

Abstract
NF‐κB is a major transcription factor that mediates a number of cellular signaling pathways. Crystal structure analysis gives an incomplete picture of the behavior of the protein, particularly in the free state; free monomers or dimers of NF‐κB have never been crystallized. NMR analysis gives insights into the structure and dynamics of the protein in solution, but a necessary first step is the assignment of resonances. The size of the heterodimer of the Rel homology regions of the NF‐κB monomers p65 and p50 (72 kDa) prohibits the straightforward use of triple‐resonance spectroscopy to obtain the assignments. However, the dynamic nature of the free heterodimer, in particular the independence of the DNA‐binding and dimerization domains of each monomer, allows the assignments made on differentially labeled smaller domains to be mapped successfully onto the spectrum of the larger full‐length RHR. Problematic areas such as the p65 nuclear localization sequence, which is disordered in the free protein, can be approached by residue‐specific labeling and comparison with previously‐published spectra of a short peptide with the same sequence. Overall, this NMR analysis of NF‐κB has given valuable insights into the highly dynamic nature of the free state, which is likely to play an important role in the functional cycle of NF‐κB in the cell.
Keywords: structure, dynamics, NMR, NF‐κB, IκBα
Introduction
The family of transcription factors collectively named NF‐κB consists of a series of homo‐ and heterodimers of five monomer units, termed p50, p52, p65 (RelA), RelB, and c‐Rel.1 Each of these monomer units contains a well‐conserved DNA binding Rel Homology region (RHR) which in turn comprises a pair of immunoglobulin‐like domains, of which the N‐terminal is concerned primarily with DNA binding, while the C‐terminal is a dimerization domain [Fig. 1(A)]. The most commonly encountered form of NF‐κB is the heterodimer of p50 and p65, which forms, together with its specific inhibitor IκBα, a highly stable complex in the cytoplasm of a resting cell.2 Upon receipt of a signal, the IκBα is phosphorylated, ubiquitinated, and degraded by the 26S proteasome, freeing the nuclear localization signal sequence at the C‐terminus of the p65 dimerization domain and allowing the translocation of NF‐κB to the nucleus.3 Transcription of NF‐κB–responsive genes includes the nfkbia gene which encodes for the IκBα protein. Following NF‐κB activation, newly synthesized IκBα enters the nucleus to strip NF‐κB from its cognate κB DNA to turn off NF‐κB signaling.4 In order to explore the mechanism of the stripping interaction, we wished to characterize NF‐κB by NMR, necessitating the development of a strategy to obtain resonance assignments for a large (72 kDa) heterodimer.
Figure 1.

A: Schematic representation of domain organization of p65 and p50. The residue numbering corresponds to mouse NF‐κB. RHR: Rel homology region; DBD: DNA‐binding domain; dd: dimerization domain; TAD: C‐terminal trans‐activation domain; NLS: nuclear localization sequence. B: Backbone representations of X‐ray crystal structures of the p65/p50 RHR heterodimer in complex with (left, PDB 1VKX) the κB DNA sequence and (middle, PDB 1NFI; right, PDB 1IKN) IκBα. Domains of p65 are colored the same as in part A. The domains of p50 (DBD and dd on the left, dd alone on the middle and right structures) are colored gray, signifying that these domains are deuterated and hence invisible in all of the NMR spectra reported here. The two strands of the duplex κB DNA sequence are shown in magenta and gold, and the 6 ankyrin repeats of IκBα are shown in gold.
The p50/p65 RHR heterodimer forms stable complexes in vitro with partners such as the κB DNA sequence and IκBα, and these complexes have been analyzed by X‐ray crystallography,5, 6, 7 as illustrated in Figure 1(B). All four domains (the two N‐terminal DNA‐binding domains, DBD, and the two C‐terminal dimerization domains, dd) form part of the DNA complex.5 The two X‐ray structures of the IκBα complex contain the p50 and p65 dimerization domains and the p65 DNA‐binding domain; the p50 DNA‐binding domain was omitted from the complex to aid in crystallization.6, 7 There have been no published crystal structures of the free states of NF‐κB or IκBα, probably because of difficulties in crystallization: NMR and other studies indicate that the C‐terminus of IκBα is not fully folded.8, 9 In the present work, we show that the DBD and dd domains of p65 (and presumably also of p50), while well‐folded in themselves, are dynamically disordered in the free state and make no substantial inter‐domain contacts. We have used this dynamic disorder to advantage in obtaining NMR resonance assignments for a protein which, at 72 kDa, would normally be impossible using the simple techniques we employ.
Resonance assignments for molecules of biological and pharmacological interest provide a useful resource for the testing of interactions of partners or potential inhibitors or drugs. Where the molecule of interest is large, transverse relaxation‐optimized (TROSY) techniques10 can be used to address the resonance line broadening caused by slow molecular tumbling, but the problems of resonance overlap of the many nuclei in the molecule remain. Resonance overlap can be addressed by systems of differential isotopic labeling, including residue‐specific labeling,11 SAIL labeling,12 and similar methods, and by the use of split inteins13 or other methods to conjugate parts of the protein that are distinctively labeled. The NF‐κB family provides a good example of a naturally modular set of proteins that are amenable to the simple application of differential isotopic labeling without the use of intein or similar technology. Here we describe a simple labeling approach that makes use of the dynamic disorder between otherwise well‐folded domains to obtain resonance assignments for the full‐length protein.
Results
Assignment of p65 domains
The initial steps in the assignment process involved the characterization of individual domains of NF‐κB. We decided to focus on one of the members of the p50/p65 heterodimer, the p65 protein. Genes for the individual N‐ and C‐terminal domains of p65 RHR were cloned from a mouse cDNA library into pET vectors for expression in E. coli. Over‐expression of the N‐terminal DNA‐binding domain (DBD) p65(19‐191) yielded a well‐folded protein that could readily be labeled with 15N and 13C. Resonance assignments for this domain were made using standard triple resonance methods, including HNCA, HNCACB, CBCA(CO)NH, HNCO, and HN(CO)CA spectra. Assignments are shown mapped on the 15N‐HSQC spectrum of p65DBD in Figure 2(A).
Figure 2.
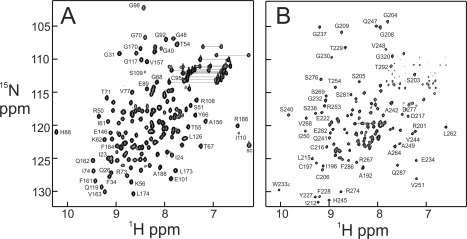
1H–15N correlation spectra of domains of p65. A: 600 MHz HSQC spectrum of p65DBD (residues 19–191) B: 900 MHz TROSY spectrum of p65dd (residues 190–321) in complex with perdeuterated p50dd (residues 245–350). Selected assignments, obtained from 3D triple‐resonance spectra acquired with 15N, 13C‐labeled p65, are mapped onto the spectra.
The dimerization domain (dd) of p65, p65(190‐321), is not stable in isolation, but forms a stable heterodimer with the corresponding domain from p50 (residues 245–350). Resonance assignments for the p65dd in complex with p50dd were made by separately preparing p50dd extensively labeled with 2H, and p65 labeled with 2H, 15N, and 13C. The differentially labeled heterodimer was formed by mixing the components, each partly ion‐exchange purified, followed by gel filtration. The heterodimer of p65dd/p50dd yields excellent NMR spectra for p65dd that show the presence of a well‐folded domain together with a disordered region [Fig. 2(B)]. Resonance assignments were readily made for the well‐structured portion of the protein, but the disordered domain, corresponding to the C‐terminal nuclear localization signal of p65, proved problematical due to resonance overlap and the multiplicity of the same residue types.
Published X‐ray crystal structures of p65RHR/p50RHR in complex with DNA (PDB 1VKX)5 and IκBα (PDB 1IKN, 1NFI)6, 7 show very little contact between the DBD and dd domains of either p50 or p65. Indeed, the DBD of p50 is missing from the two structures of the IκBα complex, due to crystallization of a proteolysed form of the complex. The angle between the domains of p65 in the IκBα complex compared with that in the DNA complex is quite different, a point that was invoked to explain the mechanism of DNA displacement by IκBα in the NF‐κB complex.6, 7 NMR measurements subsequently showed that the C‐terminal PEST sequence and the 6th ankyrin repeat domain of IκBα make intimate dynamic contact with the DNA‐contacting arginine residues of the p65DBD, indicating that X‐ray structures were locally incorrect in this region, possibly due to the absence of the p50DBD as well as disorder leading to high B‐factors in this region of the structure.14 Nevertheless, it does appear that the dd and DBD domains of p65 do not show a consistent inter‐domain angle or structure in any of the published crystal structures, and thus likely do not form intimate or permanent contacts with each other. The dynamic nature of the linkage between the DBD and dd domains of each NF‐κB monomer is further attested by the absence of any X‐ray structural data on the monomer or free homo‐ or heterodimer forms of any of the NF‐κB proteins. We therefore felt justified in attempting to map the assignments of the dd and DBD domains onto the spectrum of the full‐length protein.
Assignment of full‐length p65
The full‐length Rel‐homology domains of p65 and p50 were prepared separately using published protocols, again including the mixing of crude extracts of differentially labeled p65(19‐321) and p50(39‐363).9 The heterodimer of the RHR domains is too large at 72 kDa to be amenable to standard triple‐resonance techniques. We therefore prepared only 2H, 15N‐labeled p65RHR and formed the heterodimer by mixing it with fully deuterated p50RHR, in order to minimize T 2 relaxation in the final heterodimer samples. Back‐exchange of backbone amide protons was achieved by buffer exchange of the crude extract of the deuterated p65 before mixing with the p50 extract.
Most of the expected cross peaks from the 303‐amino acid p65 construct are observed in the TROSY spectrum [Fig. 3(A), full spectrum shown in Supporting Information Fig. S1]. An overlay of the spectra of the dd and DBD domains of p65 shows that the positions of the cross peaks closely correspond in the spectrum of the full‐length p65RHR, justifying the assumption that the domains do not differ in structure between the partial and full length RHR constructs, and that they do not make significant inter‐domain contacts. The resonance assignments for p65RHR in the p65RHR/p50RHR complex were further confirmed by TROSY based HNCA experiments incorporating non‐uniform sampling.
Figure 3.

A: Portion of an 800 MHz 1H–15N TROSY spectrum of 15N‐labeled, perdeuterated p65RHR (residues 19–325) in complex with perdeuterated p50RHR (residues 39–363) (black) superimposed on the 600 MHz HSQC spectrum of p65DBD (residues 19–191) (red) and the 900 MHz TROSY spectrum of p65dd (residues 190–321) in complex with deuterated p50dd (residues 245–350) (blue). Select resonances from the two domains are labeled in the corresponding colors. The complete spectra are shown in Supporting Information Figure S1. B: 600 MHz HSQC spectrum of p65DBD (black) superimposed on the HSQC spectrum of p65 DBD specifically labeled with 15N‐tyrosine (red). The tyrosine residues are labeled; the red cross peaks correspond to the tyrosines assigned in the spectra of the uniformly 15N‐labeled protein. C: 900 MHz TROSY spectrum of p65dd (residues 190–321) in complex with deuterated p50dd (residues 245–350) (black) superimposed on the HSQC spectrum of p65dd specifically labeled with 15N‐tyrosine and 15N‐methionine in complex with deuterated p50dd (residues 245–350) (red). The tyrosine and methionine residues are labeled; the cross peaks of Y227, M270, M284, and Y288 correspond to those assigned in the spectra of the uniformly‐labeled protein. The assignments of Y257, Y306, and M313 were made using information from the spectrum of the specifically‐labeled protein.
The assignments of the p65 NLS remain problematical, since this region clearly remains disordered in the full‐length protein, as indicated by the concentration of cross peaks in the central region of the TROSY spectrum (Supporting Information Fig. S1). Critical residues in this region were assigned by utilizing residue‐specific labeling. HSQC spectra of p65DBD specifically labeled with 15N tyrosine [Fig. 3(B)], and p65dd (in complex with deuterated p50dd) specifically labeled with 15N tyrosine and methionine [Fig. 3(C)] are shown superimposed on the spectra of the corresponding uniformly labeled proteins. The same spectra of the specifically labeled p65DBD and p65dd fragments are shown superimposed on the spectrum of the p65RHR in complex with the p50RHR in Supporting Information Figure S2. The tyrosine backbone amides seen in the spectrum of specifically labeled p65DBD map exactly to those already assigned in the triple resonance spectra; the spectrum shows an extra cross peak corresponding to the backbone amide of Tyr20, the second residue in the construct employed here. This resonance was not observed in the uniformly‐labeled spectrum, which is normal for the next‐to‐N‐terminal residue. Assignments for the NLS residues Tyr306 and Met313 were made using the heterodimeric p65dd sample specifically labeled with 15N‐Tyr and Met (in complex with deuterated p50dd).
Assignment of the p65NLS resonances in the IκBα complex
As mentioned above, the disordered p65NLS resonances were difficult to assign unambiguously by triple resonance experiments in the free p65dd/p50dd complex, due to resonance overlap. Addition of deuterated IκBα(67–287) yielded a spectrum that was very similar to that of the free p65dd/p50dd, with the exception of a few resonances. The 1H–15N TROSY spectrum of p65dd in the 51 kDa p65dd/p50dd/IκBα complex could be assigned using HNCA, HNCO, and HNCOCA spectra, although once again the resonances of the NLS proved difficult to assign, due to low intensity and the prevalence of the same residue types. Several of these resonances were assigned by referring to the spectrum of a peptide containing only the NLS sequence, in the presence and absence of a truncated form of IκBα (residues 67–206).15 The assigned resonances of the p65dd in the complex of p65dd/p50dd and p65dd/p50dd/IκBα are shown in Figure 4(A), and the areas of the p65 molecule where large chemical shift differences are observed between the free and bound states are shown mapped onto the X‐ray structure (1NFI, omitting the p65 DBD7) in Figure 4(B).
Figure 4.

A: 900 MHz TROSY spectrum of p65dd in complex with perdeuterated p50dd (black) superimposed on the spectrum of p65dd in complex with perdeuterated p50dd and perdeuterated IκBα (residues 67–287) (red). Significantly shifted resonances are labeled in green, and those with lesser shifts in blue. Residues for which the shifted resonances in the IκBα complex were identified from those of the NLS peptide complex15 are labeled in magenta. Residues for which resonances could be identified in the free p65dd/p50dd spectrum but which did not show readily‐identifiable resonances in the IκBα‐bound spectrum are labeled in black. B: X‐ray crystal structure of the complex of p65RHR/p50dd with IκBα (1NFI)7 (the p65DBD has been omitted for clarity). The backbone of IκBα is shown in gold, the backbone of p65dd in blue and the backbone of p50dd in gray. Residues with large chemical shift changes upon addition of IκBα, as shown in blue and green in part A, are mapped onto the structure in red. Residues in the NLS, for which large chemical shift changes were inferred from the positions of the assigned cross peaks in the spectrum of the NLS peptide alone15 are shown in magenta.
Discussion
We have shown that the NMR spectrum of the full‐length p65RHR of the NF‐κB heterodimer (p50/p65) can be assigned almost completely by analysis of the spectra of smaller domains. Besides the potential utility of the assigned NMR spectrum of a large signaling molecule for further studies into its interactions, this study also gives valuable insights into the nature of free NF‐κB that are not available from other sources. Structural data on the free protein have not been forthcoming from X‐ray studies, likely because of the inter‐domain flexibility that we have used to advantage in the present study. Flexibility and non‐rigid structure has been a constant theme in the NF‐κB/IκB system: like the free NF‐κB monomers and unpartnered dimers, free IκBα has never been crystallized. Further, hydrogen–deuterium exchange measured either by mass spectrometry8 or by NMR9 clearly show that parts of IκBα are molten globule‐like in the free state. Only in complex with NF‐κB do the NMR spectrum and the HD exchange results indicate that the C‐terminal part of IκBα is folded into a stable structure. NF‐κB and IκBα are mutually stabilized in their complex, an important factor in the persistence of this complex in the cytoplasm, where it has a lifetime in excess of 12 hr in vivo.16 By contrast, free IκBα has a lifetime of < 10 min in vivo, due to ubiquitin‐independent degradation by the 20S proteasome.16 Questions remain: after a signal is received by a cell, the NF‐κB‐dependent transcriptional machinery is activated through a phosphorylation cascade that results in the phosphorylation of IκBα, which leads to ubiquitination and degradation of the IκBα by the 26S proteasome.17 Degradation of IκBα in the complex frees the nuclear localization sequence of the NF‐κB, allowing it to be translocated to the nucleus to undertake the activation of transcription of its appropriate target genes.2 However, given that NF‐κB itself is clearly quite flexible in the free state, one may question whether there is some risk of degradation such as occurs for free IκBα. The answer may lie in the presence of as‐yet‐undiscovered chaperone or transport machinery that ensures that intact NF‐κB is able to enter the nucleus.
The functional relevance of the flexibility of NF‐κB and IκBα in their free states is also a question that has not yet been resolved. Since stable complexes are formed between NF‐κB and its partners, including IκBs and the cognate DNA sequences, it seems likely that, as with a number of other signaling molecules, the flexibility of the components aids in the transfer of the signaling molecule from one partner to another. For example, the flexibility of the C‐terminus of IκBα appears to play a role in stripping NF‐κB from DNA, thus “turning off” the switch that had been activated by the original extracellular signal.
Materials and Methods
Protein expression and purification
Plasmids containing various lengths of the RHR of murine p50 and p65 were prepared by subcloning from the full‐length genes, obtained from the laboratory of G. Ghosh (UCSD). The N‐terminal DNA‐binding domain of p65 (DBD) contained residues 19–191; the C‐terminal dimerization domain (dd) contained residues 190–321, including the C‐terminal nuclear localization sequence, residues 290–321. The p50dd contained residues 245–350. The p65RHR contained residues 19–325, and that of p50 contained residues 39–363. The ankyrin repeat domain of IκBα contained residues 67–287. Resonance assignments were made only for p65 and its fragments, which were 15N, 13C labeled and partially deuterated; all other proteins were prepared perdeuterated without other labels (Table 1). The proteins were expressed in M9 medium containing 15NH4Cl and 12C, 13C or [13C, 2H]‐glucose in E. coli and purified using variations of published methods.9, 18 Perdeuterated proteins were expressed in M9 minimal medium made using 2H2O instead of 1H2O; partially‐deuterated proteins were expressed in recycled 2H2O for reasons of economy. For the production of amino acid specific labeled p65, M9 medium was supplemented with 100 mg/L of 15N‐labeled tyrosine (for p65DBD) and 15N‐labeled tyrosine (Aldrich cat. No. 332151) and 15N‐labeled methionine (Cambridge Isotope Laboratories, cat. No NLM‐752) (for p65dd in complex with p50dd) and a total of 50 mM of all other unlabeled amino acids.
Table 1.
Description of Protein Samples
| Protein sample | Sample concentration (mM) | Labeling strategy |
|---|---|---|
| p65DBD | 0.80 | u‐[15N,13C]partially deuterated |
| p65DBD (15N‐Tyr) | 0.50 | [15N‐Tyr] |
| p65dd/p50dd | 0.69 | u‐[15N,13C, 2H]p65dd + [2H] p50‐dd |
| p65dd(15N‐Tyr, Met)/p50dd | 0.25 | [15N‐Tyr, Met]partially deuterated p65dd + partially deuterated p50dd |
| p65dd/p50dd/IκBα | 0.69 (dimer) | u‐[15N,13C, 2H] p65dd + [2H] p50dd + [2H] IκBα |
| p65dd(15N‐Tyr, Met)/p50dd/IκBα | 0.25 (dimer) | [15N‐Tyr, Met]partially deuterated p65dd + partially deuterated p50dd + protonated IκBα |
| p65RHR/p50RHR | 0.54 | u‐[15N,13C,2H] p65RHR + [2H] p50RHR |
All cells were lysed by sonication in lysis buffer (20 mM Tris pH 7.5, 50 mM NaCl, 0.5 mM EDTA, 10% glycerol, 0.1% Triton‐X100, 20 mM β‐ME, protease inhibitor cocktail, 0.5 mM PMSF) and centrifuged to remove the cell debris. For p65DBD, the supernatant was loaded on a tandem Q (Q Sepharose GE Healthcare Life Sciences) followed by S (SP Sepharose GE Healthcare Life Sciences) column pre‐equilibrated with the lysis buffer. The S‐column was then eluted with a NaCl gradient. The fractions containing the p65DBD were pooled and concentrated and subjected to size exclusion chromatography using Superdex‐75 column. Cells expressing the p65dd and p50dd proteins were grown separately in M9 medium containing the appropriate stable isotope labels. The supernatant after sonication of p50dd and p65dd‐expressing cells was loaded onto Q and S columns, respectively. Both the proteins were eluted using a NaCl gradient and the pooled protein fractions were mixed together and further purified by size‐exclusion chromatography using Superdex‐75 column (GE Healthcare Life Sciences). For the p65RHR/p50RHR heterodimer, the separately grown cells with respective labeling were mixed together and lysed by sonication in the lysis buffer followed by centrifugation. The supernatant was loaded on a tandem Q and S column pre‐equilibrated with the lysis buffer. The S‐column was then eluted with a NaCl gradient. The fractions containing the p65RHR/p50RHR heterodimer were pooled and concentrated and subjected to size exclusion chromatography using Superdex‐75 column. For IκBα, cells were lysed by sonication in lysis buffer. The supernatant collected was then loaded on to pre‐equilibrated Q‐sepharose column and eluted with a NaCl gradient. The IκBα fractions were pooled, concentrated, and further purified by size exclusion chromatography using Superdex‐75 column. Typical yields of purified protein were between 5 and 15 mg/L; yields were typically lower in the presence of 13C and 2H. Purity was assessed by SDS‐PAGE (examples shown in Supporting Information Fig. S3) and by examination of the 1H–15N HSQC or TROSY spectra.
For the NMR experiments, all the purified proteins were buffer exchanged with NMR buffer (20 mM deuterated Tris (pH 6.8), 50 mM NaCl, 2 mM deuterated DTT and 10% D2O) using desalting columns pre‐equilibrated in NMR buffer. Protein concentrations were calculated using absorbance at wavelength of 280 nm.
NMR spectroscopy
All NMR spectra were acquired using Bruker 600, 750, 800, and 900 MHz spectrometers (all experiments used triple‐axis gradient triple resonance probes; 600, 750, and 800 MHz spectrometers were equipped with cryoprobes) and analyzed using NMRPipe,19 NMRView,20 and CARA.21 Resonance assignments for 15N,13C‐labeled p65DBD (173 amino acids, 19.6 kDa) were made using standard triple resonance experiments, HNCA, HN(CO)CA, HNCACB, HNCO, and HN(CO)CACB. For the p65dd in complex with deuterated p50dd (131 labeled amino acids, 26.7 kDa), TROSY versions of the same spectra were acquired, using non‐uniform sampling (NUS) schemes as previously described.22 For the p65RHR in complex with deuterated p50RHR (303 labeled amino acids, 72 kDa), all standard triple resonance experiments are not practical; backbone amide resonances were assigned by transferring the assignments made for the individual domains which were further conformed using TROSY‐based HNCA incorporating NUS. For the [15N, 13C, 2H]‐p65dd/[2H]‐p50dd in complex with [2H]‐IκBα (131 labeled amino acids, 51 kDa), assignments for most of the fully structured p65dd could be made using the same set of triple resonance spectra, although the quality of the 3D spectra was lower for this larger complex. The resonances of the NLS, which was disordered in the free p65dd, were greatly shifted due to the formation of helical structure in the IκBα complex, but were difficult to assign due to low signal intensity and missing resonances. Some of these resonances were assigned by referring to the spectrum of the NLS peptide (residues 293–321) in complex with truncated IκBα (residues 67–206, corresponding to ankyrin repeats 1–4).15
Data Deposition
1H, 13C and 15N chemical shifts for the p65DBD and for the p65dd in complex with perdeuterated p50dd have been deposited in the BMRB (www.bmrb.wisc.edu) under the accession numbers 26647 and 25792.
Supporting information
Supporting Information
Acknowledgments
Authors thank Maria Martinez‐Yamout and Euvel Manlapaz for helpful discussions on the preparation of proteins, Gerard Kroon and Phillip Aoto for help with NMR experiments, and Peter Wright and members of the Wright and Dyson groups for helpful discussions.
References
- 1. Hoffmann A, Natoli G, Ghosh G (2006) Transcriptional regulation via the NF‐kB signaling module. Oncogene 25:6706–6716. [DOI] [PubMed] [Google Scholar]
- 2. Beg AA, Baldwin AS, Jr (1993) The IkB proteins: multifunctional regulators of Rel/NF‐kB transcription factors. Genes Dev 7:2064–2070. [DOI] [PubMed] [Google Scholar]
- 3. Beg AA, Finco TS, Nantermet PV, Baldwin AS, Jr (1993) Tumor necrosis factor and interleukin‐1 lead to phosphorylation and loss of IkBa: a mechanism for NF‐kB activation. Mol Cell Biol 13:3301–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown K, Park S, Kanno T, Franzoso G, Siebenlist U (1993) Mutual regulation of the transcriptional activator NF‐kappa B and its inhibitor, I kappa B‐alpha. Proc Natl Acad Sci USA 90:2532–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen FE, Huang DB, Chen YQ, Ghosh G (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF‐kB bound to DNA. Nature 391:410–413. [DOI] [PubMed] [Google Scholar]
- 6. Huxford T, Huang DB, Malek S, Ghosh G (1998) The crystal structure of the IkBα/NF‐kB complex reveals mechanisms of NF‐kB inactivation. Cell 95:759–770. [DOI] [PubMed] [Google Scholar]
- 7. Jacobs MD, Harrison SC (1998) Structure of an IkBα/NF‐kB complex. Cell 95:749–758. [DOI] [PubMed] [Google Scholar]
- 8. Truhlar SM, Torpey JW, Komives EA (2006) Regions of IkBα that are critical for its inhibition of NF‐kB.DNA interaction fold upon binding to NF‐kB. Proc Natl Acad Sci USA 103:18951–18956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sue SC, Cervantes C, Komives EA, Dyson HJ (2008) Transfer of flexibility between ankyrin repeats in IkBa upon formation of the NF‐kB complex. J Mol Biol 380:917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pervushin K, Riek R, Wider G, Wüthrich K (1997) Attenuated T2 relaxation by mutual cancellation of dipole‐dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA 94:12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE (1999) A robust and cost‐effective method for the production of Val, Leu, Ile (d1) methyl‐protonated 15N‐, 13C‐, 2H‐labeled proteins. J Biomol NMR 13:369–374. [DOI] [PubMed] [Google Scholar]
- 12. Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Mei Ono A, Güntert P (2006) Optimal isotope labelling for NMR protein structure determinations. Nature 440:52–57. [DOI] [PubMed] [Google Scholar]
- 13. Southworth MW, Adam E, Panne D, Byer R, Kautz R, Perler FB (1998) Control of protein splicing by intein fragment reassembly. EMBO J 17:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sue SC, Dyson HJ (2009) Interaction of the IkBa C‐terminal PEST sequence with NF‐kB: insights into the inhibition of NF‐kB DNA binding by IκBα. J Mol Biol 388:824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cervantes CF, Bergqvist S, Kjaergaard M, Kroon G, Sue SC, Dyson HJ, Komives EA (2011) The RelA nuclear localization signal folds upon binding to IκBα. J Mol Biol 405:754–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mathes E, O'Dea E, Hoffmann A, Ghosh G (2008) NF‐kB dictates the degradation pathway of IkB. EMBO J 27:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baldwin AS (1996) The NF‐kB and I‐kB proteins: new discoveries and insights. Ann Rev Immunol 14:649–683. [DOI] [PubMed] [Google Scholar]
- 18. Chen FE, Kempiak S, Huang DB, Phelps C, Ghosh G (1999) Construction, expression, purification and functional analysis of recombinant NFkB p50/p65 heterodimer. Protein Eng 12:423–428. [DOI] [PubMed] [Google Scholar]
- 19. Delaglio F, Grzesiek S, Vuister GW, Guang Z, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 20. Johnson BA, Blevins RA (1994) NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR 4:603–614. [DOI] [PubMed] [Google Scholar]
- 21. Keller RLJ. (2005) Optimizing the process of NMR spectrum analysis and computer aided resonance assignment Chemistry. Zurich: Eidgenossische Technische Hochschule; p 150. [Google Scholar]
- 22. Aoto PC, Fenwick RB, Kroon GJA, Wright PE (2014) Accurate scoring of non‐uniform sampling schemes for quantitative NMR. J Magn Reson 246:31–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information