 drugs are reported as quantitative drug signatures.[1, 2] Comparative analyses of these drug signatures allow for the identification of novel drug-drug similarities, and hence, novel drug-target pairings; a paradigm that has been extensively exploited.[3–6] In addition to comparative analyses of drug signatures, complementary approaches based on chemical similarities of drugs (most notably the Similarity Ensemble Approach) have also been used for inferring novel drug-target pairings.[7–11] However, despite the large amount of these excellent studies on the identification of novel drug-target pairings, relatively less focus has been dedicated to the identification of novel pathological contexts for known drug-target pairs (disease repositioning). Effective identification of such novel off- and on-target pathological contexts of drugs requires efficient integration of multi-binding properties of drugs with molecular data from different disease contexts, which would allow prioritizing of diseases to drugs.
We and others have shown that integration of molecular data with the prior network of molecular interactions can help prioritize context-specific pathways.[12–16] Although hybrid networks of functional interactions between biological molecules as well as drug-target interactions have been studied for their properties,[17] to our knowledge, such an approach has not been used for integrated drug repositioning. Here, we propose that integration of disease-specific molecular (genomic) data with the network of functional and drug-target interactions can help prioritize drug-target pairings that are most relevant to the studied disease context. For this purpose, we make use of our previously developed random walk-based data integration and network scoring algorithm, NetWalk. NetWalk allows for seamless integration of molecular data with the network of binary interactions to score each network node (e.g. gene, drug) based on the combined assessment of the data and the network structure. Thereby, NetWalk is able to assign scores to each drug in the network based on the combined assessment of the data values of their targets as well as their connectivity patterns in the network neighborhood. We have incorporated the drug-target network along with the NetWalk algorithm in the new version of our previously published software NetWalker, which is freely available for academic use (http://netwalkersuite.org).
Here, we demonstrate the use of gene expression, shRNA and drug screening data for different subsets of breast cancers as contextual cues for drug prioritization using NetWalk. In addition to retrieving expected and best-known drug-target pairings that are currently in use in the clinic for ER+ (estrogen receptor positive) and HER2+ (epidermal growth factor receptor 2) subtypes of breast cancer, our analyses also identify novel drug-target pairings for HER2+ and TNBC (triple-negative) subtypes, some of which we have verified experimentally.
Results
We previously developed NetWalk; an algorithm aimed to integrate experimental (genomic, phenotypic, etc.) data with networks of interactions between genes to score the relevance of each interaction based on both the data values of the genes as well as their local network connectivity [16]. NetWalk relies on the principle of data-biased random walks, where every node in the network is visited by a random walk process depending on the inter-node transition probability values reflecting local connectivity and the data values of nodes. NetWalk is implemented in a user-friendly software package NetWalker, which also features a comprehensive molecular interaction knowledgebase (NetWalker Interactome Knowledgebase, NIK) and a suite of user-friendly utilities for data integration and network analyses.[15] We have extended the NIK to include drug-target interaction data, consisting of 8,553 unique interactions among 1,610 genes and 4,325 drugs (see Methods). Prioritization of nodes by NetWalk is dependent on their visitation frequencies by the random walker during an infinite random walk process, which are driven by the data values attached to nodes (e.g. gene expression), data values attached to their neighbors in the network neighborhood and their connectivity patterns in the network neighborhood. Thus, the main concept behind NetWalk-based prioritization of drugs is that drugs connected to highly visited network nodes (genes) will also be highly visited by the random walker, while drugs connected to network nodes with low visitation will also be rarely visited by the random walker (Figure 1a). In order to demonstrate the use of NetWalk for network-based analyses of drug-target interaction data, we conducted an analysis in the context of different chemical, biological and genomics datasets.
Figure 1.
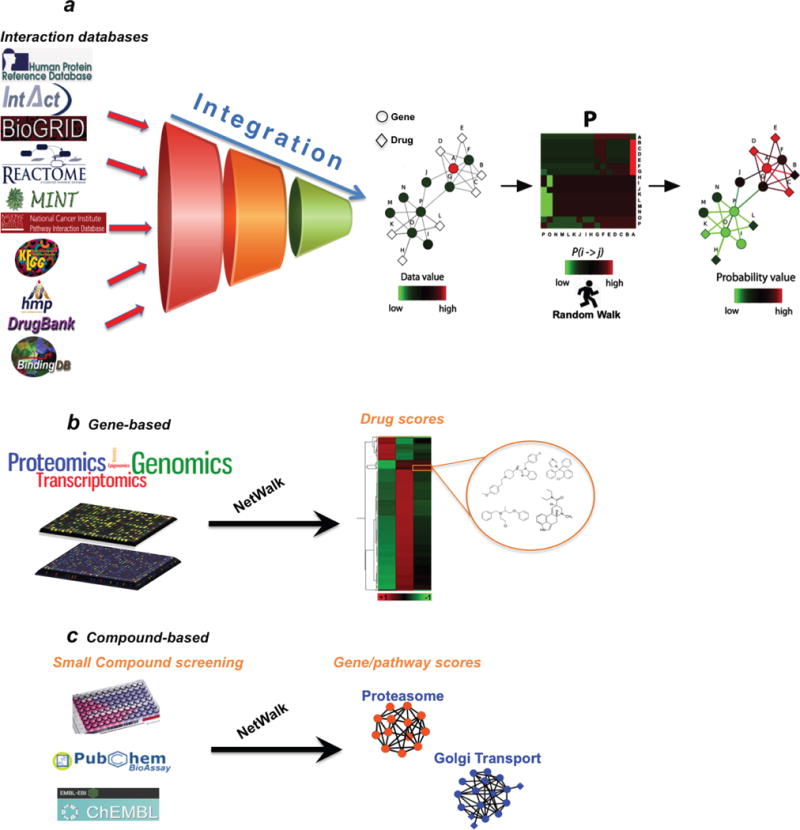
a) An imaginary drug-target network with simulated experimental data values is shown (e.g. relative gene expression values) on the left. Node A was assigned a value of 5, and all the other nodes were assigned 1. A transition probability matrix P was constructed using the input data values and the network, with transition probabilities between adjacent nodes reflecting their data values (colors in the matrix reflect transition probabilities P(i > j) according to the color key). Final visitation and flux values reflect the level of coherence between the experimental data of genes and drugs, and their relative positioning within the network. Note that node colorings in the network on the right reflect relative visitation probabilities of nodes, and line colors of edges reflect the flux values according to the same color scale. b) Scoring drugs based on gene-centric data (e.g. transcriptomics, epigenomics and proteomics). c) Scoring genes and pathways based on drug-centric data (e.g. phenotypic drug screens).
Prioritization of drugs and targets: proof of concept
An important utility of NetWalk-based scoring of a hybrid drug-gene network (Figure 1b and 1c) is the ability to score drugs based on gene-centric data (e.g. gene expression), or do a reverse analysis to score genes and pathways based on drug data (e.g. from a phenotypic drug screen). Although the former is the utility that is more intuitive and that we will stress most in this study, the latter may be a novel approach to determine the most important molecular processes in the cell that are being targeted by the active drugs in a phenotypic drug screen (see later). Indeed, in addition to helping identify the best potential drugs/compounds to modulate a cell phenotype, we suggest that phenotypic drug screens also have the potential to reveal the most important molecular processes involved in the studied phenotype through a pathway-based analysis of drug-target networks. Therefore, as a proof of concept, we will first demonstrate the performance of NetWalk in scoring drugs from gene-based data (gene-to-drug scoring), and then, scoring of genes from drug-based data (drug-to-gene scoring).
Initially, we generated a simulated dataset, where a value of 10 (a random high number indicating a high score) was assigned to the EGFR (epidermal growth factor receptor) node, and all the other nodes were assigned 1. EGFR is an oncogene that is targeted by several kinase inhibitors currently in use in the clinic, such as gefitinib and erlotinib. As expected, running NetWalk over the network using these values assigned high scores to known EGFR and related family inhibitors (Figure 2a). However, cellular (or disease) phenotypes are not necessarily defined by the direct drug targets, and can involve genes that function in the same pathway/complex as the direct drug target. In such a case, the prioritization of a drug-target pairing to the studied cellular context has to be scored based on the data from the neighboring genes of the direct drug target in the molecular network (Figure 2b). To test if NetWalk can accurately score drug-target pairings based on the data values of the network neighbors of a direct drug target, we used the proteasome as an example. Bortezomib is a proteasomal inhibitor that targets the PSMD2, PSMD1, PSMB1, PSMB2 and PSMB5 proteasomal subunits.[18, 19] We assigned a value of 10 to PSMD5, another gene in the proteasomal complex that is not directly targeted by bortezomib, and performed the NetWalk analysis over the integrated hybrid network. As expected, all proteasomal subunits and the known proteasomal inhibitors were ranked at the top (Figure 2b), suggesting that NetWalk analysis is able to correctly prioritize the most relevant drugs and targets based on direct or indirect scoring. These results reflect the coherence with the high value (red nodes on the right of Fig. 1a) used as input in comparison to the other nodes (green ones on the right of Fig. 1a) in the whole NIK. Using any value higher than 1 (i.e. the default value for all other nodes) for PSMD5 here will result in the same ranking by NetWalk.
Figure 2.
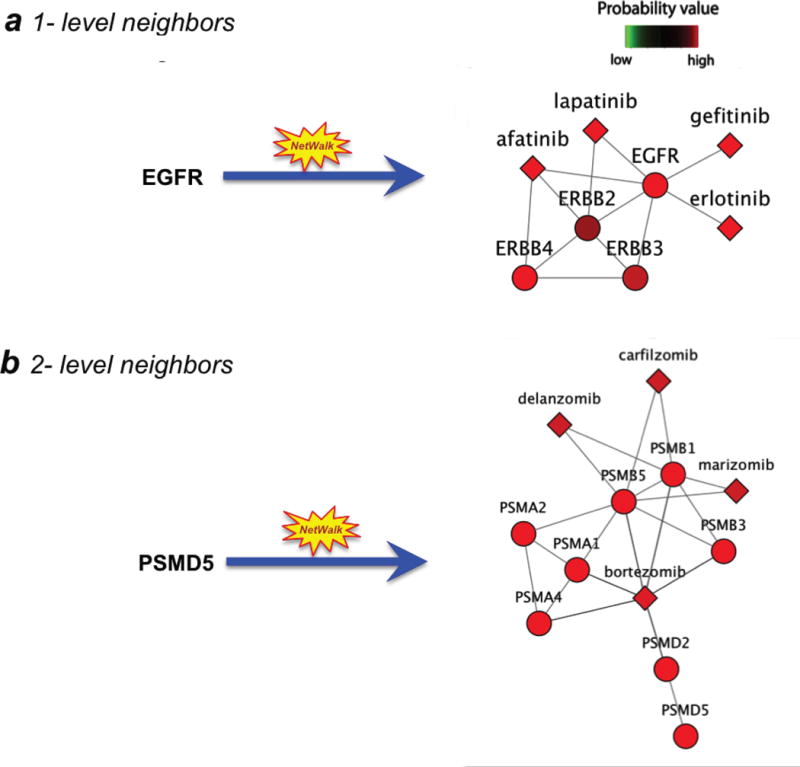
Scoring drugs based on a gene-centric simulated dataset. a) Drug scoring based on the direct neighbors. Here, the EGFR gene was assigned a value of 10, and all the other nodes were assigned 1. Then, NetWalk analysis was conducted to score the drug-target sub-network associated with the EGFR gene. b) Drug scoring based on the data from the neighboring genes of the direct drug target in the network. Here, the highest value was assigned to the PSMD5 gene, a member of the proteasomal complex that is not directly targeted by drugs. Shown is a NetWalk analysis to score the drug-target sub-network associated with the proteasome complex.
To illustrate the utility of NetWalk analysis of our hybrid network to perform gene-to-drug as well as drug-to-gene scoring, we chose phenotypic (cell lethality) drug screens over 6 cell lines from the NCI60 drug screen [20] with matching shRNA-based genetic screens of cell lethality from a different study [21]. We reasoned that if our NetWalk-based approach is useful in gene-to-drug scoring, NetWalk-based drug scoring using the shRNA screen data should prioritize the drugs that also scored significantly in the phenotypic drug screen. Therefore, scores assigned to drugs by NetWalk based on the shRNA data is expected to correlate with the experimental values from the drug screen experiments. Similarly, NetWalk-based drug-to-gene scoring should correlate with the experimental values from shRNA screens. Indeed, NetWalk-based gene-to-drug and drug-to-gene scores significantly correlated with the data from drug and shRNA screens from matching conditions, respectively (Figure 3a and 3b). Therefore, NetWalk-based scoring of drugs using gene data and vice versa is a useful method to prioritize the drugs or genes, respectively, that are most relevant to the studied context. The heatmap with some of the highest and lowest drug and gene scores from NetWalk analyses of these shRNA and drug screen data from three cancer cell lines along with the associated representative sub-networks is shown in Figure 3c. For example, pemetrexed was identified as a potential most relevant drug by NetWalk based on the analyses of shRNA lethality data; and it was also associated with significant lethality in MCF7 and MDAMB231 cells in the drug screen (Figure 3c). Similarly, its target, DHFR, was identified by NetWalk as a likely relevant target in these cell lines based on the analysis of drug screen data; and DHFR knock-down by shRNA was also associated with significant lethality in these cells in the shRNA screen. These findings are consistent with several reports and phase II clinical trials that have been conducted to evaluate the use of pemetrexed in BC.[22–26] On the other hand, bortezomib and its interacting partners, the proteasome subunits, tend to be specific for the MDAMB231 cell line. Similar results were found for entinostat, a HDAC inhibitor, and its interacting partners.
Figure 3.
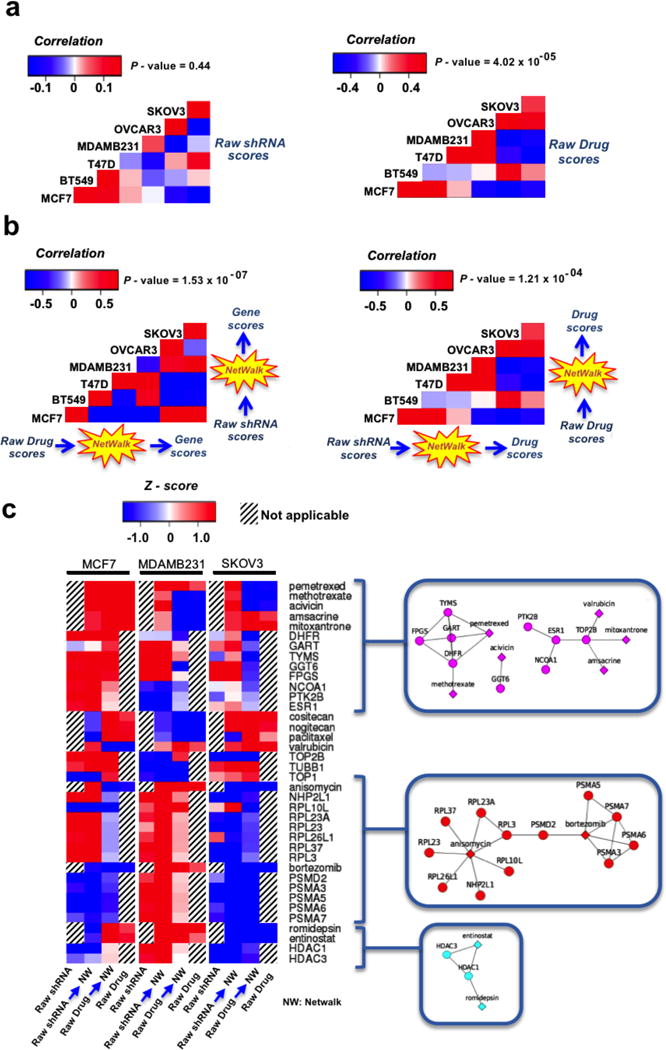
Correlation analysis of gene-to-drug and drug-to-gene scores from drug and shRNA screens from matching conditions, respectively. a) Correlation analysis by using raw scores. b) Correlation analysis by using NetWalk-based scores. c) Heatmap of drug and gene scores from NetWalk analyses of shRNA and drug screen data from three cancer cell lines and their representative sub-networks.
Drug repositioning based on the functional context
Next, we wanted to test the use of our platform for disease repurposing of drugs: assigning drug-target pairings to different subtypes of breast cancers. Breast cancers are usually classified into three subtypes based on the expression of the estrogen receptor or the HER2 oncogene; ER+ for those expressing the estrogen receptor, HER2+ for those expressing the HER2 oncogene, and triple negative (TNBC) for those expressing neither.
Since drug prioritization by NetWalk will be driven by the gene values to be used as input, it is crucial that we identify the appropriate genomic parameters to drive our analysis. In other words, the gene values used as input into NetWalk analysis should reflect the potential of those genes to be therapeutically targeted in the respective breast cancer subtype. For this purpose, we considered shRNA-based lethality scores from the shRNA screens of breast cancer cell lines (lethality profile), which provide important information about the most essential pathways sustaining breast cancer cell survival in a subtype-specific manner. In addition, to measure subtype-specific expression of genes in breast cancers, we also incorporated extensive gene expression profiles from breast cancer clinical samples (transcriptional profile). Finally, to integrate into our analysis the potential of a gene to play a role in breast cancer malignancy, we measured the correlation of expression of each gene with poor outcome in each of the three breast cancer subtypes using COX regression (survival profile). While the data from shRNA screens indicate essentiality of a gene for survival, the subtype-specific expression indicates whether the targeted pathway is specifically expressed in a subtype-selective manner; and the COX regression scores of genes indicate whether the gene has a role in conferring a more malignant phenotype to breast cancers. Therefore, if a drug-target pairing scores high within the context of shRNA lethality, gene expression and COX regression, it would indicate that the given drug-target pairing is likely to be therapeutically relevant for the given BC subtype as its target(s) are likely to be specifically expressed in, and confer survival and higher tumorigenic potential to, the corresponding breast cancer cells.
NetWalk analysis of each of the three functional profiles for the three breast cancer subtypes revealed three distinct subtype-specific clusters of drugs (Figure 4a). Interestingly, we found that each cluster is significantly enriched with certain families of drugs, as defined by their ATC codes (3-level), in comparison with the proportion of ATC codes found on randomly created clusters of the same size (P-value < 2.2 × 10−16), suggesting potential new applications for the BC treatment. For example, the ER+ cluster is enriched for blood glucose lowering drugs, while TN cluster is enriched for anti-inflammatory drugs, and the HER2+ cluster is enriched for calcium channel blockers. The prioritized drugs and some relevant subnetworks for each BC molecular subtype are shown in Figure 4a and 4b.
Figure 4.
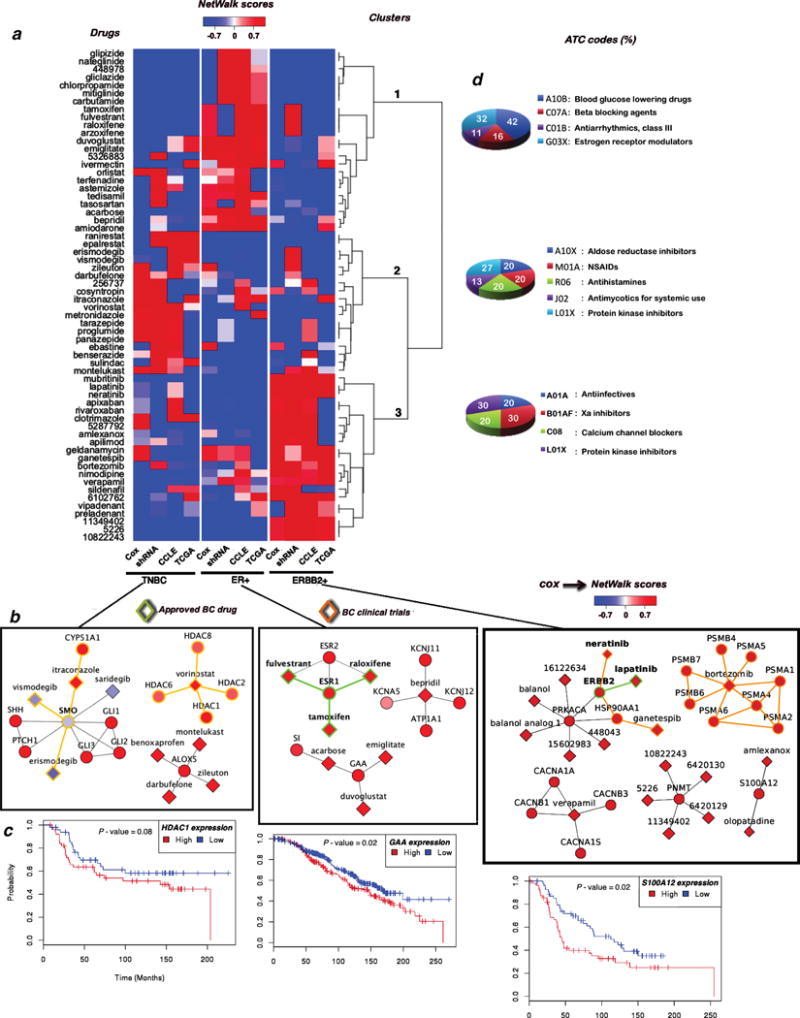
a) Heatmap of normalized NetWalk-based scores of the survival, lethality and transcriptional profiles for the three breast cancer subtypes. b) Relevant sub-networks for each BC molecular subtype. The clinical status of the drug-target pairs is coded by the edge color. c) Kaplan Meier plots for relevant targets for each BC molecular subtype. d) ATC codes distribution for the prioritized drugs for each BC molecular subtype.
Importantly, NetWalk was able to correctly prioritize several drugs to their current indications in breast cancer. For example, lapatinib and neratinib, two small molecule inhibitors of the HER2 kinase, have been assigned to the HER2+ subtype, while tamoxifene and raloxifene, the estrogen receptor antagonists, have been assigned to the ER+ subtype. These results serve as proof-of-principle validations that NetWalk can correctly prioritize the most relevant drug-target pairs.
In addition to the known BC drugs, some of the drugs were prioritized to the subtypes where they are currently undergoing clinical trials (Figure 4b). For example, vorinostat, a histone deacetylase (HDAC) inhibitor that is approved for cutaneous T cell lymphoma, is currently in clinical trials for TNBC (ClinicalTrials.gov IDs: NCT00368875, NCT00616967). Supporting the assignment of vorinostat to TNBC subtype by NetWalk based on the genomic parameters, HDAC1 expression significantly correlates with poor survival in TNBC patients (Figure 4c). Vimosdegib, erismodegib and itraconazole, inhibitors of smoothened (SMO), a critical component of the hedgehog pathway, are another set of high ranked compounds for TNBC. Erismodegib is also undergoing clinical trials for this subtype (ClinicalTrials.gov IDs: NCT01576666, NCT02027376). Interestingly, although SMO expression does not correlate with poor survival in TNBC, its upstream and downstream components in the hedgehog pathway do correlate with poor survival in TNBC (Figure 4a), showcasing the ability of NetWalk to prioritize drugs based on their indirect targets in a pathway.
For the HER2+ subtype, two drugs that were particularly of interest are bortezomib and ganetespib, both of which are in clinical trials for this BC subtype (ClinicalTrials.gov IDs: NCT00199212, NCT01497626, NCT02060253). Bortezomib is a proteasome inhibitor and is approved for multiple myeloma, while ganetespib is an experimental drug against the heat shock protein 90 (Hsp90) ATPase. Importantly, we experimentally verified the selective toxicity of bortezomib to HER2+ breast cancer cells (Figure 5), indicating that the assignment of bortezomib-proteasome pairing to HER2+ breast cancer subtype may be clinically relevant.
Figure 5.
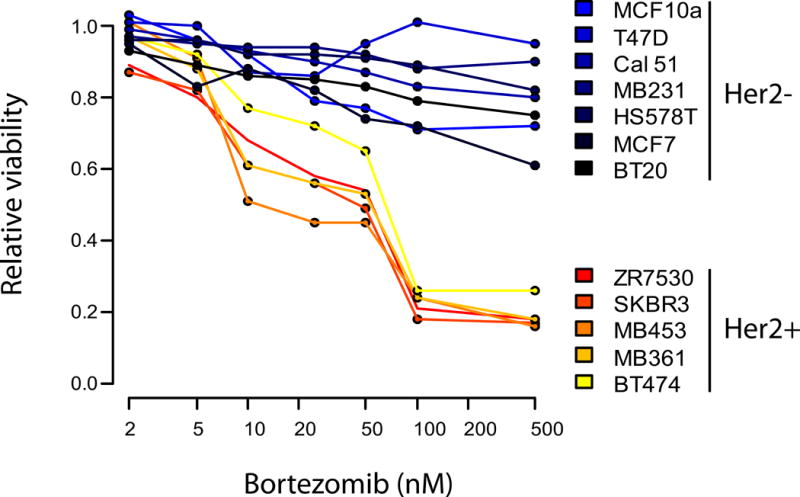
Dose survival curves of a panel of HER2+ and HER2- breast cancer cell lines in response to increasing concentrations of bortezomib, a proteasomal inhibitor.
In addition to the known and experimental indications, there were surprising results for each BC molecular subtype. For example, a set of inhibitors of the Arachidonate 5-Lipoxygenase (ALOX5) was prioritized to the TNBC cluster. These drugs, zileuton, darbufelone and montelukast, are classified as anti-inflammatory drugs in ATC (Figure 4d). Even though there are no reports for the role of ALOX5 or their inhibitors in TNBC, there is evidence for each of these in other cancers. For example, zileuton, darbufelone and montelukast have been implicated in the growth inhibition of prostate, lung and colon cancer cells, respectively.[27–29]
Other interesting drug-target pairs were those including the 5226, 11349402, 6420130, 6220129 compounds and the Phenylethanolamine N-methyltransferase (PNMT) gene, which were prioritized to the HER2+ subtype (Figure 4b). Interestingly, this gene is co-amplified with the ERBB2 (HER2) gene within the same amplicon in HER2+ breast cancers, suggesting that this gene may be a valid target in HER2+ breast cancers.[30, 31]
Discussion
Identifying and prioritizing drug targets are some of the most challenging tasks in the post-genomic era. The elucidation and analysis of interactions between drugs and their targets in the context of functional genomics data is critical for understanding the mechanisms of drug action, drug repositioning, off-target effects and speed up the development of effective and safer therapies for human diseases. Although several approaches for predicting novel drug-target interactions have been developed, methods to prioritize drugs to diseases based on functional genomics data are limited. The growing number of annotated drug-target interactions and the extensive collection of cancer genomic datasets from patient and cell line samples provides an unprecedented opportunity for the query of systemic underpinnings and context-specific relationships between drugs and their targets [32–37].
In this study, we proposed an effective approach to reposition diseases and drugs for novel, and sometimes unexpected, pathological contexts. The novelty of our approach stems from the use of biased random walks on graphs to score drugs based on gene-based data and vice versa. By using this approach and our hybrid drug-gene network, we have integrated and analyzed disease-specific genomic data to infer new uses for existing drugs in breast cancers. In addition to identifying known and experimental drugs currently in clinical use for the treatment of Her2+ and ER+ breast cancer subsets, we also identified several novel groups of subtype-specific drugs for BC with a potential clinical utility. In HER2+ BC, our analyses prioritized a group of drugs associated with targets from the unfolded protein response (UPR). Moreover, we were able to verify that bortezomib was specifically toxic to HER2+ BC cells in vitro, possibly highlighting the robustness of our approach, and the clinical potential of targeting the ubiquitin proteasome system in HER2+ breast cancers.
It is worth noting that in contrast to current drug repositioning approaches [1, 3–7, 9, 38–41], we did not use any similarity metrics or gene signatures to establish drug-target relationships, but rather elected to exploit the extensive gene- and drug-centric datasets as context cues to efficiently prioritize drugs and pathways. Central to our approach is the appropriate use of proper gene-based values to base the drug prioritization on. In our case, we used shRNA-based lethality scores of genes, as well as transcriptional and clinical survival parameters to use as cues for drug scoring, which helped prioritize genes/pathways of pharmacological interest and drugs with high potential for therapeutic interventions in BC. We believe that our approach that is incorporated into a freely available user-friendly software will enable hypothesis generation and drug repositioning from the data integration of the chemical, pharmacological and genomic spaces.
Methods
The hybrid network
We identified and gathered information from DrugBank,[42] KEGG drug,[43] Pubchem bioassay,[44] and BindingDB[45] databases to create a comprehensive repository of annotated drug-target interactions. Only human drug-target pairings data were selected from al the databases. Entries containing inorganic compounds, non-covalent complexes, biotechnology drugs and mixtures were excluded from DrugBank dataset. Only drug-target pairings with Ki values less than 10 μM were extracted from Pubchem and BindingDB databases as suggested by Cheng et al.[10]
Functional interactions between human gene products were collected and assembled from online databases. Protein-protein interactions, including signaling relationships were obtained from HPRD,[46] MINT,[47] Reactome,[48] BIND,[49] BioGRID,[50] Nature Pathway Database (http://pid.nci.nih.gov/), Biocarta (http://www.biocarta.com/) and PathwayCommons;[51] transcription factor – gene target relationships were obtained from TRANSFAC,[52] ORegAnno,[53] ENCODE,[54] and MSigDB.[55] Metabolic relationships between gene products were defined such that genes whose products catalyze consecutive reactions (that is, product of the reaction catalyzed by one is used as a reactant in the reaction catalyzed by the other gene product) were assigned an interaction; metabolic reactions catalyzed by human gene products were obtained from HMDB,[56] BiGG [57] and KEGG.[58] To increase the coverage of our knowledgebase, we also assigned interactions between pairs of genes if they shared GeneRIFs assigned to them in Entrez Gene.[59]
The NetWalker software and availability
Overall, our knowledgebase consisted of 452,005 unique interactions (444,828 gene-gene and 7,177 drug-gene interactions) including 18,722 genes and 4,755 drugs, and is available together with the the updated NetWalker software (version 2) for download at https://netwalkersuite.org/download. At the present moment, only the Windows installer is available for the version 2. Sample data (Supplementary Table 1) and detailed steps to reproduce some of the drug scoring results are provided in the Supplementary Text.
Breast Cancer Genomics Datasets
Gene expression (RNAseq v2 and Agilent) datasets from patient samples were obtained from the TCGA (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp). METABRIC datasets [60] were obtained from European Genome-Phenome Archive (https://www.ebi.ac.uk/ega/studies/EGAS00000000083). The METABRIC dataset contains clinical traits, gene expression and CNV profiles derived from breast tumors collected from participants of the METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) trial. Details about the METABRIC cohort have been published by Curtis et al [60]. Cancer Cell Line Encyclopedia (CCLE) datasets were obtained from its web site (http://www.broadinstitute.org/ccle/home). The CCLE provides public access to DNA copy number, mRNA expression and mutation data for more than a thousand cancer cell lines. shRNA screens of breast cancer cell lines were obtained from the COLT-Cancer database [21] (http://dpsc.ccbr.utoronto.ca/cancer/help.html). The COLT-Cancer database is a collection of shRNA dropout signature profiles of ~16,000 human genes in 72 cancer cell lines.
Analysis of BC genomic datasets
Previously, we developed a knowledge-based linear modeling approach coupled to network/pathway analysis to identify genotype-specific pathway profiles from cancer genomic datasets.[61] Here, by employing this approach we analyzed each breast cancer dataset to generate BC transcriptional, lethality and survival profiles associated with each molecular subtype of BC. To calculate the transcriptional profile of a BC subtype, we measure the correlation t-statistic of every gene’s expression with the given BC subtype using multiple linear regression as described previously.[61] The lethality profile is defined the same way, only using the GARP (Gene Activity Ranking Profile) scores of genes, instead of gene expression data, from the genome-wide shRNA screens. GARP score quantifies the shRNA dropout rate of a gene, based on the GARP scores; lower GARP scores (i.e. more negative) depict higher essentiality. In order to get all the values in the same scale, the GARP scores were multiplied by −1.
For a survival profile, we calculated the correlation of each gene’s expression with patient death rates (poor prognosis) using COX proportional hazards model for each molecular subtype in patient populations from the METABRIC dataset.
Network analyses
To prioritize drug-target interactions from gene-based data values, we used NetWalk, a random-walk method for the scoring of functional pathways and network interactions. The NetWalk method has been described previously.[16] Briefly, the gene values (t-statistic values from above) are used as weights (w = et: weights must be positive) in the transition probability matrix P in NetWalk:
where wj is the weight (transformed data value) assigned to node j, Ni is the set of network neighbors of node i, and s the transition probability from node i to node j. We define visitation probabilities of nodes, π, in the random walk as the dominant eigenvector of the extended transition probability matrix:
where P is the transition probability matrix, q is the restart probability for the random walk and 1n is a unit vector of length n (total number of network nodes). The second term on the right-hand side is a matrix with rank one that (1) adds a restart probability to the random walker depending on the weights of nodes and (2) ensures that the equation converges to a unique π. Visitation probability of the network interaction between nodes i and j, μij, is defined as
The vector μ reflects the probabilities of the interactions at the end of the random walk process, and each μij reflects the weights (t-values) of immediate nodes i and j, and the weights and connectivity of nodes in the local network neighborhood. To control for topological bias in the network, we also calculate , which is calculated by setting all w = 1 (that is, all t = 0). Finally, every edge in the network, including drug-gene interactions, is assigned a final Edge Flux (EF) score defined as the log-likelihood
Different edge types (drug-gene, gene-gene, etc…) can be analyzed separately or together in the NetWalker software (see accompanying manual in the web site). All of the NetWalk analyses were performed in NetWalker, a stand-alone software suite for network-based genomic data analyses.
Cell viability analyses
Cell viability was assessed using crystal violet assay (20% methanol, 0.5% crystal violet (Sigma) in 1xPBS) as previously described [62]. Briefly, equal numbers of cells in 96-well culture plates were treated with Bortezomib as indicated. After 72 h, dead cells were removed by washing in PBS and the attached cells were stained and fixed with crystal violet (Sigma) for 30 minutes at room temperature. After 30 minutes, excess stains were removed with tap water and the plates dried at room temperature. Once dried, crystal violet crystals were re-dissolved in Triton (Amresco) and the cell density was determined by measuring the absorbance at 570 nm in a microplate reader (Biotek Instruments).
Supplementary Material
Acknowledgments
ASC acknowledges CONACYT-México for support from “Estancias Posdoctorales al Extranjero para la Consolidación de Grupos de Investigación” (grant number 238386).
Funding: This work was supported by the Susan G. Komen for the Cure award to KK (CCR13263034).
References
- 1.Lamb J, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 2.Lamb J. The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer. 2007;7(1):54–60. doi: 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- 3.Hassane DC, et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood. 2008;111(12):5654–62. doi: 10.1182/blood-2007-11-126003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hieronymus H, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10(4):321–30. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Hu G, Agarwal P. Human disease-drug network based on genomic expression profiles. PLoS One. 2009;4(8):e6536. doi: 10.1371/journal.pone.0006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Noort V, et al. Novel drug candidates for the treatment of metastatic colorectal cancer through global inverse gene-expression profiling. Cancer Res. 2014;74(20):5690–9. doi: 10.1158/0008-5472.CAN-13-3540. [DOI] [PubMed] [Google Scholar]
- 7.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25(2):197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 8.Keiser MJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–81. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng F, et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput Biol. 2012;8(5):e1002503. doi: 10.1371/journal.pcbi.1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng F, et al. Prediction of chemical-protein interactions network with weighted network-based inference method. PLoS One. 2012;7(7):e41064. doi: 10.1371/journal.pone.0041064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X, Liu MX, Yan GY. Drug-target interaction prediction by random walk on the heterogeneous network. Mol Biosyst. 2012;8(7):1970–8. doi: 10.1039/c2mb00002d. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeman TC, et al. Construction, visualisation, and clustering of transcription networks from microarray expression data. PLoS Comput Biol. 2007;3(10):2032–42. doi: 10.1371/journal.pcbi.0030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim HS, et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell. 2013;155(3):552–66. doi: 10.1016/j.cell.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komurov K, et al. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komurov K, White MA, Ram PT. Use of data-biased random walks on graphs for the retrieval of context-specific networks from genomic data. PLoS Comput Biol. 2010;6(8) doi: 10.1371/journal.pcbi.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yildirim MA, et al. Drug-target network. Nat Biotechnol. 2007;25(10):1119–26. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 18.Bonvini P, et al. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21(4):838–42. doi: 10.1038/sj.leu.2404528. [DOI] [PubMed] [Google Scholar]
- 19.Zheng B, et al. Induction of cell cycle arrest and apoptosis by the proteasome inhibitor PS-341 in Hodgkin disease cell lines is independent of inhibitor of nuclear factor-kappaB mutations or activation of the CD30, CD40, and RANK receptors. Clin Cancer Res. 2004;10(9):3207–15. doi: 10.1158/1078-0432.ccr-03-0494. [DOI] [PubMed] [Google Scholar]
- 20.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6(10):813–23. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 21.Koh JL, et al. COLT-Cancer: functional genetic screening resource for essential genes in human cancer cell lines. Nucleic Acids Res. 2012;40:D957–63. doi: 10.1093/nar/gkr959. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez HL, et al. A phase II trial of pemetrexed in advanced breast cancer: clinical response and association with molecular target expression. Clin Cancer Res. 2006;12(3 Pt 1):832–8. doi: 10.1158/1078-0432.CCR-05-0295. [DOI] [PubMed] [Google Scholar]
- 23.Martin M, et al. Phase II study of pemetrexed in breast cancer patients pretreated with anthracyclines. Ann Oncol. 2003;14(8):1246–52. doi: 10.1093/annonc/mdg339. [DOI] [PubMed] [Google Scholar]
- 24.Miles DW, et al. A phase II study of pemetrexed disodium (LY231514) in patients with locally recurrent or metastatic breast cancer. Eur J Cancer. 2001;37(11):1366–71. doi: 10.1016/s0959-8049(01)00117-4. [DOI] [PubMed] [Google Scholar]
- 25.O’Shaughnessy JA, et al. Phase II study of pemetrexed in patients pretreated with an anthracycline, a taxane, and capecitabine for advanced breast cancer. Clin Breast Cancer. 2005;6(2):143–9. doi: 10.3816/CBC.2005.n.016. [DOI] [PubMed] [Google Scholar]
- 26.Schneeweiss A, et al. A randomized phase II trial of doxorubicin plus pemetrexed followed by docetaxel versus doxorubicin plus cyclophosphamide followed by docetaxel as neoadjuvant treatment of early breast cancer. Ann Oncol. 2011;22(3):609–17. doi: 10.1093/annonc/mdq400. [DOI] [PubMed] [Google Scholar]
- 27.Meng Z, et al. Inhibitor of 5-lipoxygenase, zileuton, suppresses prostate cancer metastasis by upregulating E-cadherin and paxillin. Urology. 2013;82(6):1452e7–14. doi: 10.1016/j.urology.2013.08.060. [DOI] [PubMed] [Google Scholar]
- 28.Ye X, et al. Darbufelone, a novel anti-inflammatory drug, induces growth inhibition of lung cancer cells both in vitro and in vivo. Cancer Chemother Pharmacol. 2010;66(2):277–85. doi: 10.1007/s00280-009-1161-z. [DOI] [PubMed] [Google Scholar]
- 29.Savari S, et al. CysLT(1)R antagonists inhibit tumor growth in a xenograft model of colon cancer. PLoS One. 2013;8(9):e73466. doi: 10.1371/journal.pone.0073466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dressman MA, et al. Gene expression profiling detects gene amplification and differentiates tumor types in breast cancer. Cancer Res. 2003;63(9):2194–9. [PubMed] [Google Scholar]
- 31.Kauraniemi P, et al. Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am J Pathol. 2003;163(5):1979–84. doi: 10.1016/S0002-9440(10)63556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arrell DK, Terzic A. Network systems biology for drug discovery. Clin Pharmacol Ther. 2010;88(1):120–5. doi: 10.1038/clpt.2010.91. [DOI] [PubMed] [Google Scholar]
- 33.Azmi AS, et al. Proof of concept: network and systems biology approaches aid in the discovery of potent anticancer drug combinations. Mol Cancer Ther. 2010;9(12):3137–44. doi: 10.1158/1535-7163.MCT-10-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4(11):682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 35.Leung EL, et al. Network-based drug discovery by integrating systems biology and computational technologies. Brief Bioinform. 2013;14(4):491–505. doi: 10.1093/bib/bbs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pujol A, et al. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol Sci. 2010;31(3):115–23. doi: 10.1016/j.tips.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Xie L, et al. Drug discovery using chemical systems biology: identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput Biol. 2009;5(5):e1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiang AP, Butte AJ. Systematic evaluation of drug-disease relationships to identify leads for novel drug uses. Clin Pharmacol Ther. 2009;86(5):507–10. doi: 10.1038/clpt.2009.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen NT, Brunak S, Altman RB. Generating genome-scale candidate gene lists for pharmacogenomics. Clin Pharmacol Ther. 2009;86(2):183–9. doi: 10.1038/clpt.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iorio F, et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci U S A. 2010;107(33):14621–6. doi: 10.1073/pnas.1000138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, et al. Drug repositioning by kernel-based integration of molecular structure, molecular activity, and phenotype data. PLoS One. 2013;8(11):e78518. doi: 10.1371/journal.pone.0078518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Law V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–7. doi: 10.1093/nar/gkt1068. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, et al. PubChem’s BioAssay Database. Nucleic Acids Res. 2012;40:D400–12. doi: 10.1093/nar/gkr1132. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu T, et al. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–201. doi: 10.1093/nar/gkl999. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mishra GR, et al. Human protein reference database–2006 update. Nucleic Acids Res. 2006;34:D411–4. doi: 10.1093/nar/gkj141. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chatr-aryamontri A, et al. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–4. doi: 10.1093/nar/gkl950. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joshi-Tope G, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–32. doi: 10.1093/nar/gki072. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bader GD, et al. BIND–The Biomolecular Interaction Network Database. Nucleic Acids Res. 2001;29(1):242–5. doi: 10.1093/nar/29.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breitkreutz BJ, et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008;36:D637–40. doi: 10.1093/nar/gkm1001. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cerami EG, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–90. doi: 10.1093/nar/gkq1039. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wingender E, et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28(1):316–9. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griffith OL, et al. ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic Acids Res. 2008;36:D107–13. doi: 10.1093/nar/gkm967. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerstein MB, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberzon A, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wishart DS, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–10. doi: 10.1093/nar/gkn810. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schellenberger J, et al. BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics. 2010;11:213. doi: 10.1186/1471-2105-11-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maglott D, et al. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2007;35:D26–31. doi: 10.1093/nar/gkl993. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lane A, Segura-Cabrera A, Komurov K. A comparative survey of functional footprints of EGFR pathway mutations in human cancers. Oncogene. 2014;33(43):5078–89. doi: 10.1038/onc.2013.452. [DOI] [PubMed] [Google Scholar]
- 62.Komurov K, et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol. 2012;8:596. doi: 10.1038/msb.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.