

Abstract
A novel family of compounds derivative of 1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))-bispyridinium or –bisquinolinium bromide (10a-l) containing a pair of oxygen atoms in the spacer of the linker between the biscationic moieties, were synthesized and evaluated as inhibitors of choline kinase against a panel of cancer-cell lines. The most promising compounds in this series were 1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(dimethylamino)pyridinium) bromide (10a) and 1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))-bis(7-chloro-4-(pyrrolidin-1-yl)quinolinium) bromide (10l), which inhibit human choline kinase (ChoKα1) with IC50 of 1.0 and 0.92 μM, respectively, in a range similar to that of the previously reported biscationic compounds MN58b and RSM932A. Our compounds show greater antiproliferative activities than do the reference compounds, with unprecedented values of GI50 in the nanomolar range for several of the cancer-cell lines assayed, and more importantly they present low toxicity in non-tumoral cell lines, suggesting a cancer-cell-selective antiproliferative activity. Docking studies predict that the compounds interact with the choline-binding site in agreement with the binding mode of most previously reported biscationic compounds. Moreover, the crystal structure of ChoKα1 with compound 10a reveals that this compound binds to the choline-binding site and mimics HC-3 binding mode as never before.
Cancer is a worldwide health threat and the second leading cause of mortality in developed countries1,2. Since many of the current treatments still prove toxic and/or lead to drug resistance, there is a strong demand for the discovery and development of effective new cancer therapies3.
Protein kinases have emerged as one of the most important types of targets in cancer-drug discovery due to their major roles in regulating cell growth and survival and many other cell functions4,5. An abnormal kinase signaling network underlies the development and progression of tumors, and thus the targeted inhibition of protein kinases has become an attractive strategy in cancer treatment (for a recent review see Gross et al.6) and in the last decade the intense development in the field has led to different kinase inhibitors that have been approved for use in clinical therapy.
Choline kinase (ChoK) (EC 2.7.1.32) catalyzes the phosphorylation of choline by ATP in the presence of Mg2+ to yield phosphocholine (PCho) and ADP7,8. This step introduces choline to the so-called Kennedy or CDP-choline pathway for the biosynthesis of phosphatidylcholine, which represents the most abundant class of phospholipids in eukaryotic cells, constituting 40–60% of the phospholipids content in cell membranes9. In addition to forming the major structural component of the membrane bilayer, phosphatidylcholine also serves as a precursor for the production of lipid second messengers10.
Mammalian ChoK exists as three isoforms, encoded by two separate genes11,12. In humans, ChoKα1 (457 amino acids) and ChoKα2 (439 amino acids) derive from a single gene (chk-α) by alternative splicing, while ChoKβ (395 amino acids) is the product of a different gene (chk-β). The amino acid sequence identity is 56% between ChoKα and ChoKβ, and both chk-α and chk-β mRNAs, as well as their encoded protein isoforms, are ubiquitously expressed in diverse tissues13. Each isoform is present as either dimers (homo- or hetero-) or as tetramers in solution and is not active in monomeric form7, suggesting that, for higher eukaryotes, dimeric ChoK is the minimum functional form.
Choline kinase is overexpressed in many tumors such as breast, lung, bladder, colon, prostate, ovary, and liver carcinomas14,15,16 and recently elevated enzymatic activity has also been shown in T-lymphoma17. This increasing expression leads to abnormal choline metabolism, resulting in higher phosphocholine levels, which refer to a cholinic phenotype associated with oncogenesis and tumor progression18. As a result, ChoKα, has become an attractive target for novel anticancer therapies.
The determination of the crystal structures of ChoK proteins from Caenorhabditis elegans and human, in which two monomers were dimerized in each asymmetric unit19,20 and the correct identification of ATP and choline binding sites into crystal structure of human ChoKα2 isoform, have enabled the design and the synthesis of a series of asymmetrical molecules as potential ChoK inhibitors and antiproliferative compounds21,22. Figure 1 shows HC-3, the first inhibitor of choline kinase described, MN58b and RSM923A, which belong to the first generation of Chok inhibitors23,24,25,26,27,28, and the most promising compounds developed by our group. Note that RSM932A (also called TCD-717) has evidenced a low-toxicity profile with improved tolerability in mice29 and a Phase I clinical trial has just been completed for the treatment of advanced solid tumors (http://clinicaltrials.gov/ct2/show/NCT01215864).
Figure 1. Structure of compounds HC-3, MN58b, RSM932A and structures of symmetrical and non-symmetrical inhibitors of choline kinase previously published (compounds 1–5).

General structure of compounds 10a-l described in this paper.
For compounds 1 and 2, we identified the adenine and 1-benzyl-4-(dimethylamino)pyridinium as the most efficient fragments of these molecules by the deconvolution approach based on the ChoKα1/1 (PDB ID: 3ZM9)30 and ChoKα1/2 (PDB ID: 4BR3)31 crystal structures, demonstrating that the adenine fragment occupies the ATP binding site and that the pyridinium fragment, through its positive charge delocalized over the nitrogen atom, mimics the positive charge present in choline or in HC-3.
The second generation of inhibitors (compounds 3 and 4), asymmetrical bispyridinium compounds, proved to be good inhibitors and provide the discovery of a new inhibitory binding site on ChoKα1 Compound 4 (Fig. 1), which induced the opening of new adjacent binding site where the 4-Chloro-N-methylaniline fragment is located, adopting an unprecedented modality of binding to ChoKα1(ChoKα1/4, PDB ID: 4CG8)32, while compound 3 (with biphenyl group as a linker) adopts a binding mode similar to the one observed for compound 2.
In an effort to produce additional highly active compounds, we focused on longer spacers between biphenilic or bipyridinic rings, which have electron donor or acceptor groups necessary to increase the binding to the enzyme through of hydrogen bonds and the solubility, while retaining some inhibition properties.
Deep modeling and virtual screening studies32,33,34 have suggested the interaction with the choline binding site in the ChoKα1/4 complex while keeping the biscationic structure unchanged. Thus a classical bioisosteric exchange between carbon and oxygen atoms could increase, on one hand, the polarity and the solubility of these compounds and, on the other hand, the affinity for the enzyme due to the synclinal conformation of the linker of these molecules. In this way, in the present study, we reconfigured the substitution pattern around linker moiety by the preparation of 1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))-bis[4-pyridinium or quinolinium] bromide derivatives with general structures 10a-l (Fig. 1). MN58b, RSM932A, 1, 2, 3, 4, our recently published compound 535 (Fig. 1) and the most active compounds described by S. Trousil36, were taken as patterns to improve the polarity and solubility, while also improving inhibition by the enzyme and consequently enhancing the antiproliferative effect. This series was obtained by interchanging the substitution pattern of linker by the introduction of two oxygen atoms in the linker, in order to determine the influence of these groups on the antiproliferative and inhibitory activity of ChoKα1, using various cationic heads previously synthesized by our group in similar compounds. We fixed the most successful cationic heads described previously (pyridinium and quinolinium salts)32,35,36,37,38,39,40 and examined several 4-substitutions with alkylamines or phenylamines on the arylmoiety. Also, we introduced a quinuclidinium salt, which mimics the trimethylammonium of the choline, but potentially prevents interactions with the cholinergic system41.
Results and Discussion
Chemistry
Microwave-assisted reactions present several advantages, such as a remarkable reduction in reaction times compared to those of the conventionally heated reactions and often lead to improved yields42,43. In the present work, we describe the use of microwave irradiation as an energy source for the synthesis of the intermediate (7 and 8) of twelve 1,2-bis(p-methylphenoxy)ethane derivatives 10a-l, substituted in the methylphenoxy group with different cationic heads as moieties. These compounds can be also considered as more polar analogues of choline kinase inhibitor derivatives than those previously synthesized.
The syntheses of compounds 10a-l is shown in Fig. 2, and follows three easy steps. The first is the treatment of the 4-methylphenol (6) in ethanol with NaOH (1.1 equiv) stirring at room temperature for 30 min, followed by the addition of 1,2-dibromoethane (0.5 equiv), under microwave irradiation (130 °C, 28 min) to provide the 1,2-bis(p-methylphenoy)ethane (7)44,45. Then, bromination in the methylene of 7 with NBS and dibenzoylperoxide in CCl4 also under microwave irradiation (120 °C, 21 min), to give the 1,2-bis(4-bromomethylphenoy)ethane (8)45,46. In comparison with conventional (thermal) heating, the microwave heating reduced the reaction time in both reactions (30 min vs. 8 h and 21 min vs. 5 h, respectively), but we also noted some yield improvement (35% vs. 21% and 65% vs. 39%, respectively)45,46.
Figure 2.

General synthetic pathway followed in the preparation of compounds 10a-l i) NaOH, EtOH, RT 30 min. ii) Br-(CH2)2-Br, MW, 130 °C, 28 min. iii) NBS, CCl4, dbp, MW 130 °C, 21 min. iv) 10a-l, CH3CN, 72 hours.
We conducted different experiments to achieve these successful results with the MW (Table S1 of Supplementary information). Although in the second step, we were restricted by the quantity to use, since using only 100 mg of derivative 7 gave the best yields, while more quantity of 7 led to diminished yields. This result was due to the volume of the reactor, which allows only 5 mL of the mixture, while more than 100 mg of 7 derivative would need more solvent to dissolve it.
Finally, the last step is the introduction of cationic heads (previously synthesized using the procedure reported32,35,36,37,38,39,40 by means of a simple SN2 reaction in acetonitrile under argon atmosphere for 72 h at reflux of 1,2-bis(4-bromomethylphenoxy)ethane (8) and the 4-substituted pyridine derivative (9a-c), quinuclidine derivative (9d-e) or 4-substituted quinoline or 7-Chloro-4-substituted quinoline (9f-l) to afford 10a-l with moderate or good yields46.
Docking studies
Docking studies were made in order to design the new ChoK inhibitors. The crystal structures of greatest interest for docking studies are those of ChoKα1 isoenzyme in complex with compounds 2 (PDB ID: 4BR3)31 and 4 (PDB ID: 4CG8)32, since the cationic heads of compounds described in this paper are similar to those of compounds 2 and 4.
Figure 3A shows compound 2 (carbon atoms in yellow color) inserted into the Cho binding site, being stabilized by cation-π interactions with Tyr333, Tyr354, Tyr440, Trp420, Trp423, and Phe435 (carbon atoms in cyan color). In particular, the biphenyl group shows optimal parallel hydrophobic stacking interactions with Tyr354, and the 4-(dimethylamino)pyridinium moiety interacts through parallel cation-π interaction with Trp420. The orientation of this compound inside the Cho binding site was accommodated by a conformational change of Tyr333, which moved back to create an extra space30,31. The adenine fragment of compound 2 inserted into the Cho binding site was outside the enzyme and showed no interaction with the protein (Figure S1 of Supplementary information), 1-(biphenyl-4-ylmethyl)-4-(dimethylamino)pyridinium being the key fragment of this compound for the interaction at the Cho binding site30,31. Figure 3B shows compound 4 (carbon atoms in orange color) inserted into the ChoKα1 crystal structure. This compound adopts a new different binding mode, inducing a conformational change in some amino acids. Tyr256, Tyr333, and Trp420 are the residues that undergo the major changes, and the rotation of these side chains is critical to allow the insertion of the 4-chloro-N-methylaniline fragment into an additional binding site (carbon atoms in magenta color), being stabilized by hydrophobic interactions with Trp248, Tyr256, Tyr333, Leu419, Trp420, and Trp423. The rest of the molecule is located inside the Cho binding site (carbon atoms in cyan color), the pyridinium moieties being stabilized through cation-π interactions with Tyr333, Tyr354, Trp420, and Tyr44032. Compound 4 is more inserted into the Cho binding site in comparison to compound 2 (Figure S1 of Supplementary Information) since this compound makes the complete opening of this site.
Figure 3.

(A) Crystal structure of ChoKα1/2 complex (PDB ID: 4BR3). Compound 2 (carbon atoms in yellow color) is inserted into the Cho binding site (carbon atoms in cyan color). (B) Crystal structure of ChoKα1/4 complex (PDB ID: 4CG8). Compound 4 (carbon atoms in orange color) is inserted into the Cho binding site (carbon atoms in cyan color) and in an additional binding site (carbon atoms in magenta color) that has been open by a conformational change of Tyr256, Tyr333, and Trp420 sidechains, induced by the insertion of compound 4 into the enzyme. (C) Resulting pose of compound 10a (carbon atoms in green color) in the Cho binding site of the ChoKα1/2 crystal structure, and D) Resulting pose of compound 10l (B, carbon atoms in yellow color) in the ChoK binding site of the ChoKα1/4 crystal structure.
Docking studies have been performed in both crystal structures and the analysis of the resulting poses indicates which compounds could be similar to compound 2 or to compound 4. In fact, compounds 10a, 10b, 10d, and 10e have shown good poses in the crystal structure of compound 2, while the correct poses of compounds 10c, 10f, 10g, 10h, 10i, 10j, 10k, and 10l resulted in the crystal structure of compound 4.
As an example, Fig. 3C,D show the resulting pose of compounds 10a and 10l. Compound 10a (carbon atoms in green color) has two 4-(dimethylamino)pyridinium cationic heads, similarly to compound 2 (Fig. 3A). One 1-benzyl-4-(dimethylamino)pyridinium fragment is inserted in a way very similar to that of compound 2: i) the cationic head is situated close to Trp420, being stabilized by π-cation interactions with Trp420, Tyr333, and Trp423; and: ii) the benzyl fragment is also optimized by hydrophobic stacking interactions with Tyr354. The linker of compound 10a is extremely long, and the second 1-benzyl-4-(dimethylamino)pyridinium fragment is situated outside of the enzyme, an additional hydrophobic interaction occurring between the second phenyl fragment and Ile433. Compound 10l (carbon atoms in orange color) has two 7-chloro-4-(pyrrolidin-1-yl)quinolinium) cationic heads, one of which is inserted very similarly to compound 4 (Fig. 3B): the 4-pyrrolidin fragment is inserted into the additional binding site and stabilized by hydrophobic interactions, while the 7-chloroquinolinium moiety is situated into the Cho binding site and stabilized by cation-π interactions. The second cationic head is also inserted into the protein, being stabilized by hydrophobic interactions with Ile433 and Arg117, and the phenyl group connected to this cationic head is also stabilized by cation-π interaction with Phe435. The most notable effect in these molecules is the conformation of the linker, since the 1,2-dioxoethane fragment adopts a synclinal conformation due to the gauche effect of the O-C-C-O bonds. This conformation of the linker allows the total insertion of compound 10l inside the Cho binding site, and also favors the insertion of compound 10a.
Figure S2 of Supplementary Information shows the resulting pose of compounds 10b, 10d, and 10e. The pose of compound 10b is very similar to that of compound 10a, the pyridinium moiety being slightly more separated from Tyr333 due to the higher volume of the pyrrolidine fragment, but the interaction of the whole molecule with the Cho binding site is very similar to that of compound 10a. Compound 10d shows also a similar pose to that of compound 10a, though slightly more inserted into the Cho binding site due to the smaller volume of the quinuclidine cationic head. The resulting pose of compound 10e is also similar, being slightly outside compound 10a due to the establishment of two H-bond between the 3-OH groups and Asn305 and Glu434, respectively. Figure S3 of Supplementary Information shows the resulting pose of compounds 10f-k and 10c. Compounds 10f-k have shown a pose very similar to that of compound 10l (Fig. 3D), one of the cationic heads being inserted inside the additional binding site and into the Cho binding site. The second cationic head is also inserted into the protein being stabilized by hydrophobic interactions with Arg117 and Ile433, and the phenyl group of this cationic head is also stabilized by π-cation interaction with Phe435. The resulting pose of compound 10c shows a slight difference. This compound has two 4-((4-chlorophenyl)(methyl)amino)pyridinium cationic heads. One cationic head is also inserted into the additional binding site and into the choline binding site, as in compound 4, and the second cationic head is also inserted into the enzyme, but with a different orientation. Nevertheless, the most noteworthy effect is that in the resulting pose of these compounds the 1,2-dioxoethane fragment also adopted a synclinal conformation and, for this reason, all these compounds should be completely inserted into the enzyme and probably will show good ChoKα1 inhibition.
Inhibition of ChoKα1 enzymatic activity
It has been reported that a potent anticancer effect inducing maximal apoptosis is achieved only when ChoKα1 expression is specifically knocked down, without affecting ChoKβ levels47. Thus, in an initial step we evaluated whether these compounds have a selective behaviour on ChoKα1.
We selected the most representative compounds of each family, (10a for pyridinium compounds and 10f, 10g, 10k, and 10l for quinolinium compounds) and, since selectivity may be explained by a reduced flexibility of Trp353 in ChoKβ compared to its homologue Trp420 in ChoKα1, as was proposed for HC-331, tryptophan fluorescence assays were performed (Figure S4 of Supplementary information). As expected from their chemical structures similar to that of compound 4, the results of the spectroscopy analysis, depicted in Table 1, showed that the Kd values of these compounds for ChoKα1 were in the low μM range (0.241–0.700 μM), indicating high affinity for the enzyme. These results agree with the first experimental validation of the docking studies described above.
Table 1. Kd values of selected compounds evaluated by tryptophan fluorescence quenching.
| Compound | HsChoKα1 Kd (μM)a |
|---|---|
| 10a | 0.700 ± 0.080 |
| 10f | 0.295 ± 0.127 |
| 10g | 0.517 ± 0.099 |
| 10k | 0.241 ± 0.03 |
| 10l | 0.357 ± 0.039 |
| 4 | 0.110 ± 0.01 |
| HC-3 | 0.18 ± 0.03 |
aKd values of indicated compounds for ChoKα1 are expressed as mean ± S.D. of at least three independent experiments.
Table 2 summarizes the clog P calculated by Pallas (3.8.1.1. PrologP) and the inhibitory effect on purified human ChoKα1 activity.
Table 2. In vitro inhibitory effects of compounds 10a-l.
| Comp. | clogP Ann 2005 | aIC50 (μM) ChoKα1 purified | Antiproliferative activity bGI50 (μM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HeLa | HT−29 | Jurkat | HL-60 | RS4,11 | MCF-7 | PC-3 | A549 | MDA-MB-231 | |||
| 10a | −0.36 | 1.00 ± 0.01 | 0.079 ± 0.024 | 0.11 ± 0.01 | 0.12 ± 0.08 | 0.10 ± 0.04 | 0.045 ± 0.005 | 0.092 ± 0.019 | 0.051 ± 0.01 | 0.027 ± 0.010 | 0.10 ± 0.05 |
| 10b | 0.36 | 9.56 ± 1.45 | 0.082 ± 0.041 | 4.3 ± 0.42 | 0.068 ± 0.016 | 0.042 ± 0.005 | 1.12 ± 0.22 | 0.17 ± 0.032 | 4.5 ± 1.1 | 2.3 ± 1.1 | 0.09 ± 0.02 |
| 10c | 1.8 | 1.63 ± 0.14 | 5.6 ± 0.25 | 4.3 ± 1.1 | 7.3 ± 0.50 | 2.1 ± 0.6 | 2.7 ± 0.13 | 3.5 ± 0.72 | 3.1 ± 0.7 | 4.9 ± 0.7 | 2.2 ± 0.27 |
| 10d | −1.01 | 37.54 ± 4.45 | 16.9 ± 2.5 | >100 | >100 | 84.1 ± 7.4 | 37.5 ± 4.2 | 91.4 ± 7.6 | 94.3 ± 12.4 | 61.3 ± 16.7 | 74.3 ± 2.6 |
| 10e | −2.42 | 9.51 ± 1.14 | >100 | 94.0 ± 4.2 | >100 | >100 | 66.0 ± 6.6 | 50.9 ± 11.7 | 48.0 ± 1.1 | >100 | 63.2 ± 3.9 |
| 10f | 3.02 | 6.85 ± 0.81 | 0.15 ± 0.031 | 0.12 ± 0.017 | 0.060 ± 0.002 | 0.063 ± 0.013 | 0.24 ± 0.05 | 0.17 ± 0.065 | 0.47 ± 0.12 | 0.21 ± 0.06 | 0.001 ± 0.001 |
| 10g | 3.56 | 3.27 ± 0.66 | 0.25 ± 0.06 | 0.75 ± 0.12 | 0.098 ± 0.031 | 0.71 ± 0.24 | 0.15 ± 0.04 | 0.46 ± 0.063 | 0.26 ± 0.01 | 0.11 ± 0.018 | 0.19 ± 0.009 |
| 10h | 3.82 | 2.79 ± 0.17 | 0.32 ± 0.062 | 0.35 ± 0.07 | 0.31 ± 0.07 | 0.92 ± 0.06 | 0.026 ± 0.006 | 0.32 ± 0.04 | 0.85 ± 0.32 | 0.18 ± 0.08 | 0.23 ± 0.04 |
| 10i | 4.23 | 16.22 ± 0.44 | 1.5 ± 0.73 | 1.0 ± 0.15 | 0.76 ± 0.26 | 0.87 ± 0.28 | 0.50 ± 0.09 | 0.66 ± 0.07 | 0.72 ± 0.09 | 0.29 ± 0.10 | 0.15 ± 0.05 |
| 10j | 2.27 | 1.66 ± 0.09 | 0.17 ± 0.084 | 0.15 ± 0.08 | 0.11 ± 0.026 | 0.42 ± 0.22 | 0.16 ± 0.012 | 0.11 ± 0.047 | 0.09 ± 0.04 | 0.30 ± 0.068 | 0.01 ± 0.005 |
| 10k | 2.76 | 2.02 ± 0.08 | 0.26 ± 0.074 | 0.27 ± 0.05 | 0.072 ± 0.013 | 0.18 ± 0.06 | 0.036 ± 0.008 | 0.28 ± 0.09 | 0.11 ± 0.028 | 0.52 ± 0.02 | 0.061 ± 0.003 |
| 10l | 2.36 | 0.92 ± 0.01 | 0.37 ± 0.18 | 0.56 ± 0.21 | 0.007 ± 0.003 | 0.16 ± 0.07 | 0.42 ± 0.14 | 0.022 ± 0.007 | 0.8 ± 0.2 | 0.14 ± 0.06 | 0.05 ± 0.02 |
| MN58b | 0.01 | 0.78 ± 0,03 | 1.9 ± 0.1 | 1.9 ± 0.4 | 0.35 ± 0.1 | 0.32 ± 0.03 | 1.0 ± 0.3 | 1.8 ± 0.06 | n.d. | 0.54 ± 0.2 | 0.31 ± 0.12 |
| RSM932A | 2.92 | 1.92 ± 0,06 | 0.83 ± 0.1 | 0.4 ± 0.2 | 0.41 ± 0.1 | 0.93 ± 0.1 | 0.17 ± 0.04 | 0.18 ± 0.10 | n.d. | 0.45 ± 0.09 | 0.17 ± 0.05 |
aIC50 = Compound concentration required to inhibit ChoKα1 enzyme by 50%.
bGI50 = Compound concentration required to inhibit tumor-cell proliferation by 50%.
Values are the mean ± SEM for three independent experiments. n.d. not determined.
Of all tested compounds, the ones that present an alkylamine or a cycloalkylamine substituted at position 4 of the pyridinium or quinolinium system, 10a-b and 10j-l, offer the best results in terms of enzyme inhibition and antiproliferative assays.
Regarding ChoKα1 inhibitory effect, all compounds showed a micromolar activity, comparable to that of the two reference compounds RSM932A and MN58b. The docking studies indicated that all compounds could be inserted into the choline binding site and could be grouped into two families on the basis of their different insertion modes. Compounds 10a, 10b, 10d, and 10e could be inserted similarly to compound 2. Compound 10a showed good HsChoKα1 inhibition (IC50 = 1.0 μM), thanks to the presence of the 1-benzyl-4-(dimethylamino)pyridinium fragment, which performs a strong π-cation interaction with the Cho binding site (Fig. 3C), this having been described as one of the most efficient moieties for ChoKα1 inhibition33. Compound 10b showed a reduced ChoKα1 inhibition (IC50 = 9.56 μM) attributable to the volume of the 4-(pyrrolidin-1-yl)pyridinium cationic head, causing a decrease in the π-cation interaction (Figure S2A). In this family, compound 10d showed very poor ChoKα1 inhibition (IC50 = 37.54 μM). This result may be due to the presence of the quinuclidine and the consequently low lipophilicity (clog P = −1.01) cationic head that probably prevented the interactions with Tyr333, Tyr354, Trp420, and Tyr440 (Figure S2B). However, although compound 10e had a 3-hydroxyquinuclidine cationic head and low lipophilicity (clog P = −2.42), it showed better ChoKα1 inhibition (IC50 = 9.51 μM) than could be explained by the establishment of two additional H-bonds with the enzyme (Figure S2C).
In summary, when compounds bind to the choline site, such as compound 2 (compound 10a-b, d-e), low lipophilicity values are sufficient to achieve good inhibition of the enzyme, the pydidinic ring being the most appropriate moiety, and the quinuclidine ring decreases the activity unless the lack of aromatic ring is offset by the formation of a H bond provided by the hydroxyl group.
The second group interacted with the choline binding site as compound 4. Compounds 10c 10j-l showed a good ChoKα1 inhibition (IC50 = 1.63, 1.66, 2.02 and 0.92 μM respectively), while compounds 10f-i showed a slightly reduced ChoKα1 inhibition (IC50 = 6.85, 3.27, 2.79 and 16.22 μM, respectively). All these compounds had a 4-substituted and 7-substituted quinolinium cationic head, 10f-l, except 10c, which had the rest of the N-methylanilino at the 4 position of the pyridinium cationic head. The structure of the 4-substituted fragment conditions the inhibitory activity of these molecules. In fact, compounds 10j-l had a 4-cycloalkylamino fragment, while compounds 10c and 10f-i had a 4-N-methylanilino substituent. The resulting IC50 values indicated that the 4-cycloalkylamino favored the ChoKα1 inhibition, probably promoting a more effective insertion into the additional binding site. In this group the chloro atom increased the volume of molecule and the lipophilicity. Thus, the compound 10l offered the right balance between volume and lipophilicity. The perhydroazepine group (10i) provided the volume to be inserted into the choline binding site properly and the addition of a choro atom slightly depressed the activity (10j).
Conversely, the compounds with an N-methylanilino system at the 4 position of the pyridinium or quinolinium cationic heads showed lower IC50 than did those in the alkylamine system. In these compounds, the chloro atom in para position of 4-(methyl(phenyl)amino)quinolinium or pyridinium fragment (10c, 10g and 10i) or in position 7 of the quinolinium ring (10h and 10i) seemed to play an essential role in the enzyme inhibition. In fact, the presence of the chloro atom allowed the cationic head to be accommodated at the choline-binding site, likely by an increase in the lipophilicity provided by the halogen atom, regardless of where the halogen was located (10c, 10g, and 10h IC50 = 1.63, 3.27, and 2.79 μM, respectively). A direct correlation between volume-lipophilic activity was found in these compounds (10c, 10f-i), so that the less bulky and lipophilic compounds offered the best values (10c, ChoKα1, IC50 = 1.66 μM, clog P = 1.8) while adding a second aromatic ring slightly diminished the activity of 10f-h. On the other hand, the two chloro atoms present in compound 10i made the molecule too bulky to bind to the choline site, lowering its inhibition activity (ChoKα1, IC50 = 16.22 μM), and raising its lipophilicity (clog P = 4.23). However, the absence of chloro atom in 10f also considerably decreased ChoKα1 inhibition (IC50 = 6.85 μM), highlighting the important role of the chloro atom.
Regardless the binding mode to the enzyme, the best inhibitory activities were found when a fragment of alkylamine or cycloalkylamine was present at the 4 position in pyridinium or quinolinium cationic heads, such as 10a, 10j, 10k, and 10l (IC50 = 1.0, 1.66, 2.02, and 0.92 μM, respectively), These results reveal that the volume of the cationic head probably fits properly into the choline binding site. The only exception was 10b (IC50 = 9.56 μM), and this was a consequence of the higher volume of the 4-(pyrrolidin-1-yl)pyridinium cationic head that diminished the π-cation interaction (Figure S2A) mentioned above.
Cancer-cell growth inhibition
All compounds were evaluated for their antiproliferative activity against a panel of nine different human tumor-cell lines (Table 2). All were given GI50 values generally lower than 1 μM, some of them even at nanomolar concentrations. Only two compounds, i.e. 10d and 10e, registered GI50 values higher than 10 μM against all tested lines.
Compounds 10a, 10b, 10f, and 10l offered the best antiproliferative activities against all cell lines. In particular, 10a gave GI50 values ranging from 0.027 to 0.12 μM, although the best value was by 10f in MDA-MB-231 cell line (GI50 = 0.001 μM). Compared to quinolinium derivatives, in general the pyridinium moiety provided better results in all tested cell lines.
In the pyridinium family, the best results were found when the substitution was an alkyl or cycloalkylamine (10a-b). The switching of dimethylamine to pyrrolidine caused a curious decline in activity for the majority of cell lines (10b, GI50 = 0.042 μM to 4.5 μM). The replacement of this tertiary amine by a conjugate aromatic system (4-(4-chloro-N-methylanilino)pyridinium, 10c) resulted in lower activity (GI50 = 2.1 μM to 7.3 μM). It bears noting that despite of their low lipophilicity (10a, clog P = −0.36 and 10b clog P = 0.36), the compounds 10a-b provided better results than did compound 10c (clog P = 1.8). This finding can be explained by the different mode to bind to enzymes of these compounds, as mentioned above.
Replacing the pyridinium system with quinuclidine as cationic head, such as compounds 10d and 10e, led to a total loss of activity in all cell lines. This result may be due to the very low lipophilicity of 10d-e (clog P = −1.01 and −2.42 respectively) and to the low IC50 value.
Regarding the quinolinium family (10f-l), all compounds had GI50 in the range of submicromolar values, 10l being the best compound (0.007 μM for Jurkat cells). No differences in the activity were detected between the different substituents in the 4 position upon the quinolinium system, since 10f and 10l (with N-methylanilino and 4-pyrrolidine in the 4 position, respectively) offer the best antiproliferative activity over nearly all the cell lines. However, the presence or not of a chloro atom over the quinolinium system, seems to play a crucial role in inhibiting cell growth. The chloro atom provided not only a higher lipophilicity to these compounds, improving the passage through the plasma membrane, but also a larger volume that impaired the insertion in the choline binding site. In fact, compound 10f, which has an N-methylanilino group at the 4 position without any chloro atom, exhibited better antiproliferative activity than did compound 10h or 10g, which have a chloro atom in the quinolinium.
However, 10f registered a lower value of ChoKα1 inhibition (IC50 = 6.86 μM) than 10g and 10h (IC50 = 3.27 and 2.79 μM, respectively) with chloro in para position of the N-methylanilino system (10g) or in 7 position of quinolinium fragment (10h). The chloro atom provided lipophilicity and good results of enzyme inhibition but this lipophilicity can allow binding to other targets in the cancer cells. On the other hand, the presence of two chloro atoms make compound 10i too bulky to inhibit the enzyme (IC50 = 16.22 μM), strongly reducing the antiproliferative activity, and it is also more lipophilic (clog P = 4.23), and thus does not bind to other targets. In conclusion, among the quinolinium family, the compounds with an N-methylanilino system upon the 4 position (10f-i), 10f offers the best antiproliferative values but lower inhibition of the enzyme (6.85 μM). This could be due to a greater affinity of 10f by the enzyme in a whole cancer cell while the higher lipophicility of 10g-h could help them to be more suitable for binding to other targets. This highlights the need for a balance between lipophilicity, inhibitory activity of the enzyme isolated (affinity and selectivity), and antiproliferative activity for achieving successful results.
The insertion of chloro in position 7 as in compound 10k improves the activity. This suggests that although the enzymatic inhibitory activity is almost the same for these compounds (IC50 from 0.92 to 2.02 μM), the chloro leads to an appreciable increase in the antiproliferative activity (10j vs. 10k) in nearly all the cell lines.
Finally, 10a and 10l were the compounds with the best GI50 values in almost all the cell lines. The two belong to different subfamilies. The first one, 10a, has a residue of dimethylaminopyridinium as a cationic head which could offer the best volume to fit in a choline binding site, while the compound 10l has a residue of 7-Chloro-4-pyrrolidinequinolinium which provides more lipophilicity to the quinolinium moiety, allowing the compound to cross the plasma membrane more easily.
Trypan blue exclusion assay
The cell viability in the presence of 10a was also evaluated by the trypan blue exclusion assay. The results depicted in Fig. 4 reflect that 10a significantly inhibited cell growth in three cell lines (Fig. 4 panel A Jurkat, Panel B = HeLa; panel C = MDA-MB-231) tested in a concentration-dependent manner, roughly confirming the results found with the MTT test (see Table 2). Notably, we observed that the inhibition of cell proliferation was not dependent on the presence of the molecule in the incubation medium. In fact, experiments in which the cells treated with 10a were harvested, washed, and incubated with fresh medium without 10a proliferation continued to be inhibited, suggesting that the catalytic activity of the enzyme may have been irreversibly inhibited (Figure S5 of Supplementary information).
Figure 4.

Cell viability evaluated by trypan blue count in Jurkat cells (A) HeLa cells (B) and MDA-MB-231 (C) after incubation with the indicated concentrations of compound 10a. Data are presented as mean±SEM of three independent experiments.
Effect of 10a in non-tumoral cells
We investigated the effect of the most active compound (10a) in non-tumoral cells e.g. human lymphocytes and human umbilical-vein endothelial cells (HUVEC) from healthy donors. As shown in Table 3, in general, in resting lymphocytes, fibroblasts, and HUVEC, compound 10a had lower toxicity compared to tumor cells. Instead, in lymphocytes stimulated with a mitogen (e.g. phytohematoagglutinin, Pha), the compound had cytotoxicity comparable to that seen in tumor cells, indicating a certain preference only towards proliferating cells. In this context, it bears noting that other choline kinase inhibitors such as MN58b or RSM932A presented low or reduced cytotoxicity in oncogene-transformed cells and in tumor cells, in agreement with previously published data44,48.
Table 3. In vitro inhibitory effects of selected compounds in non-tumoral cells.
| Antiproliferative activitya GI50 (μM) | ||||
|---|---|---|---|---|
| Comp | PBL(resting) | PBL(+Pha)b | Human fibroblasts | HUVEC |
| 10a | 1.5 ± 0.64 | 0.034 ± 0.007 | 5.8 ± 1.3 | 5.1 ± 0.43 |
| 10b | 34.8 ± 15.6 | 1.88 ± 0.71 | 30.5 ± 9.6 | n.d. |
| 10f | 1.4 ± 0.6 | 0.10 ± 0.03 | 9.8 ± 2.5 | n.d. |
| 10g | 0.98 ± 0.25 | 0.55 ± 0.11 | 7.4 ± 2.4 | 10.4 ± 3.5 |
| 10h | 1.0 ± 0.42 | 0.32 ± 0.03 | 3.7 ± 0.43 | 16.0 ± 6.6 |
| 10j | 2.0 ± 0.24 | 0.095 ± 0.021 | 14.3 ± 1.2 | n.d. |
| 10k | 2.1 ± 0.80 | 0.034 ± 0.007 | 3.2 ± 0.86 | n.d. |
| 10l | 3.8 ± 0.55 | 0.49 ± 0.15 | 9.6 ± 1.3 | 11.4 ± 5.9 |
| RSM932A | 1.1 ± 0.09 | 0.23 ± 0.05 | n.d | 0.46 ± 0.047 |
| MN58b | 2.0 ± 0.4 | 0.15 ± 0.05 | n.d. | 2.1 ± 0.58 |
aGI50 = Compound concentration required to inhibit tumor-cell proliferation by 50% Values are the mean ± SEM for three independent experiments. n.d. not determined.
bPha, Phytohematoagglutini.
The crystal structure of the complex ChoKα1/10a shows that the compound binds to the choline binding site
Although the docking studies clearly suggested that the compounds bind to the choline binding site (Fig. 3, Figures S2 and S3 in the Supplementary information), we carried out further crystallization experiments with the most active ChoKα1 inhibitors, 10a and 10l, in order to compile more consistent data concerning their binding mode (Fig. 5).
Figure 5.

(a) Active site of ChoKα1 in complex with compound 10a. Unbiased difference electron density maps are shown at 2.2 σ. (b) Superimposition of 10a with HC-3 (PDB ID: 3G15) and compound 2 (PDB ID: 3ZM9). (c) Residues that stabilize the positive charge of the cationic head in the three superimposed ligands. (d) Residues that undergo the most notable conformational changes when compounds 2 or 10a bind to the choline binding site.
Despite the large number of trials, we managed to solve only the crystal structure of ChoKα1 in complex with compound 10a at 1.45 Å (see Methods and Supplementary information, Table S2). The other compound (10l) was too hydrophobic (clog P = 2.36, Table 2) and therefore insoluble in the mother liquor.
For ChoKα1/10a complex, successive iterative model building and refinement cycles were carried out to produce a final model with good refinement statistics (R = 0.197, Rfree = 0.218, Table S2).
The structure is a monomer formed by a small N-terminal and a large C-terminal domain. Whereas the ATP binding site is located in a cleft formed by N- and C-terminal domains, the choline binding site is found in a deep hydrophobic pocket. One molecule of compound 10a was visualized at the choline binding site with good electron density (Fig. 5a). One 1-benzyl-4-(dimethylamino)pyridinium fragment was deeply positioned within the pocket and established π-cation interactions with Trp423 and Trp420, whereas it set π-π interactions with Tyr333, Tyr354, Phe435, and Tyr440 (Fig. 5a). The second 1-benzyl-4-(dimethylamino)pyridinium moiety was directed towards the outside part of the choline binding site, where it established π-π and hydrophobic interactions with residues Phe361 and Ile433, respectively (Fig. 5a).
When the crystal pose was compared with the docked one, few differences were found between the two, especially regarding the conformation that the first 1-benzyl-4-(dimethylamino)pyridinium fragment adopted (Supporting information, Figure S6).
This moiety was completely superimposed in the two poses, indicating the accuracy of the theoretical predictions. Nevertheless, the second 4-(dimethylamino)pyridinium moiety was flipped almost 90° towards the residue Phe361 but not towards the residue Ile433, as the docking had predicted. The reason for this difference is that the pyridinium ring set π-π interactions with Phe361 in the crystal pose, increasing the stability of the ligand-protein complex.
Remarkably, the positive charge of the quaternary ammonium of the first 1-benzyl-4-(dimethylamino)pyridinium fragment was positioned at the choline binding site in the same place as one of the previously reported ChoKα1 inhibitors, such as compound 2 (PDB ID:4BR3) and HC-3 (PDB ID: 3G15) (Fig. 5b). This indicates that common residues should participate in the positive-charge stabilization regardless of the chemical nature of the spacer groups. As reflected in Fig. 5c, these residues are Tyr333, Tyr354, Trp420, and Trp423, which set parallel π-cation and π-π interactions with the quaternary ammonium and the aromatic rings of the 1-benzyl-4-(dimethylamino)pyridinium fragment. Nevertheless, depending on the spacer and the cationic head of the compounds, some conformational changes in some of these residues were observed. For instance, residues Trp420 and Tyr333 underwent a noticeable retraction to open the hydrophobic cavity when compound 10a or compound 2 bound the protein in order to accommodate the positive charge of the cationic head (Fig. 5d).
10a and G1 phase cell-cycle arrest
Compound 10a induced a G1 arrest of the cell cycle, which occurred in a concentration-dependent manner in the three cell lines tested (Jurkat, MCF-7 and MDA-MB-231). Together with the G1 increase, a concomitant reduction was found in the S phase (Fig. 6). Notably, cells with hypodiploid DNA content, suggestive of activation of apoptotic signaling, were not detected (data not shown). Similar results were also found with the two lead compounds RSM932A and MN58b, suggesting a common mechanism of action. Our results agree with the data of Granata et al.16 which showed that ChoKα downregulation in ovary-cancer cells inhibits cell proliferation without affecting survival signaling pathways whereas, a reduction in the S-phase, proportional to growth inhibition, was observed in cells knocked down for ChoKα1. The cell cycle also showed a slight G1 cell-cycle arrest in silenced cells compared with controls. On the contrary, Sanchez-Lopez et al.15 showed that RSM932A and MN58b induce a significant decrease of the G1 phase in breast- and colon-cancer cells without any alteration of the other phases of the cell cycle. It should be noted that these data correspond to a concentration (15 μM) higher than that used in the present study.
Figure 6.

Percentage of cells in each phase of the cell cycle in Jurkat (Panels A), MCF-7 (Panel B) and MDA-MB231 cells (Panels C–E) treated with the indicated compounds at the indicated concentrations for 24 h. Cells were fixed and labeled with PI and analyzed by flow cytometry as described in the experimental section.
10a induces low levels of apoptosis
To evaluate whether 10a causes cell death, we conducted a biparametric cytofluorimetric analysis using PI, which stains DNA and enters only dead cells, together with fluorescent immunolabeling of the protein annexin-V, which binds to PS in a highly selective manner. Dual staining for annexin-V and with PI enables the discrimination between live cells (annexin-V−/PI−), early apoptotic cells (annexin-V+/PI−), late apoptotic cells (annexin-V+/PI+), and necrotic cells (annexin-V−/PI+). For a positive control, we used two well-known anticancer drugs such as Cis-Pt and Etoposide that in all cell lines tested induce significant apoptosis.
As depicted in Fig. 7, 10a after 72 h of incubation induced a modest increment in apoptotic cells only in Jurkat cells while both in MCF-7 and MDA-MB-231 the increase appeared not to be significantly different from that of the untreated cells.
Figure 7.

Flow cytometric analysis of apoptotic cells after treatment of Jurkat (panel A), MCF-7 (panel B) and MDA-MB-231 (panel C) cells with 10a at the indicated concentrations after incubation for 72 h. The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. As positive controls Cis-Pt and Etoposide (Eto) were used at the concentration of 50 μM and 5 μM, respectively. Data are represented as mean ±SEM of three independent experiments.
Conclusions
In conclusion, the synthesis of a novel family of 1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))-bispyridinium or –bisquinolinium bromide (10a-l) and their evaluation as inhibitors of choline kinase against a panel of cancer-cell lines are described. These compounds were efficiently synthesized in three steps, starting from the building block 6. The chemistry used was appropriate to obtain the designed target compounds, and both the yield and the time of reaction were improved considerably with microwave irradiation.
Preliminary docking studies performed on both crystal structures, ChoKα1/2 (PDB ID: 4BR3) and (ChoKα1/4 PDB ID: 4CG8), and the analysis of the resulting poses, indicated that these compounds (10a-l) could adopt similar behaviour to that of compound 2 or to compound 4 thanks to the synclinal conformation of the linker that allowed the insertion of these molecules inside the Cho binding site and consequently enhanced the antiproliferative effect. The first experimental validation of the docking studies is shown with the results of tryptophan assays for these compounds, which offer very good Kd values. Among all the compounds, those belonging to the families of pyridinium and quinolinium offered similar or better IC50 ChoKα1 than did the lead compounds MN58b and RSM932A. In fact, the best inhibitors were 10a and 10l, and the crystal structure of ChoKα1/10a showed that the compound binds to the choline binding site, indicating the accuracy of the theoretical predictions, wherein the first one-benzyl- 4- (dimethylamino) pyridinium represents the appropriate fragment to inhibit the enzyme33.
Also, we have shown these compounds to have an excellent antiproliferative profile, better than that of the two lead compounds RSM932A and MN58b in a panel of human tumor-cell lines. More importantly, our compounds presented lower or reduced toxicity in some non-tumor-cell lines in comparison to transformed cells. Indeed our results agree with previous observations indicating increased ChoKα1 activity in cell cultures treated with growth factors or insulin23,49,50.
In this context it is important to note that our results indeed confirm these previous findings, and compound 10a in fact presented higher activity only in rapidly proliferating cells such as mitogen-stimulated lymphocytes in comparison to quiescent cells.
Another important finding is that 10a significantly arrested the cell cycle in G1 together with a sharp reduction of the S phase, confirming their ability to inhibit cell growth. Curiously, despite their ability to block cell proliferation, 10a induced a low proportion of cell death, as reflected by a low level of Annexin-V positive cells (Fig. 7). Notably, this occurred also for the two reference compounds RSM932A and MN58b, which even at the maximum concentration used showed a negligible percentage of apoptotic cells, according to the analysis of the cell cycle. It is important to note that, although some reports26 indicate these two compounds may induce apoptosis, this takes place at concentrations much higher than the IC50 (15 μM), which can have an off-target effect.
This intriguing aspect is under active investigation by our group. Nevertheless, our data demonstrate that 10a is a highly promising new Chokα1 inhibitor and is worthy of further preclinical evaluation as a potential anticancer drug.
Methods
Chemistry
General procedure C for the synthesis of the final compounds 10a-l
A solution of 1eq 1,2-bis(4-bromomethylphenoy)ethane (8) in dry CH3CN was added drop by drop to a solution of 9a-l (2 eq) in dry CH3CN under argon conditions. The mixture was heated under reflux for a 3 additional days and, after cooling down to room temperature, washed with diethyl ether and hexane, filtered, and dried under vacuum to afford 10a-l as a solid product.
Characterization data for final products are described below
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(dimethylamino)pyridinium) bromide (10a)
Following general procedure C furnished 10a as a yellow solid, yield 42%, mp: 62–63 °C. 1H NMR (300 MHz, CD3OD) δ: 8.20 (d, J = 7.86 Hz, 4H), 7.35 (d, J = 8.73 Hz, 4H), 7.02 (d, J = 8.73 Hz, 4H), 6.99 (d, J = 7.86 Hz, 4H), 5.30 (s, 4H), 4.33 (s, 4H), 3.24 (s, 12H). 13C RMN (75 MHz, CD3OD) δ: 161.80 × 2, 158.87 × 2, 143.76 × 4, 131.98 × 4, 129.20 × 2, 117.30 × 4, 109.93 × 4, 68.86 × 2, 62.22 × 2, 41.20 × 4. HRMS (m/z): [M]2+ calcd for C30H36N4O2: 242.1419, found: 242.1409.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(pyrrolidin-1-yl)pyridinium) bromide (10b)
Following general procedure C furnished 10b as a brown solid, yield 48%, mp: 129–130 °C. 1H NMR (300 MHz, CD3OD) δ: 8.17 (d, J = 7.77 Hz, 4H), 7.34 (d, J = 8.73 Hz, 4H), 7.01 (d, J = 8.73 Hz, 4H), 6.84 (d, J = 7.77 Hz, 4H), 5.28 (s, 4H), 4.32 (s, 4H), 3.54 (t, J = 6.84 Hz, 8H), 2.11 (t, J = 6.86 Hz, 8H), 13C NMR (75 MHz, CD3OD) δ: 161.79 × 2, 156.05 × 2, 143.68 × 4, 131.92 × 4, 129.31 × 2, 117.29 × 4, 110.51 × 4, 68.86 × 2, 62.26 × 2, 50.28 × 4, 26.98 × 4. HRMS (m/z): [M]2+ calcd for C34H40N4O2: 268.1576, found: 268.1568.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-((4-chlorophenyl)(methyl)amino)pyridinium) bromide (10c)
Following general procedure C furnished 10c as a white solid, yield 30%, mp: >300 °C. 1H NMR (600 MHz, CD3OD) δ: 8.28 (d, J = 8.5 Hz, 4H), 7.58 (d, J = 8.5 Hz, 4H), 7.37 (m, 8H), 7.02 (d, J = 8.4 Hz, 4H), 6.92 (m, 4H), 5.35 (s, 4H), 4.33 (s, 4H), 3.53 (s, 6H). 13C NMR (75 MHz, CD3OD) δ: 160.85 × 2, 158. 31 × 2 143.52 × 4, 135.52 × 2, 131.94 × 4, 131.13 × 4, 129.28 × 4, 127.95 × 2, 116.27 × 8, 110.27 × 2, 67.81 × 2, 61.64 × 2, 30.55 × 2. HRMS (m/z): [M]2+ Calcd for C20H19N2OCl: 338.1186, found: 338.1194.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(quinuclidinium) bromide (10d)
Following general procedure C furnished 10d as a white solid, yield 56%, mp: >300 °C. 1H NMR (300 MHz, CD3OD) δ: 7.47 (d, J = 8.78 Hz, 4H), 7.13 (d, J = 8.78 Hz, 4H), 4.43 (s, 4H), 4.35 (s, 4H), 3.48 (m, 12H), 2.18 (dt, J = 6.44, 3.23 Hz, 2H), 2.01 (dt, J = 8.23, 3.23 Hz, 12H). 13C NMR (75 MHz, CD3OD) δ: 162.80 × 2, 136.48 × 4, 121.54 × 2, 117.14 × 4, 69.52 × 2, 68.87 × 2, 56.39 × 6, 25.81 × 6, 22.33 × 2. HRMS (m/z) [M]2+ calcd for C30H42N2O2: 231.1623, found: 231.1628.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(3-hydroxyquinuclidinium) bromide (10e)
Following general procedure C furnished 10e as a white solid, yield 63%, mp: 268–270 °C. 1H NMR (300 MHz, DMSO-d6) δ: 7.48 (d, J = 8.66 Hz, 8H), 7.13 (d, J = 8.66 Hz, 8H), 4.43 (s, 8H), 4.41 (s, 8H), 4.08, 3.64, 3.35, 3.04, 2.27, 2.10 (6m, 52H), 13C NMR (75 MHz, DMSO-d6) δ: 159.52 × 4, 134.43 × 8, 119.63 × 4, 114.77 × 8, 65.61 × 4, 62.26 66.37, 63.27, 53.46, 52.52, 26.83, 20.87, 17.29 × 4. HRMS (m/z) [M]2+ calcd for C30H42N2O4: 247.1572, found: 247.1565.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(methyl(phenyl)amino)quinolinium) bromide (10f)
Following general procedure C furnished 10f as a yellow solid, yield 64%, mp: 169–170 °C. 1H NMR (300 MHz, CD3OD) δ: 8.86 (d, J = 7.44 Hz, 2H), 8.13 (d, J = 8.37 Hz). 7.81 (dt, J = 5.57, 1.43 Hz, 2H), 7.62 (dd, J = 8.8, 1.3 Hz, 2H), 7.53 (m, 4H), 7.46 (t, J = 7.36 Hz, 2H), 7.40–7.29 (m, 12H), 7.03 (d, J = 8.76 Hz, 4H), 5.89 (s, 4H), 4.33 (s, 4H), 3.84 (s, 6H). 13C NMR (75 MHz, CD3OD) δ: 161.47 × 2, 160.80 × 2, 150.14 × 2, 148.54 × 2, 141.68 × 2, 135.51 × 2, 132.75 × 4, 130.63 × 4, 130.28 × 2, 130.15 × 2, 128.64 × 2, 127.85 × 2, 127.73 × 4, 122.29 × 2, 120.97 × 2, 117.30 × 4, 107.73 × 2, 68.85 × 2, 59.94 × 2, 46.75 × 2. HRMS (m/z) [M]2+ calcd for C48H44N4O2: 354.1732, found: 354.1736.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-((4-chlorophenyl)(methyl)amino)quinolinium) bromide (10g)
Following general procedure C furnished 10g as a yellow solid, yield 70%, mp: 178–180 °C. 1H NMR (400 MHz, CD3OD) δ: 8.90 (d, J = 7.40 Hz, 2H), 8.16 (dd, J = 8.9, 0.6 Hz, 2H), 7.84 (dt, J = 5.64, 1.36 Hz, 2H), 7.67 (dd, J = 8.8, 1.2 Hz, 2H), 7.52 (d, J = 8.91 Hz, 4H), 7.41–7.37 (m, 8H), 7.33 (d, J = 8.77 Hz, 4H), 7.03 (d, J = 8.77 Hz, 4H), 5.91 (s, 4H), 4.33 (s, 4H). 3.82 (s, 6H), 13C NMR (75 MHz, CD3OD) δ: 161.48 × 2, 160.92 × 2, 148.81 × 2, 148.77 × 2, 141.68 × 2, 135.68 × 2, 135.58 × 2, 132.72 × 4, 130.69 × 4, 130.00 × 2, 129.28 × 4, 128.52 × 2, 128.20 × 2, 122.41 × 2, 121.13 × 2, 117.30 × 4, 108.36 × 2, 68.84 × 2, 60.09 × 2, 46.53 × 2. HRMS (m/z) [M]2+ calcd for C48H42N4O2Cl2: 388.1342, found: 388.1338.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(7-chloro-4-(methyl(phenyl)amino)quinolinium) bromide (10h)
Following general procedure C furnished the crude residue which was purified by flash chromatography using CH2Cl2: MeOH (9:1 v/v) as eluent to obtain 10h as a yellow-green solid, yield 61%, mp: 181–183 °C. 1H NMR (300 MHz, CD3OD) δ: 8.82 (d, J = 7.50 Hz, 2H), 8.15 (d, J = 1.89 Hz, 2H), 7.56–7.53 (m, 6H), 7.47 (t, J = 7.4 Hz, 2H), 7.41–7.40 (d, J = 7.48 Hz, 4H), 7.34–7.31 (m, 6H), 7.29 (dd, J = 9.3, 1.9 Hz, 2H), 7.06–7.03 (d, J = 8.70 Hz, 4H), 5.85 (s, 4H), 4.34 (s, 4H), 3.82 (s, 6H, CH3). 13C NMR (75 MHz, CD3OD) δ: 161.58 × 2, 160.51 × 2, 149.76 × 2, 148.95 × 2, 142.51 × 2, 141.90 × 2, 132.92 × 4, 131.80 × 2, 130.77 × 4, 130.59 × 2, 128.35 × 2, 128.24 × 2, 127.72 × 4, 128.24 × 2, 120.68 × 2, 120.45 × 2, 117.41 × 4, 107.93 × 2, 68.86 × 2, 59.91 × 2, 46.87 × 2. HRMS (m/z): [M]2+ calcd for C48H42N4O2Cl2: 388.1342, found: 388.1331.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(7-chloro-4-((4-chlorophenyl)(methyl)amino)quinolinium) bromide (10i)
Following general procedure C furnished the crude residue which was purified by flash chromatography using CH2Cl2: MeOH (9:1 v/v) as eluent to obtain 10i as a yellow solid, yield 39%, mp: 185–186 °C, 1H NMR (300 MHz, CD3OD) δ: 8.87 (d, J = 7.46 Hz, 2H), 8.20 (d, J = 1.93 Hz, 2H), 7.63 (d, J = 9.31 Hz, 2H), 7.55 (d, J = 8.78 Hz, 4H), 7.42 (d, J = 8.78 Hz, 4H), 7.40 (d, J = 1.96 Hz, 2H), 7.38 (d, J = 7.47 Hz, 2H), 7.35 (d, J = 8.73 Hz, 4H), 7.06 (d, J = 8.73 Hz, 4H, H-2), 5.88 (s, 4H), 4.35 (s, 4H), 3.82 (s, 6H, CH3). 13C NMR (75 MHz, CD3OD) δ: 161.59 × 2, 160.65 × 2, 149.22 × 2, 148.45 × 2, 142.50 × 2, 142.06 × 2, 135.92 × 2, 132.88 × 4, 131.68 × 2, 130.85 × 4, 129.33 × 4, 128.71 × 2, 128.15 × 2, 120.82 × 2, 120.61 × 2, 117.40 × 4, 108.57 × 2, 68.85 × 2, 60.05 × 2, 46.69 × 2. HRMS (m/z) [M]2+ calcd for C48H40N4O2Cl4: 422.0953, found: 422.0952.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(azepan-1-yl)quinolinium) bromide (10j)
Following general procedure C furnished 10j as a yellow solid, yield 67%, mp: 75–77 °C. 1H RMN (300 MHz, CD3OD) δ: 8.54 (d, J = 7.73 Hz, 2H), 8.42 (dd, J = 8.62, 1.28 Hz, 2H), 8.05 (dd, J = 8.83, 1.05 Hz, 2H), 7.90 (dt, J = 5,67, 1,33 Hz, 2H), 7.66 (dt, J = 5.77, 1.15 Hz, 2H), 7.27 (d, J = 8.80 Hz, 4H), 7.10 (d, J = 7.74 Hz), 7.00 (d, J = 8.80 Hz, 4H), 5.74 (s, 4H), 4.30 (s, 4H), 4.09 (m, 8H), 2.09 (m, 8H), 1.75 (dt, J = 5.40, 2.54 Hz, 8H), 13C NMR (75 MHz, CD3OD) δ: 161.91 × 2, 161.34 × 2, 146.48 × 2, 142.10 × 2, 135.52 × 2, 130.48 × 4, 130.44 × 2, 128.98 × 2, 127.04 × 2, 121.41 × 2, 120.33 × 2, 117.22 × 4, 104.55 × 2, 68.84 × 2, 58.99 × 2, 56.06 × 4, 29.37 × 4, 29.23 × 4. HRMS (m/z) [M]2+ calcd for C46H52N4O2: 346.6700, found 346.2039.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(4-(azepan-1-yl)-7-chloroquinolinium) bromide (10k)
Following general procedure C furnished the crude residue which was purified by flash chromatography using CH2Cl2: MeOH (8:1 v/v) as eluent to obtain 10k as a white solid, yield 41%, mp: 87–88 °C. 1H RMN (400 MHz, CD3OD) δ: 8.51 (d, J = 7.75 Hz, 2H), 8.40 (d, J = 9.21 Hz, 2H), 8.07 (d, J = 1.82 Hz, 2H), 7.29 (d, J = 8.64 Hz, 4H), 7.65 (dd, J = 9.2, 1.9 Hz, 2H), 7.11 (d, J = 7.78 Hz, 2H), 7.03 (d, J = 8.64 Hz, 4H), 5.72 (s, 4H), 4.33 (s, 4H), 4.08 (m, 8H), 2.08 (m, 8H), 1.74 (m, 8H). 13C NMR (75 MHz, CD3OD) δ :161.56 × 2, 161.44 × 2, 146.77 × 2, 142.95 × 2, 141.82 × 2, 132.31 × 2, 130.59 × 4, 128.57 × 2, 127.45 × 2, 119.88 × 2, 119.78 × 2, 117.32 × 4, 105.00 × 2, 68.85 × 2, 59.00 × 2, 56.12 × 4, 29.28 × 4, 29.16 × 4. HRMS (m/z) [M-Br]+ calcd for C46H50N4O2Cl2Br : 839.24949, found: 839.2494.
1,1′-(((ethane-1,2-diylbis(oxy))bis(4,1-phenylene))bis(methylene))bis(7-chloro-4-(pyrrolidin-1-yl)quinolinium) bromide (10l)
Following general procedure C furnished the crude residue which was purified by flash chromatography using CH2Cl2: MeOH (9:1 v/v) as eluent to obtain 10l as a white solid, yield 48%, mp: 118–120 °C. 1H RMN (300 MHz, CD3OD) δ: 8.63 (d, J = 9.23 Hz, 2H), 8.51 (d, J = 7.69 Hz, 2H), 8.04 (d, J = 2.02 Hz, 2H), 7.66 (dd, J = 9.2, 2.0 Hz, 2H), 7.27 (d, J = 8.75 Hz, 4H), 7.01 (d, J = 8.75 Hz, 4H), 6.90 (d, J = 7.69 Hz, 2H), 5.71 (s, 4H), 4.32 (s, 4H), 4.02 (m, 8H), 2.20 (m, 8H). 13C NMR (75 MHz, CD3OD) δ: 161.37 × 2, 157.92 × 2, 146.91 × 2, 142.45 × 2, 141.76 × 2, 131.76 × 2, 130.45 × 4, 128.68 × 2, 127.71 × 2, 120.16 × 2, 119.73 × 2, 117.29 × 4, 104.64 × 2, 68.83 × 2, 59.06 × 2, 55.43 × 4, 24.64 × 4. HRMS (m/z) [M]2+ calcd for C42H42N4O2Cl2: 352.1337, found: 352.1353.
Docking Studies
Molecular-modeling studies were performed by using Sybyl program51. Crystal structures of human ChoKα1 in complex with compounds 2 (PDB entry 4BR3) and 4 (PDB entry 4CG8) were used for docking studies. In both cases, using the Structure Preparation Tool module of Sybyl refined protein structure. Missing side chains of those residues situated far away from the binding sites were added and protein N-terminal and C-terminal were fixed with ACE and NME, respectively. Hydrogens and charges were also added and protonation type of Glu, Asp, Gln and Asp was analyzed and fixed. Hydrogen orientations were also checked in order to maintain intramolecular hydrogen bonds within the protein. Finally, the molecules of compounds 2 and 4 inserted into the ATP binding site were carefully checked to assure the correction of these molecules. Structures of compounds 10a-l were constructed from standard fragments of the Libraries of the Sybyl program, and used as ligands for docking studies. As previously described52, a new type of atom was necessary to define in order to build the molecules: N.ar4, the quaternary nitrogen of the pyridinium fragments. Additional parameters were also developed from initio calculations to optimize the geometry of these molecules Atomic charges were calculated by means of Gaussian Program53 and optimizations were undertaken using the BFGS method.
The Surflex-Dock54, module implemented in the Sybyl program was used for docking studies. Surflex Dock Protomol was prepared using compound 2 or 4 inserted into the ChoK binding site, with a threshold value of 0.5 and a Bloat of 0 A. Surflex-Dock GeomX (SFXC) protocol was used, the search grid was expanded in 5 Å, 50 additional starting conformations were used for each molecule and 30 conformations per fragment. The results were analyzed using the Sybyl program and the most stable pose for each molecule was chosen as the preferred one inside the ChoK enzyme. Figures were built using the PyMOL program55.
Biological experiments
Materials and Methods
[methyl-14C]choline chloride (55 mCi/mmol) was supplied by Perkin Elmer (Massachusetts, USA). Fetal bovine serum (FBS) was from The Cell Culture Company (Pasching, Austria). Minimal essential medium (MEM) was from Sigma-Aldrich (Madrid, Spain). Cell proliferation reagent WST-1 was from Roche Applied Sciences (Mannhein, Germany). Thin-layer chromatography (TLC) plates of Silica Gel 60 A were acquired from Analtech (Newark, DE, USA). Microwell plates and culture dishes were obtained from NuncTM (Langenselbold, Germany). All other reagents were of analytical grade.
Determination of Human choline kinase α1 (ChoKα1) activity
The effect of the different inhibitors on human choline kinase (ChoK) was assayed in ChoKα1 purified as previously described32. In each experiment DMSO-assays were always run in parallel as a control. DMSO in no case exceeded a concentration of 0.1% in order to avoid unspecific ChoK inhibition. ChoK activity was assayed by measuring the rate of incorporation of 14C from [methyl-14C]choline into phosphocholine both in the presence or absence of different inhibitor concentrations. Briefly, the final reaction mixture contained 100 mM Tris (pH 8.5), 10 mM MgCl2, and 10 mM ATP, and 20 ng of purified ChoKα1. After the samples were preincubated at 37 °C for 5 min, the reaction was initiated with 1 mM [methyl-14C]choline (4500 dpm/nmol) and incubated at 37 °C for 10 min, the final volume being 55 μl. The assay was stopped by immersing the reaction tubes in boiling water for 3 min. Aliquots of the reaction mixture were applied to the origin of Silica Gel plates in the presence of phosphocholine (0.1 mg) and choline (0.1 mg) as carriers. The chromatography was developed in methanol/0.6% NaCl/28% NH4OH in water (50:50:5, v/v/v) as solvent. Phosphocholine was visualized under exposure to iodine vapor and the corresponding spot was scraped and transferred to scintillation vials for measurement of radioactivity by a Beckman 6000-TA (Madrid, Spain) liquid-scintillation counter. At least three experiments were performed in all assays. The 50% inhibitory concentrations (IC50 values) were determined from the % activity of the enzymes at different concentrations of synthetic inhibitors by using a sigmoidal dose-response curve (the ED50plus v1.0 software).
Cloning and purification of ChoKα1
Details on cloning and purification of human ChoKα1 have been previously reported32.
Tryptophan fluorescence quenching
All compounds were prepared in 100% DMSO. Their Kds against human ChoKα1 were measured by monitoring the quenching of tryptophan fluorescence. All experiments were performed in a Cary Eclipse spectrofluorometer (Varian) at 25 °C with the enzymes at 1 μM, and concentrations of compounds varying from 0.1 to 5 μM for ChoKα1 in 25 mM Tris, 150 mM NaCl, pH 7.5. Fluorescence emission spectra were recorded in the 300–400 nm range with an excitation wavelength of 280 nm, with slit width of 5 nm. Controls were determined by incubating the enzymes with equivalent amounts of DMSO. As indicated previously, data analysis was performed in Prism (GraphPad software) considering a model with a single binding site (Eq. 1), where F0 is the intrinsic fluorescence of the enzyme in the absence of quencher (Q), F1 is the observed fluorescence at a given quencher concentration, fa is the fractional degree of fluorescence, and Kd is the dissociation constant.
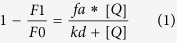 |
Protein crystallography
ChoKα1 at 20 mg/ml in buffer 25 mM Tris/HCl, 150 mM NaCl pH 7.5 was used as the protein solution. The sitting-drop vapor-diffusion method was used to produce apo-crystals by mixing 0.5 μl of the protein solution and an equal volume of mother liquor (crystals appeared in 20% polyethylene glycol [PEG] 3350 and 0.25 M potassium isothiocyanate). Tetragonal crystals (space group P43212) grew within in 3–4 days and were soaked in 2 μL of the mother liquor with 0.2 μL of a dilution 500 mM of compounds 10a, 10g, 10h, 10k, and 10l in DMSO (DMSO was at 10% final concentration in the mix whereas compounds were at 50 mM) for two days. Only crystals soaked with compound 10a contained the compound. The crystals used in this study were cryoprotected in mother-liquor solutions containing 20% ethylenglycol and frozen in a nitrogen gas stream cooled to 100 K.
Diffraction data of the binary complexes were collected at beamline I04-1 (Diamond, Oxford). The data was processed and scaled using the XDS package and CCP4 software, relevant statistics are given in Table S2 of Supplementary information
Structure determination and refinement
The structure of the binary complex was solved by molecular replacement using PDB ID 3G15 as a template. Initial phases were further improved by cycles of manual model building in Coot and refinement with REFMAC5. The final model was validated with PROCHECK, model statistics are given in Table S2 of the supplemental information section. Coordinates and structure factors have been deposited in the Worldwide Protein Data Bank (wwPDB, and see Table S2 for the pdb code).
Antiproliferative assays in cancer cells
Human T-cell leukemia (Jurkat), human B-cell leukemia (RS 4,11) and human promyelocytic leukemia (HL-60) cells were grown in RPMI-1640 medium (Gibco, Milan, Italy). Breast adenocarcinoma (MCF-7 and MDA-MB-231), human non-small cell lung carcinoma (A549), human cervix carcinoma (HeLa), human prostate adenocarcinoma (PC-3), and human colon adenocarcinoma (HT-29) cells were grown in DMEM medium (Gibco, Milan, Italy). Both media were supplemented with 115 units/mL of penicillin G (Gibco, Milan, Italy), 115 μg/mL of streptomycin (Invitrogen, Milan, Italy) and 10% fetal bovine serum (Invitrogen, Milan, Italy). Stock solutions (10 mM) of the different compounds were obtained by dissolving them in DMSO. Individual wells of 96-well tissue-culture microtiter plates were inoculated with 100 μL of complete medium containing 8 × 103 cells. The plates were incubated at 37 °C in a humidified 5% CO2 incubator for 18 h prior to the experiments. After medium removal, 100 μL of fresh medium containing the test compound at different concentrations was added to each well and incubated at 37 °C for 72 h. The percentage of DMSO in the medium in no case exceeded 0.25%. Cell viability was assayed by the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide test as previously described56. The GI50 was defined as the compound concentration required to inhibit cell proliferation by 50%, in comparison with cells treated with the maximum amount of DMSO (0.25%) and considered as 100% viability. In additional experiments, cell viability was also determined by calculating the values of trypan blue positive cells (dead cells) and trypan blue negative (live cells) from a mixture of the cell suspension and 0.4% trypan blue solution.
Antiproliferative assays in non-tumoral cells
Peripheral mononuclear cells (PBMC) from healthy donors were obtained by separation on Lymphoprep (Fresenius KABI Norge AS) gradient. After extensive washing, cells were resuspended (1 × 106 cells/mL) in RPMI-1640 with 10% fetal bovine serum and incubated overnight. For cytotoxicity evaluations in proliferating PBL cultures, non-adherent cells were resuspended at 5 × 105 cells/mL in growth medium, containing 2.5 μg/mL PHA (Irvine Scientific). Different concentrations of the test compounds were added, and viability was determined 72 h later by the MTT test. For cytotoxicity evaluations in resting PBL cultures, non-adherent cells were resuspended (5 × 105 cells/mL) and treated for 72 h with the test compounds, as described above.
Human Umbilical Vein Endothelial cells (HUVEC), were prepared from human umbilical-cord veins, as previously described57. The adherent cells were maintained in M200 medium added by LSGS (low serum growth supplement), containing FBS, hydrocortisone, hEGF, bFGF, heparin, gentamycin/amphotericin (Life Technologies, Monza, Italy). Once confluent, the cells were detached by trypsin–EDTA solution and used in experiments from the first to sixth passages.
Human fibroblasts from foreskin were isolated as previously described58 and maintained in DMEM medium with 10% fetal bovine serum added.
Cell-cycle distribution analysis
For flow-cytometric analysis of the DNA content, 5 × 105 HeLa or Jurkat cells in exponential growth were treated with different concentrations of the test compounds for 24 and 48 h. After the incubation period, the cells were collected, centrifuged, and fixed with ice-cold ethanol (70%). The cells were then treated with lysis buffer containing RNAse A and 0.1% Triton X-100, and then stained with propidium iodide (PI). Samples were analyzed in a Cytomics FC500 flow cytometer (Beckman Coulter). DNA histograms were analyzed using MultiCycle for Windows (Phoenix Flow Systems).
Annexin-V/PI assay
Surface exposure of PS on apoptotic cells was measured by flow cytometry with Cytomics FC500 (Beckman Coulter) by adding simultaneously annexin-V conjugated to fluorescein isothiocyanate (FITC) and PI to cells according to the manufacturer′s instructions (Annexin-V Fluos, Roche Diagnostic).
Additional Information
How to cite this article: Schiaffino-Ortega, S. et al. Design, synthesis, crystallization and biological evaluation of new symmetrical biscationic compounds as selective inhibitors of human Choline Kinase α1 (ChoKα1). Sci. Rep. 6, 23793; doi: 10.1038/srep23793 (2016).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the ‘Consejería de Innovación, Ciencia y Empresa, Junta de Andalucía’ (Excellence Research Project: P07-CTS-03210), the ‘Diputación General de Aragón (B89)’ and the ‘Ministerio de Ciencia e Innovación’ (SAF2009-11955, BFU2010-19504 and CTQ2013-44367-C2-2-P) for the financial support, the award of grants from ‘Ministerio de Educación’ to P.R.-M. and S.S.-E. is gratefully acknowledged, and the ‘Centro de Servicios de Informática of the University of Granada (Spain) for the use of their computers and scientific software. G.V.,R.B., R.M. and G.B. We thanks also the Fondazione Cariparo by the “Progetto Ricerca Pediatrica”. We thank synchrotron radiation sources ALBA (Barcelona), and in particular the beamline XALOC. The research leading to these results has also received funding from the FP7 (2007–2013) under BIOSTRUCTX-7687.
Footnotes
Author Contributions L.C.L.-C. and A.E. designed the compounds and their synthesis L.C.L.-C., S.S.O. and E.B. synthesized and characterized the compounds L.S.A. crystallized the compound and 10a and Quenching of intrinsic ChoKα1 tryptophan fluorescence. L.C.L.-C. analyzed the chemical data. M.G. and A.E. funded the chemical synthesis. R.H.G funded the crystallization and tryptophan fluorescence assays. M.P.C., P.R.M. and C.M. carried out the ChoKα1 inhibition assays. R.B. and E.M, Performed the biological experiments. L.C.L.-C., G.B., G.V., A.E. interpreted the biological data and wrote the work.
References
- Adje A. A. et al. Novel anticancer agents in clinical development. Cancer Biol. Ther. 2, S5–S15, (2003). [PubMed] [Google Scholar]
- Neidle S. et al. Chemical approaches to the discovery and development of cancer therapies. Nat. Rev. Cancer. 5, 285–296 (2005). [DOI] [PubMed] [Google Scholar]
- Kamb S. et al. Why is cancer drug discovery so difficult? Nat. Rev. Drug Discov. 6, 115–120 (2007). [DOI] [PubMed] [Google Scholar]
- Brognard J. & Hunter T. Protein Kinase Signalling Networks in Cancer Curr Opin Genet Dev. 21, 4–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Wagih O. & Bader G. D. The mutational landscape of phosphorylation signaling in cancer. Sci Rep. 3, 2651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S., Rahal R., Stransky N., Lengauer C. & Hoeflich K. P. Targeting cancer with kinase inhibitors J Clin Invest. 125, 1780–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishidate K. Choline⁄ethanolamine kinase from mammalian tissues. Biochim Biophys Acta. 1348, 70–78 (1997). [DOI] [PubMed] [Google Scholar]
- Aoyama H. et al. Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid. Res. 43, 266–281 (2004). [DOI] [PubMed] [Google Scholar]
- Kent C. Regulation of phosphatidylcholine biosynthesis. Prog. Lipid. Res. 29, 87–105 (1990). [DOI] [PubMed] [Google Scholar]
- Exton J. H. & Phospholipase D. Ann N Y Acad Sci. 905, 61–68 (2000). [DOI] [PubMed] [Google Scholar]
- Aoyama C. et al. Molecular cloning of mouse choline kinase and choline/ethanolamine kinase: their sequence comparison to the respective rat homologs. Biochim. Biophys. Acta. 1393, 179–185 (1998). [DOI] [PubMed] [Google Scholar]
- Aoyama C. et al. Complementary DNA sequence for a 42 kDa rat kidney choline/ethanolamine kinase. Biochim. Biophys. Acta. 1390, 1–7 (1998). [DOI] [PubMed] [Google Scholar]
- Aoyama C. et al. Expression and characterization of the active molecular forms of choline/ethanolamine kinase-alpha and -beta in mouse tissues, including carbon tetrachloride-induced liver. Biochem. J. 363, 777–784 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez de Molina A. et al. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem. Biophys. Res. Commun. 296, 580–583 (2002). [DOI] [PubMed] [Google Scholar]
- Hernández-alcoceba R., Fernández F. & Lacal J. C. In Vivo Antitumor Activity of Choline Kinase Inhibitors : A Novel Target for Anticancer Drug Discovery Cancer Res. 59, 3112–3118 (1999). [PubMed] [Google Scholar]
- Granata A. et al. Choline kinase-alpha by regulating cell aggressiveness and drug sensitivity is a potential druggable target for ovarian cancer. Br J Cancer. 110, 330–340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J. et al. Dysregulated choline metabolism in T-cell lymphoma: role of choline kinase-α and therapeutic targeting. Blood Cancer J 5, 287 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glunde K., Bhujwalla Z. M. & Ronen S. M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 11, 835–848 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisach D. et al. The crystal structure of choline kinase reveals a eukaryotic protein kinase fold. Structure. 11, 703–713 (2003). [DOI] [PubMed] [Google Scholar]
- Malito E. et al. Elucidation of human choline kinase crystal structures in complex with the products ADP or phosphocholine. J. Mol. Biol. 364, 136–151. (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Ruiz B. et al. Design, Synthesis, Theoretical Calculations and Biological Evaluation of New Non Symmetrical Choline Kinase Inhibitors. Eur. J. Med. Chem. 50, 154–162 (2012). [DOI] [PubMed] [Google Scholar]
- Schiaffino-Ortega S. et al. New non-symmetrical choline kinase inhibitors. Bioorg. Med. Chem. 21, 7146–7154 (2013). [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A. et al. Inhibition of choline kinase as a specific cytotoxic strategy in oncogene-transformed cells. Oncogene. 22, 8803–8812 (2003). [DOI] [PubMed] [Google Scholar]
- Ramírez de Molina A. et al. From Ras signalling to ChoK inhibitors: a further advance in anticancer drug design. Cancer Lett. 206, 137–148 (2004). [DOI] [PubMed] [Google Scholar]
- Al-Saffar N. M. et al. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of the choline kinase inhibitor MN58b in human carcinoma models. Cancer Res. 66, 427–434 (2006). [DOI] [PubMed] [Google Scholar]
- Hernández-Alcoceba R. et al. In vivo antitumor activity of choline kinase inhibitors: a novel target for anticancer drug discovery. Cancer Res. 59, 3112–3118 (1999). [PubMed] [Google Scholar]
- Lloveras J. et al. Action of hemicholinium-3 on phospholipid metabolism in Krebs II ascites cells. Biochem. Pharmacol. 34, 3987–3993 (1985). [DOI] [PubMed] [Google Scholar]
- Ramirez de Molina A. et al. Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 64, 6732–6739 (2004). [DOI] [PubMed] [Google Scholar]
- Lacal J. C. & Campos J. M. Preclinical Characterization of RSM-932A, a Novel Anticancer Drug Targeting the Human Choline Kinase Alpha, an Enzyme Involved in Increased Lipid Metabolism of Cancer Cells. Mol. Canc Ther 14, 31–40 (2015). [DOI] [PubMed] [Google Scholar]
- Sahún-Roncero M. et al. The Mechanism of Allosteric Coupling in Choline Kinase α1 Revealed by the Action of a Rationally Designed Inhibitor. Angew Chem. 52, 4582- 4586 (2013). [DOI] [PubMed] [Google Scholar]
- Sahún-Roncero M. et al. Determination of Potential Scaffolds for Human Choline Kinase α1 by Chemical Deconvolution Studies. ChemBioChem. 14, 1291–1295 (2013). [DOI] [PubMed] [Google Scholar]
- Rubio-Ruiz B. et al. Discovery of a New Binding Site on Human Choline Kinase α1: Design, Synthesis, Crystallographic Studies, and Biological Evaluation of Asymmetrical Bispyridinium Derivative. J. Med. Chem. 57, 507–515 (2014). [DOI] [PubMed] [Google Scholar]
- Serrán-Aguilera L. et al. Pharmacophore-based virtual screening to discover new active compounds for human choline kinase α1. Molecular Informatics, 34, 458–466 (2015). [DOI] [PubMed] [Google Scholar]
- Serrán-Aguilera L. et al. Choline Kinase Active Site Provides Features for Designing Versatile Inhibitors. Current Topic In Medicinal Chemistry, 14, 2684–2693 (2014). [DOI] [PubMed] [Google Scholar]
- Castro-Navas F. F. et al. New more polar symmetrical bipyridinic compounds: new strategy for the inhibition of choline kinase α1. Future Medicinal Chemistry. 7, 417–436 (2015). [DOI] [PubMed] [Google Scholar]
- Trousil S., et al. Design of symmetrical and nonsymmetrical N,N-dimethylaminopyridine derivatives as highly potent choline kinase alpha inhibitors. Med. Chem. Commun. 4, 693–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Pérez V. et al. Novel 4-Amino Bis-pyridinium and Bis-quinolinium Derivatives as Choline Kinase Inhibitors with Antiproliferative Activity against the Human Breast Cancer SKBR-3 Cell Line. Chem. Med. Chem. 7, 663–669 (2012). [DOI] [PubMed] [Google Scholar]
- Campos J. M. et al. Quantitative Structure Activity Relationships for a Series of Symmetrical Bisquaternary Anticancer compounds. Bioorg. & Med. Chem. 10, 2215–2231 (2002). [DOI] [PubMed] [Google Scholar]
- Conejo-García A. et al. Bispyridinium cyclophanes: novel templates for human choline kinase inhibitors. J. Med. Chem. 46, 3754–3757 (2003). [DOI] [PubMed] [Google Scholar]
- Sánchez-Martín R. M. et al. Symmetrical bis-quinolinium compounds: New human choline kinase inhibitors with antiproliferative activity against the HT-29 cell line. J. Med. Chem. 48, 3354–3363 (2005). [DOI] [PubMed] [Google Scholar]
- Everitt S. et al., Inventors; Vertex Pharmaceuticals Inc., assignee. Compounds useful as inhibitors of choline kinase. European Patent EP, 2, 758, 395, A1, 2014 Jul 30.
- Kappe C. O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 43, 6250–6284 (2004). [DOI] [PubMed] [Google Scholar]
- Loupy A. Microwaves in organic synthesis, Wiley-VCH: Weinheim. 253–290. (2002). [Google Scholar]
- Cantrill S. J. et al. The influence of macrocyclic polytether constitution upon ammmonium ion/crown ether recognition proceses Chem. Eur. J. 6, 2274–2287 (2000). [DOI] [PubMed] [Google Scholar]
- Xiao J. et al. Binuclear titanocenes linked by the bridge combination of rigid and flexible segment: Synthesis and their use as catalysts for ethylene polymerization. Journal of Molecular Catalysis A: Chemical. 267, 86–91 (2007). [Google Scholar]
- Cabezón B. et al. Self-Complementary [2]catenanes and Their Related [3]Catenanes. Chem. Eur. J. 6, 2262–2273 (2000). [DOI] [PubMed] [Google Scholar]
- Gruber J. et al. Balance of human choline kinase isoforms is critical for cell cycle regulation. Implications for the development of choline kinase-targeted cancer therapy. FEBS Journal. 279, 1915–1928 (2012). [DOI] [PubMed] [Google Scholar]
- Báñez-Coronel M. et al. A novel 4,4′-bispyridyl-5,5′-perfluoroalkyl-2,2′-bisoxazol with antitumoral activity via cell cycle arrest and induction of apoptosis. Int. J. Oncol. 25, 1097–1103 (2004). [PubMed] [Google Scholar]
- Uchida T. et al. Purification and properties of choline kinase from rat brain. Biochim Biophys Acta 1043, 281–288 (1990). [DOI] [PubMed] [Google Scholar]
- Warden C. H. et al. Regulation of choline kinase activity and phosphatidylcholine biosynthesis by mitogenic growth factors in 3T3 fibroblasts. J Biol Chem 260, 6006–6011(1985). [PubMed] [Google Scholar]
- SYBYL-X 2.0, Tripos International, 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA. http://www.tripos.com.
- Conejo-García A. et al. Conformational dynamics of a bispyridinium cyclophane. J. Org. Chem. 68, 8697–8699 (2003). [DOI] [PubMed] [Google Scholar]
- Frisch M. J. et al. Gaussian, Inc. (2004). Wallingford CT. USA. URL http: //www.gaussian.com.
- Jain A. N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 21, 281–306 (2007). [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.4, Schrödinger, LLC.
- Romagnoli R. et al.; Concise Synthesis and Biological Evaluation of 2-aroyl-5-amino benzo[b]thiophene derivatives as a novel class of potent antimitotic agents. J. Med. Chem. 56, 9296–9309 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcù E. et al. TR-644 a novel potent tubulin binding agent induces impairment of endothelial cells function and inhibits angiogenesis. Angiogenesis 16, 647–662 (2013). [DOI] [PubMed] [Google Scholar]
- Panula S. et al. Preparation of human foreskin fibroblasts for human embryonic stem cell culture. CSH Protoc pdb.prot5043. (2008). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.