

Abstract
Biological outcomes are governed by multiple genetic and environmental factors that act in concert. Determining multifactor interactions is the primary topic of interest in recent genetics studies but presents enormous statistical and mathematical challenges. The computationally efficient multifactor dimensionality reduction (MDR) approach has emerged as a promising tool for meeting these challenges. On the other hand, complex traits are expressed in various forms and have different data generation mechanisms that cannot be appropriately modeled by a dichotomous model; the subjects in a study may be recruited according to its own analytical goals, research strategies and resources available, not only consisting of homogeneous unrelated individuals. Although several modifications and extensions of MDR have in part addressed the practical problems, they are still limited in statistical analyses of diverse phenotypes, multivariate phenotypes and correlated observations, correcting for potential population stratification and unifying both unrelated and family samples into a more powerful analysis. I propose a comprehensive statistical framework, referred as to unified generalized MDR (UGMDR), for systematic extension of MDR. The proposed approach is quite versatile, not only allowing for covariate adjustment, being suitable for analyzing almost any trait type, for example, binary, count, continuous, polytomous, ordinal, time-to-onset, multivariate and others, as well as combinations of those, but also being applicable to various study designs, including homogeneous and admixed unrelated-subject and family as well as mixtures of them. The proposed UGMDR offers an important addition to the arsenal of analytical tools for identifying nonlinear multifactor interactions and unraveling the genetic architecture of complex traits.
Introduction
Genes act together in interconnected networks to generate organismal phenotypes under a developmental environment. The network's architecture strongly influences its overall behavior in ways that cannot be predicted based on the parts alone and are filled with nonintuitivity and nonlinearity (Nijhout, 2003). Consequently, the presence of interactions among genes, gene–gene or epistatic interactions, and between genes and environmental factors (broadly defined as all non-genetic exposures), gene–environment interactions, is a rule rather than an exception (Carlson et al., 2004). Pervasive interactions usually result in a weak marginal correlation between a factor and the phenotype, making the factor rather difficult to be tracked down (Phillips, 2008). Traditional univariate hunting strategies have been proved to bring only limited success, giving rise to the mystery of missing heritability (Manolio et al., 2009). Failure to take account of the context dependence also leads to a ‘flip-flop' phenomenon in gene discoveries (Lin et al., 2007). It remains a daunting task in genetics fields to dissect genetic architecture of a complex biological trait.
It necessitates a multifactorial strategy to identify highly mutually dependent factors underlying a trait. However, such a search has to face a significant obstacle called ‘the curse of dimensionality', a problem caused by the exponential increase in volume of possible interactions with the number of factors to consider (Moore and Ritchie, 2004). The conventional regression methods, established by the extension under the concept of univariate approaches, are hardly appropriate for tackling ubiquitous yet elusive interactions because of several problems: heavy (usually intractably) computational burden, increased type I and II errors, and reduced robustness and potential bias as a result of highly sparse data in a multifactorial model (Carlborg and Haley, 2004). Novel approaches such as data mining and machine learning have been explored for various kinds of phenotypes (Cordell, 2009).
Among these methods emerged recently, data reduction approaches (a constructive induction strategy), such as the multifactor dimensionality reduction (MDR) method (Ritchie et al., 2001) and the restricted partition method (Culverhouse et al., 2004), are promising to address the multidimensionality problems. A data-reduction strategy seeks for a pattern in a combination of factors/attributes of interest that maximizes the phenotypic variation it explains. It treats the joint action as a whole, offering a solution that avoids the exponential growth in the number of parameters as each new variable is added. It also has a straightforward correspondence to the concept of the phenotypic landscape that unifies biological, statistical genetics and evolutionary theories (Wolf, 2002). Notably, the pioneering MDR method has sustained its popularity in detection of interactions since its launch.
There are, however, a few limitations in the original MDR and the other data-reduction methods that may restrict their practical use, including not allowing for covariates, being unable to accommodate various kinds of phenotypes and being confined to specific study designs, such as unrelated samples. To overcome these weaknesses, MDR has been extended to survival analysis (Gui et al., 2011; Lee et al., 2012), case–control study of structured populations (Niu et al., 2011), family study (Martin et al., 2006) and inclusion of covariates (Lee et al., 2007). The existing generalized MDR (GMDR) (Lou et al., 2007, 2008) is still limited in tackling diverse phenotypes and samples, specifically being unable to analyze ordinal or polytomous nominal data, survival data and multivariate phenotypes, correct for population stratification in unrelateds and unify both unrelated and family samples into a joint analysis. Recently, Chen et al. (2014) developed a GMDR method for unifying analyses of both unrelateds and families for dichotomous and continuous traits based on an adjustment with the principal components on the phenotypes but not on the markers. I propose a comprehensive and versatile framework called unified GMDR (UGMDR) for extension of MDR with applicability to continuous, dichotomous, polytomous, ordinal, event-count, time-to-an-event and multivariate response variables and various kinds of samples, including unrelated-subject, family and admixed individual, as well as a better adjustment strategy for population stratification.
Materials and methods
The original MDR method for a case–control study proceeds as follows (Ritchie et al., 2001): each subject is allocated into a cell in a space spanned by a set of m attributes of either genes or discrete environmental factors based on the attribute observations; every nonempty cell in this m-dimensional space is labeled as either ‘high-risk' or ‘low-risk' according to whether or not the ratio of cases to controls in the cell is larger than a preset threshold; and then a new dichotomous attribute (that is, a classification model) is formed by pooling the high-risk cells into the high-risk group and the low-risk cells into the low-risk group, thus changing the representation space of the data from originally higher dimensions to one dimension. The new attribute is evaluated for its ability to classify or predict the phenotype; accuracy, defined as the proportion of the correct classifications (that is, cases in the high-risk group and controls in the low-risk group), is a commonly used measure. Cross-validation and/or permutation technique can be integrated into the above process of defining the new attribute for evaluation of model, and the optimal subset(s) of features can be selected in terms of the classification ability measured by accuracy or its derivatives, such as P-value. In essence, MDR is a constructive induction approach for feature subset selection (Michalski, 1983).
GMDR uses the same variable construction algorithm as in MDR. The generalization lies in that GMDR substitutes a general statistic for affection status, to classify the two divergent groups. Thereby, GMDR offers a flexible framework for addressing diverse phenotypes, allowing for covariate adjustment, accommodating various study designs, controlling for cryptic population structure and unifying analyses for unrelated and family samples. In a nutshell, the statistic of individual i with respect to cell k in a given contingency table is expressed as the product of its membership coefficient πik and residual ri, respectively, defined in subsections ‘Residuals in the null model' and ‘Membership coefficient',
As summarized in Figure 1, the conceptual framework is composed of three components: (1) to compute the residuals in a fitted model under the null hypothesis; (2) to determine the membership coefficients of a subject belonging to given cells in the space spanned by the putative factors; and (3) to implement the data-reduction algorithm using the statistic as the classification criterion. The three components are elaborated as follows.
Figure 1.

An overview of GMDR.
Residuals in the null model
Assuming no effects of any target factors, the residuals are computed from an appropriate statistical model corresponding to data type and plausible data-generation mechanism. The residuals need not re-compute for different combinations of target factors. To make the presentation self-contained, the relevant statistical models and estimation theory are recapitulated in Supplementary Appendix A.
Generalized linear model (GLM) for dichotomous, count or continuous phenotypes
Many phenotypes can be represented by a GLM in the exponential family of distributions (Nelder and Wedderburn, 1972). Supposing for a response variable Y generated from a GLM, a set of explanatory variables influence the outcome only through a linear function and the linear predictor can be written as,
where μ= E(Y) is the expectation, l(·) is an invertible link function relating the mean μ to the linear predictor η, β0 is the intercept, βt is the vector of the ‘target effects' that we wish to infer, xt is the target predictor variable vector that, in the genetics context, codes for gene–gene and/or gene–environment interactions of interest and is composed of the membership coefficients defined in Equation (2.2), βc is the covariate effect vector and xc is the corresponding vector coding for covariates. When the linear predictor η is the same as the location parameter, also known as the canonical parameter, θ, l(·) is called the canonical link.
The general procedure for model fitting is shown in Supplementary Appendix A1. Briefly, the maximum likelihood estimator for β can be derived by setting the score equal to zero and solving the resulting estimating equations, and when necessary, the scale parameter φ can be estimated via the moments method. In the GMDR, fit β̂0 and β̂c as well as φ̂0 when necessary to data under the null hypothesis of no target effects (that is, βt=0). Then, the score-contributed residual can be computed by Equation (A1) in Supplementary Appendix A. The statistic of an individual can be computed via Equation (2.1) that is, in essence, the score residual in a GLM.
Quasi-likelihood model (QLM) for dichotomous, count or continuous phenotypes
It is not always possible as in GLMs to establish a certain probability model for data because of insufficient information on data nature, even if some of the data features can be specified: how the mean is related to the explanatory variables and how the variance of an observation is related to its mean. In such cases, quasi-likelihood functions can be constructed that mimic proper likelihood functions and have the same properties as log-likelihood function (McCullagh, 1983). A QLM only specifies the link function and the relationship between the first two moments but does not necessarily specify the complete distribution of the response variable and thus can model a broader class of phenotypes than a GLM.
As shown in Supplementary Appendix A2, with a known function between the expectation of a response variable and a set of predictor variables as in equation (2.2), the quasi-score function can be formulated by differentiating the quasi-likelihood function. The quasi-score behaves like the score in GLMs. QLMs can be fitted using a straightforward extension of the algorithms used to fit GLMs. As in GLMs, after the residual under the null hypothesis is computed by Equation (A2) in Supplementary Appendix A, the statistic can be computed via Equation (2.1).
Generalized estimating equations (GEEs) model for correlated observations and multiple complex traits
In GLMs and QLMs, all observations are assumed to be independent. This assumption does not necessarily hold true in a repeated measurement experiment in which there are several measurements on the same observational unit, a longitudinal study in which there are multiple observations over time and a clustered design in which subjects are not sampled independently but in a group. Further, many complex disorders such as asthma, mental disorders and drug addictions are generally a multifaceted phenotype, measured by a set of scales and/or intermediate phenotypes that can be correlated as the outputs of a metabolic network with many interwoven pathways and governed by pleiotropic determinants. The GEE model can be used to handle a GLM or QLM with a possible unknown correlation (Liang and Zeger, 1986).
GEE model requires only to specify a functional form for relationships between the outcome variable and the explanatory factors and between the mean and the variance of the marginal distribution, avoiding the need to model the multivariate distribution for data. Specifically, letting Y=(Y1, Y2, ⋯, YK)T be a group of response variables, suppose that (1) there is a link function relating the expectation of Y, μ, to a linear predictor, l(μ)=η=Xβ, where β consists of β0, βt and βc for the intercept, the target effects and the covariate effects, respectively; and (2) the variance is a function of the mean, Var(Yj)=aj(φj)Vj(μj), where φj is the scale parameter and aj(·) and Vj(·) are some known functions. Parameters β(j) and φj, functions aj(·) and Vj(·) and regressor values xj corresponding to component Yj may be either the same or different to characterize GEE models for diverse scenarios.
Considering the data consisting of a number of strata that are uncorrelated with each other, the estimating function is formed via a set of score or quasi-score functions. The function behaves like the derivative of a log-likelihood. The estimates of β can be found typically by solving the estimating equation. Supplementary Appendix A3 outlines parameter estimation in GEE models. After fitting the model under the null hypothesis, the residuals can be computed via Equations (A3) or (A4) for different purposes. In the case where the components of a stratum have the same target parameter βt whether or not the predictor vectors of the components are distinct, the statistic of component j in stratum i with respect to cell k in a given contingency table can be computed by (treating as individual ij and cell k),
where rij is the residual and πijk is the membership coefficient of component j in stratum i pertaining to cell k; all rijs of stratum i (j=1, 2, ⋯, Ki) are the same in a repeated measurement study; and all πijks of stratum i (j=1, 2, ⋯, Ki) for cell k are the same in a repeated measurement study and probably in a longitudinal study. In application to multivariate phenotypes with a goal to detect the overall and/or pleiotropic effects of genetic determinants, the statistic is considered as (treating as individual i and cell jk),
where rij is the residual and πik is the membership coefficient denoting individual i belonging to cell k in a given contingency table.
Multinomial logistic model for polytomous data
Many complex phenotypes of medical importance such as disease severity are neither continuous nor binary but in the form of multi-categories (either ordinal or nominal). To use the traditional logit model, the common strategy is to collapse the categories into two mutually exclusive groups or to limit the analysis to pairs of categories. Such a strategy that ignores observations or combines categories will lead to loss in efficiency.
The dichotomous logit model can be extended to a polytomy by employing the multivariate logistic distribution (Begg and Gray, 1984). Considering a multinomial response variable with K categories, denote the outcome Y=(Y1, Y2, ⋯, YK)T where Yj is an indicator variable taking value 1 if the observed category is j and 0 otherwise. A polytomous logit model can be formed by nominating one of the response categories as a baseline and then formulating a set of K−1 logits for all other categories relative to the baseline. Without loss of generality, using category K as the baseline, the multinomial density has a multivariate exponential form with a canonical link, ηj=l(μj)=xTβ(j), j=1, 2, ⋯, K−1, where β(j) consists of β0, βt βc for the intercept, the target effects and the covariate effects, respectively.
Solving the score equations leads to the maximum likelihood estimation. As an alternative, the GEE approach in subsection ‘Generalized estimating equations (GEEs) model for correlated observations and multiple complex traits' can also be used to fit the polytomous logit model (Sutradhar and Kovacevic, 2000). Having fitted parameters to data under the null hypothesis where βt(j) = 0 (j = 1, 2,..., K−1), the residual can be calculated by Equation (A5) in Supplementary Appendix A. As there are various sets of effect parameters for response categories, it is proper to treat the residual as a (K−1)-dimensional vector as in the case of multivariate phenotype. The statistic is computed by Equation (2.4).
The polytomous logistic model does not utilize the ordering of response categories. It is applicable to analysis of both unordered and ordered categorical outcomes.
Proportional odds model for ordinal data
Commonly, outcomes of interest are measured in an ordinal scale, which have natural ordering of severity or certainty (Ananth and Kleinbaum, 1997). Such response categories are no longer purely qualitative or nominal, rather than ordered. Although the polytomous logit model may be applied to ordinal data by assuming cardinality instead of ordinality as well, it is often less desirable to ignore the ordering of categories. One of the most popular models for ordinal responses is the proportional odds model (Agresti, 1999). This model uses logits of cumulative probabilities, and assumes an identical effect of the predictors for each cumulative probability, thus being a more parsimonious model (McCullagh, 1980).
Consider an ordinal response consisting of K ordered categories in a decreasing order of severity or certainty, denoted by 1, 2, ⋯, K. Define a new response, Zj (j=1, 2, ⋯, K), taking value 1 if the observed category is ⩽j and 0 otherwise. Then, the observation vector, Z=(Zj)=LY, and its expectation, E(Z)=γ=Lμ, where Y and μ are defined in subsection ‘Multinomial logistic model for polytomous data' and L is a lower triangular matrix with element 1. The proportional odds model with the restriction that only the intercepts of the regression equations differ has the following representation, formed by K−1 logit equations,
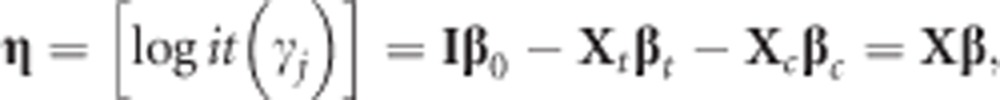 |
where I is a unit matrix.
Parameter estimation is relegated to Supplementary Appendix A5. Fisher's scoring or Newton–Raphson iterative procedure can be employed to find the maximum likelihood estimates. The residual is calculated via Equation (A6) in Supplementary Appendix A. As the set of effect parameters are the same for different response categories, it is appropriate to treat the residual as a scalar. The statistic can be computed via Equation (2.1).
Proportional hazards model for survival data
In many medical studies, an outcome of interest is the time to an event, conventionally termed this kind of phenotypes survival outcomes regardless of the nature of the event (Altman and Bland, 1998). The distinguishing feature of survival data is that the observations are probably censored because the observation period expires before the event occurs or the subjects are lost to follow-up. Further, the survival times are more unlikely to be normally distributed and the probability distribution of time is difficult to model. Moreover, in addition to the status of event, the ascertainment time and the time to event can also carry valuable information. Thus the analysis of survival data requires specialized techniques that focus on the distribution of survival times.
Recently, Gui et al. (2011) proposed the surv-MDR method for identifying gene–gene interactions by using log-rank statistics instead of case–control ratios within the MDR frame. Similar to the log-rank test, the surv-MDR method does not allow for inclusion of other explanatory variables. It also involves intensive computations for log-rank statistics of all possible factor combinations. To overcome these drawbacks, Lee et al. (2012) developed the Cox-MDR by incorporating the martingale residual into the GMDR framework. Both the surv-MDR and the Cox-MDR, however, are suitable only for unrelated samples from a homogeneous population; no population and family structures are permitted in samples. This article is to advance a unified frame for handling diverse sources of survival data.
Hazard rate is a crucial parameter to characterize survival data. The best known proportional hazards model assumes the hazard as a product of the time-related baseline hazard and the covariates-related component. Typically in such a model, the baseline hazard can ‘cancel out', and the effects of predictor variables can be estimated by maximizing the remaining partial likelihood, thus being reported as hazard ratios. The Cox proportional hazards model with stationary coefficients is used here to illustrate the proposed method. The further extensions are straightforward, for example, to time-dependent effects and to parametric proportional hazards models by specifying a baseline hazard function.
The Cox proportional hazards model (Cox, 1972) is a semi-parametric model where the dependence of time-to-event on explanatory variables is precisely modeled, but the actual survival distribution, that is, the baseline hazard function, is not specified and can take any form. The Cox model (Cox, 1972) is written as,
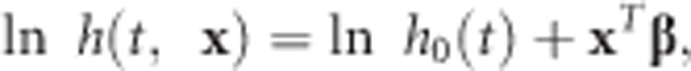 |
where h(t, x) is the hazard rate at time t, h0(t) is the baseline hazard rate, β consists of βt and βc for the target effects and the covariate effects, respectively, and x is the predictor vector.
Using the score function and Hessian matrix in Supplementary Appendix A6 the partial likelihood can be maximized via the Newton–Raphson algorithm. After fitting a null proportional hazards model assuming no target effects, the residuals and the statistics can be computed via Equation (A7) in Supplementary Appendix A and Equation (2.1), respectively.
Membership coefficient
Membership coefficients are used to characterize to which cell(s) a subject can be allocated in the space spanned by a set of target factors and a fractional membership is also allowed for adjusting population stratification. The membership coefficient is applied for various study designs and purposes. Family-based and unrelated subject-based designs are commonly used in genetic studies. Sometimes, there are both kinds of data available in a single study, and a combined analysis is required. The principle of transmission disequilibrium is used here to formulate a unified framework for samples in different designs. In sexual reproduction, corresponding to a real individual, there exists a pseudo individual, called the nontransmitted sib, who is composed of two gametids complementary, respectively, to the egg and the sperm that unite to develop into the zygote. The nontransmitted sib can serve as an internal control to test the null hypothesis (that is, equi-probable transmission) when his/her genotype can be inferred from the genetic information on the real individual and other pedigree member(s) (Lou et al., 2008). When no genetic information on the pedigree member(s) is available (for example, singletons or founders in pedigrees), the nontransmitted sib is considered as being missing.
Unrelated samples without population stratification
For an unrelated subject sampled from a homogeneous population, the membership coefficient is defined as an indicator variable, πik, coded as 1 if subject i is allocated to cell k and 0 otherwise.
Unrelated samples with population stratification
When the population homogeneity does not hold true, it is necessary to correct for the population structure in unrelated samples in order to eliminate the potential spurious results (false positives or false negatives). Principal components analysis (PCA) method has been proved to be effective in correcting for population structure (Price et al., 2006). PCA can be implanted into the GMDR framework to rule out the effects of population stratification. The PCA-based procedure is summarized in Supplementary Appendix B.
Let gGi be the coding genotypic value at loci of interest that takes 1 when the multilocus genotype is G or 0 otherwise and ri be the residual, for unrelated individual i. The genotypic values and the residual values can be adjusted with the first L principal components, assuming the population structure can be represented by them. Then the membership coefficient of individual i, πik, that may be fractional, is 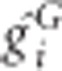 if the subject is allocated to cell k and φ(k)=G, where
if the subject is allocated to cell k and φ(k)=G, where 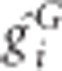 is adjusted values and φ(k) is an operation of taking the genotype for cell k and 0 otherwise. When adjustment is applicable, using adjusted
is adjusted values and φ(k) is an operation of taking the genotype for cell k and 0 otherwise. When adjustment is applicable, using adjusted 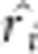 in place of ri computes the score statistic. In the previous report (Chen et al., 2014), an adjustment strategy only on the phenotypes, but not on the genetic markers, was used for correcting population structure. Although the resulting GMDR is valid in the sense of controlling correct type I error rates, as shown in Supplementary Appendix B, the current strategy of adjustment both on the phenotypes and on the markers is more theoretically sound from the perspective of statistical power.
in place of ri computes the score statistic. In the previous report (Chen et al., 2014), an adjustment strategy only on the phenotypes, but not on the genetic markers, was used for correcting population structure. Although the resulting GMDR is valid in the sense of controlling correct type I error rates, as shown in Supplementary Appendix B, the current strategy of adjustment both on the phenotypes and on the markers is more theoretically sound from the perspective of statistical power.
Family samples
The principle of transmission is used to tackle pedigree structure. As in (Lohmueller et al., 2003), it is algebraically equivalent to contrasting the observed genotype with the control being from a population with equal numbers of transmitted and nontransmitted genotypes. For a nonfounder in a pedigree, the membership coefficient, πik, can be defined as 0.5 if subject i is allocated to but his nontransmitted sib is not allocated to cell k, 0 if neither or both of subject i and his nontransmitted sib are allocated to cell k and −0.5 otherwise. It is equivalent to that in the pedigree-based GMDR (Lou et al., 2008) when only nonfounders are considered.
Pooled nonfounder and unrelated samples
When both family and unrelated samples are available, founders in pedigrees and singletons can be considered as unrelateds, while nonfounders can be treated as a contrast of transmission with nontransmission. Similar to the literature (Lohmueller et al., 2003), both nonfounders and unrelated samples can be pooled into a unified framework through the adjusted or unadjusted membership coefficients. Usually, such a strategy can boost statistical power substantially.
Once all membership coefficients are determined, they can serve as the design or incidence matrix for the target effects in statistical models. The statistic for the score residuals can be computed. The potential bias due to population stratification and family structure can be guarded against through the PCA adjustment technique and the principle of transmission equilibrium.
Multifactor dimensionality reduction algorithm
The statistic defined above reflects a putative association between the phenotype(s) of interest and the target factor(s), offering a possibility for variable construction to create new attributes that maximize the residual phenotypic correlation. The dimensionality reduction process is illustrated with the c-fold cross-validation procedure although such a cross-validation is not always necessary as other techniques such as permutation testing may determine whether a classification model is beyond chance. The MDR process is outlined in Figure 2 and can be briefly described as follows.
Figure 2.

Summary of the steps involved in implementing GMDR method. A balanced case–control study with no covariate is assumed for illustrative purpose in which a case has a score of 0.5 while a control does a score of −0.5. In step 3, bars represent hypothetical distributions of cases (left, dark shading) and controls (right, light shading); the numbers not in parentheses above bars are the numbers of cases/controls and those in parentheses above bars are sums of the scores. In steps 4 and 6, the numbers in parentheses are the average scores. Assuming an average score of 0.0 as the threshold, ‘high-risk' cells are indicated by dark shading, ‘low-risk' cells by light shading and ‘empty' cells by no shading. A full color version of this figure is available at the Heredity journal online.
In Step 1, the data are randomly split into c equal or nearly equal parts for cross-validation (c=10 in Figure 2). One subset is used as the testing set and the remaining as the independent training set. Then, Steps 2 through 5 are run for the training set to construct a new dichotomous attribute and Step 6 for the testing set to evaluate the fitness of the new attribute(s). In Step 2, a subset of m factors is selected from all M genetic and/or discrete environmental factors, giving rise to a total of C(M,m) distinct subsets. In Step 3, each such subset spans into an m-way contingency table and each component membership of a subject corresponds to one cell in the table. The statistic value can be averaged over each nonempty cell, for example, for cell k, 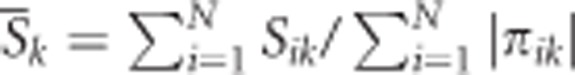 . Each nonempty cell is labeled either high-valued if its average statistic value is larger than some threshold T or low-valued otherwise. In Step 4, a new attribute is created by pooling high-valued and low-valued cells into two contrasting (that is, high-valued and low-valued) groups, representing the classification that best captures the correlation between this set of factors and the phenotype(s). In Step 5, the classification accuracy can be assessed for each contingency table. The best model(s) can be identified among all the possible m-way contingency tables based on classification accuracy. In Step 6, the independent testing set is used to evaluate the testing accuracy for the best model among those with different dimensionalities identified in Step 5. If the null hypothesis holds true and the classification model is formed purely by chance, it will give a null testing accuracy of 0.5 as the testing set is independent of the training set in which the model selection is involved. The significance test can be implemented based on permutation testing, nonparametric sign testing and asymptotical z-test of testing accuracy (Chen et al., 2014). The test procedures are briefly described as follows. The empirical P-value can be determined according to how extreme the observed accuracy is in its null distribution that is generated by permutations. Specifically, after analyzing a set of real data, a certain number of sets of pseudo samples are generated by randomly shuffling either the genotypes or the residual statistic among the real samples so that any potential relationship of interest between the genotypes and the phenotypes was disrupted in the sets of permuted samples. The same analysis as in the real data set is applied to each set of pseudo samples, and the resulting accuracies form the null distribution of accuracy. The P-value is then estimated by the proportion of the pseudo samples resulting in larger accuracy than the observed one in the real data set. When the null hypothesis holds true, the testing accuracy will be equi-probably either ⩾0.5 (positive sign) or <0.5 (negative sign). The frequencies of positive and negative signs can be used to evaluate P-values. When the sample size is sufficiently large, as the result of the central limit theory, testing accuracy asymptotically has a normal distribution and the P-value can be assessed from the z-test.
. Each nonempty cell is labeled either high-valued if its average statistic value is larger than some threshold T or low-valued otherwise. In Step 4, a new attribute is created by pooling high-valued and low-valued cells into two contrasting (that is, high-valued and low-valued) groups, representing the classification that best captures the correlation between this set of factors and the phenotype(s). In Step 5, the classification accuracy can be assessed for each contingency table. The best model(s) can be identified among all the possible m-way contingency tables based on classification accuracy. In Step 6, the independent testing set is used to evaluate the testing accuracy for the best model among those with different dimensionalities identified in Step 5. If the null hypothesis holds true and the classification model is formed purely by chance, it will give a null testing accuracy of 0.5 as the testing set is independent of the training set in which the model selection is involved. The significance test can be implemented based on permutation testing, nonparametric sign testing and asymptotical z-test of testing accuracy (Chen et al., 2014). The test procedures are briefly described as follows. The empirical P-value can be determined according to how extreme the observed accuracy is in its null distribution that is generated by permutations. Specifically, after analyzing a set of real data, a certain number of sets of pseudo samples are generated by randomly shuffling either the genotypes or the residual statistic among the real samples so that any potential relationship of interest between the genotypes and the phenotypes was disrupted in the sets of permuted samples. The same analysis as in the real data set is applied to each set of pseudo samples, and the resulting accuracies form the null distribution of accuracy. The P-value is then estimated by the proportion of the pseudo samples resulting in larger accuracy than the observed one in the real data set. When the null hypothesis holds true, the testing accuracy will be equi-probably either ⩾0.5 (positive sign) or <0.5 (negative sign). The frequencies of positive and negative signs can be used to evaluate P-values. When the sample size is sufficiently large, as the result of the central limit theory, testing accuracy asymptotically has a normal distribution and the P-value can be assessed from the z-test.
An open source GMDR package is developed in Java for implementing the proposed methods. Software note is presented in Supplementary Appendix C. The software is available at http://www.soph.uab.edu/ssg/software.
Results
Real data analysis
To demonstrate the increased power, the UGMDR method was applied to detect interactions among single-nucleotide polymorphisms in three genes that were revealed responsible for smoking-related phenotypes in the literature (Li et al., 2010; Chen et al., 2012): 8 in CHRNA5, 12 in CHRNA3, and 20 in CHRNB4, for nicotine dependence in the cohort for Study of Addiction: Genetics and Environment (SAGE) composed of three subsamples: the Collaborative Study on the Genetics of Alcoholism (COGA), the Collaborative Study on the Genetics of Nicotine Dependence (COGEND), and the Family Study of Cocaine Dependence (FSCD). A majority of the SAGE are unrelated samples plus a few families. After quality control, a total of 2793 individuals, including 695 in the COGA, 1353 in the COGEND, and 745 in the FSCD were used for analysis. Sex and age were modeled as covariates for the Fagerström Test for Nicotine Dependence score, and the five principal components were used to correct the potential population stratification. As a contrast, the benchmark meta-analysis was also conducted with the Fisher's combining P-value method. Figure 3 depicts the Manhattan plots on the P-values of 1920 trigenic interactions from the two methods. As compared with the meta-analysis, the unified strategy substantially increased the statistical power, supporting that a pooled analysis is likely more powerful than a meta-analysis of combining P-values from individual analyses (Skol et al., 2006).
Figure 3.

Manhattan plots for trigenic interactions. The dummy coordinates along the x axis index the 1920 interactions while the negative logarithm of the P-value associated with each interaction is displayed on the y axis. SAGE represents the unified analysis for three subsamples: FSCD, COGEND, and COGA, while the meta-analysis is implemented with the Fisher's combining P-value method for individual analyses of the three subsamples. The brown triangle represented the interaction, rs6495306–rs8040868–rs12905641, which had the highest P-value in the SAGE samples. A full color version of this figure is available at the Heredity journal online.
Simulation study
To further verify the power gain, a series of simulations was conducted based on the genotypic data of the same set of SAGE samples analyzed in the real case study. A trigenic (that is, three functional loci involved) interaction model was used to simulate the phenotypic data, in which the genotypes with three uppercase were set as a high-valued phenotypic groups and the rest as a low-valued group. The phenotypic value of an individual was generated under the following linear model,
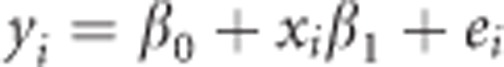 |
where β0=0 is the intercept, β1=0.3 is the regression coefficient, xi is the indicator variable taking 1 if the genotype belongs to the high-valued group or taking 0 otherwise and ei~N(0,1) is the residual error. One single-nucleotide polymorphism in each of the CHRNA5, CHRNA3 and CHRNB4 genes was set as a causal variant, resulting in a total of 1920 trigenic scenarios. The heritability varied from a trigenic scenario to another because of different allelic frequencies and linkage disequilibrium structures across these single-nucleotide polymorphism loci, and the upper limit of the heritability was ~0.07 when the high-valued group and the low-valued group were nearly equi-frequent. A total of 200 simulations were run to evaluate the statistical power.
As in the real data analysis, the unified analysis and meta-analysis were performed for simulated phenotypes. The P-value was determined by permutation testing with 1000 replicates in each simulation, and statistical power was calculated at the significance level of 0.05. Figure 4 displays the Manhattan plots on the statistical power of 1920 trigenic interactions from the two methods. As shown in Figure 4, the averaged power of the unified analysis and the meta-analysis were 0.706 and 0.566, respectively, supporting a substantial power gain in the unified analytical strategy.
Figure 4.

Manhattan plots of power for trigenic interactions. The dummy coordinates along the x axis index the 1920 interactions while the power associated with each interaction is displayed on the y axis. SAGE represents the unified analysis for three subsamples: FSCD, COGEND, and COGA, while the meta-analysis is implemented with the Fisher's combining P-value method for individual analyses of the three subsamples. The reference line in red is the average of power over the 1920 trigenic scenarios. A full color version of this figure is available at the Heredity journal online.
Discussion
No gene or environmental factor is an island, entire of itself, in shaping a biological phenotype; every one is a piece of the interactive genome and epigenome. Thus it is pivotal in unveiling genetic basis of such polygenic and multifactorial traits to identify background-specific factors among genes in combination with lifestyles and environmental exposures. The MDR approach is one of the most prevailing methods and has considerable appeal as a feasible strategy to attack the formidable analytical and computational difficulties arising from the tremendous volume of potential interactions. However, the existing extensions of MDR are still underpowered in applications to diverse types of phenotypes and various study designs. Complex traits take various forms, for example, continuous in distribution such as height and blood pressure, dichotomous such as affection status ‘well' and ‘affected', ordinal such as disease severity, meristic such as tumor number, time-to-event such as survival time, multifaceted such as substance addictions, or other, and each type of data may have its own nature corresponding to the specific developing mechanism. MDR is also subject to both false positives and false negatives in the presence of population stratification and cryptic structure. MDR is not able to handle family-based samples that are immune to confounding and biasedness due to population heterogeneity and pooled unrelated and family samples.
The proposed GMDR offers a conceptual framework for a comprehensive extension of MDR to handling the breadth of data types and addressing statistical issues associated with study design and sampling scheme. The generalization is based on use of two flexible coding schemes, one related to the attributes of interest and the other related to the phenotypic outcome. The former takes care of the issues on the study design and sample structure while the latter accounts for different types and multiplicity of phenotypes. Different combinations of coding schemes well serve for multi-purposes in genetic studies. Although retaining the advantage of MDR being computationally efficient, the proposed UGMDR can be used for many scenarios in identifying multifactor interaction, not only allowing for covariate adjustment, being suitable for the analysis of almost any type of phenotypic data, for example, real-valued, binary-valued, categorical-valued, count-valued, as well as combinations of those, but also being applicable to various study designs, including unrelated-subject, admixed sample and family as well as mixtures of them. The proposed conceptual framework will pave the way toward more tailored and effective analysis and broaden the use of GMDR approach. There is an expanding list of applications of GMDR to a number of complex disorders for detection of gene–gene and/or gene–environment interactions since the appearance of GMDR (see an incomplete summary of applications in Supplementary Appendix D). These support that GMDR is having an increasingly important role in tracking down interacting contributors and mapping complex genotype–phenotype relationship.
The proposed UGMDR can promote statistical power, theoretically arisen from the covariate adjustment to eliminate the noise attributable to confounders, a unified analytical strategy to entertain unrelated and related samples as well as those from structured populations, capturing information from correlated phenotypes and better choice for model fitting. The power gain has been extensively demonstrated in the real data analyses and the simulations in the present study and other reports (Lou et al., 2007, 2008; Chen et al., 2011, 2014).
In addition to the models presented in the Methods section, there are many alternative statistical models used in genetics such as log-linear model, probit model and complementary log-log model for dichotomous and polytomous phenotypes, continuous latent response model and continuation-ratio model for ordinal phenotypes and parametric survival model. It is straightforward to integrate these models into the proposed GMDR framework for a more relevant choice of phenotypic models. Moreover, the statistic reflecting a subject-specific contribution to the component residual score/quasi-score is used as it is coherent with the estimating equations. Several other types of residuals can be also considered in GMDR, including response residual, working residual, Pearson residual and deviance residual. The validity of GMDR does not depend on the choice of residuals in the sense of correct type I error rates, although the power may.
Data archiving
The UGMDR software is available at http://www.soph.uab.edu/ssg/software.
Acknowledgments
I thank Jason Moore for making the MDR Java source code available to this project and Guo-Bo Chen, Xi-Wei Sun, Hai-Ming Xu and Lei Yan for their contributions in the development of GMDR. This project was supported in part by an NIH Grant DA025095 to X-YL. The data set used was obtained from the database of Genotypes and Phenotypes (dbGaP) through dbGaP accession number phs000092.v1.p.
The author declares no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Agresti A (1999). Modelling ordered categorical data: recent advances and future challenges. Stat Med 18: 2191–2207. [DOI] [PubMed] [Google Scholar]
- Altman DG, Bland JM (1998). Statistics notes - time to event (survival) data. Br Med J 317: 468–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananth CV, Kleinbaum DG (1997). Regression models for ordinal responses: a review of methods and applications. Int J Epidemiol 26: 1323–1333. [DOI] [PubMed] [Google Scholar]
- Begg CB, Gray R (1984). Calculation of polychotomous logistic regression parameters using individualized regressions. Biometrika 71: 11–18. [Google Scholar]
- Carlborg O, Haley CS (2004). Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5: 618–625. [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Kruglyak L, Nickerson DA (2004). Mapping complex disease loci in whole-genome association studies. Nature 429: 446–452. [DOI] [PubMed] [Google Scholar]
- Chen GB, Liu N, Klimentidis YC, Zhu X, Zhi D, Wang X et al. (2014). A unified GMDR method for detecting gene-gene interactions in family and unrelated samples with application to nicotine dependence. Hum Genet 133: 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GB, Xu Y, Xu HM, Li MD, Zhu J, Lou XY (2011). Practical and theoretical considerations in study design for detecting gene-gene interactions using MDR and GMDR approaches. PLoS ONE 6: e16981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LS, Baker TB, Piper ME, Breslau N, Cannon DS, Doheny KF et al. (2012). Interplay of genetic risk factors (CHRNA5-CHRNA3-CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry 169: 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ (2009). Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet 10: 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DR (1972). Regression models and life-tables. J R Stat Soc Ser B (Methodological) 34: 187–220. [Google Scholar]
- Culverhouse R, Klein T, Shannon W (2004). Detecting epistatic interactions contributing to quantitative traits. Genet Epidemiol 27: 141–152. [DOI] [PubMed] [Google Scholar]
- Gui JA, Moore JH, Kelsey KT, Marsit CJ, Karagas MR, Andrew AS (2011). A novel survival multifactor dimensionality reduction method for detecting gene-gene interactions with application to bladder cancer prognosis. Hum Genet 129: 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kwon MS, Oh JM, Park T (2012). Gene-gene interaction analysis for the survival phenotype based on the Cox model. Bioinformatics 28: i582–i588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Chung Y, Elston RC, Kim Y, Park T (2007). Log-linear model-based multifactor dimensionality reduction method to detect gene gene interactions. Bioinformatics 23: 2589–2595. [DOI] [PubMed] [Google Scholar]
- Li MD, Xu Q, Lou XY, Payne TJ, Niu T, Ma JZ (2010). Association and interaction analysis of variants in CHRNA5/CHRNA3/CHRNB4 gene cluster with nicotine dependence in African and European Americans. Am J Med Genet B Neuropsychiatr Genet 153B: 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KY, Zeger SL (1986). Longitudinal data-analysis using generalized linear-models. Biometrika 73: 13–22. [Google Scholar]
- Lin PI, Vance JM, Pericak-Vance MA, Martin ER (2007). No gene is an island: the flip-flop phenomenon. Am J Hum Genet 80: 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003). Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33: 177–182. [DOI] [PubMed] [Google Scholar]
- Lou XY, Chen GB, Yan L, Ma JZ, Mangold JE, Zhu J et al. (2008). A combinatorial approach to detecting gene-gene and gene-environment interactions in family studies. Am J Hum Genet 83: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou XY, Chen GB, Yan L, Ma JZ, Zhu J, Elston RC et al. (2007). A generalized combinatorial approach for detecting gene-by-gene and gene-by-environment interactions with application to nicotine dependence. Am J Hum Genet 80: 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ et al. (2009). Finding the missing heritability of complex diseases. Nature 461: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, Ritchie MD, Hahn L, Kang S, Moore JH (2006). A novel method to identify gene-gene effects in nuclear families: the MDR-PDT. Genet Epidemiol 30: 111–123. [DOI] [PubMed] [Google Scholar]
- McCullagh P (1980). Regression models for ordinal data. J R Stat Soc Ser B (Methodology) 42: 109–142. [Google Scholar]
- McCullagh P (1983). Quasi-likelihood functions. Ann Stat 11: 59–67. [Google Scholar]
- Michalski RS (1983). A theory and methodology of inductive learning. Artif Intell 20: 111–161. [Google Scholar]
- Moore JH, Ritchie MD (2004). The challenges of whole-genome approaches to common diseases. J Am Med Assoc 291: 1642–1643. [DOI] [PubMed] [Google Scholar]
- Nelder JA, Wedderburn RWM (1972). Generalized linear models. J R Stat Soc Ser A-G 135: 370–384. [Google Scholar]
- Nijhout HF (2003). On the association between genes and complex traits. J Investig Dermatol Symp Proc 8: 162–163. [DOI] [PubMed] [Google Scholar]
- Niu A, Zhang S, Sha Q (2011). A novel method to detect gene-gene interactions in structured populations: MDR-SP. Ann Hum Genet 75: 742–754. [DOI] [PubMed] [Google Scholar]
- Phillips PC (2008). Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 9: 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Hahn LW, Roodi N, Bailey LR, Dupont WD, Parl FF et al. (2001). Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet 69: 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skol AD, Scott LJ, Abecasis GR, Boehnke M (2006). Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38: 209–213. [DOI] [PubMed] [Google Scholar]
- Sutradhar BC, Kovacevic M (2000). Analysing ordinal longitudinal survey data: generalised estimating equations approach. Biometrika 87: 837–848. [Google Scholar]
- Wolf JB (2002). The geometry of phenotypic evolution in developmental hyperspace. Proc Natl Acad Sci USA 99: 15849–15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.