

Abstract
Molecular advances support the existence of an alternative pathway of colorectal carcinogenesis that is based on the hypermethylation of specific DNA regions that silences tumor suppressor genes. This alternative pathway has been called the serrated pathway due to the serrated appearance of tumors in histological analysis. New classifications for colorectal cancer (CRC) were proposed recently based on genetic profiles that show four types of molecular alterations: BRAF gene mutations, KRAS gene mutations, microsatellite instability, and hypermethylation of CpG islands. This review summarizes what is known about the serrated pathway of CRC, including CRC molecular and clinical features, prognosis, and response to chemotherapy.
Keywords: Colorectal cancer, Methylator phenotype, Serrated pathway, Chemotherapy, CIMP
Core tip: Recently, the implication among colorectal cancers with methylator phenotype has burst into the gastroenterology literature. In this review, we analyze the correlation between serrated cancers, the methylator phenotype and other genetic features in order to assess their prognosis and response to adjuvant chemotherapy.
INTRODUCTION
Colorectal cancer (CRC) is considered a major health issue: it is the most prevalent cancer and the second largest cause of cancer death in Western countries[1]. Traditionally, colorectal carcinogenesis research has focused on the chromosomal instability (CIN) pathway, in which the APC gene mutation is the first pathogenic event that leads to allelic losses and to somatic gene amplification and translocation. This classical carcinogenetic model is responsible for about 70%-80% of all CRC cases[2-5]. A second carcinogenic pathway was described in the last decades of the 20th century. This pathway is related to inactivation of the mismatch repair (MMR) gene system, which in turn leads to inactivation of mutated tumor suppressor genes, and is called MMR or MSI pathway. Lynch syndrome is the paradigm of this alternative carcinogenetic model; this syndrome leads to diploid tumors that have a microsatellite instability (MSI) phenotype[6].
Lastly, molecular advances have identified a third pathway of colorectal carcinogenesis. This pathway does not cause changes at the chromosomal level or in the MMR system; rather, it involves hypermethylation of specific DNA regions near the promoter genes: the CpG islands. This alternative pathway is called the serrated pathway due to the serrated appearance of tumors in histological analysis. The main molecular feature of these tumors is the variable degree of methylation in promoter gene regions[7]. Like MSI tumors, serrated cancers are diploid tumors, and some genetic variability has been described for tumors caused by the serrated pathway. New classifications were proposed recently for CRC based on genetic profiles that show four types of molecular alterations: BRAF gene mutations, KRAS gene mutations, MSI, and hypermethylation of CpG islands[8,9].
The aim of this review is to summarize what is known about the serrated pathway and CRC, including CRC molecular and clinical features, prognosis, and response to chemotherapy (CT).
SERRATED LESIONS
Serrated polyps constitute a heterogeneous group of lesions that include four types of polyps: sessile serrated adenomas (SSAs), traditional serrated adenomas (TSAs), mixed polyps (MPs), and hyperplastic polyps (HPs)[10]. HPs are the most prevalent serrated lesion type, accounting for around 80%-90% of serrated polyps. HPs are subdivided into three subtypes: microvesicular HPs (MVHPs), goblet cell HPs (GCHPs), and mucin-poor HPs (MPHPs)[11]. Although only a minority of HPs will progress to CRC, mainly when they are right-sided, there can be progression to other serrated lesions that can evolve into CRC[11]. MVHPs are more likely to localize to the right colon and may progress more frequently to SSA. Conversely, when GCHPs evolve into other lesions, they are more likely to be left-sided TSAs (Figure 1)[12].
Figure 1.
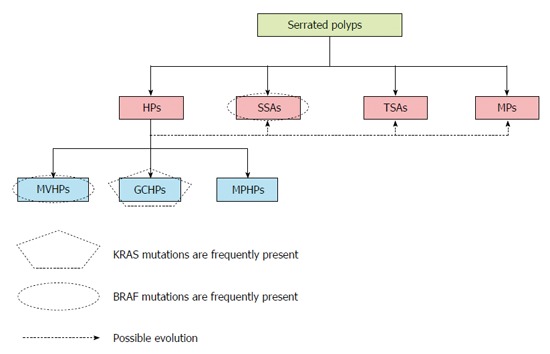
Types of serrated polyps. Serrated polyps can be divided into four types: hyperplastic polyps (HPs), sessile serrated adenomas (SSAs), traditional serrated adenomas, and mixed polyps (MPs). The polyps can also be genetically characterized according to their CIMP, BRAF, KRAS, and MSI status, and certain patterns are found frequently for each category of polyp. Both microvesicular hyperplastic polyps (MVHPs), which are an HP subset, and SSAs have BRAF mutations more often than KRAS mutations. Conversely, goblet cell hyperplastic polyps (GCHPs), another HP subset, more frequently show KRAS mutations. BRAF mutations often correlate with high-grade CIMP CRC, whereas KRAS-mutated serrated polyps are more commonly found in low-grade CIMP. MPHP: Mucin-poor hyperplastic polyp.
SSAs are less common than HPs, accounting for about 10%-25% of serrated polyps. Located mainly in the right colon, they can be original tumors or can evolve from HPs[13]. TSAs account for about 1%-2% of serrated polyps and are more frequent on the left colon[13]. Finally, MPs constitute around 1%-4% of serrated polyps[14]. Mixed tumors usually comprise a dysplastic lesion (TSA or conventional adenoma) plus a non-dysplastic one, usually an HP or SSA[15].
GENETIC AND MOLECULAR FEATURES
The serrated pathway is complex and is currently poorly understood. This pathway has two key characteristics, namely aberrant hypermethylation of certain promoter regions in the genome and alterations in MAP kinase signaling pathway genes.
Aberrant hypermethylation
Functions related to the human genome are tightly regulated, and enzymatic systems control processes such as DNA mismatch repair, DNA transcription and replication. DNA transcription is the result of the regulation of genomic expression. Specifically, the transcription machinery interacts with the start codon ATG via DNA methyltransferases and forms methyl cytosine[16]. In humans and other mammals, only cytosine residues that precede a guanosine in the DNA sequence are modified to form CpG dinucleotides. These can change the three-dimensional configuration of the DNA and consequently affect its interaction with transcription factors. Methylation of these CpG dinucleotides usually leads to genetic silencing. CpG dinucleotides are located throughout the DNA sequence, and approximately half of all human gene promoter sequences are embedded in these CpG clusters, which are termed CpG islands[17]. The CpG island sequence is at least 200 bases long and is usually > 500 bases; the CG content is > 50%, and the ratio of observed-to-expected CpGs is > 60%[18,19]. In the genomes of cells in healthy tissue, the CpG islands are usually unmethylated, especially those associated with gene promoters. Conversely, about 80% of the CpG dinucleotides that are not part of CpG islands (i.e., that are in DNA non-coding regions) are heavily methylated to prevent the expression of viral sequences that are integrated into the genome as well as the expression of many other DNA elements[20]. On the one hand, hypomethylation of these CpG dinucleotides could result in harmful gene expression; on the other hand, methylation of the promoter regions could have undesirable adverse effects.
Notably, some methylated promoters do not play any role in tumor development-these genes with methylated promoters are called “methylated in tumor” or MINT genes[16]. However, other methylated CpG islands in promoter regions silence the expression of known tumor suppressor genes by interrupting the interactions between transcription factors and the start codon. When this happens, it is considered a new phenotype of CRC that is caused mainly by epigenetic alterations rather than by DNA mutations and is called the CpG island methylator phenotype or CIMP. This term was introduced in 1999 when it was first suggested that CRC could be initiated by genetic silencing in the absence of DNA sequence modifications[21]. There are two types of CIMPs that are closely related and that are sometimes difficult to distinguish. Whereas CIMP-A is more related to aging, CIMP-C is more related to cancer[22,23]. In addition, some authors have suggested different ways to categorize CIMPs[24]. Shen et al[25] defined three subgroups of CIMP CRCs: CIMP1, CIMP2, and CIMP-negative. KRAS mutations are reported to be the strongest predictor for CIMP2 (92%). Later, Ogino et al[26]. Proposed the use of 8 markers rather than 5 to classify methylation. They defined CRCs as CIMP-0 when none of the markers were methylated; as CIMP-low (CIMP-L) when 1 to 5 markers were methylated; and as CIMP high (CIMP-H) when 6-8 markers were methylated. The new cluster of methylated markers proposed by Weisenberger suggests that CIMP-L tumors might have high levels of methylation at other unknown loci rather than at a just a few loci. In fact, more CIMP CRCs were identified when the test panel of methylated markers was expanded[7]. Although the Weisenberg panel of loci is currently used more often than other panels, it is not yet clear which markers are the most appropriate for establishing the CIMP status of tumors.
The prevalence of CIMP tumors varies in different types of serrated lesions. CIMP-H is present in around 41%-73.3% of MVHPs but only in about 8%-18.2% of GCHPs[27]. The proportion of CIMP-H in evolved serrated lesions is similar: the proportion is about 44%-76.8% in TSAs and about 80% in SSAs[28]. Nonetheless, low grade CIMP is more often associated with TSAs than with SSAs (Figures 2 and 3)[13,27-32].
Figure 2.
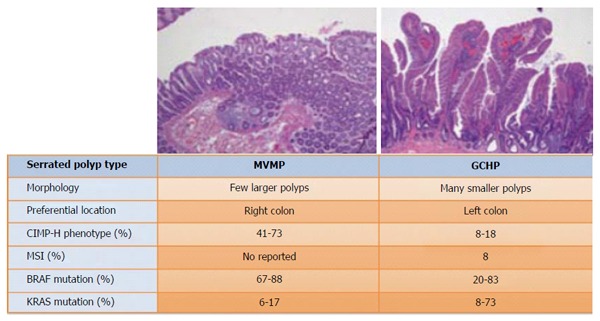
Features of microvesicular hyperplastic polyps and goblet cell hyperplastic polyps. Microvesicular hyperplastic polyps (MVHPs) and goblet cell hyperplastic polyps (GCHPs) are premalignant lesions that differ mainly in their location and morphology. MVHPs are more likely to occur as a few large polyps in the ascendant colon, and GCHPs are more likely to occur as multiple small polyps in the left colon. Each can become senescent or evolve into another type of polyp. Although they can transform into other adenomas, MVHPs more often evolve into sessile serrated adenomas (SSAs), and GCHPs more often evolve into traditional serrated adenomas (TSAs). The prevalence of KRAS mutations is higher in TSAs, while BRAF mutations are more prevalent in SSAs.
Figure 3.
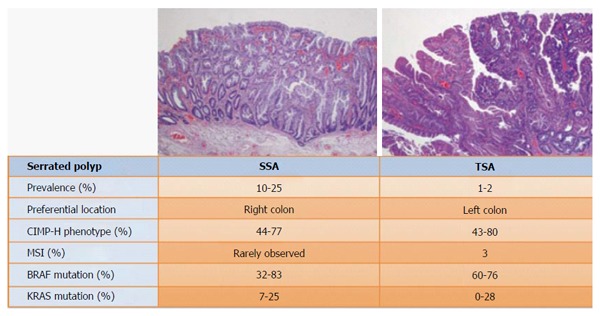
Features of sessile serrated adenoma and traditional serrated adenomas. Pathological analysis shows that sessile serrated adenoma (SSAs) are more common than traditional serrated adenomas (TSAs). While TSA is preferentially located on the left colon, SSA is preferentially located on the right side. In addition, both SSAs and TSAs are found in high-grade CIMP tumors, although several studies show a relationship between low-grade CIMP tumors and TSAs. BRAF mutations are often observed in SSA, whereas KRAS mutations are often observed in TSAs. Contrary to what is found in serrated adenocarcinomas, MSI is almost never present in serrated polyps.
The MAPK pathway
When the serrated pathway was first described, investigators considered it a unique and linear carcinogenesis pathway. However, the current view is that the early events in this pathway include both an alteration of the MAPK pathway plus concomitant DNA epigenetic alterations[4]. The MAP kinase pathway is a via of intercellular signaling transmission. Mutations in their constituent proteins mainly involve the Raf and RAS families (Figure 4). There are 3 types of Raf kinases, termed types A, B, and C. B-Raf (or BRAF) is located on 7p34, is involved in serrated CRC, and is mutated in approximately 10% of CRC cases[33]. A BRAF mutation is rare in CIN tumors, and its presence almost completely excludes a Lynch syndrome diagnosis[34,35]. Therefore BRAF can be considered to be specific to CRCs arising via the serrated pathway[7,33,35]. The BRAF V600E mutation, in which valine is substituted for glutamate at codon 600 on chromosome 7[36], is the most common and the best characterized mutation. V600E leads to constitutive gene activation, thereby inducing cell proliferation and inhibiting apoptosis.
Figure 4.
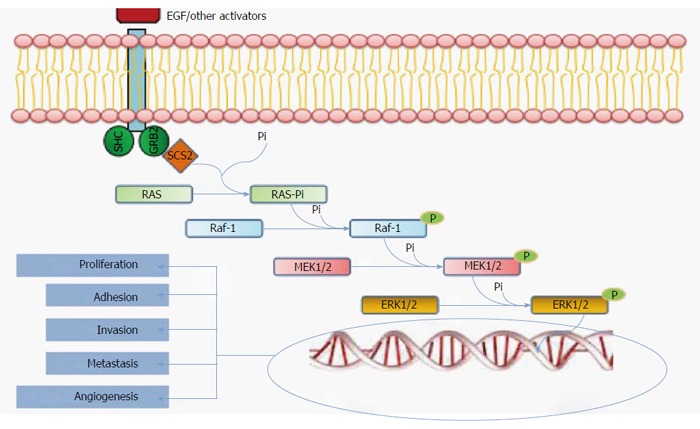
MAPK pathway. This simplified diagram of the MAPK pathway shows the RAS/Raf-1/MEK/ERK pathway. RAS mutations are found in 36% of serrated polyps, and Raf-1 mutations are found in 9%-11%. These mutations promote gene transcription and cellular growth that results in cellular adhesion, invasion, metastasis, and angiogenesis.
RAS mutations can also lead to dysfunctional MAPK pathway signaling. There are at least three RAS genes, namely H-RAS, N-RAS, and K-RAS or KRAS[37]. The consequences of KRAS gene mutations are similar to those of BRAF mutations in that the mutations can induce proliferation and inhibit apoptosis. Classically, RAS mutation has been linked to the CIN pathway, but RAS is also impaired in some serrated cancers. KRAS mutations are found in 30%-40% of CRCs[38,39]. Activating KRAS mutations are most common (up to 80%) in codon 12 but are also found in codon 13; these include the G12D, G12V, and G13D KRAS point mutations[40]. Notably, the combination of KRAS mutation plus low grade CIMP in many serrated lesions constitutes an alternative subset of CRC that is established via the serrated pathway. For example, an increase in MGMT methylation rather than MLH1 methylation is associated with CIMP-L and KRAS mutations[41,42].
Serrated pathway
In the serrated pathway, BRAF or KRAS mutation initially induces a burst of cellular proliferation in the normal colorectal epithelium, and serrated aberrant hyperplastic crypt foci seem to be the earliest histological lesions[43]. Mutations in both genes, although more frequently mutations in BRAF, can lead to upregulation of p16INK4a and to an increase in insulin-like growth factor binding protein 7 (IGFBP7) secretion at aberrant crypt foci[44]. p16INK4a and IGFBP7 are tumor suppressor proteins that prevent polyp growth and that drive these proliferative cells to form small senescent lesions. The silencing of these genes, for example by methylation, allows the cells to escape from cellular senescence and permits the progression to MVHPs or GCHPs and then to serrated polyps[37]. Some mRNAs have also been linked to serrated carcinogenesis. One example is microRNA-31, located at 9p21.3, which seems to correlate with mutated BRAF tumors. MicroRNA-31 may be involved in the progression from HPs to SSAs, since it has been detected in a high proportion of these lesions as well as in the CRCs that evolved from them[45,46].
Some genes are inhibited in the pathway to malignancy in CRC. Among them, the methylguanine methyltransferase gene, MGMT, and the MLH1 gene are the best characterized. MGMT is frequently involved in the progression of TSA[42]. While MLH1 silencing is linked to SSA evolution. MLH1 is one of the main genes in the MMR system, and its inactivation leads to an accumulation of mutations in microsatellite sequences. This results in an MSI phenotype in these tumors, which is a hallmark of CRC in Lynch syndrome. However, only 3% of all CRCs and 20% of MSI CRCs are due to Lynch syndrome[18]. The remaining 80% of MSI CRCs are considered sporadic CRCs that are caused by the epigenetic inactivation of MLH1, which is present in approximately 15% of all CRCs[47]. These tumors follow the serrated pathway of carcinogenesis, especially when CIMP is present[24,48]. In fact, most of the sporadic MSI CRCs are CIMP tumors, although only half of CIMP CRCs show methylation of the MMR genes (Figure 5)[49,50]. “the natural course from serrated polyps to advanced cancers can be followed in this figure”.
Figure 5.
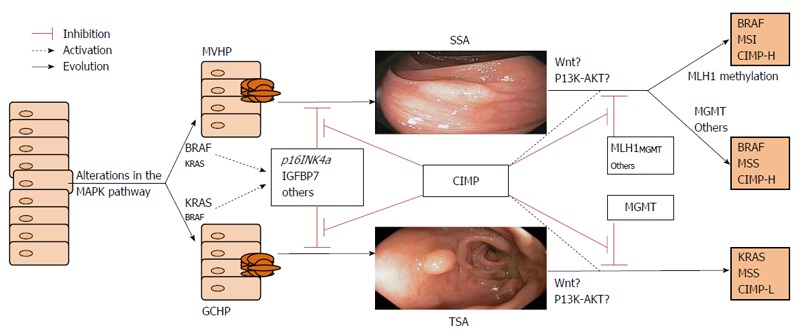
Serrated pathway. This diagram shows the chronological changes that occur during the development of neoplasia. MAPK mutations do not affect just one gene; rather, they affect some genes more than others. Gene methylation is successive rather than simultaneous and contributes to the progression of the serrated lesion. Depending on which genes are most affected, serrated cancers can be classified into three subtypes. GCHP: Goblet-cell hyperplastic polyp; MVHP: Microvesicular hyperplastic polyp; SSA: Sessile serrated adenoma; TSA: Traditional serrated adenoma.
CLINICAL FEATURES
The serrated pathway leads to a broad spectrum of CRCs. It is important to view each CRC as the result of both genetic and epigenetic alterations. In a review of the literature, serrated adenocarcinoma was frequently located on the cecum (52% of the time) and constituted around of 16% of all proximal CRCs[51-53]. Serrated adenocarcinoma is more common in Caucasians than in Hispanics or African Americans[54]. Some studies have suggested that there is an association between specific genetic profiles and certain clinical characteristics. Based on this premise, several studies have reported the clinical profiles of serrated adenocarcinoma cases[8,9].
Some studies distinguish between serrated adenocarcinoma with vs without mutant BRAF. Serrated adenocarcinoma with mutant BRAF seems to be more frequent in older patients who are heavy smokers and in female patients[55]. It is more prevalent on the right colon in larger tumors that are usually diagnosed at an advanced stage (either pT4 or N2)[56,57]. In fact, in the right colon, there seems to be a close relationship between these features and the loss of expression of the CDX2 gene and increased levels of annexin A10[58,59]. In addition, serrated adenocarcinoma cases show a low frequency of liver and lung involvement at the metastatic stage, but there is a higher prevalence of peritoneal involvement at the onset of metastasis[55,60]. From a pathological viewpoint, BRAF-mutated serrated adenocarcinomas are more frequently high grade tumors than other serrated adenocarcinomas[61].
Mutated KRAS tumors, represent a subset of CRCs that arise via the serrated pathway. Clinically, these tumors are more variable than tumors with mutated BRAF, probably due to the difficulties of differentiating mutated KRAS tumors that arise via the serrated pathway from those that arise via the CIN pathway. KRAS mutated serrated tumors are not linked with proximal location, and they are associated with higher body weight, higher body fat percentage, and with female sex[62]. Although one study found a higher prevalence in men[63]. KRAS-mutated serrated adenocarcinomas frequently show both low grade tumor differentiation and low grade CIMP.
The clinical features of CIMP and MSI tumors frequently overlap. CIMP tumors with MSS have not been described in great detail, but one study showed that they are associated with proximal CRC location, female sex, and the presence of lymph node metastasis and that they tend to present with liver metastasis (Figure 6)[64].
Figure 6.
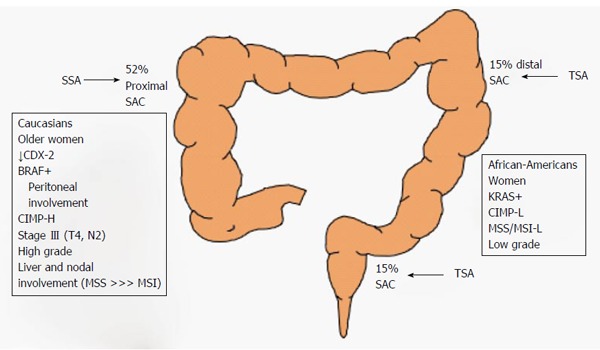
Polyp features according to location. Half of serrated adenocarcinomas (SACs) are located on the right colon. KRAS-mutated polyps are preferentially left-sided. However, polyps with BRAF mutations dominate those found on the right side of the colon. TSA: Traditional serrated adenoma; SSA: Sessile serrated adenoma.
PROGNOSIS
Until now, the prognosis and management of CRCs has been based on the TNM staging system. However, the genetic and molecular profiles of the different CRC types should be used to improve the classification and management of these tumors. Few studies have addressed the question of the prognoses of the different CRC subtypes.
The BRAF mutation was the first biomarker to be investigated. It has been described as a marker of poor survival, due to its association with remodeling of the extracellular matrix, a process that is important for tumoral invasion and metastasis. Notably, its prognostic role was investigated in a meta-analysis of 26 CRC studies[65]. In general, the presence of BRAF mutation is an independent marker of a poor prognosis and is related to a decrease in 5-year disease-free survival (DFS)[66]. Its predictive value remains after stratification by age, disease stage, and degree of differentiation. However, other studies found that mutant BRAF does not affect the intrinsically good prognosis of MSI CRC. For example, Popovici et al[67] found BRAF to be a marker of poor prognosis only in subpopulations with microsatellite-stable left-sided tumors. Similarly, others report worse prognosis in MSS cancers in men but not in women[68]. As noted above, there seems to be a strong relationship between the CIMP status and BRAF mutations, with the prognostic value of CIMP status being related to the presence of BRAF mutations and, less frequently, to the presence of KRAS mutations[69]. Finally, regulation of SOX2 gene expression and its role as a biomarker of poor survival has been linked to tumors with BRAF mutations. This may be related to cellular migration and invasion, since an increase in SOX2 expression also correlates with CRC liver metastasis[70].
In contrast to BRAF, the MSI status is closely related to good prognosis in CRC, including in serrated tumors. The predictive power of MSI is independent of CIMP, BRAF, and KRAS status. However, it is clear that MSS tumors are associated with better DFS at any stage when no CIMP, KRAS, or BRAF mutations are found; in such cases, DFS is similar to that in familial or sporadic MSI-CRC[9] Some clinical trials have reported poor outcomes in patients with CIMP-H MSS CRC. Comparing the two MMR states, stage II and III CRC with MSI has a better prognosis than MSS CRC[47]. Moreover, combined MSS and CIMP CRC with mutant BRAF or KRAS frequently correlates with liver metastasis at diagnosis and has the worst prognosis of all CRCs[8,9,64]. The exactly role of CIMP status in metastatic stage has not been elucidated. It’s possible that the worst prognosis of those tumors could be due to the presence of BRAS mutations as well.
The prognostic value of KRAS mutations has also been investigated. KRAS mutation is proposed to be a predictor of lower 5-year DFS in almost all studies, without any prognostic differences between right- and left-sided CRCs. However, some point mutations have been described that impact prognosis. For example, a KRAS codon 13 mutant is associated with more aggressive CRC[68].
The prognostic value of CIMP status remains controversial. There are few doubts about the relatively good prognosis of sporadic CIMP-H MSI CRC. But for sporadic MSS CRC, some studies show no effect of CIMP-H on survival prediction, while others show either a negative or a positive impact[70]. Ogino et al[71] described CIMP-H as a specific marker of low CRC mortality, and it appeared to counter the adverse prognostic effect of BRAF mutation in serrated cancers. Perhaps, the hypermethylation of certain genes avoided the proliferative effect of BRAF mutations. One clinical trial found it to be an independent predictor of cancer survival[72]. Despite this finding, many researchers consider CIMP a predictor of short survival, especially in proximal stage III CRCs[69,73]. These authors discussed some possible reasons for these controversial findings, namely differences in cohorts, the use of different panels of methylated markers, the use of different criteria to define CIMP, and confounding factors such as BRAF mutation[71]. In a recent meta-analysis of CIMP prognostic value, both DFS and overall survival (OS) were shorter in patients with CIMP CRC after adjusting for age, sex, disease stage, and treatment; it was not possible to adjust for BRAF or KRAS mutation or MMR status since not all the studies assessed these possible confounders)[70]. It seems likely that when a CIMP serrated tumor shows MSI, the MSI acts to confer a good prognosis[71].
Recent studies have revealed a link between some methylated genes and patient prognosis. MLH1 and MGMT methylation have been suggested to be predictors of good prognosis, in part due to the frequent absence of BRAF mutations in these subgroups of tumors. In contrast, hypermethylation of the p14 (CDKN2AINK4a), RASSF1A, and p16 (CDKN2AARF) genes have been suggested to predict poorer prognosis independent of both stage and histological differentiation grade[74-76]. Aberrant methylation of genes encoding proteins involved in extracellular matrix remodeling also confers a worse prognosis, as does hypomethylation of LINE-1 in sporadic MSI CRC[77]. Hypomethylation of the IGF-2 gene and RAC1b overexpression are proposed markers of poor prognosis in KRAS/BRAF wild-type metastatic CRCs treated with FOLFOX as first-line therapy[78]. Finally, a mutated P1K3CA gene is associated with a worse survival rate in CRC with wild-type BRAF. Alteration of P1K3CA is found in 10%-20% of cases with CIMP-H. It is currently unknown whether P1K3CA mutation defines a new subset of CRC[79].
Genetic variability can strongly influence patient outcome, highlighting the need for a new or more fine-tuned CRC classification system. Recently, two publications established 5 subtypes of CRC and described differences in survival according to the subtypes’ phenotypic characteristics. First, based on the classification system of Jass[80], Phipps et al[8] divided CRCs into MSI-H and MSS/MSI-L CRC. Later, this group further divided cases according to CIMP status and then by BRAF and KRAS mutation status. The final system describes 5 well-defined CRC subtypes (Table 1). Among the subtypes, the highest survival was found for subtype 5, which is characterized by non-CIMP MSI CRC with wild-type BRAF and KRAS; this is also known as familial MSI CRC or Lynch syndrome. Conversely, the worst prognosis was found for subtype 2, which is CIMP-positive MSS/MSI-L CRC with BRAF mutations and wild-type KRAS. Subtype 2 has the lowest probability of being diagnosed as stage I disease and shows the worst 5-year DFS. Interestingly, subtypes 1 and 2 are very similar, with the MSI status of subtype 1 being the only difference between them.
Table 1.
Proposed[80] colorectal cancer classification
| MMR | CIMP | BRAF | KRAS | |
| Phipps | ||||
| Subtype 1 | MSI-H | + | + | - |
| Subtype 2 | MSS or MSI-L | + | + | - |
| Subtype 3 | MSS or MSI-L | - | - | + |
| Subtype 4 | MSS or MSI-L | - | - | - |
| Subtype 5 | MSI-H | - | - | - |
| Sinicrope | ||||
| Subtype 1 | MSI | + | + | +/- |
| Subtype 2 | MSS | +/- | + | - |
| Subtype 3 | MSS | +/- | - | + |
| Subtype 4 | MSS | +/- | - | - |
| Subtype 5 | MSI | - | - | +/- |
Sinicrope et al[9] proposed a similar classification system based on patients with stage III CRC. This group also divided CRC into 5 subgroups, each of which was compared to subtype 4 (MSS CRC with wild-type BRAF and KRAS, representing the classic CIN pathway). The subtypes with MSI have the best prognosis, followed by subtype 5 (which is identical to the Phipps subtype 5) and subtype 1. Regardless of BRAF mutation status, MSI confers relatively good DFS in subtype 1 CRC. In contrast, subtypes 2 and 3 do not differ in terms of prognosis, but their prognosis differs from that of subtype 4. As in Phipps’ classification, subtype 2 (CIMP-H MSS CRC with wild-type KRAS and mutant BRAF) shows the worst survival rate. KRAS mutation alone, with no other apparent genetic alterations, is the major alteration in subtype 3, representing an alternative subset of CRC that arises via the serrated pathway. Its prognosis is 1.5-times worse than that of subtype 4 (Table 1).
These findings illustrate the importance of classification based on genetic profiling and could improve our understanding of patient prognosis and disease management.
RESPONSE TO CHEMOTHERAPY
The current trend is towards offering treatment that is individualized according to CRC subtype. The most recent advances in CRC are leading to novel approaches and classification schemes, and it is attractive to think that a particular therapy can be used to target a particular type of CRC, including serrated tumors. Unfortunately, CT is not yet fully guided by the combination of genetic alterations in a CRC, and even the most recent studies have not assessed specific therapeutic management strategies for each subtype. Additional clinical trials are needed to address this. As of now, decisions must be based on studies that evaluated the responses according to individual markers (Table 2).
Table 2.
Summary of the treatment implications of genetic alterations in colorectal cancer
| Molecular marker | Implications for CRC treatment |
| KRAS mutation | Broadly studied in metastatic CRC |
| The most predictive biomarker for no response to anti-EGFR, either alone or with CT | |
| Worse OS when oxaliplatin is the first-line treatment | |
| Irinotecan efficacy is controversial; it may have better effects in stage II and III CRC | |
| BRAF mutation | Overall, no predictive power for CT response |
| No predictive power for response to 5-FU plus irinotecan/oxaliplatin or to 5-FU alone in stage II disease | |
| A trend toward better survival with 5-FU plus irinotecan in stage III disease | |
| Effects of anti-EGFR therapy are controversial, although most studies show a poor response | |
| Some studies show no differences in OS/DFS with FOLFOX-panitumumab or with FOLFIRI-cetuximab treatment | |
| Resistance to BRAF inhibitors | |
| CIMP | CT results are controversial |
| 5-FU improves DFS and OS in some studies; in others, survival is reduced | |
| One study shows the benefits of 5-FU plus irinotecan in CIMP tumors after stratification by MMR status. CIMP was more strongly associated than MMR status with a better response to irinotecan | |
| The use of 5-FU in CIMP tumors is not currently recommended | |
| To date, no clinical trials have evaluated the response of CIMP tumors to anti-EGFR therapy | |
| MMR | Prognosis is intrinsically better for MSI CRC, but MSS tumors show a better response to CT |
| 5-FU improves both DFS and OS in stage II and III MSS CRC but not in MSI CRC | |
| CT should only be given for stage II MMS tumors if a high risk factor such as T4 local extension is present | |
| MSI CRC shows a good response to irinotecan if BAX expression has been lost |
CT: Chemotherapy; DFS: Disease-free survival; anti-EGFR: Epidermal growth factor receptor antibodies; OS: Overall survival; CRC: Colorectal cancer; MMR: Mismatch repair.
KRAS mutations have mainly been studied in metastatic CRCs. In the QUASAR study, KRAS was not a negative predictive factor for the response to standard CT based on 5-Fluoruracil (5-FU) and folinic-acid[81]. However, the major characteristic of mutant KRAS remains its resistance to anti-EGFR monoclonal antibodies: it is the most important predictive marker in terms of the response to panitumumab and cetuximab, either alone or in combination with CT. This finding, which was confirmed in a meta-analysis[82], is valid for CRC with any inactivating KRAS mutation. It is also possible that the presence of KRAS mutations plus other alterations in NRAS, exons 3 and 4, PIK3CA, PTEN, and BRAF may influence CT resistance. In fact, Tian et al[83] found that a significant response to anti-EGFR was possible when KRAS, BRAF, and PIK3CA were all wild-type, but not when any one of them was mutated. It is currently standard practice to investigate KRAS status in order to predict the response to anti-EGFR in metastatic CRC.
BRAF mutation has also been widely studied, but its role as a predictive marker remains unclear. BRAF has repeatedly been described as a non-predictive biomarker in CRC treated using standard CT. When CRCs with V600EBRAF were treated with 5-FU plus irinotecan or oxaliplatin, BRAF did not correlate with either a positive or negative response[84]. Nonetheless, there was a slightly non-significant trend toward better survival in stage III CRC when irinotecan was added to a 5-FU/leucovorin regimen[85]. Notably, regarding the response to other drugs, both a null predictive value and a poor response to anti-EGFR have been reported for CRC with mutant BRAF[65]. BRAF did not show predictive power in the OPUS or CRYSTAL trials, which used FOLFIRI ± cetuximab[86,87]. The PRIME study reported similar results with FOLFOX4 ± panitumumab treatment[88]. Regarding the poor response, Di Nicolantonio et al[89] found that KRAS-/BRAF+ CRC showed a 0% response rate to anti-EGFR in second or subsequent lines of treatment; in contrast, the response rate was 32% in KRAS-/BRAF- CRC. Loupakis et al[90] reported a similar response rate with irinotecan plus cetuximab. A meta-analysis that included these last two studies as well as other studies comparing metastatic CRC with mutated vs wild-type BRAF concluded that the response to anti-EGFR could be considered poor in tumors with mutant BRAF[91]. However, other studies did not find differences in the responses of CRCs with BRAF mutation vs wild-type BRAF after treatment with cetuximab, capecitabine, oxaliplatin, and bevacizumab[92,93]. Thus, to summarize, the use of BRAF mutation status as a predictive marker of the response to EGFR-targeted treatment remains controversial.
BRAF inhibitors such as vemurafenib and dabrafenib show good efficacy in melanoma. Silencing of V600EBRAF correlates with an increase in epithelial differentiation, in CDX-2 and claudin-1 expression[94,95], and in cellular adhesion. However, experiments in cell lines demonstrate that CRCs are intrinsically resistant to BRAF inhibition. This resistance may be dependent or independent of the ERK MAPK signaling pathway. Activating PIK3CA and AKT1 mutations and the loss of PTEN function have been proposed as possible mechanisms that underlie this resistance[82,96,97]. CRAF may also be involved. Recently, better growth inhibition was observed when BRAF inhibitors were used in combination with anti-EGFR monoclonal antibodies. Treatment with an anti-EGFR agent (like cetuximab) or a tyrosine-kinase inhibitor (such as erlotinib or gefitinib) plus a BRAF inhibitor results in sustained MAPK pathway suppression[98,99]. Different combinations of vemurafenib, erlotinib, capecitabine and/or bevacizumab, and cetuximab and/or irinotecan have shown better responses than vemurafenib alone[100]. Moreover, using vemurafenib plus anti-EGFR could improve clinical efficacy[101]. In fact, a pilot trial with 15 patients is ongoing, and the results look promising[102]. Simultaneous inhibition of the BRAF and PIK3 cascades shows a trend towards tumor regression in both mouse and human cell cultures. Ongoing studies are assessing triple combination therapy (i.e., inhibition of BRAF, EGFR, and PIK3CA).
A different question regarding serrated CRCs concerns the response of CIMP tumors to the different CT options. To date, several studies have reported conflicting results. The hypothesis that CIMP cancers inhibit gamma-glutamyl hydrolase, thus enhancing intracellular folate levels and modulating a better response to 5-FU, suggested that these CRCs would benefit from adjuvant CT regimens[103-107]. Juo et al[70] published the first meta-analysis to address this matter. The meta-analysis identified 7 studies that evaluated the response of serrated CRC to CT as adjuvant therapy after surgical resection. Some of the studies described the benefits of 5-FU in both stage II and III CRC. Van Rijnsoever et al[108] found higher DFS in patients treated with 5-FU compared with patients that received surgical resection alone, as did Donada et al[109] who looked at stage II CRC. However, Jover et al[110] described a significant response to 5-FU in terms of DFS in stage II and III non-CIMP CRCs but not in CIMP CRCs. Two studies found that CIMP status had non-significant predictive value in terms of 5-FU treatment. Kim et al[111] compared a FOLFIRI regimen in non-CIMP vs CIMP metastatic CRC, while Han et al[112] investigated patients with stage II and III disease treated with a FOLFOX combination. A total of 4 studies reported that CT was beneficial in these patients, whereas 2 found non-significant results and 1 concluded the opposite. In a study that was not part of the meta-analysis, Wang et al[113] also found worse DFS rates when 5-FU was given to CIMP-positive tumors, in agreement with the findings of Jover’s study. Shiovitz et al[114] also analyzed OS with irinotecan therapy. They compared 5-FU plus leucovorin with and without irinotecan in stage III CRCs and found a non-significant trend towards better survival with the triple combination. In addition, patients with CIMP tumors seemed to benefit more after the tumors were stratified according to MSI status. It could be due to the better prognosis associated to unstable tumors, but further explanations regarding this are needed. Moreover, in this study, CIMP status was more strongly associated with the response to irinotecan than was MMR status.
These conflicting results may be due in part to differences in definitions, gene panels, CIMP marker thresholds, and laboratory techniques used to assess CIMP status. Currently, the National Comprehensive Cancer Network (NCCN) treatment guidelines for colon cancer (version 3) do not recommend the use of CT for stage II (T2N0M0) CIMP cancers[115]. Because of these controversial results, the predictive value of CIMP status regarding treatment with 5-FU remains unclear, and more randomized clinical trials are needed.
Finally, the role of MMR status in predicting the response to CT has been studied extensively[116,117]. Several studies have established that MSI tumors have an intrinsically better prognosis than MSS tumors. Jover et al[118] analyzed OS and DFS in a cohort of 505 patients with stage II or III CRC according to their MMR status after receiving 5-FU. MSS CRCs showed improved DFS and OS, but no improvement was seen in MSI CRCs[102,119]. These differences remained after multivariate analysis controlled for the TNM stage, age, and sex. A prospective study by Sargent et al[119] had similar findings and suggested that cancers with MSI do not benefit from 5-FU adjuvant CT. However, the decision to treat stage II MSS or MSI-L cancers should be based on high risk factors such as T4 tumor status, perforation, and obstruction, among others. The response to irinotecan was also assessed in MSI CRC. There was a better response when the tumor had lost BAX expression, and the author proposed that this was by far the best criterion for predicting efficacy[120]. Current management may include the addition of irinotecan or oxaliplatin, but more studies are needed to evaluate different combinations of CT for MSI CRC.
FUTURE DIRECTIONS
This review enhances the current change of mind about CRC. The concept of a unique model of carcinogenesis is obsolete and the consideration of pathogenesis of CRC as only three possible pathways is changing. In the recent years, a lot of publications about several aspects, from molecular and genetic discoveries to pathologic classifications, have seen the light. Current trends seem to guide to new approaches of CRC in many aspects. Great disparity in clinical trials in prognosis and response to treatment has taken place among last years. In fact, it seems to have more than twenty-five genetic alterations to date, and increasingly. Some authors have understood the problem and are making new classifications basing on prognostic implications. Even, it’s possible that CRCs following the serrated pathway are divided into more genotypes in the near future. The complete understanding of molecular pathways is the first necessary step.
Many efforts are necessary to get many consensuses, like about the definition and method of assessing CIMP phenotype and the possibility of treating CRCs with mutant BRAF when no lymph nodes are found. Randomized clinical trials evaluating response to different CTs, drugs as EGFR-ab and other biologic treatments should take place to clarify their paper on serrated cancers. After that, assessing response to each subtype, it shall be possible to establish a more individualized prognosis and therapy to each patient.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest relevant to this article were reported.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: November 12, 2015
First decision: November 27, 2015
Article in press: January 30, 2016
P- Reviewer: Daniel F, HuangYB, Kodaz H, Nishida T, Pellicano R, Uppara M S- Editor: Qi Y L- Editor: A E- Editor: Wang CH
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Cottrell S, Bicknell D, Kaklamanis L, Bodmer WF. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet. 1992;340:626–630. doi: 10.1016/0140-6736(92)92169-g. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 6.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 7.Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138:2088–2100. doi: 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 8.Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148:77–87.e2. doi: 10.1053/j.gastro.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sinicrope FA, Shi Q, Smyrk TC, Thibodeau SN, Dienstmann R, Guinney J, Bot BM, Tejpar S, Delorenzi M, Goldberg RM, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haque T, Greene KG, Crockett SD. Serrated neoplasia of the colon: what do we really know? Curr Gastroenterol Rep. 2014;16:380. doi: 10.1007/s11894-014-0380-6. [DOI] [PubMed] [Google Scholar]
- 11.Yamane L, Scapulatempo-Neto C, Reis RM, Guimarães DP. Serrated pathway in colorectal carcinogenesis. World J Gastroenterol. 2014;20:2634–2640. doi: 10.3748/wjg.v20.i10.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawkins NJ, Bariol C, Ward RL. The serrated neoplasia pathway. Pathology. 2002;34:548–555. [PubMed] [Google Scholar]
- 13.Spring KJ, Zhao ZZ, Karamatic R, Walsh MD, Whitehall VL, Pike T, Simms LA, Young J, James M, Montgomery GW, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology. 2006;131:1400–1407. doi: 10.1053/j.gastro.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 14.Carr NJ, Mahajan H, Tan KL, Hawkins NJ, Ward RL. Serrated and non-serrated polyps of the colorectum: their prevalence in an unselected case series and correlation of BRAF mutation analysis with the diagnosis of sessile serrated adenoma. J Clin Pathol. 2009;62:516–518. doi: 10.1136/jcp.2008.061960. [DOI] [PubMed] [Google Scholar]
- 15.Tadepalli US, Feihel D, Miller KM, Itzkowitz SH, Freedman JS, Kornacki S, Cohen LB, Bamji ND, Bodian CA, Aisenberg J. A morphologic analysis of sessile serrated polyps observed during routine colonoscopy (with video) Gastrointest Endosc. 2011;74:1360–1368. doi: 10.1016/j.gie.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 17.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 18.Goel A, Boland CR. Epigenetics of colorectal cancer. Gastroenterology. 2012;143:1442–1460.e1. doi: 10.1053/j.gastro.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011;8:686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 21.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahuja N, Issa JP. Aging, methylation and cancer. Histol Histopathol. 2000;15:835–842. doi: 10.14670/HH-15.835. [DOI] [PubMed] [Google Scholar]
- 23.Worthley DL, Whitehall VL, Buttenshaw RL, Irahara N, Greco SA, Ramsnes I, Mallitt KA, Le Leu RK, Winter J, Hu Y, et al. DNA methylation within the normal colorectal mucosa is associated with pathway-specific predisposition to cancer. Oncogene. 2010;29:1653–1662. doi: 10.1038/onc.2009.449. [DOI] [PubMed] [Google Scholar]
- 24.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 25.Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogino S, Cantor M, Kawasaki T, Brahmandam M, Kirkner GJ, Weisenberger DJ, Campan M, Laird PW, Loda M, Fuchs CS. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–1006. doi: 10.1136/gut.2005.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang S, Farraye FA, Mack C, Posnik O, O’Brien MJ. BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol. 2004;28:1452–1459. doi: 10.1097/01.pas.0000141404.56839.6a. [DOI] [PubMed] [Google Scholar]
- 28.Kim KM, Lee EJ, Ha S, Kang SY, Jang KT, Park CK, Kim JY, Kim YH, Chang DK, Odze RD. Molecular features of colorectal hyperplastic polyps and sessile serrated adenoma/polyps from Korea. Am J Surg Pathol. 2011;35:1274–1286. doi: 10.1097/PAS.0b013e318224cd2e. [DOI] [PubMed] [Google Scholar]
- 29.Kim YH, Kakar S, Cun L, Deng G, Kim YS. Distinct CpG island methylation profiles and BRAF mutation status in serrated and adenomatous colorectal polyps. Int J Cancer. 2008;123:2587–2593. doi: 10.1002/ijc.23840. [DOI] [PubMed] [Google Scholar]
- 30.Konishi K, Yamochi T, Makino R, Kaneko K, Yamamoto T, Nozawa H, Katagiri A, Ito H, Nakayama K, Ota H, et al. Molecular differences between sporadic serrated and conventional colorectal adenomas. Clin Cancer Res. 2004;10:3082–3090. doi: 10.1158/1078-0432.ccr-03-0334. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien MJ, Yang S, Mack C, Xu H, Huang CS, Mulcahy E, Amorosino M, Farraye FA. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol. 2006;30:1491–1501. doi: 10.1097/01.pas.0000213313.36306.85. [DOI] [PubMed] [Google Scholar]
- 32.Sandmeier D, Benhattar J, Martin P, Bouzourene H. Serrated polyps of the large intestine: a molecular study comparing sessile serrated adenomas and hyperplastic polyps. Histopathology. 2009;55:206–213. doi: 10.1111/j.1365-2559.2009.03356.x. [DOI] [PubMed] [Google Scholar]
- 33.Kambara T, Simms LA, Whitehall VL, Spring KJ, Wynter CV, Walsh MD, Barker MA, Arnold S, McGivern A, Matsubara N, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53:1137–1144. doi: 10.1136/gut.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, Truta B, Sleisenger MH, Kim YS. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:191–195. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]
- 35.McGivern A, Wynter CV, Whitehall VL, Kambara T, Spring KJ, Walsh MD, Barker MA, Arnold S, Simms LA, Leggett BA, et al. Promoter hypermethylation frequency and BRAF mutations distinguish hereditary non-polyposis colon cancer from sporadic MSI-H colon cancer. Fam Cancer. 2004;3:101–107. doi: 10.1023/B:FAME.0000039861.30651.c8. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki H, Igarashi S, Nojima M, Maruyama R, Yamamoto E, Kai M, Akashi H, Watanabe Y, Yamamoto H, Sasaki Y, et al. IGFBP7 is a p53-responsive gene specifically silenced in colorectal cancer with CpG island methylator phenotype. Carcinogenesis. 2010;31:342–349. doi: 10.1093/carcin/bgp179. [DOI] [PubMed] [Google Scholar]
- 37.Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–684. doi: 10.1093/jnci/90.9.675. [DOI] [PubMed] [Google Scholar]
- 38.Ogino S, Meyerhardt JA, Irahara N, Niedzwiecki D, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, Hantel A, et al. KRAS mutation in stage III colon cancer and clinical outcome following intergroup trial CALGB 89803. Clin Cancer Res. 2009;15:7322–7329. doi: 10.1158/1078-0432.CCR-09-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, Garner RC, Steele RJ, Wolf CR. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci USA. 2002;99:9433–9438. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 41.Huang CS, Farraye FA, Yang S, O’Brien MJ. The clinical significance of serrated polyps. Am J Gastroenterol. 2011;106:229–240; quiz 241. doi: 10.1038/ajg.2010.429. [DOI] [PubMed] [Google Scholar]
- 42.Whitehall VL, Walsh MD, Young J, Leggett BA, Jass JR. Methylation of O-6-methylguanine DNA methyltransferase characterizes a subset of colorectal cancer with low-level DNA microsatellite instability. Cancer Res. 2001;61:827–830. [PubMed] [Google Scholar]
- 43.Rosenberg DW, Yang S, Pleau DC, Greenspan EJ, Stevens RG, Rajan TV, Heinen CD, Levine J, Zhou Y, O’Brien MJ. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer Res. 2007;67:3551–3554. doi: 10.1158/0008-5472.CAN-07-0343. [DOI] [PubMed] [Google Scholar]
- 44.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 45.Aoki H, Nosho K, Igarashi H, Ito M, Mitsuhashi K, Naito T, Yamamoto E, Tanuma T, Nomura M, Maguchi H, et al. MicroRNA-31 expression in colorectal serrated pathway progression. World J Gastroenterol. 2014;20:12346–12349. doi: 10.3748/wjg.v20.i34.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nosho K, Igarashi H, Nojima M, Ito M, Maruyama R, Yoshii S, Naito T, Sukawa Y, Mikami M, Sumioka W, et al. Association of microRNA-31 with BRAF mutation, colorectal cancer survival and serrated pathway. Carcinogenesis. 2014;35:776–783. doi: 10.1093/carcin/bgt374. [DOI] [PubMed] [Google Scholar]
- 47.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 48.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 49.Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, O’Connor T, Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–1387. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 50.Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 51.Mäkinen MJ, George SM, Jernvall P, Mäkelä J, Vihko P, Karttunen TJ. Colorectal carcinoma associated with serrated adenoma--prevalence, histological features, and prognosis. J Pathol. 2001;193:286–294. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH800>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 52.Patai AV, Molnár B, Tulassay Z, Sipos F. Serrated pathway: alternative route to colorectal cancer. World J Gastroenterol. 2013;19:607–615. doi: 10.3748/wjg.v19.i5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tuppurainen K, Mäkinen JM, Junttila O, Liakka A, Kyllönen AP, Tuominen H, Karttunen TJ, Mäkinen MJ. Morphology and microsatellite instability in sporadic serrated and non-serrated colorectal cancer. J Pathol. 2005;207:285–294. doi: 10.1002/path.1850. [DOI] [PubMed] [Google Scholar]
- 54.Wallace K, Grau MV, Ahnen D, Snover DC, Robertson DJ, Mahnke D, Gui J, Barry EL, Summers RW, McKeown-Eyssen G, et al. The association of lifestyle and dietary factors with the risk for serrated polyps of the colorectum. Cancer Epidemiol Biomarkers Prev. 2009;18:2310–2317. doi: 10.1158/1055-9965.EPI-09-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coppedè F, Lopomo A, Spisni R, Migliore L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J Gastroenterol. 2014;20:943–956. doi: 10.3748/wjg.v20.i4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clancy C, Burke JP, Kalady MF, Coffey JC. BRAF mutation is associated with distinct clinicopathological characteristics in colorectal cancer: a systematic review and meta-analysis. Colorectal Dis. 2013;15:e711–e718. doi: 10.1111/codi.12427. [DOI] [PubMed] [Google Scholar]
- 57.Kalady MF, Dejulius KL, Sanchez JA, Jarrar A, Liu X, Manilich E, Skacel M, Church JM. BRAF mutations in colorectal cancer are associated with distinct clinical characteristics and worse prognosis. Dis Colon Rectum. 2012;55:128–133. doi: 10.1097/DCR.0b013e31823c08b3. [DOI] [PubMed] [Google Scholar]
- 58.Dawson H, Galván JA, Helbling M, Muller DE, Karamitopoulou E, Koelzer VH, Economou M, Hammer C, Lugli A, Zlobec I. Possible role of Cdx2 in the serrated pathway of colorectal cancer characterized by BRAF mutation, high-level CpG Island methylator phenotype and mismatch repair-deficiency. Int J Cancer. 2014;134:2342–2351. doi: 10.1002/ijc.28564. [DOI] [PubMed] [Google Scholar]
- 59.Zlobec I, Bihl MP, Schwarb H, Terracciano L, Lugli A. Clinicopathological and protein characterization of BRAF- and K-RAS-mutated colorectal cancer and implications for prognosis. Int J Cancer. 2010;127:367–380. doi: 10.1002/ijc.25042. [DOI] [PubMed] [Google Scholar]
- 60.Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH, Agarwal A, Maru DM, Sieber O, Desai J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yokota T, Ura T, Shibata N, Takahari D, Shitara K, Nomura M, Kondo C, Mizota A, Utsunomiya S, Muro K, et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br J Cancer. 2011;104:856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brändstedt J, Wangefjord S, Nodin B, Eberhard J, Sundström M, Manjer J, Jirström K. Associations of anthropometric factors with KRAS and BRAF mutation status of primary colorectal cancer in men and women: a cohort study. PLoS One. 2014;9:e98964. doi: 10.1371/journal.pone.0098964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn. 2006;8:582–588. doi: 10.2353/jmoldx.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee S, Cho NY, Choi M, Yoo EJ, Kim JH, Kang GH. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol Int. 2008;58:104–113. doi: 10.1111/j.1440-1827.2007.02197.x. [DOI] [PubMed] [Google Scholar]
- 65.Thiel A, Ristimäki A. Toward a Molecular Classification of Colorectal Cancer: The Role of BRAF. Front Oncol. 2013;3:281. doi: 10.3389/fonc.2013.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phipps AI, Buchanan DD, Makar KW, Burnett-Hartman AN, Coghill AE, Passarelli MN, Baron JA, Ahnen DJ, Win AK, Potter JD, et al. BRAF mutation status and survival after colorectal cancer diagnosis according to patient and tumor characteristics. Cancer Epidemiol Biomarkers Prev. 2012;21:1792–1798. doi: 10.1158/1055-9965.EPI-12-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Popovici V, Budinska E, Bosman FT, Tejpar S, Roth AD, Delorenzi M. Context-dependent interpretation of the prognostic value of BRAF and KRAS mutations in colorectal cancer. BMC Cancer. 2013;13:439. doi: 10.1186/1471-2407-13-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wangefjord S, Sundström M, Zendehrokh N, Lindquist KE, Nodin B, Jirström K, Eberhard J. Sex differences in the prognostic significance of KRAS codons 12 and 13, and BRAF mutations in colorectal cancer: a cohort study. Biol Sex Differ. 2013;4:17. doi: 10.1186/2042-6410-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lundberg IV, Löfgren Burström A, Edin S, Eklöf V, Öberg Å, Stenling R, Palmqvist R, Wikberg ML. SOX2 expression is regulated by BRAF and contributes to poor patient prognosis in colorectal cancer. PLoS One. 2014;9:e101957. doi: 10.1371/journal.pone.0101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, Yu T, Easwaran H, Baylin S, van Engeland M, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25:2314–2327. doi: 10.1093/annonc/mdu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, Giovannucci EL, Fuchs CS. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC, Kim NK, Issa JP. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2011;117:1847–1854. doi: 10.1002/cncr.25737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA, Zali MR. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol Bed Bench. 2013;6:120–128. [PMC free article] [PubMed] [Google Scholar]
- 74.Jensen LH, Rasmussen AA, Byriel L, Kuramochi H, Crüger DG, Lindebjerg J, Danenberg PV, Jakobsen A, Danenberg K. Regulation of MLH1 mRNA and protein expression by promoter methylation in primary colorectal cancer: a descriptive and prognostic cancer marker study. Cell Oncol (Dordr) 2013;36:411–419. doi: 10.1007/s13402-013-0148-2. [DOI] [PubMed] [Google Scholar]
- 75.Nilsson TK, Löf-Öhlin ZM, Sun XF. DNA methylation of the p14ARF, RASSF1A and APC1A genes as an independent prognostic factor in colorectal cancer patients. Int J Oncol. 2013;42:127–133. doi: 10.3892/ijo.2012.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xing X, Cai W, Shi H, Wang Y, Li M, Jiao J, Chen M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: a meta-analysis. Br J Cancer. 2013;108:2542–2548. doi: 10.1038/bjc.2013.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Fuchs CS. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2008;122:2767–2773. doi: 10.1002/ijc.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alonso-Espinaco V, Cuatrecasas M, Alonso V, Escudero P, Marmol M, Horndler C, Ortego J, Gallego R, Codony-Servat J, Garcia-Albeniz X, et al. RAC1b overexpression correlates with poor prognosis in KRAS/BRAF WT metastatic colorectal cancer patients treated with first-line FOLFOX/XELOX chemotherapy. Eur J Cancer. 2014;50:1973–1981. doi: 10.1016/j.ejca.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 79.Rosty C, Young JP, Walsh MD, Clendenning M, Sanderson K, Walters RJ, Parry S, Jenkins MA, Win AK, Southey MC, et al. PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PLoS One. 2013;8:e65479. doi: 10.1371/journal.pone.0065479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 81.Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, Richman S, Chambers P, Seymour M, Kerr D, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–1270. doi: 10.1200/JCO.2010.30.1366. [DOI] [PubMed] [Google Scholar]
- 82.Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014;53:852–864. doi: 10.3109/0284186X.2014.895036. [DOI] [PubMed] [Google Scholar]
- 83.Tian S, Simon I, Moreno V, Roepman P, Tabernero J, Snel M, van’t Veer L, Salazar R, Bernards R, Capella G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut. 2013;62:540–549. doi: 10.1136/gutjnl-2012-302423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, Taylor G, Barrett JH, Quirke P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–5937. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- 85.Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, Saltz LB, Mayer RJ, Schaefer P, Whittom R, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clin Cancer Res. 2012;18:890–900. doi: 10.1158/1078-0432.CCR-11-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, Celik I, Köhne CH. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–1475. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 87.Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 88.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25:1346–1355. doi: 10.1093/annonc/mdu141. [DOI] [PubMed] [Google Scholar]
- 89.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 90.Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–721. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yuan ZX, Wang XY, Qin QY, Chen DF, Zhong QH, Wang L, Wang JP. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: a meta-analysis. PLoS One. 2013;8:e65995. doi: 10.1371/journal.pone.0065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–99. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 93.Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink-Börger ME, van Cleef PH, van Krieken JH, Punt CJ, Nagtegaal ID. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer. 2010;46:1997–2009. doi: 10.1016/j.ejca.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 94.Caruso M, Fung KY, Moore J, Brierley GV, Cosgrove LJ, Thomas M, Cheetham G, Brook E, Fraser LM, Tin T, et al. Claudin-1 Expression Is Elevated in Colorectal Cancer Precursor Lesions Harboring the BRAF V600E Mutation. Transl Oncol. 2014;7:456–463. doi: 10.1016/j.tranon.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Herr R, Köhler M, Andrlová H, Weinberg F, Möller Y, Halbach S, Lutz L, Mastroianni J, Klose M, Bittermann N, et al. B-Raf inhibitors induce epithelial differentiation in BRAF-mutant colorectal cancer cells. Cancer Res. 2015;75:216–229. doi: 10.1158/0008-5472.CAN-13-3686. [DOI] [PubMed] [Google Scholar]
- 96.Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, Wang WV, Vecchione L, de Vriendt V, Weinstein BJ, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res. 2013;19:2688–2698. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R, Dayyani F, Gopal YN, Jiang ZQ, Wistuba II, Tang XM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 100.Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, Schostack K, Simcox ME, Kopetz S, Heimbrook D, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72:779–789. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 101.Tie J, Desai J. Targeting BRAF mutant metastatic colorectal cancer: clinical implications and emerging therapeutic strategies. Target Oncol. 2015;10:179–188. doi: 10.1007/s11523-014-0330-0. [DOI] [PubMed] [Google Scholar]
- 102.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iacopetta B, Kawakami K, Watanabe T. Predicting clinical outcome of 5-fluorouracil-based chemotherapy for colon cancer patients: is the CpG island methylator phenotype the 5-fluorouracil-responsive subgroup? Int J Clin Oncol. 2008;13:498–503. doi: 10.1007/s10147-008-0854-3. [DOI] [PubMed] [Google Scholar]
- 104.Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Watanabe G, Iacopetta B. The folate pool in colorectal cancers is associated with DNA hypermethylation and with a polymorphism in methylenetetrahydrofolate reductase. Clin Cancer Res. 2003;9:5860–5865. [PubMed] [Google Scholar]
- 105.Min BH, Bae JM, Lee EJ, Yu HS, Kim YH, Chang DK, Kim HC, Park CK, Lee SH, Kim KM, et al. The CpG island methylator phenotype may confer a survival benefit in patients with stage II or III colorectal carcinomas receiving fluoropyrimidine-based adjuvant chemotherapy. BMC Cancer. 2011;11:344. doi: 10.1186/1471-2407-11-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Raghunathan K, Priest DG. Modulation of fluorouracil antitumor activity by folic acid in a murine model system. Biochem Pharmacol. 1999;58:835–839. doi: 10.1016/s0006-2952(99)00157-4. [DOI] [PubMed] [Google Scholar]
- 107.Sakamoto E, Tsukioka S, Oie S, Kobunai T, Tsujimoto H, Sakamoto K, Okayama Y, Sugimoto Y, Oka T, Fukushima M, et al. Folylpolyglutamate synthase and gamma-glutamyl hydrolase regulate leucovorin-enhanced 5-fluorouracil anticancer activity. Biochem Biophys Res Commun. 2008;365:801–807. doi: 10.1016/j.bbrc.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 108.Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–2903. [PubMed] [Google Scholar]
- 109.Donada M, Bonin S, Barbazza R, Pettirosso D, Stanta G. Management of stage II colon cancer - the use of molecular biomarkers for adjuvant therapy decision. BMC Gastroenterol. 2013;13:36. doi: 10.1186/1471-230X-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jover R, Nguyen TP, Pérez-Carbonell L, Zapater P, Payá A, Alenda C, Rojas E, Cubiella J, Balaguer F, Morillas JD, et al. 5-Fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancer. Gastroenterology. 2011;140:1174–1181. doi: 10.1053/j.gastro.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim JY, Kim JS, Baek MJ, Kim CN, Choi WJ, Park DK, Namgung H, Lee SC, Lee SJ. Prospective multicenter phase II clinical trial of FOLFIRI chemotherapy as a neoadjuvant treatment for colorectal cancer with multiple liver metastases. J Korean Surg Soc. 2013;85:154–160. doi: 10.4174/jkss.2013.85.4.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Han SW, Lee HJ, Bae JM, Cho NY, Lee KH, Kim TY, Oh DY, Im SA, Bang YJ, Jeong SY, et al. Methylation and microsatellite status and recurrence following adjuvant FOLFOX in colorectal cancer. Int J Cancer. 2013;132:2209–2216. doi: 10.1002/ijc.27888. [DOI] [PubMed] [Google Scholar]
- 113.Wang Y, Long Y, Xu Y, Guan Z, Lian P, Peng J, Cai S, Cai G. Prognostic and predictive value of CpG island methylator phenotype in patients with locally advanced nonmetastatic sporadic colorectal cancer. Gastroenterol Res Pract. 2014;2014:436985. doi: 10.1155/2014/436985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shiovitz S, Bertagnolli MM, Renfro LA, Nam E, Foster NR, Dzieciatkowski S, Luo Y, Lao VV, Monnat RJ, Emond MJ, et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage III colon cancer. Gastroenterology. 2014;147:637–645. doi: 10.1053/j.gastro.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Benson AB, Bekaii-Saab T, Chan E, Chen YJ, Choti MA, Cooper HS, Engstrom PF, Enzinger PC, Fakih MG, Fenton MJ, et al. Localized colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2013;11:519–528. doi: 10.6004/jnccn.2013.0069. [DOI] [PubMed] [Google Scholar]
- 116.Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126:394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 117.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Balaguer F, Sempere L, Xicola RM, Bujanda L, et al. The efficacy of adjuvant chemotherapy with 5-fluorouracil in colorectal cancer depends on the mismatch repair status. Eur J Cancer. 2009;45:365–373. doi: 10.1016/j.ejca.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 119.Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fallik D, Borrini F, Boige V, Viguier J, Jacob S, Miquel C, Sabourin JC, Ducreux M, Praz F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003;63:5738–5744. [PubMed] [Google Scholar]