

Innate immune responses play a critical role in atherosclerosis progression. Beside the well-known implication of macrophages,1 plasmacytoid dendritic cells (PDC) emerge as cells present in atherosclerotic lesions that may participate in or regulate atherosclerosis.2 These cells can exert both pro- and anti-inflammatory functions.3 This is the case of other cellular messengers, namely microparticles (MP),4 also implicated in atherosclerosis.4 Depending on their cellular origin, MP could either activate or inhibit PDC inflammatory responses.5 However, signaling pathways involved in these PDC responses still have to be identified. As membrane-derived by-products, MP after internalization can be a source of intracellular cholesterol derivatives6 that trigger liver X receptors (LXR).7 LXR are nuclear receptors regulating innate immune responses,8 including cell-derived by-product internalization, such as apoptotic bodies9 that belong together with MP to extracellular vesicles. Here, implication of the LXR pathway in MP engulfment by PDC and the subsequent PDC inflammatory responses were investigated.
Human PDC were isolated by magnetic cell separation (Diamond PDC Isolation Kit II, Miltenyi Biotec) from blood of healthy donors after the signature of informed consent and were stimulated with 3 different LXR agonists: 22-R-Hydroxycholesterol (22RHC), T0901317 and GW3965, LXR antagonist 22-S-Hydroxycholesterol (22SHC, Sigma Aldrich), MP or toll-like receptor (TLR)7 ligand, R848 (1 μg/mL, or TLR9 ligand, CpG-ODN2216 [class A CpG-containing oligodeoxynucleotides (ODN), 2 μmol/L; Invivogen]. Endothelial-derived MP (EMP) were isolated from a TNF-α-stimulated endothelial cell line and platelet-derived MP (PMP) from platelet concentrate supernatants of healthy donors as described in the Online Supplementary Methods. Microparticle size (using Megamix plus™ microbeads, BioCytex), count (using Cytocount® microbeads, Dako) and phenotype [(CD31, CD41, CD235a expression or Annexin V (AnxV) staining)] were determined by cytometry (Navios cytometer with the CXP software, and analyzed with the Kaluza 1.2 software (Beckman Coulter). PDC expression of CD123, BDCA-2/CD303, BAI-1 (brain-specific angiogenesis inhibitor-1) and intracellular TNF-α, as well as detection of eFluor+ PDC after incubation with eFluor-labeled MP, were determined by cytometry (FACS Canto II cytometer with DIVA 6.1 software, BD Biosciences). Unconjugated monoclonal antibodies and their control isotypes were used to block BAI-1 or detect CD123, ABCA1 (ATP-binding cassette transporter A1) and phosphorylated NF-κB subunit RelA (pRelA) after staining with a fluorochrome-conjugated anti-Ig secondary antibody (Life Technologies). Fluorescent images were acquired on an Olympus FV1000 confocal microscope and analyzed with Olympus FV-viewer software. Culture supernatants were collected after PDC treatment to measure the following cytokines: TNF-α, IL-6 and IL-8 (DIAplex technology, Diaclone), or interferon (IFN)-α (Platinum ELISA kit; eBioscience). Expression of LXR target gene, cytokine or endocytic receptor mRNA was assessed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) using either the Power SYBR Green PCR Master Mix or Taqman Universal Buffer II (Applied Biosystems) on a CFX96 Real-Time System (Biorad). Further details are available in the Online Supplementary Appendix.
Freshly isolated PDC were treated with either LXR agonists (T0901317, GW3965, 22RHC) or LXR antagonist (22SHC) for 24 h, and expression of LXR target genes (srebp1c and abca1) was assessed by qRT-PCR. 22SHC inhibited srebp1c mRNA expression, while LXR agonists increased both srepb1c and abca1 mRNA (Figure 1A), suggesting LXR pathway activation in PDC. LXR (lxra and lxrb) isoform mRNA expression was assessed by qRT-PCR in freshly isolated PDC. Preferential expression of the ubiquitous lxrb isoform mRNA by PDC was shown (Online Supplementary Figure S1). Increased expression of ABCA1 at protein level after LXR agonist treatment was confirmed by induction of ABCA1 expression on PDC membrane, as assessed by confocal microscopy (Figure 1B). In other immune cells, LXR activation has been described to alter TLR stimulation8,10 and promote phosphatidylserine (PtdSer) positive apoptotic body internalization.9 We evaluated whether LXR stimulation modulates PDC responses to TLR signaling. PDC were first treated with LXR agonists for 24 h, and stimulated with TLR ligands, R848 (TLR7) and CpG-ODN2216 (TLR9) for 18 h. Secretion of TNF-α and IL-6 was significantly reduced when PDC were pre-treated with LXR agonists (Figure 1C). PDC CAL-1 cells expressed reduced intracellular TNF-α levels when pre-treated with LXR agonists before TLR7 stimulation (Figure 1C). However, LXR agonist pre-treatment did not affect TLR9-induced cytokine production by PDC (Figure 1C). In contrast to TLR9, TLR7 stimulation involves predominantly the canonical NF-κB signaling pathway in PDC.11 Pre-treatment of PDC for 24 h with LXR agonists, T0901317 or GW3965 followed by R848 stimulation for 45 min significantly decreased pRelA staining analyzed by confocal microscopy (Figure 1D). This demonstrates that the LXR pathway is fully functional in human PDC and its activation inhibits TLR7-mediated NF-κB activation and pro-inflammatory cytokine secretion. This was shown using three different LXR agonists, including GW3965 considered as a pure LXRα/β agonist.12 As described in macrophages or conventional DC,8,10 LXR triggering in PDC blocks TLR signaling pathway. NF-κB inhibition by LXR triggering involves stabilization of a corepressor/NF-κB complex in target gene promoter,8 or prevention of NF-κB p50 subunit nuclear translocation.10 Here, we observed an inhibition of RelA phosphorylation.
Figure 1.
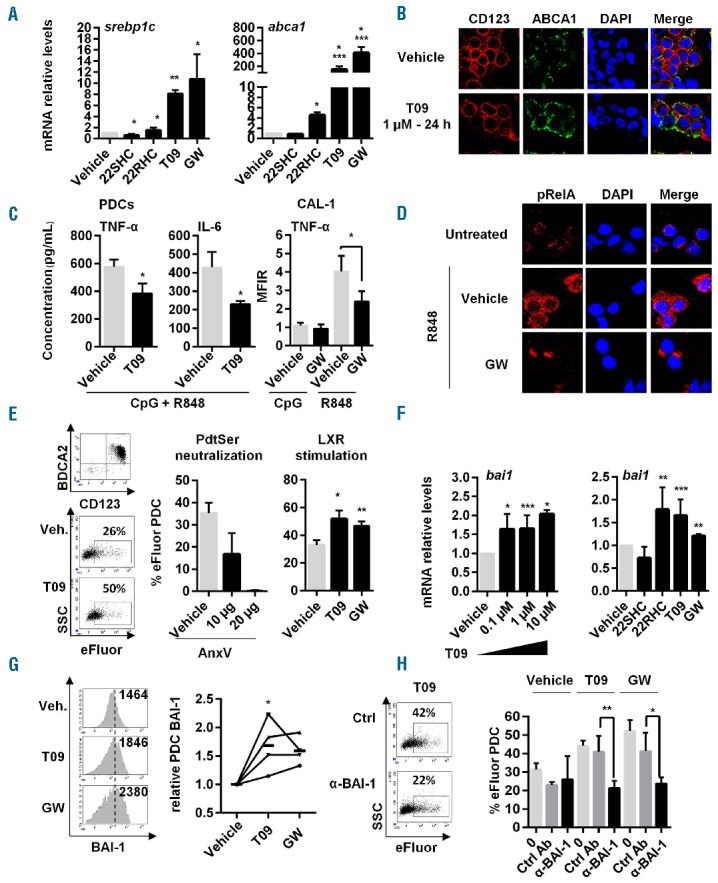
Stimulation of LXR pathway in PDC inhibits TLR7-induced NF-κB activation, pro-inflammatory cytokine secretion and up-regulates MP internalization via BAI-1. (A) Different LXR agonists, T0901317 (T09), GW3965 (GW), 22RHC or LXR antagonist 22SHC were added (1 μM) to PDC from healthy donors for 24 h and LXR target gene (srebp1c, abca1, lxra [not depicted] data not shown) expression was assessed by qRT-PCR (n=7, meaning PDC isolated from 7 different donors). (B) Freshly isolated PDC were stimulated with 1 μM of T0901317 (T09) for 24 h. Then CD123 and ABCA1 cell surface expression was evaluated by confocal microscopy analysis. Results from a representative experiment out of 3 using PDC from 3 different donors. (C) Freshly isolated PDC (left panels) or PDC cell line CAL-1 cells (right panel) were treated with 1 μM of T0901317 (T09) or GW3965 (GW) for 24 h, followed by a 18-h stimulation using CpG-ODN2216 (CpG, 2 μM) + R848 (1 μg/mL) (left panels) representing TLR9 and TLR7 ligand, respectively, or each ligand separately (right panel). Supernatants were then collected and assessed for TNF-α and IL-6 concentration by multiplex assays (n=5). Intracellular TNF-α levels in CAL-1 cells were also assessed by cytometry. Cumulated mean fluorescent intensity ratio (MFIR=MFI of treated cells/MFI of untreated vehicle cells) is shown (right panel) as mean±S.E.M. of the 5 independent experiments. (D) Freshly isolated PDC were treated for 24 h with 1 μM of GW3965 (GW), followed by TLR7 ligand, R848 (1 μg/mL) for 45 min. Phosphorylation of the NF-κB p65 (pRelA) subunit and its cellular localization were evaluated by confocal microscopy analysis. Results from a representative experiment out of 3 using PDC from 3 different donors. (E) PDC were treated with 1 μM of T0901317 (T09) or GW3965 (GW), washed and cultured with eFluor-labeled MP for 4 h at a 1/40 PDC/MP ratio. MP uptake by PDC was evaluated by measuring eFluor+ PDC by cytometry (n=3). Left panel shows a representative experiment. Middle panel shows that MP uptake requires PdtSer/PdtSer receptor interactions since blockade of these interactions using unlabeled Annexin-V (AnxV at 10 μg or 20 μg) incubated for 30 min with e-Fluor labeled MP decreases the percentage of eFluor+ PDC (n=3). This shows that free eFluor released from MP was not responsible for PDC labeling. (Right) Cumulative data of 3 independent experiments using LXR agonist-treated PDC from 3 different donors expressed as mean±S.E.M. are shown. (F) PDC were treated with increasing concentrations of T09 (0.1 μM, 1 μM or 10 μM) for 24 h (left panel) or LXR agonists, T0901317 (T09), GW3965 (GW), 22RHC or LXR antagonist 22SHC (for a single dose of 1 μM, then bai1 gene expression was quantified by qRT-PCR (n=5). (G) PDC were treated 24 h with 1 μM of T0901317 (T09) and GW3965 (GW) and BAI-1 expression was assessed by cytometry (1 representative experiment out of 4, left panel). Numbers represents mean fluorescent intensity (MFI). Data of 4 independent experiments using PDC from 4 different donors expressed as relative PDC BAI expression obtained using the following formula [% of PDC stained with anti-BAI-1 mAb after treatment] / [% of PDC stained with anti-BAI-1 mAb in vehicle condition], bold lines represent the mean of the 4 experiments (mean±SEM;*P<0.05, Wilcoxon) (right panel). (H) PDC were treated with 1 μM of T0901317 (T09) or GW3965 (GW) for 24 h. Then, cells were cultured for 1 h with neutralizing anti-BAI-1 antibody (α-BAI-1) or irrelevant control antibody (Ctrl Ab), before adding eFluor-labeled MP for 4 h and analyzing by cytometry (n=4). Dot plot from one experiment representative of 4 (left panel). Cumulative data of 4 independent experiments expressed as mean±S.E.M. of eFluor+ PDC are shown. Unless specified, all data were expressed as mean±S.E.M. from n independent experiments. *P<0.05, **P<0.001, ***P<0.005 (Mann-Whitney).
Despite the fact that PDC are not professional phagocytes participating in apoptotic cell/body removal like macrophages,13 PDC are able to internalize membrane-derived MP.5 We then evaluated the role of LXR in PtdSer-dependent MP internalization by PDC. After showing that MP uptake by PDC was PtdSer-dependent using AnxV, we observed that LXR stimulation significantly increased eFluor-labeled MP internalization by PDC (Figure 1E). T0901317 treatment showed a concentration-dependent upregulation of bai1 mRNA levels. The same effect was obtained with the two other LXR agonists, while 22SHC did not change bai1 mRNA expression (Figure 1F). Increased BAI-1 membrane expression on PDC was confirmed after T0901317 or GW3965 treatment by cytometry (Figure 1G). Finally, blockade of BAI-1 using an anti-BAI-1 antibody significantly reduced MP uptake by LXR-stimulated PDC (Figure 1H). While LXR activation in macrophages induces Mer-tk expression,9 neither stabilin2, tim1, tim4 nor mertk PtdSer receptor mRNA expression were modulated in PDC after LXR activation (data not shown). Moreover, Mer-tk blockade in PDC has no effect (data not shown). Overall, LXR activation in PDC significantly enhances MP uptake through upregulation of the Ptdser receptor BAI-1. This was observed whatever the MP origin, EMP or PMP (Online Supplementary Figure S2).
Since MP contain potential cholesterol-derived LXR ligands,6 we evaluated LXR activation in PDC after MP co-culture. PMP or EMP were previously reported to exert anti- or pro-inflammatory effects, respectively.5 We confirmed here that PMP decreased TNF-α and IL-8 secretion in a PMP number-dependent manner (Figure 2A and B). Looking at the LXR pathway, we observed a significant induction of LXR target gene (lxra, srebp-1c and abca1) and bai1 transcription, while tnfa mRNA levels were significantly diminished in PDC, depending again on the number of PMP (Figure 2C). PDC pre-treatment with the LXR antagonist 22SHC before PMP addition tend to prevent LXR target gene expression upregulation, while significantly increasing tnfa mRNA expression (Figure 2D). This identifies a novel anti-inflammatory mechanism of PMP: the activation of the LXR pathway.
Figure 2.
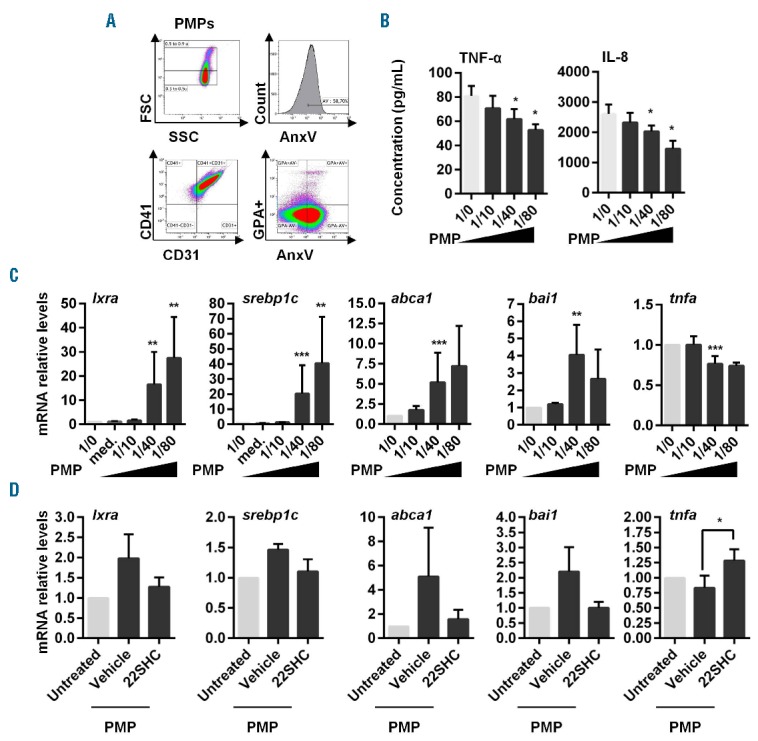
Platelet-derived microparticles reduce PDC pro-inflammatory cytokine secretion and stimulate LXR activation in PDC. (A) Flow cytometric characterization and quantification of PMP. MP isolated from platelet concentrates of healthy donors were characterized for PdtSer expression [(annexin V [AnxV] staining)]. In addition, to confirm that PMP are derived from platelets and not red blood cells, expression of CD31, CD41 and CD235a was assessed using Navios cytometer (Beckman Coulter). PMP are positive for CD31 and CD41 but negative for red blood cell marker Glycophorin A (CD235a). Data from one representative experiment out of 7 independent productions of PMP. (B) Freshly isolated PDC were cultured with PMP at different PDC/PMP ratio (i.e. without, 1/0; one PDC for 10 PMP, 1/10; one PDC for 40 PMP, 1/40; 1 PDC for 80 PMP, 1/80) for 18 h. Supernatants from PDC/PMP culture were then collected and assessed for TNF-α and IL-8 using multiplex assays (n=7). (C) LXR target gene (lxra, srebp1c, abca1), bai1 and tnfa mRNA transcripts were quantified by qRT-PCR (n=9). The last wash supernatant of PMP preparation in an equivalent volume of PMP suspension was added to PDC culture as control of lxra and srebp1c mRNA transcript quantification (med. condition). (D) PDC were treated with the LXR antagonist 22SHC (1 μM) or vehicle (DMSO) for 24 h. Then, cells were washed and cultured with PMP at a 1/40 PDC/PMP ratio for 18 h (PMP) or no PMP (Untreated). LXR target gene (lxra, srebp1c, abca1), bai1 and tnfa mRNA transcripts were quantified by qRT-PCR (n=4). Data from n independent experiment were expressed as mean±S.E.M. *P<0.05, **P<0.001, ***P<0.005 (Mann-Whitney).
Endothelial-derived MP are endowed with pro-inflammatory properties.5 After having confirmed that TNF-α and IL-8 secretion was increased after EMP incubation, we observed that EMP induced NF-κB activation in PDC associated with an increase of tnfa mRNA levels (Figure 3A). A downregulation of LXR target gene (srebp1c and abca1) transcription in PDC in an EMP number-dependent manner was also observed (Figure 3B). This effect of EMP was prevented by the prior LXR activation and associated with a significant decreased tnfa mRNA expression (Figure 3C). These data suggest an inhibition of the LXR pathway by NF-κB. To confirm this hypothesis, PDC CAL-1 cells were treated simultaneously with the LXR agonist GW3965 and TLR7 ligand R848 in the presence of the NF-κB inhibitor, JSH-23 or not. R848 treatment down-regulated LXR target gene expression induced by GW3965, while addition of JSH-23 totally reversed this effect (Figure 3D). Thus, the TLR signaling pathway dominates the LXR pathway, and NF-κB blockade is sufficient to restore LXR pathway activation. To translate this observation into EMP, freshly isolated PDC co-cultured with EMP were simultaneously treated with JSH-23. While JSH-23 led to a slight increase in lxra mRNA expression, we observed a reduction of il6 and tnfa mRNA levels, statistically significant for tnfa mRNA (Figure 3E). Restoration of LXR pathway activation in response to EMP by NF-κB blockade was confirmed by increased ABCA1 membrane expression, assessed by confocal microscopy (Figure 3F). Finally, PDC treatment with R848 simultaneously to PMP significantly decreased lxra mRNA expression induced by PMP and increased il6 mRNA expression (Online Supplementary Figure S3). These results confirm the critical role of NF-κB and LXR signaling pathway activation during MP engulfment by PDC, determining the pro- versus anti-inflammatory response of these cells.
Figure 3.
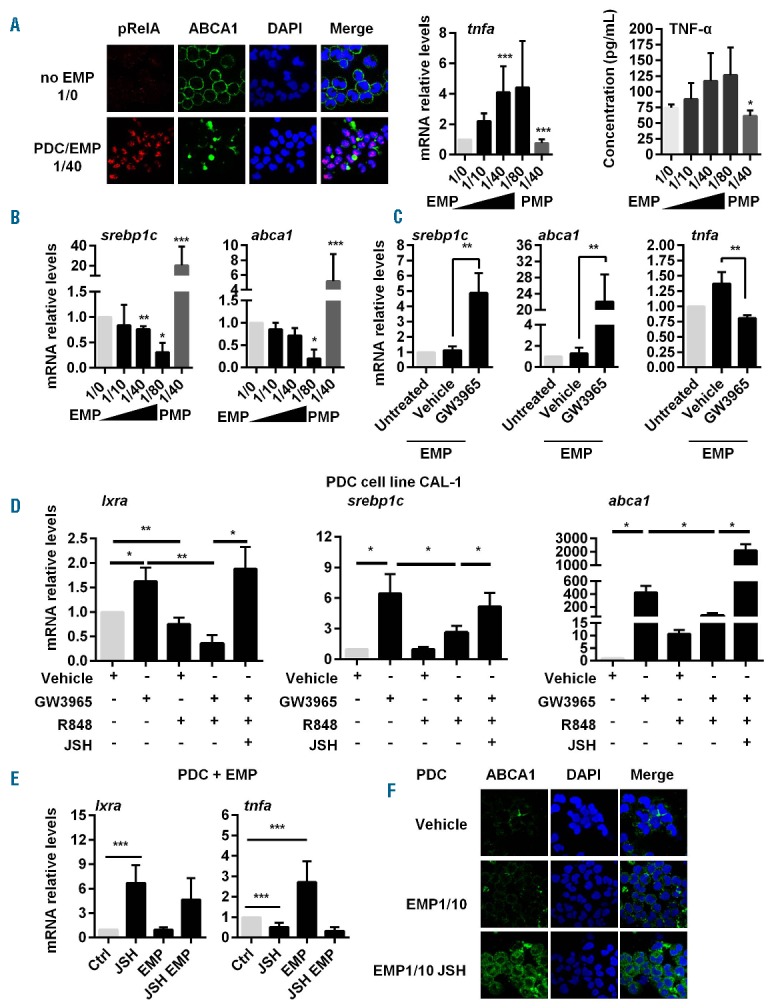
NF-κB triggering by EMP stimulation or TLR7 activation prevents LXR activation in PDC. (A) Freshly isolated PDC were incubated with EMP at different PDC/EMP ratio (i.e. without, 1/0; one PDC for 10 EMP, 1/10; one PDC for 40 EMP, 1/40; one PDC for 80 EMP, 1/80). (Left) Nuclear translocation of phosphorylated NF-κBp65 (pRelA) subunit was determined in PDC cultured at a 1/40 ratio for 2.5 h by confocal microscopy. Nuclei were stained with DAPI. EMP stimulation of PDC decreases also membrane ABCA-1 expression. Results from a representative experiment out of 3. (Middle) tnfa mRNA levels from PDC cultured with EMP at the different ratio or with PMP (1/40 ratio) for 18 h, were quantified by qRT-PCR (n=5). (Right) TNF secretion from PDC cultured with EMP at the different ratio or with PMP (1/40 ratio) for 24 h were quantified by multiplex assays (n=5 for EMP and n=3 for PMP). (B) PDC were cultured with EMP at different PDC/EMP ratio (i.e. without, 1/0; one PDC for 10 EMP, 1/10; one PDC for 40 EMP, 1/40; one PDC for 80 EMP, 1/80) for 18 h. LXR target gene (srebp1c, abca1) mRNA transcripts were quantified by qRT-PCR (n=5). (C) PDC were treated with the LXR agonist GW3965 (1 μM) or vehicle (DMSO) for 24 h. Then, cells were washed and cultured with EMP at a 1/40 PDC/EMP ratio for 18 h (EMP). Untreated PDC were used as control. Expression of LXR target genes (lxra, abca1), and tnfa gene were quantified by qRT-PCR (n=5). (D) Cells from PDC cell line CAL-1 were treated with GW3965 (1 μM), R848 (1 μg/mL), JSH-23 (JSH, 25 μM), or a combination of these treatments for 18 h. LXR target gene (lxra, srebp1c, abca1) mRNA transcripts were quantified by qRT-PCR (n=5). (E) Freshly isolated PDC were treated for 18 h with the NF-κB inhibitor, JSH-23 (JSH), with or without EMP at a PDC/EMP ratio of 1:40. Expression of LXR target gene (lxra) and inflammatory cytokine [tnfa, il6 (not depicted)] gene mRNA was quantified by qRT-PCR (n=3). (F) PDC were treated with the NF-κB inhibitor JSH-23 (JSH) with or without EMP (at a PDC/EMP ratio of 1/10, EMP/1/10) for 2.5 h. Membrane ABCA1 expression was analyzed by confocal microscopy (one representative experiment out of 3 using PDC from 3 different donors). Data from n independent experiments were expressed as mean±S.E.M. *P<0.05, **P<0.001, ***P<0.005 (Mann-Whitney).
Overall, this study demonstrates a functional LXR pathway in PDC and a unique LXR/NF-κB balance that controls the non-inflammatory versus inflammatory status of MP engulfed by PDC. This remains to be confirmed in pathological settings. However, since increased circulating EMP levels and pro-inflammatory PDC may be implicated in atherosclerosis, we may hypothesize that NF-κB signaling dominates the LXR pathway in this setting. Whether LXR activation could be a potential therapeutic target in atherosclerosis and other pathologies involving PDC and EMP dysregulation3 (including lupus or psoriasis9,14,15) remains to be investigated.
Acknowledgments
The authors would like to thank Dr Maeda (Nagasaki University, Japan) for kindly providing us with the human PDC line CAL-1, Sarah Odrion for editorial assistance, Yassin Tachikart and Aurore Gaillardet for technical assistance, Prof M de Carvalho Bittencourt (Université de Lorraine, France) for stimulating discussions.
Footnotes
Funding: this study was supported by the Etablissement Français du Sang (EFS, grant #2011–11 to PS), the Agence Nationale de la Recherche (Labex LipSTIC, ANR-11-LABX-0021), the Conseil Régional de Franche-Comté (“soutien au LabEX LipSTIC” 2014 & 2015 to PS), and the Fondation de Coopération Scientifique Bourgogne Franche-Comté (BQR BFC to DM & PS).
The online version of this letter has a SupplementaryAppendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Zeller I, Srivastava S. Macrophage functions in atherosclerosis. Circ. Res. 2014;115(12):e83–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Döring Y, Zernecke A. Plasmacytoid dendritic cells in atherosclerosis. Front Physiol. 2012;3:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chironi GN, Boulanger CM, Simon A, et al. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335(1):143–151. [DOI] [PubMed] [Google Scholar]
- 5.Angelot F, Seilles E, Biichle S, et al. Endothelial cell-derived microparticles induce plasmacytoid dendritic cell maturation: potential implications in inflammatory diseases. Haematologica. 2009;94(11):1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biró É, Akkerman JWN, Hoek FJ, et al. The phospholipid composition and cholesterol content of platelet-derived microparticles: a comparison with platelet membrane fractions. J Thromb Haemost. 2005;3(12):2754–2763. [DOI] [PubMed] [Google Scholar]
- 7.Janowski BA, Grogan MJ, Jones SA, et al. Structural requirements of ligands for the oxysterol liver X receptors LXRa and LXRb. Proc Natl Acad Sci USA. 1999;96(1):266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiss M, Czimmerer Z, Nagy L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. J Allergy Clin Immunol. 2013;132(2):264–286. [DOI] [PubMed] [Google Scholar]
- 9.A-Gonzalez N, Bensinger SJ, Hong C, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of LXR. Immunity. 2009;31(2):245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canavan M, McCarthy C, Larbi NB, et al. Activation of liver X receptor suppresses the production of the IL-12 family of cytokines by blocking nuclear translocation of NF-κBp50. Innate Immun. 2014; 20(7):675–687. [DOI] [PubMed] [Google Scholar]
- 11.Bao M, Liu Y-J. Regulation of TLR7/9 signaling in plasmacytoid dendritic cells. Protein Cell. 2013;4(1):40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito A, Hong C, Rong X, et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saas P, Kaminski S, Perruche S. Prospects of apoptotic cell-based therapies for transplantation and inflammatory diseases. Immunotherapy. 2013;5(10):1055–1073. [DOI] [PubMed] [Google Scholar]
- 14.Pelletier F, Garnache-Ottou F, Angelot F, et al. Increased levels of circulating endothelial-derived microparticles and small-size platelet-derived microparticles in psoriasis. J Invest Dermatol. 2011;131(7):1573–1576. [DOI] [PubMed] [Google Scholar]
- 15.Parker B, Al-Husain A, Pemberton P, et al. Suppression of inflammation reduces endothelial microparticles in active systemic lupus erythematosus. Ann Rheum Dis. 2014;73(6):1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]