

Abstract
Disulfide cleavage is one of the major causes underlying ultraviolet (UV) light-induced protein damages. While previous studies have provided strong evidence to support the notion that this process is mediated by photo-induced electron transfer from the excited state of an aromatic residue (e.g., tryptophan) to the disulfide bond, many mechanistic details are still lacking. For example, we do not know how quickly this process occurs in a protein environment. Herein, we design an experiment, which uses the unfolding kinetics of a protein as an observable, to directly assess the kinetics and mechanism of photo-induced disulfide cleavage. Our results show that this disulfide bond cleavage event takes place in ∼2 μs via a mechanism involving electron transfer from the triplet state of a tryptophan (Trp) residue to the disulfide bond. Furthermore, we find that one of the photoproducts of this reaction, a Trp-SR adduct, is formed locally, thus preventing the protein from re-cross-linking. Taken together, these findings suggest that a Trp-disulfide pair could be used as a photo-trigger to initiate protein folding dynamics and to control biological activities of disulfide-containing peptides.
Keywords: Disulfide, Photocleavage, IR spectroscopy, Tryptophan, Protein folding, Photodamage
1. Introduction
The effect of ultraviolet (UV) radiation on protein structure and function has been an area of active research for over a century.1-5 Although UV light is necessary for vitamin D synthesis in the cell6 and offers other therapeutic benefits,7 it can also cause DNA8,9 and protein damage,10-13 and hence, diseases.14,15 For proteins, such photodamage is linked to the strong molar absorptivity of three aromatic amino acids, i.e., tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe), in the UVB region. Among the known protein photodegredation pathways, one arises from disulfide bond cleavage or reduction. For example, the study of Correia et al.16 showed that the UV-induced damage of human insulin, characterized by a decrease in β-sheet and α-helical content and also a reduction in the antibody binding ability and hormonal function, is correlated with an increase in the number of free thiols present in the protein. Additionally, Wang and coworkers17,18 have utilized UV-induced disulfide bond cleavage as a mean to form nanoparticles with free thiol groups that can be modified to load small molecules for drug-delivery. While a disulfide bond can be broken by directly absorbing a UV photon, this mechanism is unlikely the main cause of photoinduced disulfide cleavage in proteins due to the weak absorbance of a disulfide bond around 280 nm (ε = 125 M-1 cm-1).19 Instead, disulfide bond cleavage has been connected to UV excitation of aromatic residues, especially Trp. It is well known that upon photoexcitation, the indole ring of Trp is capable of donating an electron to either the solvent or a nearby acceptor.20 Thus, it has been proposed that the photoinduced disulfide bridge cleavage in proteins is mediated by Trp,21 which acts as an electron donor. The transient absorption experiments of Hayon and coworkers22 on disulfide model systems suggested that the Trp-mediated disulfide cleavage could occur via two mechanisms. The first one, as indicated in Schemes (1) – (4), involves transfer of an electron from the triplet state of Trp (3Trp), generated from its excited singlet state (1Trp*), to the disulfide (RSSR), leading to formation of a disulfide radical anion (RSSR•−), which further reacts to cause the disulfide linkage to break apart:
| (1) |
| (2) |
| (3) |
| (4) |
The second one (Schemes (5) – (6)) involves production of a solvated electron upon photoexcitation of Trp, which then reacts with the disulfide to generate the precursor RSSR•−, as shown below:
| (5) |
| (6) |
While studies on small molecules suggest that both mechanisms are plausible, mechanistic studies on proteins are few and have not provided a complete picture of the individual processes involved in disulfide bond cleavage. For example, to the best of our knowledge the only kinetics study of a Trp-mediated disulfide bond cleavage process in a protein environment is that of Petersen and coworkers.23 By performing a flash photolysis experiment on the protein cutinase, they were able to monitor the formation kinetics and lifetimes of several transient species involved in this process, including those of and RSSR•−. However, due to spectral congestion, they were unable to directly determine every key kinetic event leading to disulfide cleavage nor were they able to confirm the aforementioned mechanisms.
To provide further insight into the kinetics and mechanism of this important biological process, herein we designed and carried out an experiment to directly assess the Trp-mediated cleaving kinetics of a disulfide bond in a protein. The strategy is to induce the protein in question to unfold via photocleavage of a key disulfide cross-linker, allowing the disulfide cleavage kinetics to be assessed by measuring the protein unfolding kinetics. Specifically, we used a 34-residue mini-protein (referred to as Z34C) designed by Wells and coworkers.24 Z34C forms a helix-turn-helix (HTH) structure (Figure 1) that is stabilized by a disulfide bond near the termini.25 Thus, removal of this disulfide cross-linker leads to unfolding of the HTH structure, allowing for direct assessment of its cleavage by monitoring the kinetics of unfolding. Since Z34C does not contain Trp, we inserted this electron donor next to the disulfide bridge through a double mutation with the resulting sequence: FNMQCWRRFY-EALHDPNLNE-EQRNAKIKSI-RDWC (hereafter referred to as Trp-Z34C).
Figure 1.
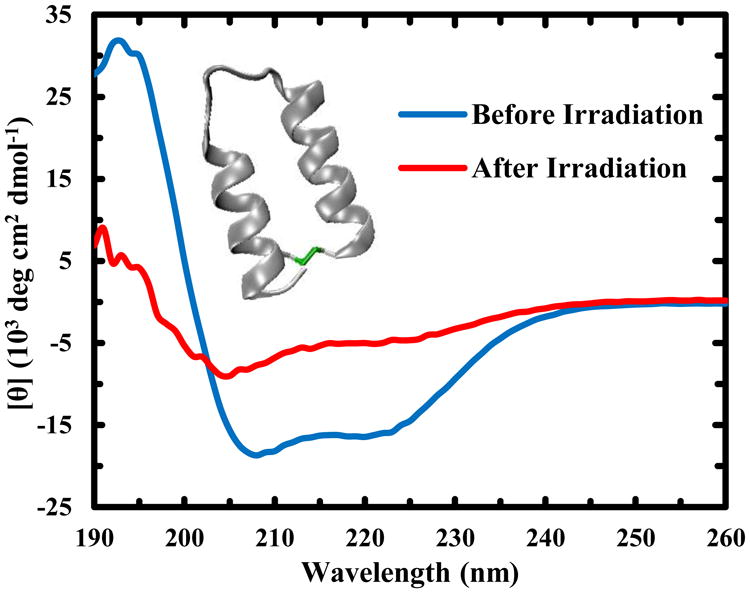
CD spectra of a 67 μM Trp-Z34C sample taken at 25 °C before and after irradiation with 266 nm light for 23 hrs, as indicated. Inset: NMR structure of Z34C (PDB code: 1ZDC), showing the location of the disulfide cross-linker (green).
Photoinduced release of a conformational constraint is a very useful strategy to trigger a protein folding or unfolding event.26 In comparison to other commonly used triggering methods, such as laser-induced temperature-jump (T-jump) techniques,27 the advantage of this strategy is that it offers site-specific control. Thus, an additional goal of this study is to show that the Trp-mediated disulfide cleavage can be used to trigger protein folding/unfolding kinetics,28 while preventing geminate recombination.29-32
Materials and Methods
Sample preparation
The Z34C and Trp-Z34C peptides were synthesized on a Liberty Blue microwave peptide synthesizer (CEM Corporation, NC) and a PS3 automated peptide synthesizer (Protein Technologies, MA), respectively, using Fmoc-protocols. They were then compartment purified by reverse-phase chromatography, and verified by matrix assisted laser desorption ionization (MALDI) mass spectroscopy. For each peptide, the purified sample was dissolved in a 20% DMSO/water solution and stirred overnight, allowing oxidation of the thiol groups to form a disulfide bond. The oxidized sample was subject to a second round of purification and the peptide product was further verified by MALDI. Trifluoroacetic acid (TFA) removal and H-D exchange for IR measurements were achieved by multiple rounds of lyophilization in acidic D2O solution. Peptide samples used in the experiments were prepared by dissolving an appropriate amount of lyophilized peptide in a 20 mM sodium phosphate buffer in D2O and the final pH of the solution was 7.
Irradiation
Irradiation was performed at 25 °C by placing the peptide sample in either a 1 mm quartz cuvette (for CD and UV-Vis measurements) or an IR cell with a 120 μm spacer (for FTIR measurements) in a Fluorolog 3.10 spectrofluorometer (Jobin Yvon Horiba, NJ), and illuminating it with 266 ± 15 nm light derived from the excitation source of the fluorometer for the amount of time reported. The intensity of the irradiation light was ∼2.5 – 5.0 mW cm-2.
Steady-state measurements
CD spectra were obtained on an Aviv 62A DS spectropolarimeter (Aviv Associates, NJ) using a 1 mm sample holder. A 67 μM sample of Trp-Z34C was used, and the sample was irradiated for 23 hrs. UV-Vis spectra were collected on a Lambda 25 UV-Vis spectrometer (Perkin Elmer, MA) using a 1 mm quartz cuvette at room temperature with a peptide concentration of ∼100 μM and an irradiation time of 2 hrs. Fourier transform infrared (FTIR) spectra were collected on a Magna-IR 860 spectrometer (Nicolet, WI) using a two-CaF2 sample cell of 56 μm pathlength with 1 cm-1 resolution and at room temperature.
Time-resolved IR measurements
The transient IR setup is similar to the T-jump IR setup described previously.33 In brief, a Minilite II Nd:YAG laser (Continuum, CA) was used to produce the pump pulse of 0.05 mJ, 3 ns, and 266 nm pulse which was focused to a spot size of ∼60 μm at the sample. A continuous wave (CW) quantum cascade (QC) mid-IR laser (Daylight Solutions, CA) was used as the probe. The peptide sample was held in a sample cell consisting of two CaF2 windows with an optical pathlength of 120 μm. The concentration of the peptide samples used in the time-resolved IR measurements was ∼500 μM, which led to an absorbance of ∼0.1 at 266 nm.
Results and Discussion
As shown (Figure 1), upon irradiation with a low power (∼2.5–5.0 mW cm-2), continuous wave (CW), 266 nm light source (derived from a Xenon arc lamp) for a long period of time, the CD spectrum of Trp-Z34C lost intensity at 208 and 222 nm, both of which are characteristic of α-helical conformations. However, under the same irradiation conditions, wild-type Z34C does not show the same degree of spectral changes (Figure S1). Measurements of the amide I′ (amide I in D2O) band of Trp-Z34C also show that UV irradiation results in a decrease in its α-helical content (Figure S2). As the reduced form (or uncross-linked) of Z34C (and Trp-Z34C) has significantly less α-helical character than its oxidized (or cross-linked) counterpart,25 these results, taken together, not only confirm the notion that UV radiation can break the disulfide cross-linker in Trp-Z34C, but also indicate that the underlying process is mediated by photoexcitation of Trp.
To directly assess the timescale of this disulfide cleavage process, we measured the relaxation kinetics of Trp-Z34C in response to photoexcitation of the Trp residue by a 3 ns, 266 nm laser pulse using transient infrared (IR) spectroscopy.34 As indicated (Figure S2), photocleavage of the disulfide in Trp-Z34C leads to a change in its amide I′ band that is characterized by a loss/gain of absorbance at 1630 cm-1/1664 cm-1. In addition, the folding-unfolding of Z34C has been shown to follow two-state or single-exponential kinetics on the μs timescale.25 Thus, if the UV pump-induced transient signal contained contribution only from the unfolding process of the HTH motif, we would expect to observe a single, negative-going kinetic component on the μs timescale at 1630 cm-1 due to decreased helical content upon unfolding.35 However, the transient kinetics obtained at this frequency show very different behaviors (Figure 2 – blue curve), exhibiting a negative-going spike followed by a positive-going signal evolving on a slower (ns and μs) timescale. In particular, the negative-going component is fully developed within the time resolution of the instrument, and hence, cannot arise from the unfolding kinetics of Trp-Z34C. Since the indole ring of Trp (i.e., the chromophore absorbing the 266 nm light in the current case) does not have any appreciable absorbance between 1600-1700 cm-1 (except a weak C=C band of indole at 1618 cm-1),36 this initial kinetic phase must manifest as a spectral change in the amide I′ band of Trp-Z34C induced by photoexcitation of the Trp residue. This assessment is corroborated by the kinetics obtained at 1664 cm-1 (Figure 2 – red curve), the initial kinetic phase of which is positive-going, as expected for a spectral change that results in a decrease/increase of the absorbance at 1630 cm-1/1664 cm-1. A previous study by Huang et al.37 showed that photoexcitation of a Ru-complex covalently attached to the N-terminus of an α-helical peptide produces a transient IR signal that arises instantaneously in the amide I′ band region of the peptide. They attributed this signal to a Stark effect,38,39 arising from a large difference (∼8 D) between the dipole moments of the ground and excited states of the chromophore. We believe that the transient IR signals in the current case are also caused by a Stark effect, induced by either a dipole moment change or generation of a charged species upon excitation of the Trp chromophore (see below).
Figure 2.

IR transient kinetic traces of Trp-Z34C obtained at two representative probing frequencies, 1630 cm-1 (blue) and 1664 cm-1 (red). The smooth lines are global fits of these traces to a three-exponential function plus an offset with the following time constants: 7 ns, 150 ns, and 2.0 μs. Shown in the inset are the same data with a logarithm time axis.
To further substantiate this assignment, we globally analyzed the kinetic traces obtained at 1630 cm-1 and 1664 cm-1 and found that both curves can be satisfactorily fit by convoluting the instrument response function (IRF) with a three-exponential function (plus an offset) with the following time constants: 7 ns, 150 ns, and 2.0 μs. Because our IRF is about 30 ns, the 7 ns component is unresolved. This component most likely arises from the lowest excited singlet state of Trp (1La), which has a lifetime between 2–4 ns and hence cannot be time-resolved with our current time resolution. More importantly, the dipole moment (μe) of the 1La state is much larger than that (μg) of the ground state (i.e., μe − μg = 3.5 D),20 making it detectable via the aforementioned Stark effect. Because the other two decay components do not exhibit a resolvable rise time, they must originate from two longer-lived species that are not only quickly generated (i.e., within the instrument response time), but also carry a charge or a large dipole moment (thus becoming detectable). The only species that meet these requirements are Trp•+ and RSSR•−, produced via Scheme (3). This assessment is supported by the following reasons. First, 3Trp is formed during the lifetime of 1Trp* or within 2–4 ns.40 Second, the subsequent 3Trp quenching by the nearby disulfide bond is expected to occur between 0.2 ns and 2 ns, estimated based on the reported bimolecular rate constant (i.e., 1.9 × 108 M-1 s-1) for this reaction41 and the effective disulfide concentration (see ESI for details). Thus, combined, these rates lead to rapid (and unresolved) formation of Trp•+ (and RSSR•−). It is worth noting, based on previous studies,42,43 that Trp•+ could also be formed from 1Trp* via an electron transfer process to the disulfide on a picosecond timescale. However, the current study cannot distinguish between these two mechanisms. Third, the time constant (150 ns) of the second kinetic component is comparable to the lifetime of Trp•+ observed in other studies. For example, the study of Aubert et al.44 on the photoinduced electron transfer kinetics in DNA photolyase indicated that a Trp•+ species can live as long as 300 ns. Similarly, a study23 on the photophysics of free Trp in aqueous solution using UV pump and UV/visible probe spectroscopy showed that the spectral signature associated with Trp•+ decays in a multi-exponential manner, and the fastest component has a time constant of 340 ns. Fourth, this same study also indicated that while the spectroscopic signal of RSSR•− for two different proteins exhibits multi-exponential decay kinetics, one of the decay time constants is 1.5–4 μs, which matches that (2.0 μs) of the third kinetic component in the present case. Thus, based on these similarities, we attribute the 150 ns and 2.0 μs components to the decay kinetics of Trp•+ and RSSR•−, respectively. Although Tyr has also been shown to be able promote disulfide bond cleavage,45,46 it is unlikely that in the current case the single Tyr residue in Trp-Z34C is responsible for the observed kinetics because it is several residues away from the disulfide bond and its absorbance at the excitation wavelength is approximately an order of magnitude lower than that of the two Trp residues.
According to Scheme (6), one of the reaction outcomes of RSSR•− is to break up the disulfide bond, which, in the current case, would result in unfolding of the HTH structure. As discussed above, this unfolding process will result in a μs kinetic component that shows a decrease (increase) in absorbance at 1630 cm-1 (1664 cm-1) as a function of time. However, such a kinetic component was not observed. We believe that this is caused by the similarity in the rates of unfolding and disulfide cleavage, and the larger amplitude of the latter makes the former not directly observable. In support of this idea, a previous study25 revealed that the unfolding time of the uncross-linked variant of Z34C is 3.0 ± 0.5 μs at 25 °C, which is indeed very similar to the time constant (∼2.0 μs) of the third kinetic component. To provide more direct proof of this claim, we turned our attention to the offset of the transient kinetics. The basic idea is that if the disulfide bond indeed breaks up in 2.0 μs, leading to formation of an uncross-linked and also unfolded Trp-Z34C, the transient spectrum obtained at a longer delay time (e.g., the offset) should reflect the underlying structural change thus induced. To make this comparison, we collected additional transient kinetic traces at frequencies across the whole amide I′ region (Figure 3). As expected, those traces can also be globally fit by the function used to describe the kinetics obtained at 1630 cm-1 and 1664 cm-1 (Figure S3 and Table S1 in ESI). More importantly, as shown (Figure 4), upon subtracting out the signal from water, due to an increase in temperature caused by part of the photoexcitation energy that is dissipated to the solvent, which is negative and has a linear dependence on frequency in the amide I′ band region (see detail in ESI), the offsets of those data produce a transient spectrum that resembles the difference spectrum (Figure S2 in ESI) generated by the IR spectra of Trp-Z34C obtained before and after irradiation with 266 nm light. Thus, this analysis provides direct support of the notion that the 2.0 μs kinetic component originates from the disulfide bond cleavage.
Figure 3.

IR transient kinetic traces of Trp-Z34C obtained at different probing frequencies, as indicated. For clarity, these traces have been offset.
Figure 4.

Transient IR spectrum of Trp-Z34C generated using the offsets of the decay kinetics at the corresponding probing frequencies. A contribution from the solvent at each frequency, due to pump-induced temperature jump, has been removed (see detail in ESI).
Taken together, our results support a mechanism wherein the precursor that leads to cleavage of a disulfide, RSSR•−, is produced via 3Trp (Scheme (3)) but not via a solvated electron (Scheme (5)). To further validate this point, we compared the UV-induced disulfide cleaving efficiencies of two Trp-Z34C samples, with and without the presence N2O, an efficient electron scavenger,47 using CD spectroscopy as a probe. As shown (Figure S4 in ESI), upon irradiation with 266 nm light for 2 hours, the CD spectra of both samples show the same changes, thus providing strong evidence indicating that RSSR•− is not formed via the solvated electron pathway, i.e., Schemes (5) and (6).
In addition, we carried out an experiment to determine the number of free thiols generated upon UV excitation of Trp-Z34C using Ellman's reagent, which reacts with a free thiol group in a 1:1 ratio to produce 2-nitro-5-thiobenzoate dianion (TNB2-). As shown (Figure S5 in ESI), the absorbance of the TNB2- anion at 412 nm, obtained by reacting Ellman's reagent with a Trp-Z34C sample that had been illuminated with 266 nm light overnight, indicates the presence of at least one thiol in the photoproduct of the disulfide cleavage reaction. This implies that the other thiol radical, RS•, produced in Scheme (6), must undergo a different reaction. As indicated (Figure 5), the UV-Vis spectra of Trp-Z34C obtained before and after irradiation with a 266 nm CW light for 2 hours show an increase in the absorbance at both 320 nm and 280 nm, similar to the changes observed for the formation of a Trp-SR adduct in goat α-lactalbumin upon UV excitation.48 Thus, this result, in conjunction with the fact that photoexcitaion of the Trp residue induces Trp-Z34C to unfold, suggests that this adduct is formed on one side of the peptide. In other words, upon RSSR•− dissociation, the sulfur atom closer to the Trp•+ is converted to a radical (i.e., RS• in Scheme (3)). Further reaction between this radical and Trp•, formed via Trp•+ deprotonation, results in uncross-linking and hence unfolding of Trp-Z34C. However, it is worth noting that over an extended period of UV irradiation (i.e., overnight), the peptide sample showed an increase in light scattering, suggesting the existence of a slower process that leads to peptide aggregation.49
Figure 5.

Absorption spectra of Z34C-Trp before and after irradiation with 266 nm light for 2 hrs, as indicated.
Conclusions
In summary, we have designed and carried out a UV-pump and IR probe experiment on a mini-protein (Trp-Z34C), aiming to provide new insight into the mechanism of Trp-mediated disulfide cleavage in proteins. Our strategy exploits the key stabilizing role of a terminal disulfide cross-linker in this protein and uses the UV-triggered unfolding process to assess the underlying disulfide cleavage kinetics. Our results are consistent with a mechanism wherein the cleavage is initiated by an electron transfer event from the triplet state of the photoexcited Trp residue to the disulfide in question, leading to formation of a reactive disulfide species (i.e., RSSR•−), which further dissociates to break up the disulfide bond on a timescale of 2–3 μs. Furthermore, we find that one of the photoproducts of this reaction, a Trp-SR adduct, is formed locally, thus does not yield a new cross-linker. Because a disulfide can be easily incorporated into a polypeptide sequence, our finding suggests that it is possible to use a Trp and disulfide pair to site-specifically control the starting point of a folding or unfolding process. Another potential application is to use UV light to control biological functions as disulfides are commonly found in naturally occurring and designed peptides, such as antimicrobial peptides,50 that perform a wide range of biological activities.
Supplementary Material
Highlight.
A method was devised to directly assess the kinetics and mechanism of tryptophan-mediated disulfide reduction in a protein environment.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM-065978). R.M.A. is an NSF Graduate Research Fellow (DGE-1321851). The transient IR experiments were performed on instruments that were developed under an NIH Research Resource Grant (P41-GM104605).
Footnotes
Supporting Information: Electronic Supplementary Information (ESI) available: Calculation of the triplet state quenching rate, determination of the solvent contribution to the transient IR kinetics due to pump-induced temperature increase, and additional experimental details and data. See DOI: XXX
References
- 1.Bovie WT. Science. 1913;37:373–375. doi: 10.1126/science.37.949.373-c. [DOI] [PubMed] [Google Scholar]
- 2.Bernhart FW. J Biol Chem. 1939;128:289–295. [Google Scholar]
- 3.Setlow R. Radiat Res Suppl. 1960;2:276–289. [PubMed] [Google Scholar]
- 4.Baden HP, Pearlman C. J Invest Dermatol. 1964;43:71–75. [PubMed] [Google Scholar]
- 5.Pattison DI, Davies MJ. Cancer: Cell Structures, Carcinogens and Genomic Instability. 2006:131–157. [Google Scholar]
- 6.Okano T, Mizuno K, Kobayashi T. J Nutr Sci Vitaminol (Tokyo) 1978;24:511–518. doi: 10.3177/jnsv.24.511. [DOI] [PubMed] [Google Scholar]
- 7.Green C, Diffey BL, Hawk JLM. Phys Med Biol. 1992;37:1–20. doi: 10.1088/0031-9155/37/1/001. [DOI] [PubMed] [Google Scholar]
- 8.Sinha RP, Häder DP. Photochem Photobiol Sci. 2002;1:225–236. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 9.Kao YT, Saxena C, Wang L, Sancar A, Zhong D. Proc Natl Acad Sci U S A. 2005;102:16128–16132. doi: 10.1073/pnas.0506586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zigman S, Reddan J, Schultz JB, McDaniel T. Photochem Photobiol. 1996;63:818–824. doi: 10.1111/j.1751-1097.1996.tb09637.x. [DOI] [PubMed] [Google Scholar]
- 11.Bhattacharya D, Basu S, Mandal PC. J Photochem Photobiol B Biol. 2000;59:54–63. doi: 10.1016/s1011-1344(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 12.Permyakov EA, Permyakov SE, Deikus GY, Morozova-Roche LA, Grishchenko VM, Kalinichenko LP, Uversky VN. Proteins. 2003;51:498–503. doi: 10.1002/prot.10371. [DOI] [PubMed] [Google Scholar]
- 13.Xie J, Qin M, Cao Y, Wang W. Proteins. 2011;79:2505–2516. doi: 10.1002/prot.23074. [DOI] [PubMed] [Google Scholar]
- 14.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. Proc Natl Acad Sci. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zigman S. Photochem Photobiol. 1977;26:437–441. doi: 10.1111/j.1751-1097.1977.tb07511.x. [DOI] [PubMed] [Google Scholar]
- 16.Correia M, Neves-Petersen MT, Jeppesen PB, Gregersen S, Petersen SB. PLoS One. 2012;7:e50733. doi: 10.1371/journal.pone.0050733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie J, Cao Y, Xia M, Gao X, Qin M, Wei J, Wang W. Adv Healthc Mater. 2013;2:795–799. doi: 10.1002/adhm.201200285. [DOI] [PubMed] [Google Scholar]
- 18.Xie J, Li Y, Cao Y, Xu C, Xia M, Qin M, Wei J, Wang W. Biomater Sci. 2013;1:1216. doi: 10.1039/c3bm60174a. [DOI] [PubMed] [Google Scholar]
- 19.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Creed D. Photochem Photobiol. 1984;39:537–562. [Google Scholar]
- 21.Dose K. Photochem Photobiol. 1968;8:331–335. doi: 10.1111/j.1751-1097.1968.tb05829.x. [DOI] [PubMed] [Google Scholar]
- 22.Bent DV, Hayon E. J Am Chem Soc. 1975;97:2612–2619. doi: 10.1021/ja00843a004. [DOI] [PubMed] [Google Scholar]
- 23.Neves-Petersen MT, Klitgaard S, Pascher T, Skovsen E, Polivka T, Yartsev A, Sundström V, Petersen SB. Biophys J. 2009;97:211–226. doi: 10.1016/j.bpj.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starovasnik MA, Braisted AC, Wells JA. Proc Natl Acad Sci. 1997;94:10080–10085. doi: 10.1073/pnas.94.19.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du D, Gai F. Biochemistry. 2006;45:13131–13139. doi: 10.1021/bi0615745. [DOI] [PubMed] [Google Scholar]
- 26.Markiewicz BN, Culik RM, Gai F. Sci China Chem. 2014;57:1615–1624. doi: 10.1007/s11426-014-5225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips CM, Mizutani Y, Hochstrasser RM. Proc Natl Acad Sci U S A. 1995;92:7292–7296. doi: 10.1073/pnas.92.16.7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waegele MM. PhD Thesis. University of Pennsylvania; 2011. [Google Scholar]
- 29.Lu HSM, Volk M, Kholodenko Y, Gooding E, Hochstrasser RM, DeGrado WF. J Am Chem Soc. 1997;119:7173–7180. [Google Scholar]
- 30.Kolano C, Helbing J, Kozinski M, Sander W, Hamm P. Nature. 2006;444:469–472. doi: 10.1038/nature05352. [DOI] [PubMed] [Google Scholar]
- 31.Kolano C, Helbing J, Bucher G, Sander W, Hamm P. J Phys Chem B. 2007;111:11297–11302. doi: 10.1021/jp074184g. [DOI] [PubMed] [Google Scholar]
- 32.Milanesi L, Waltho JP, Hunter CA, Shaw DJ, Beddard GS, Reid GD, Dev S, Volk M. Proc Natl Acad Sci U S A. 2012;109:19563–19568. doi: 10.1073/pnas.1211764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang CY, Klemke JW, Getahun Z, DeGrado WF, Gai F. J Am Chem Soc. 2001;123:9235–9238. doi: 10.1021/ja0158814. [DOI] [PubMed] [Google Scholar]
- 34.Serrano AL, Waegele MM, Gai F. Protein Sci. 2012;21:157–170. doi: 10.1002/pro.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyer RB, Gai F, Woodruff WH, Gilmanshin R, Callender RH. Acc Chem Res. 1998;31:709–716. [Google Scholar]
- 36.Barth A. Prog Biophys Mol Biol. 2000;74:141–173. doi: 10.1016/s0079-6107(00)00021-3. [DOI] [PubMed] [Google Scholar]
- 37.Huang CY, He S, DeGrado WF, McCafferty DG, Gai F. J Am Chem Soc. 2002;124:12674–12675. doi: 10.1021/ja028084u. [DOI] [PubMed] [Google Scholar]
- 38.Volk M, Kholodenko Y, Lu HSM, Gooding EA, DeGrado WF, Hochstrasser RM. J Phys Chem B. 1997;101:8607–8616. [Google Scholar]
- 39.Oh KI, Fiorin G, Gai F. Chem phys chem. 2015;16:3595–3598. doi: 10.1002/cphc.201500777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szabo AG, Rayner DM. J Am Chem Soc. 1980;102:554–563. [Google Scholar]
- 41.Lapidus LJ, Eaton WA, Hofrichter J. Proc Natl Acad Sci. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Barkley MD. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 43.Qiu W, Li T, Zhang L, Yang Y, Kao YT, Wang L, Zhong D. Chem Phys. 2008;350:154–164. [Google Scholar]
- 44.Aubert C, Vos MH, Mathis P, Eker AP, Brettel K. Nature. 2000;405:586–590. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 45.Bent DV, Hayon E. J Am Chem Soc. 1975;97:2599–2606. doi: 10.1021/ja00843a002. [DOI] [PubMed] [Google Scholar]
- 46.Xie J, Wang H, Cao Y, Qin M, Wang W. Ann Phys (N Y) 2015;358:225–235. [Google Scholar]
- 47.Janata E, Schuler RH. J Phys Chem. 1982;86:2078–2084. [Google Scholar]
- 48.Vanhooren A, Devreese B, Vanhee K, Van Beeumen J, Hanssens I. Biochemistry. 2002;41:11035–11043. doi: 10.1021/bi0258851. [DOI] [PubMed] [Google Scholar]
- 49.Xie J, Qin M, Cao Y, Wang W. Proteins. 2011;79:2505–2516. doi: 10.1002/prot.23074. [DOI] [PubMed] [Google Scholar]
- 50.Lehrer R, Lichtenstein A, Ganz T. Annu Rev Immunol. 1993;11:105–128. doi: 10.1146/annurev.iy.11.040193.000541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.