

Abstract
RNase T is a classical member of the DEDDh family of exonucleases with a unique sequence preference in that its 3′‐to‐5′ exonuclease activity is blocked by a 3′‐terminal dinucleotide CC in digesting both single‐stranded RNA and DNA. Our previous crystal structure analysis of RNase T‐DNA complexes show that four phenylalanine residues, F29, F77, F124, and F146, stack with the two 3′‐terminal nucleobases. To elucidate if the π–π stacking interactions between aromatic residues and nucleobases play a critical role in sequence‐specific protein–nucleic acid recognition, here we mutated two to four of the phenylalanine residues in RNase T to tryptophan (W mutants) and tyrosine (Y mutants). The Escherichia coli strains expressing either the W mutants or the Y mutants had slow growth phenotypes, suggesting that all of these mutants could not fully substitute the function of the wild‐type RNase T in vivo. DNA digestion assays revealed W mutants shared similar sequence specificity with wild‐type RNase T. However, the Y mutants exhibited altered sequence‐dependent activity, digesting ssDNA with both 3′‐end CC and GG sequences. Moreover, the W and Y mutants had reduced DNA‐binding activity and lower thermal stability as compared to wild‐type RNase T. Taken together, our results suggest that the four phenylalanine residues in RNase T not only play critical roles in sequence‐specific recognition, but also in overall protein stability. Our results provide the first evidence showing that the π−π stacking interactions between nucleobases and protein aromatic residues may guide the sequence‐specific activity for DNA and RNA enzymes.
Keywords: protein–DNA interactions, protein–RNA interactions, nucleases, π–π interactions
Introduction
Protein–nucleic acid interactions are the driving forces for various biological processes, making sequence‐specific interactions critical for a protein molecule to differentiate its target from nontarget nucleic acid sequences. Sequence‐specific recognition of DNA by proteins is achieved mainly through recognition of sequence‐dependent DNA shape, and recognition of base sequences via nonbonded interactions, including hydrogen bonding and van der Waals interactions.1 Although hydrogen bonding plays a major role in protein–nucleic acid recognition, van der Waals interactions compose more than 30% of protein–DNA contacts,2, 3 and they are even more prevalent than hydrogen bonding in protein–RNA complexes.4 Among van der Waals interactions, the π–π stacking interactions between nucleobases and aromatic amino acids are frequently observed in protein–DNA and protein–RNA complexes.5 Nevertheless, it remains uncertain if π–π stacking interactions contribute to protein sequence‐specific recognition of nucleic acids.
Here we used RNase T as a model system to investigate the sequence‐specific interactions between proteins and nucleic acids via π–π stacking interactions. RNase T is a member of the well‐characterized DEDDh family of exonucleases that digest nucleic acids by removing one nucleotide at a time at 3′ end via a two metal ion dependent pathway.6, 7 RNase T was first identified as a 3′‐5′ exoRNase participating in tRNA and rRNA maturation in Escherichia coli.8, 9, 10, 11, 12, 13, 14 Recently, it was characterized as an exoDNase, processing the 3′ end of structural DNA, including bulge, bubble and Y‐structured DNA, in various DNA repair pathways.15
RNase T can process many different RNA and DNA precursors with different sequences and structures because it has a unique sequence specificity. Its exonuclease activity is inhibited by a 3′‐end C, and almost completely abolished by a 3′‐end dinucleotide CC sequence in digesting single‐stranded DNA and RNA. The crystal structures of RNase T have been co‐crystallized with a cleavable and noncleavable single‐stranded DNA with a 3′‐end CC or AA sequences.16, 17 In the cleavable AA complex, the aromatic side chains of F29 and F77 stack with the 3′‐end A, whereas F124 and F146 stack with the penultimate 3′‐end A (Fig. 1). In the noncleavable CC complex, the side chain of E73 is rotated to form hydrogen bonds with the 3′‐end C, and the four phenylalanine residues shift their positions slightly to stack with the 3′‐end CC. Together, these residues pull the scissile phosphate away from the active site [Fig. 1(C)]. As a result, the general base H181 is shifted away from the distorted active site with only a single bound Mg2+ ion, and, therefore, the DNA with a 3′‐end CC sequence cannot be hydrolyzed by RNase T.16
Figure 1.
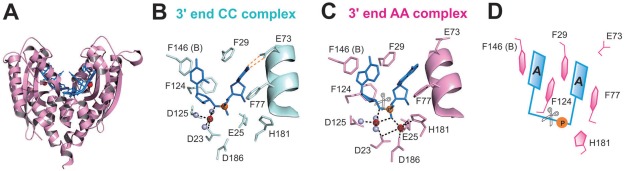
Crystal structures of RNase T in complex with the cleavable (AA complex) and noncleavable (CC complex) single‐stranded DNA. (A) Overall structure of the dimeric RNase T bound with a DNA segment with a sequence of 5′‐ACC‐3′. (B) In the CC complex (PDB entry: 3V9Z), the DNA with a 3′‐end CC is bound in an inactive conformation with only one Mg2+ ion (red sphere) in the active site. (C) In the AA complex (PDB entry: 3V9X), a DNA with a 3′‐end AA is bound in an active conformation with two Mg2+ ions in the active site. The grey spheres represent water molecules bound to Mg2+. (D) Schematic diagram showing that the 3′‐end AA bases stack with the four phenylalanine side chains in RNase T. The scissor indicates the phophodiester bond that is cleaved by RNase T.
Our previous structural analyses suggested that the four phenylalanine residues likely play a critical role in the sequence preference of RNase T. To examine the sequence‐specific activity of RNase T, we constructed a series of RNase T mutants, replacing two to four of the phenylalanine residues, F29, F77, F124, and F146, with aromatic residues tyrosine (Y mutants) or tryptophan (W mutants). We found that these Y and W mutants could not, or could only weakly, support the growth of E. coli, indicating the critical importance of these phenylalanine residues in the ability of RNase T to process DNA and RNA. DNA digestion assays further confirmed that two of the Y mutants indeed had altered sequence preferences and digested DNA with a 3′‐end CC equally well as those with a 3′‐end GG. However, most of the W and Y mutants had low exonuclease activities due to reduced thermal stability and DNA‐binding activity. Our results support that the π–π stacking interactions between protein aromatic side chains and nucleobases may guide the sequence specificity of nucleic acid enzymes.
Results
Loss of phenylalanine residues in RNase T inhibits cell growth
To investigate the role of π–π stacking interactions in sequence‐specific protein–DNA interactions, we constructed four RNase T mutants, replacing the F29 and F77 phenylalanine residues that stack with the 3′‐end nucleobase with tryptophan (W) or tyrosine (Y) and some of the F124 and F146 residues with the nonaromatic alanine, generating 2W1A (F29W/F77W/E73A), 2W3A (F29W/F77W/F124A/F146A/E73A), 2Y1A (F29Y/F77Y/E73A), and 2Y3A (F29Y/F77Y/F124A/F146A/E73A) [see Fig. 2(A)]. We also constructed four RNase T mutants with all four phenylalanine residues (F29, F77, F124, and F146) mutated to tryptophan (W) or tyrosine (Y), including 4W (F29W/F77W/F124W/F146W), 4W1A (F29W/F77W/F124W/F146W/E73A), 4Y (F29Y/F77Y/F124Y/F146Y), and 4Y1A (F29Y/F77Y/F124Y/F146Y/E73A). E73 was also mutated to A in some of these mutants to exclude its influence because E73 may form hydrogen bonds with the 3′‐end C and contribute to the sequence‐specific activity of RNase T. Moreover, we also constructed a mutant for the negative control, replacing the critical 3′‐end nucleobase‐binding residues F29, E73, and F77 to A, which we referred to as the “binding” mutant.
Figure 2.
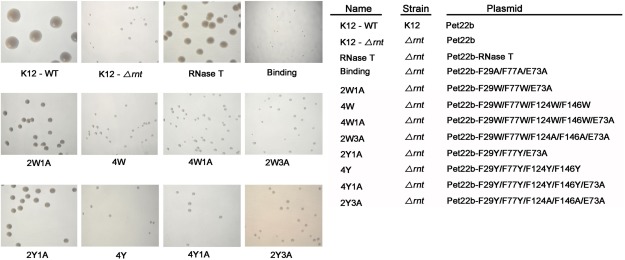
Transformation rescue experiments of the RNase T‐knockout E. coli K12 strains (Δrnt) by expression of RNase T mutants. The RNase T deletion strain (K12‐Δrnt) had a slow‐growth phenotype that can be fully rescued by introducing a plasmid expressing wild‐type RNase T as shown by the colony size variations of E. coli K12 strains on LB plates. The negative control of the “Binding” mutant (F29A/E73A/F77A) could not rescue the slow growth phenotype. The 2W1A and 2Y1A mutants moderately rescued, whereas the 4W, 4W1A, 2W3A, 4Y, 4Y1A, and 2Y3A mutants could not rescue, the slow growth phenotype of the RNase T deletion strain.
We carried out the cell transformation rescue experiments in the rnt‐deletion strain (K12‐Δrnt), which had a slow growth phenotype compared to the wild‐type K12 strain (K12‐WT) (Fig. 2). The binding mutant (F29A/E73A/F77A) with mutations at the substrate‐binding site indeed could not rescue the slow growth phenotype of the rnt deletion strain, whereas expression of RNase T successfully rescued the slow growth phenotype [Fig. 2(A)]. The RNase T mutants 2W1A and 2Y1A moderately rescued cell growth, whereas 4W, 4W1A, 2W3A, 4Y, 4Y1A, and 2Y3A did not rescue the bacteria and had a slow‐growth phenotype similar to that of the RNase T‐knockout mutant strain (see Table 1). Cell growth was improved by introducing the plasmid expressing 2W1A and 2Y1A mutants, likely because these two mutants had only three mutated residues. All the other mutants, containing four to five mutated residues, had much worse defects in cell growth. This result was consistent to the previous finding showing that the double‐mutant F124A/F146A and the triple‐mutant F29A/E73A/F77A had largely reduced DNA‐binding and exonuclease activity, and could not support the growth of E. coli.16 Taken together, these results suggest that all the four phenylalanine residues are critical for the exonuclease activity of RNase T.
Table 1.
The Cell Growth Phenotypes and Sequence‐Specific Exonuclease Activities of the RNase T Mutants
| RNase T mutant | Cell growtha | Melting Point (°C) | 3'‐end sequence preference in digesting 5'‐AGTTATGAXX−3' | |
|---|---|---|---|---|
| RNase T | Wild type | +++ | 50 | AA > GG ≫ CC |
| 2W1A | F29W/F77W/E73A | + | ||
| 4W | F29W/F77W/F124W/F146W | – | ||
| 4W1A | F29W/F77W/F124W/F46W/E73A | – | ||
| 2W3A | F29W/F77W/F124A/F146A/E73A | – | 42 | AA > GG ≫ CC |
| 2Y1A | F29Y/F77Y/E73A | + | ||
| 4Y | F29Y/F77Y/F124Y/F146Y | – | 40 | AA > GG CC |
| 4Y1A | F29Y/F77Y/F124Y/F146Y/E73A | – | 49 | AA > GG CC |
| 2Y3A | F29Y/F77Y/F124A/F146A/E73A | – |
See the transformation rescue experiments of the RNase T‐knockout E. coli K12 strains (Δrnt) by expression of RNase T mutants in Figure 2.
Y mutants of RNase T had altered sequence‐specific exonuclease activity
We suspected that these W and Y mutants had altered sequence specificity and, therefore, they prevented growth in E. coli. To determine the sequence specificity of the RNase T mutants, we further purified the recombinant RNase T mutants for the biochemical assays. The His‐tagged wild‐type and mutated RNase T were expressed and purified by chromatographic methods. Three of the mutants were not well expressed, whereas the remaining five mutants, 2W1A, 2W3A, 2Y1A, 4Y, and 4Y1A, were successfully expressed and purified with high homogeneity, as shown by SDS gel electrophoresis [Fig. 3(A)]. These RNase T mutants had molecular masses close to their estimated masses with less than ±3 Dalton difference as measured by mass spectroscopy (data not shown). All mutants retained their expected secondary structures as they had similar circular dichroism (CD) spectra as that of wild‐type RNase T [Fig. 3(B)].
Figure 3.
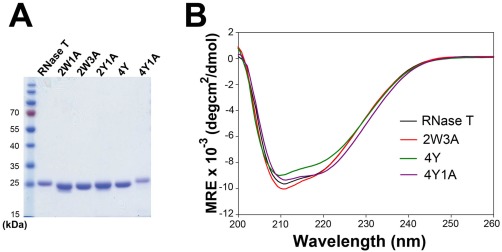
The recombinant RNase T mutants share similar secondary structures to that of wild‐type RNase T. (A) The purified recombinant RNase T mutants, 2W1A, 2W3A, 2Y1A, 4Y, and 4Y1A, had a high structural homogeneity as shown by SDS‐PAGE. (B) The circular dichroism (CD) spectra of RNase T mutants 2W3A, 4Y, and 4Y1A are similar to that of wild‐type RNase T. The spectra were represented by mean residue ellipticity (θ) in deg⋅cm2⋅dmol−1.
We next used single‐stranded DNA with different 3′‐end sequences, AA, CC, or GG (5′‐AGTTATGAXX−3′), as substrates for exonuclease digestion assays using the RNase T mutants 2W3A, 4Y, and 4Y1A, because these three mutants completely lost the ability to support growth in E. coli, which suggests altered sequence specificity. The DNA substrates were labeled at the 5′ end with [γ‐32P] ATP and then incubated with wild‐type or mutant RNase T at different concentrations. All the RNase T mutants had about 1% wild‐type activity and, therefore, we used higher concentrations (0.2–20 μM) of the mutants in the digestion assays compared to the wild‐type enzyme (0.002–0.2 μM). The 2W3A mutant had a similar sequence specificity in digesting ssDNA compared to wild‐type RNase T, with a cleavage order of AA > GG ≫ CC, such that the ssDNA with a 3′‐end CC were not digested [Fig. 4(A,B)]. However, the Y mutants, 4Y1A and 4Y, gained additional cleavage activity for DNA with a 3′‐end CC, with a cleavage order of AA>GG≃CC [Fig. 4(C,D)]. This additional activity suggests that the π–π stacking interactions via the four tyrosine residues, but not the hydrogen bonding via E73, are sufficient to change the sequence specificity of RNase T. Taken together, these results suggest that the replacement of the four phenylalanine residues to tyrosine is sufficient to change the sequence‐dependent activity of RNase T, making it less specific and able to digest ssDNA with not only 3′‐end AA and GG, but also CC.
Figure 4.
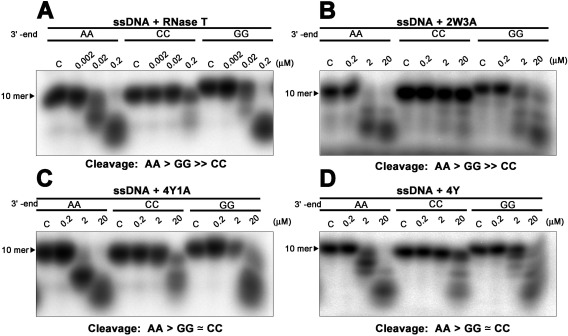
Mutating phenylalanine residues alters the cleavage preferences of RNase T. (A) RNase T was incubated with a single‐stranded DNA with a different sequence at 3'‐end: 5'‐AGTTATGAXX−3', XX = AA, GG, or CC. The exonuclease activity of the wild‐type RNase T is inhibited by the single‐stranded DNA with a 3'‐end CC, with a cleavage order of preference of AA > GG ≫ CC. (B) The RNase T mutant 2W3A (F29W/F77W/E73A/F124A/F146A) had weaker exonuclease activity but a similar sequence‐specific exonuclease activity to wild‐type RNase T. (C,D) The 4Y (F29Y/F77Y/F124Y/F146Y) and 4Y1A (F29Y/F77Y/F124Y/F146Y/E73A) mutants had an altered sequence‐dependent exonuclease activity compared to wild‐type RNase T with a cleavage preference of A > G ≃ C.
W and Y RNase T mutants had reduced DNA‐binding activities and thermal stability
The 2W3A mutant shared a similar sequence‐specific exonuclease activity to that of RNase T, but it still could not support the growth of E. coli, likely because it had a reduced exonuclease activity. To understand why RNase T mutants had reduced exonuclease activities, DNA‐binding activities of these mutants were measured by gel shift assays in the absence of Mg2+ ions. The single‐stranded DNA 5′‐AACCTTACAAA‐3′ was labeled at the 5′ end with [γ‐32P] ATP by T4 polynucleotide kinase and incubated with various concentrations of wild‐type or mutated RNase T (2W3A, 4Y, 4Y1A). Gel shift assays showed that wild‐type RNase T bound DNA at low concentrations (5–20 μM). In contrast, the three RNase T mutants, 2W3A, 4Y, and 4Y1A, bound to ssDNA only at high enzyme concentrations (100–200 μM) [Fig. 5(A)]. These results suggest that the DNA‐binding ability of these mutants was largely reduced (∼1% of the wild type) and that their decreased exonuclease activity primarily resulted from their reduced substrate‐binding activity.
Figure 5.
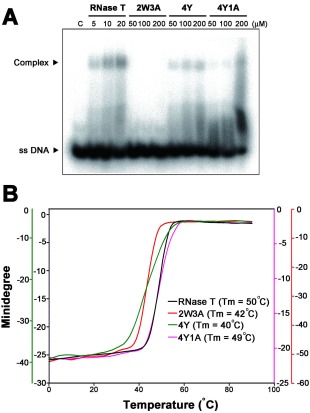
The DNA binding activity and thermal stability of wild type and phenylalanine mutation in RNase T. (A) Gel shift assay revealed that wild‐type RNase T had a higher binding affinity to the single‐stranded DNA (5'‐AACCTTACAAA‐3') than the mutated enzymes, including 2W3A, 4Y, and 4Y1A. (B) The thermal stability of wild‐type and mutated RNase T was monitored by circular dichroism from 1 to 90°C at a wavelength of 220 nm. The melting temperature was 50°C for wild‐type RNase T, 42°C for 2W3A, 40°C for 4Y, and 49°C for 4Y1A.
Aromatic tryptophan and tyrosine residues are capable of forming π–π stacking interactions with nucleobases. We were intrigued that the W and Y RNase T mutants had reduced DNA‐binding activities. To further examine this phenomenon, we measured the thermal denaturation profiles of the wild‐type and mutated RNase T. The melting point (Tm) of RNase T was monitored by CD at a wavelength of 220 nm to measure the increase of the ellipticity at this wavelength due to the disruption of the secondary structure at increased temperatures [Fig. 5(B)]. The wild‐type RNase T had a melting point of 50°C, whereas the three mutants all had reduced melting points: 42°C for 2W3A, 40°C for 4Y, and 49°C for 4Y1A [Fig. 5(B)]. The 4Y mutant had a largely reduced melting point of 40°C but interestingly 4Y1A had a higher melting point of 49°C. Presumably, the hydroxyl groups on the tyrosine side chain of the 4Y mutant interfered with Glu73 side chains and, therefore, the mutation of Glu73 to Ala produced a more stable 4Y1A mutant. This thermal denaturing result suggests that 2W3A and 4Y mutants are not as stable as the wild‐type RNase T, and this is likely one of the major causes for their low DNA‐binding and exonuclease activity.
Discussion
It is not clear if the π−π stacking interaction between aromatic amino acids and nucleobases can determine the sequence specificity of nucleic acid enzymes. Using E. coli RNase T, we examined the four phenylalanine residues that stack with the last two nucleobases in binding nucleic acids. We found that the mutants in which only two phenylalanines were replaced with tyrosine or tryptophan could moderately support the growth of E. coli, whereas the mutants in which all four phenylalanines were replaced, were unable to restore growth. This result suggests that all four phenylalanine residues are important for the exonuclease activity of RNase T. Compared to the wild‐type RNase T, the two tyrosine mutants, 4Y (F29Y/F77Y/F124Y/F146Y) and 4Y1A (F29Y/F77Y/F124Y/F146Y/E73A), had different sequence‐dependent exonuclease activities. RNase T does not digest single‐stranded DNA with a 3′‐end CC, whereas 4Y and 4Y1A gain additional activity in digesting DNA with a 3′‐end CC. This result supports that π–π stacking interactions can guide the sequence‐specific activity of DNA and RNA enzymes.
Previous analyses of 428 high‐resolution crystal structures of protein–DNA complexes reveal 344 π–π interactions between nucleobases and amino acids among 175 structures, suggesting that π–π interactions likely play prevalent roles in protein–DNA interactions.5 Among all the aromatic residues, phenylalanine is the most frequently identified in forming the π–π interactions with nucleobases, with a percentage of 44% for F, 32% for Y, 13% for W, and 11% for H.5 Previous studies on protein–RNA interactions showed that phenylalanine more frequently stacks with purines with a preferential order of A > G > C.4, 18 On the other hand, in protein–DNA complexes, phenylalanine stacks more frequently with pyrimidines with a tendency of T > C > A > G.5 The magnitude of the π–π stacking interactions between phenylalanine and a nucleobase can be up to −30 kJ/mol and vary with the relative distance and orientation. However of the known structures in the protein databank, compared to other nucleobases, cytosine seems to stack less well with phenylalanine with an optimum stacking energy of −21.3 kJ/mol, as compared to −24.4 kJ/mol for A, −25.0 kJ/mol for T, and −26.2 kJ/mol for G.5 Taken together, these statistical and computational analyses suggest that phenylalanine more frequently stacks with different nucleobases with different interaction energies based on distance, orientation, and types of nucleobases.
When a nucleic acid is bound to RNase T, it generally is bound in an active conformation with the 3′‐end nucleobase stacking with F29 and F77 and the penultimate 3′‐end nucleobase stacking with F124 and F146 [Fig. 1(C]. However, when a nucleic acid with a 3′‐end CC is bound to RNase T, likely due to the weaker stacking interactions between phenylalanine and cytosine, it induces a conformational change and the complex structure is slightly different from the ones that are bound with 3′‐end non‐CC substrates [Fig. 1(B,C)]. In the CC complex, besides the hydrogen bonds between E73 and 3′‐end C, F29 also shifts slightly to stack more closely with the 3′‐end C [Fig. 1(B)]. In such an “inactive” conformation, the scissile phosphate is pulled away from the active site, and as a result only a single Mg2+ ion is bound in the distorted active site. Therefore, due to the different stacking interactions between aromatic amino acids and nucleobases, RNase T is capable of differentiating its substrates with 3′‐end CC or non‐CC sequences.
We superimposed the structure of RNase T with a number of DEDDh exonucleases, including oligoribonuclease,19 ε186 of DNA polymerase III,20 CRN‐4,21 3′‐hExo,22 TREX1,23 and TREX2.24 The amino acid residues matched at the same positions as those involved in substrate interactions in RNase T, including F29, E73, F77, F124, and F146, are mostly hydrophobic and aromatic residues, such as leucine, isoleucine, tryptophan, and tyrosine.17 This result suggests that these residues are likely involved in forming van der Waals interactions with nucleic acids and directing the sequence specificity. In conclusion, we show here that RNase T is an ideal model system for investigating π–π stacking interactions between aromatic residues and nucleobases in protein–nucleic acid interactions. We provide mutational and biochemical evidence showing that π–π stacking interactions between nucleobases and protein aromatic residues can guide the sequence‐specific activity for DNA and RNA enzymes.
Materials and Methods
Protein expression and purification
The pET28‐RNase T plasmid bearing the gene encoding the full‐length RNase T inserted in the pET28 vector was constructed as described previously.16 The pET28‐RNase T plasmid was transformed into E. coli BL21‐CodonPlus (DE3)‐RIPL strain (Stratagene), which was cultured in LB medium supplemented with 50 μg/mL kanamycin. Cells were grown to a density of ∼0.5 OD600 and induced by 1 mM IPTG at 18°C for 20 h. The harvested cells were disrupted by a microfluidizer in 50 mM Tris–HCl (pH 8) containing 300 mM NaCl, and the cell extracts were loaded onto a cobalt‐charged HiTrap chelating affinity column (Qiagen) and washed with a step gradient of a buffer containing 0.5M imidazole, 300 mM NaCl, and 50 mM Tris–HCl (pH 8) using a standard protocol. Peak fractions were then applied to a HiTrap heparin column (GE Healthcare) and subsequently, to a gel filtration chromatography column (Superdex 200, GE Healthcare). Purified RNase T samples were concentrated to 20–30 mg/mL in 300 mM NaCl and 50 mM Tris–HCl. The RNase T mutants 2W1A (F29W/F77W/E73A), 4W (F29W/F77W/F124W/F146W), 4W1A (F29W/F77W/F124W/F146W/E73A), 2W3A (F29W/F77W/F124A/F146A/E73A), 2Y1A (F29Y/F77Y/E73A), 4Y (F29Y/F77Y/F124Y/F146Y), 4Y1A (F29Y/F77Y/F124Y/F146Y/E73A), and 2Y3A (F29Y/F77Y/F124A/F146A/E73A) were generated by using the Quik Change Site‐Directed Mutagenesis Kit (Stratagene) and purified using the same procedures as those of wild‐type RNase T.
Transformation rescue experiments
The wild‐type E. coli K‐12 and RNase T knockout (Δrnt) strains used in the transformation rescue experiments were from the Keio collection.25 The rnt gene of the wild type and mutants 2W1A, 4W, 4W1A, 2W3A, 2Y1A, 4Y, 4Y1A, and 2Y3A was inserted into the NdeI/XhoI sites of the expression vector pET‐22b (Novagen) to generate the plasmid encoding RNase T under the control of the T7 promoter. The constructed plasmids were transformed into the RNase T knockout E. coli K12 strain (Δrnt), and the cells were spotted on LB plates containing 100 μg/mL ampicillin and 50 μg/mL kanamycin overnight at 37°C.
Exonuclease activity assays
DNA oligonucleotides with a 3′‐end AA, CC and GG (5′‐AGTTATGAXX−3′) were used for nuclease activity assays. They were labeled at the 5′ end with [γ‐32P] ATP by T4 polynucleotide kinase. The isotope‐labeled DNA was purified on a Microspin G‐25 column (GE Healthcare) to remove the unincorporated nucleotides. Purified substrates were incubated with RNase T or mutants (2W3A, 4Y, 4Y1A) at various concentrations in a buffer solution containing 120 mM NaCl, 2 mM MgCl2, and 50 mM Tris–HCl (pH 7.0) at 37°C for 30 min. Reaction samples were then resolved on 20% denaturing polyacrylamide gels and visualized by autoradiography (Fujifilm, FLA‐5000).
DNA binding assays
For electrophoretic mobility shift assays, 5′‐end 32P‐labeled ssDNA (5′‐GACACTA AAA‐3′) was incubated with RNase T or mutants (2W3A, 4Y, 4Y1A) for 30 min at room temperature in a binding buffer of 250 mM KCl, 5 mM DTT, 2 mM EDTA, 500 µg/mL BSA, 40% glycerol, and 100 mM Tris–HCl, pH 7.4. For low affinity mutants, 2W3A, 4Y, and 4Y1A, higher protein concentrations (50–200 µM) were used. The samples were fractionated by native 20% PAGE at 4°C.
Circular dichroism
CD spectra for RNase T were measured on the Aviv MODEL 400 Circular Dichroism Spectrometer, with a protein concentration of 20 µM in 300 mM NaCl and 50 mM Tris–HCl (pH 7.0). CD spectra were recorded at 25°C from 260 to 195 nm using 0.1‐cm path length quartz cuvettes. The final spectra were represented by mean residue ellipticity (θ) in deg⋅cm2⋅dmol−1. Thermal denaturation curves were monitored by measuring the CD signal at 220 nm from 0 to 90°C at 1°C/min for RNase T and mutants (2W3A, 4Y, 4Y1A). All the experiments were independently repeated three times.
Acknowledgments
Additional assistance was provided by the Institute of Molecular Biology English Editing Core. The authors of this article have no conflicts of interest to declare.
References
- 1. Rohs R, Jin X, West Joshi MS, Honig RB, Mann RS (2010) Origins of specificity in protein‐DNA recognition. Annu Rev Biochem 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Luscombe NM, Laskowski RA, Thornton JM (2001) Amino acid‐base interactions: a three‐dimensional analysis of protein‐DNA interactions at an atomic level. Nucleic Acids Res 29:2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lejeune D, Delsaux N, Charloteaux B, Thomas A, Brasseur R (2005) Protein‐nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins 61:258–271. [DOI] [PubMed] [Google Scholar]
- 4. Ellis JJ, Broom M, Jones S (2007) Protein‐RNA interactions: structural analysis and functional classes. Proteins 66:903–911. [DOI] [PubMed] [Google Scholar]
- 5. Wilson KA, Kellie JL, Wetmore SD (2014) DNA‐protein p‐interactions in nature: abundance, structure, composition and strength of contacts between aromatic amino acids and DNA nucleobases or deoxyribose sugar. Nucleic Acids Res 42:6726–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zuo Y, Deutscher MP (2001) Exonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res 29:1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang W (2011) Nucleases: diversity of structure, function and mechanism. Quart Rev Biophys 44:1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deutscher MP, Marlor CW, Zaniewski R (1984) Ribonuclease T: new exoribonuclease possibly involved in end‐turnover of tRNA. Proc Natl Acad Sci USA 81:4290–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z, Deutscher MP (2002) RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Misra TK, Apirion D (1979) RNase E, an RNA processing enzyme from Escherichia coli. J Biol Chem 254:11154–11159. [PubMed] [Google Scholar]
- 11. Cormack RS, Mackie GA (1992) Structural requirements for the processing of Escherichia coli 5 S ribosomal RNA by RNase E in vitro. J Mol Biol 228:1078–1090. [DOI] [PubMed] [Google Scholar]
- 12. Ehretsmann CP, Carpousis AJ, Krisch HM (1992) Specificity of Escherichia coli endoribonuclease RNase E: in vivo and in vitro analysis of mutants in a bacteriophage T4 mRNA processing site. Genes Dev 6:149–159. [DOI] [PubMed] [Google Scholar]
- 13. Li Z, Deutscher MP (1995) The tRNA processing enzyme RNase T is essential for maturation of 5S RNA. Proc Natl Acad Sci USA 92:6883–6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Z, Pandit S, Deutscher MP (1999) Maturation of 23S ribosomal RNA requires the exoribonuclease RNase T. RNA 5:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsiao Y‐Y, Fang W‐H, Lee C‐C, Chen Y‐P, Hsu P‐C, et al. (2014) Structural insights Into DNA repair by RNase T—an exonuclease processing 3′ end of structured DNA in repair pathways. PLoS Biol 12:e1001803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsiao YY, Yang CC, Lin CL, Lin JL, Duh Y, et al. (2011) Structural basis for RNA trimming by RNase T in stable RNA 3′‐end maturation. Nat Chem Biol 7:236–243. [DOI] [PubMed] [Google Scholar]
- 17. Hsiao YY, Duh Y, Chen YP, Wang YT, Yuan HS (2012) How an exonuclease decides where to stop in trimming of nucleic acids: crystal structures of RNase T‐product complexes. Nucleic Acids Res 40:8144–8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones S, Daley DT, Luscombe NM, Berman HM, Thornton JM (2001) Protein‐RNA interactions: a structural analysis. Nucleic Acids Res 29:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chin KH, Yang CY, Chou CC, Wang AH, Chou SH (2006) The crystal structure of XC847 from Xanthomonas campestris: a 3′‐5′ oligoribonuclease of DnaQ fold family with a novel opposingly shifted helix. Proteins 65:1036–1040. [DOI] [PubMed] [Google Scholar]
- 20. Hamdan S, Carr PD, Brown SE, Ollis DL, Dixon NE (2002) Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure 10:535–546. [DOI] [PubMed] [Google Scholar]
- 21. Hsiao YY, Nakagawa A, Shi Z, Mitani S, Xue D, et al. (2009) Crystal structure of CRN‐4: implications for domain function in apoptotic DNA degradation. Mol Cell Biol 29:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cheng Y, Patel DJ (2004) Crystallographic structure of the nuclease domain of 3′hExo, a DEDDh family member, bound to rAMP. J Mol Biol 343:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, Hollis T. (2007) The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a polyproline II helix for protein partnering. J Biol Chem 282:10537–10543. [DOI] [PubMed] [Google Scholar]
- 24. Perrino FW, Harvey S, McMillin S, Hollis T (2005) The human TREX2 3′ ‐> 5′‐exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis. J Biol Chem 280:15212–15218. [DOI] [PubMed] [Google Scholar]
- 25. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. (2006) Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]