

Abstract
2-Methoxyestradiol (2-ME), a metabolite of estradiol with little affinity for estrogen receptors, inhibits proliferation of vascular smooth muscle cells; however, the molecular mechanisms underlying this effect are incompletely understood. Our previous work shows that 2-ME inhibits initiation (blocks phosphorylation of ERK and Akt) and progression (reduces cyclin expression and increases expression of cyclin inhibitors) of the mitogenic pathway and interferes with mitosis (disrupts tubulin organization). Because the RhoA/ROCK1 pathway (RhoA → ROCK1 → myosin phosphatase targeting subunit → myosin light chain) is involved in cytokinesis, herein we tested the concept that 2-ME also blocks the RhoA/ROCK1 pathway. Because of the potential importance of 2-ME for preventing/treating vascular diseases, experiments were conducted in female human aortic vascular smooth muscle cells. Microarray transcriptional profiling suggested an effect of 2-ME on the RhoA/ROCK1 pathway. Indeed, 2-ME blocked mitogen-induced GTP-bound RhoABC expression and membrane-bound RhoA, suggesting interference with the activation of RhoA. 2-ME also reduced ROCK1 expression, suggesting reduced production of the primary downstream signaling kinase of the RhoA pathway. Moreover, 2-ME inhibited RhoA/ROCK1 pathway downstream signaling, including phosphorylated myosin phosphatase targeting subunit and myosin light chain; the ROCK1 inhibitor H-1152 mimicked these effects of 2-ME; both 2-ME and H-1152 blocked cytokinesis. 2-ME also reduced the expression of tissue factor, yet another downstream signaling component of the RhoA/ROCK1 pathway. We conclude that 2-ME inhibits the pathway RhoA → ROCK1 → myosin phosphatase targeting subunit → myosin light chain, and this likely contributes to the reduced cytokinesis in 2-ME treated HASMCs.
Keywords: 2-methoxyestradiol, RhoA, ROCK1, myosin phosphatase targeting subunit, myosin light chain, cytokinesis
2-methoxyestradiol (2-me) is an endogenous metabolite of estradiol that attenuates vascular smooth muscle cell (VSMC) proliferation, migration, and extracellular matrix synthesis (5, 8, 9) and reduces injury-induced neointima formation (4), cholesterol-induced atherosclerosis (9), monocrotaline-induced vascular thickening in pulmonary hypertension (9), and injury-induced glomerosclerosis (9). Although 2-ME inhibits VSMC proliferation and is thus effective against multiple proliferative disorders (4, 9, 10), the mechanisms via which 2-ME mediates these actions remain incompletely understood.
Our previously published work shows that in human aortic vascular smooth muscle cells (HASMCs) 2-ME inhibits cell proliferation in part by blocking initiation of the mitogenic pathway (4). Specifically, 2-ME inhibits phosphorylation of both ERK1/2 and Akt (Fig. 1), which turns off the mitogenic pathway by decreasing the expression and activity of cyclins. Indeed, we (4) found that 2-ME 1) blocks cell-cycle progression in both G0/G1 and in G2/M phases, 2) reduces cyclin D1 and cyclin B1 expression, 3) inhibits Cdk-1 and Cdk-2 activity, 4) reduces hyperphosporylation of retinoblastoma protein, and 5) upregulates the cyclin-Cdk inhibitor p27Kip1. In addition, our previous work demonstrates that 2-ME also blocks mitosis by interfering with tubulin polymerization (4) (Fig. 1). Thus, we have identified two major sites of action of 2-ME that mediate its effects on cell proliferation (4).
Fig. 1.
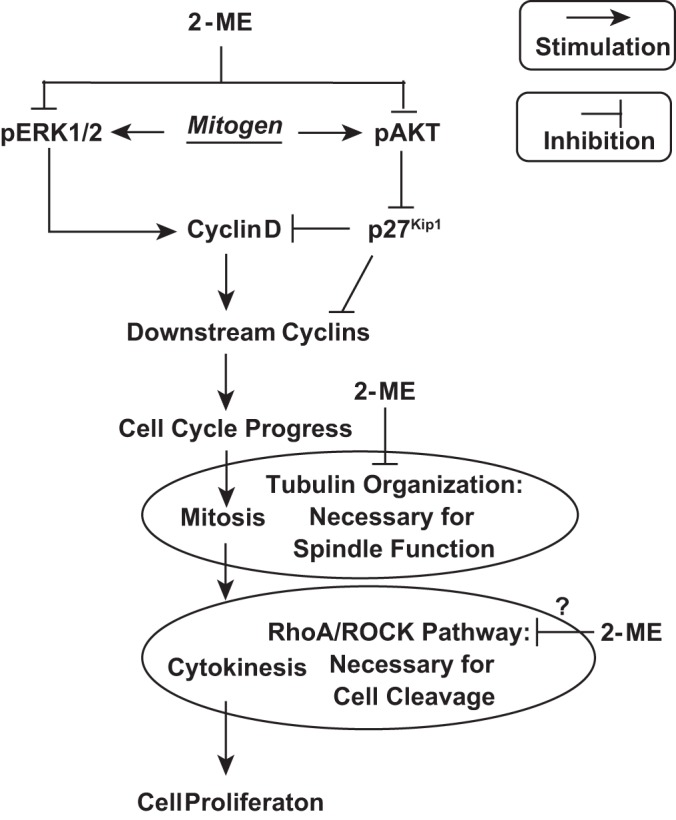
Signaling schematic depicting our hypothesis of how 2-methoxyestradiol (2-ME) regulates human aortic smooth muscle cells (HASMC) cell-cycle proliferation. Extracellular mitogens activate classical signal transduction pathways that ultimately phosphorylate (and thus activate) ERK1/2 and Akt. ERK1/2 is well known to increase expression of cyclin D (G1 phase cyclin). Phosphorylated Akt activates a signal transduction pathway that stabilizes Skp2, which is the F-box protein of SCFSkp2 ubiquitin ligase that polyubiquitinates p27Kip1 and thus accelerates the degradation of p27Kip1. Removal of p27Kip1 deinhibits cyclin D activity. Cyclin D activates cyclin-dependent kinase-4 and -6 to hyperphosphorylate retinoblastoma protein (Rb), thus releasing the transcription factor E2F and allowing increased expression of downstream cyclins, resulting in the progression of the cell cycle toward mitosis. Mitosis requires the organization of tubulin to construct the mitotic spindle and achieve proper orientation of chromosomes during metaphase and their separation during anaphase. Our previous studies show that 2-ME inhibits mitogen-induced phosphorylation of both ERK1/2 and Akt and, in addition, inhibits tubulin organization (which blocks mitosis). In the current study, we propose yet a third site of action, i.e., inhibition of the RhoA/ROCK pathway, which is required for contractile ring function leading to cytokinesis. Taken together with our past results, the present study indicates that 2-ME blocks cell proliferation by interfering with mitogenesis, mitosis, and cytokinesis.
The last step in cell proliferation is cytokinesis, which requires the organization of a contractile ring that mediates the cleavage furrow, leading to cell division. Importantly, the Ras homolog gene family member A (RhoA)/Rho-associated, coiled-coil-containing protein kinase-1 (ROCK1) pathway is involved in cytokinesis. Studies show that inactivation of RhoA leads to profound defects in cytokinesis, and in most cases cleavage furrow formation is completely blocked (29). Furthermore, there is evidence that microtubules are required for correct cytokinesis (29). In this regard, central spindle microtubules are necessary to guide the assembly on the equatorial cortex of the actomyosin contractile ring that mediates the contraction process that forms the cleave furrow resulting in formation of daughter cells (29). Indeed, disruption of microtubules by colchicine prevents the formation of the cleavage furrow (14). Importantly, RhoA accumulates at the contractile ring and is activated by ECT2, a cytokinesis-specific RhoGEF, during cytokinesis (29). Taken together, these observations indicate that RhoA acts as a microtubule-dependent signal in cytokinesis. It is known that 2-ME disrupts tubulin polymerization (4), and our previous microarray experiment shows a downregulation by 2-ME of genes necessary for mitotic spindle assembly (6). Thus, it is conceivable that 2-ME inhibits the activation of the RhoA/ROCK1 pathway via its microtubule disrupting action and thereby blocks cytokinesis (Fig. 1). If this hypothesis is correct, 2-ME is a unique antiproliferative agent because it blocks cell proliferation at three key states of the proliferative mechanism: 1) initiation and progression of the mitogenic program, 2) mitosis, and 3) cytokinesis (Fig. 1). Motivated by these facts, we decided to examine the hypothesis that 2-ME blocks the RhoA/ROCK1 pathway in HASMCs. We selected HASMCs for two reasons: 1) so we could compare the present results with past studies in HASMCs, and 2) to increase the clinical relevance of our findings.
MATERIALS AND METHODS
Overview of research strategy.
To accomplish our objective, we employed multiple approaches. First, to validate the consistence of our model system (HASMCs) compared with our previous findings, we reexamined the effects of 2-ME on cell number, DNA synthesis, proline synthesis, cell migration, and tubulin dynamics. Second, we investigated the effects of 2-ME on nuclear size (marker for lack of cytokinesis). Third, using high-density oligonucleotide microarrays, we examined the effects of 2-ME on transcripts involved in the RhoA/ROCK1 pathway. Fourth, we determine the effects of 2-ME on activity and localization of RhoA. Fifth, we examined the effects of 2-ME on ROCK1 expression. Sixth, we investigated the effects of 2-ME on classic downstream mediators of the RhoA/ROCK1 pathway (RhoA/ROCK1 → phosphorylated myosin phosphatase targeting subunit → phosphorylated myosin light chain). Seventh, we measured the effects of 2-ME on the expression of tissue factor, yet another downstream mediator of the RhoA/ROCK1 pathway (18). Eighth, we compared the effects of a ROCK inhibitor with those of 2-ME on cell cycle signaling pathways and cytokinesis. Ninth, we assessed the in vivo effects of 2-ME on ROCK1 and tissue factor expression in neointimal tissue.
Materials.
Primary antibodies, secondary antibodies, and chemicals and buffers are listed in Table 1 along with their sources.
Table 1.
List of antibodies, chemicals, and buffers used in this study
| Antibodies, Chemicals, Buffers | Host | Company | Dilution |
|---|---|---|---|
| Primary antibodies | |||
| Anti-β-actin | Mouse | Sigma | 1:5,000 |
| Anti-Akt | Rabbit | Cell Signaling | 1:1,000 |
| Anti-phospho-Akt (Thr308) | Rabbit | Cell Signaling | 1:1,000 |
| Anti-cyclin B1 | Mouse | Upstate | 1:1,000 |
| Anti-cyclin D1 | Mouse | Upstate | 1:1,000 |
| Anti-ERK1/2 | Mouse | Upstate | 1:1,000 |
| Anti-phospho-ERK1/2 (Thr202/Tyr204) | Rabbit | Upstate | 1:1,000 |
| Anti-rMLC | Rabbit | Cell Signaling | 1:1,000 |
| Anti-phospho-rMLC (Ser19) | Mouse | Cell Signaling | 1:1,000 |
| Anti-phospho-rMLC (Ser19) | Rabbit | Cell Signaling | 1:1,000 |
| Anti-MYPT | Rabbit | Santa Cruz Biotechnology | 1:200 |
| Anti-phopsho-MYPT (Thr696) | Rabbit | Santa Cruz Biotechnology | 1:200 |
| Anti-RhoA | Mouse | Santa Cruz Biotechnology | 1:500 |
| Anti-ROCK | Rabbit | Santa Cruz Biotechnology | 1:500 |
| Anti-tissue factor | Rabbit | American Diagnostic | 1:1,000 |
| Secondary antibodies: target | |||
| IRDye 800 conj. anti-mouse IgG | Goat | LI-COR Biosciences | 1:12,500 |
| IRDye 800 conj. anti-rabbit IgG | Goat | LI-COR Biosciences | 1:12,500 |
| IRDye 680 conj. anti-rabbit IgG | Goat | LI-COR Biosciences | 1:12,500 |
| IRDye 680 conj. anti-mouse IgG | Goat | LI-COR Biosciences | 1:12,500 |
| IGF-peroxidase conj. anti-mouse IgG | Goat | Pierce | 1:100,000 |
| IGF-peroxidase conj. anti-rabbit IgG | Goat | Pierce | 1:100,000 |
| FITC conj. anti-mouse IgG | Goat | Sigma | 1:1,000 |
| TRITC conj. anti-rabbit IgG | Goat | Sigma | 1:1,000 |
| Chemicals and buffers | |||
| Thrombin | Sigma | ||
| TNF-α | R&D Systems | ||
| Angiotensin II | Sigma | ||
| Aprotinin | Sigma | ||
| APS (ammoniumpersulfate) | Sigma | ||
| BCA (bicinchoninic acid) protein assay kit | Pierce | ||
| BSA (bovine serum albumin) | Sigma | ||
| DAPI (4′-6-diamidino-2-phenylindole) | Sigma | ||
| GGPP (geranylgeranyl pyrophosphate) | Sigma | ||
| H-1152 | Enzo Life Sciences | ||
| Leupeptin (hemisulfate salt) | Sigma | ||
| Loading buffer (5x) | Fermentas, H | ||
| Lysis buffer | Cell Signaling Technology | ||
| 2-ME (2-methoxyestradiol) | Steraloids | ||
| MTT (thiazolyl blue tetrazolium bromide) | Sigma | ||
| PFA (paraformaldehyde) | Merck, Darmstadt | ||
| PBS (phosphate-buffered saline) | Gibco-BRL | ||
| PDGF-BB | Sigma | ||
| PMSF (phenylmethylsulfonyl fluoride) | Sigma | ||
| Ponceau S solution (2%) | Sigma | ||
Culture of HASMCs.
HASMCs from females (Cascade Biologics, Portland, OR) between 4th and 8th passages were cultured under standard tissue culture conditions (37°C, 5% CO2) in M231 culture medium supplemented with 1× antibiotic-antimycotic (100 μg/ml streptomycin, 100 μg/ml penicillin, and 0.025 μg/ml amphotericin B) and 25× smooth muscle growth supplement [SMGS; 5% fetal calf serum (FCS), 2 ng/ml human basic fibroblast growth factor, 0.5 ng/ml human epidermal growth factor, 5 ng/ml heparin, 5 μg/ml insulin, and 0.2 μg/ml bovine serum albumin] (11). Medium was changed every 2 days. Once the culture reached ∼80% confluence, the medium was change every day. To split the culture, the medium was removed, and the cells were washed twice with HBSS without Ca2+ and Mg2+. After trypsinization with 0.25% trypsin diluted in HBSS (without Ca2+ and Mg2+), an equal volume of medium was added, and the cells were plated on culture dishes or in 75-cm2 flasks. Cell density was ∼20,000 cells/cm2. Studies were conducted using phenol red-free medium. When not specified, HASMCs were plated in normal growth medium at a density of 50,000 cells/well on 24-well plates (2 cm2) and allowed to attach overnight. The cultures were growth arrested in DMEM-F12 supplemented with 1× antibiotic-antimycotic, 0.1% sodium bicarbonate solution, and 0.4% steroid-free FCS for 24 h. HASMCs were treated in DMEM-F12 supplemented with 1× antibiotic-antimycotic, 0.1% sodium bicarbonate solution, and 0.4% steroid-free FCS in the presence or absence of the test agents in various concentrations. Treatments were changed every 48–72 h. Controls were treated with vehicle (maximal 0.1% DMSO).
Cell number and [3H]thymidine, and [3H]proline incorporation studies.
Cell number, DNA synthesis ([3H]thymidine incorporation), and collagen synthesis ([3H]proline incorporation) were conducted as previously described (4–6). After starvation, the cultures were treated with PDGF-BB (20 ng/ml) in the presence (at different concentrations) or absence of the test agents. For cell number, after different time periods, cells were dislodged by trypsinization and counted with a Coulter counter. To assess the effects of 2-ME (1–3 μmol/l) on DNA and collagen synthesis, HASMCs were pulsed with [3H]thymidine or [3H]proline, respectively, and their incorporation in acid-insoluble fraction was analyzed on a β-scintillation counter, as described before (11). These studies were conducted in triplicate.
Cell migration studies.
The effects of 2-ME on PDGF-BB-induced HASMC migration was assessed using a modified Boydens Chamber and as previously described in detail by us (12).
Analysis of 2-ME effects on protein expression and phosphorylation.
Changes in the protein expression of RhoA, RhoABC (RhoA + RhoB + RhoC), ROCK1, p-rMLC [phosphorylated (Ser19) regulatory subunit of myosin light chain], p-MYPT [phosphorylated (Thr696) myosin-targeting subunit of myosin light-chain phosphatase (MLCP)], p-ERK1/2 (phosphorylated extracellular signal regulated kinases 1 and 2), p-Akt (phosphorylated Akt), cyclin B1, cyclin D1, and tissue factor were analyzed by Western blotting. To study the effects of 2-ME on the expression of selected proteins, HASMCs plated in 60-mm culture dishes were washed twice in sterile PBS before being growth arrested for 24 h in DMEM-12 medium supplemented with 1× antibiotic-antimycotic, 0.1% sodium bicarbonate solution, and 0.4% steroid-free FCS. The cultures were treated in DMEM-F12 (as starvation) for different time periods (0–48 h). Before stimulation with PDGF-BB, cells were pretreated for 1 h with 2-ME. Controls were treated with vehicle (maximum 0.1% DMSO). After treatment, the cells were washed once with PBS and lysed by the addition of 70–100 μl of lysis buffer 1. The cell lysate was scraped from the dishes and homogenized by sonication (twice for 3 s) at low power. The protein concentration was determined as described under Protein concentration assay. Samples were kept at −20°C until use. Protein expression was analyzed by western blotting as previously described (5).
Protein concentration assay.
Protein concentrations were determined with the bicinchoninic acid (BCA) Protein Assay kit according to the manufacturer's protocol. The BCA Protein Assay is a detergent-compatible formulation based on BCA for the colorimetric detection and quantitation of total protein amounts in samples. The absorbance was measured with a SpectraFluor Plus and analyzed with the software Magellan 6.
Western blotting.
Western blot analysis was performed with whole cell lysates, with the exception of membrane fraction samples. Equal amounts of proteins (15–25 μg/lane) were diluted with 5× loading buffer and 0.1 M dithiotheritol (DTT), and water was added to a volume of 17 μl. After denaturated at 95°C for 5 min, the samples were loaded onto an SDS polyacrylamide gel (% of gel depending on molecular weight of analyzed protein). After separation on the gel, the proteins were transferred to a nitrocellulose membrane. Successful transfers were visualized by Ponceau S staining. For this the Ponceau S solution was added to the membrane. After 2 s, the Ponceau S solution was transferred back to the vial, and the membrane was washed in PBS until the red staining disappeared. If necessary, membranes were cut at the appropriate molecular weight before the Ponceau staining was washed out. Membranes were blocked in 5% nonfat dry milk in PBS either overnight at 4°C or at room temperature (RT) for 1 h. After blocking, the membrane was incubated with the primary antibody for 1–4 h at RT or overnight at 4°C. Primary and secondary antibodies were dissolved in 1% nonfat dry milk in PBS-0.2% Tween 20. To remove unbound primary antibodies, the membrane was washed 3 × 10 min with 1% nonfat dry milk in PBS-0.2% Tween 20. Incubation with peroxidase-conjugated secondary antibodies was again performed either for 1–4 h at RT or overnight at 4°C. The membrane was washed once with 1% nonfat dry milk in PBS-0.2% Tween 20 and twice with PBS-0.2% Tween 20. Labeled peroxidase activity was detected using ECL, and the membranes were exposed to X-OMAT LS films. When the membranes were analyzed with the LI-COR system, all steps were performed as described above, except that the membranes were blocked in 5% nonfat dry milk in PBS without Tween and incubated with the secondary antibody (IR Dye 680/800 conjugated goat anti-rabbit IgG) for 1 h and thereafter washed with PBS without Tween. For successive detection of different proteins on the same membrane, the membrane was washed with PBS-0.2% Tween 20 after analysis of the first protein, incubated for 15 min with stripping buffer for HPRO antibodies 1 (0.1 M glycine in PBS, pH 2–3), and washed in stripping buffer for HRPO 2 (1 M NaCl in PBS). Subsequently, the membranes were washed three times with PBS-0.2% Tween 20. When the membranes were analyzed with the LI-COR system, the membrane was incubated for 5 min with Newblot Nitro Stripping buffer and then washed 3 × 10 min in PBS. Changes in protein expression were analysed by measuring optical density using ImageJ software. Proteins of interest were normalized either to β-actin or to unphosphorylated protein. Changes in protein expression following different treatments were normalized to vehicle-treated controls, which was 100% (within the controls of each experiment, the individual values were normalized against the overall mean value of the control and presented as mean ± SE).
Separation of membrane fractions from whole cell lysate.
To study the effects of 2-ME on the translocation of RhoA to the plasma membrane, HASMCs plated in 100-mm culture dishes were washed twice in sterile PBS and growth arrested for 24 h in DMEM-F12 medium supplemented with 1× antibiotic-anti-mycotic, 0.1% sodium bicarbonate solution, and 0.4% steroid-free FCS. The cultures were treated in DMEM-F12 medium. Before stimulation with PDGF-BB for 24 h, cells were pretreated 6 h with 2-ME. Controls were treated with vehicle (maximum 0.1% DMSO). Membrane fractions from whole cell lysate were separated by centrifugation. Cells were washed once with PBS and lysed by the addition of 0.2 ml of lysis buffer 2. The cell lysates were scraped from the dishes and homogenized 5× through a 25-gauge needle. Aliquots of whole lysate were collected before centrifugation at 1,000 g for 10 min at 4°C. The pellet (nuclear fraction) was discarded and the supernatant collected. After addition of 4 ml of lysis buffer 2, the supernatant from this first spin was centrifuged at 100,000 g for 1 h at 4°C, and the cell membrane pellet was resuspended in lysis buffer 1. The supernatant remaining after the high-speed spin contained the cytosolic fraction. Samples were kept at −20°C until use. The protein expression was analyzed by Western blotting.
Synchronization of cell population in G1/S-phase of the cell cycle by double thymidine block.
Treatment with excess thymidine (2 mmol/l) causes the arrest of cells at the G1/S border owing to an inhibition of DNA synthesis that is attributable to feedback inhibition of nucleotide synthesis caused by an imbalance of the nucleotide pool. To arrest HASMCs at early S-phase, the cells were plated in standard growth medium (M231 + amino acids + SMGS) to achieve ∼40% confluence the following day. After 24 h, the standard growth medium was replaced with medium containing 2 mmol/l thymidine and incubated for at least 12 h under standard tissue culture conditions (37°C, 5% CO2). Then the cells were washed 3× with PBS, refed standard growth medium, and incubated for 12 h. Subsequently, the standard medium was replaced again with medium containing 2 mmol/l thymidine, and the cells were incubated for the next 12 h before release by 3× washing with PBS. The cells were than treated with the test agents.
Immunofluorescence microscopy.
For the analysis of p-rMLC and rMLC, HASMCs were grown on 8-well chamber slides. After 1 h of pretreatment with or without 5 μmol/l 2-ME or 1 μmol/l ROCK inhibitor H1152, cells were stimulated for 4 h with 20 ng/ml PDGF-BB. Fixation/permeabilization solution (4% paraformaldehyde + 0.5% Triton X-100 in PBS) was added, and the chamber slide was shaken for 20 min at RT. Cells were then washed 3 × 5 min with PBS before blocking with 3% BSA in PBS for 1 h at RT. Cells were incubated with primary antibodies (p-rMLC and rMLC) overnight at 4°C; control cells were kept in blocking solution. To remove unbound primary antibody, the chamber slide was washed 5× with PBS. Incubation with either FITC-conjugated anti-mouse or TRITC-conjugated anti-rabbit antibody was performed for 1 h at RT. The chamber slide was washed again 5× with PBS before addition of DAPI solution (100 ng/ml in PBS) on top of the cells. After 10 min the chamber slide was washed and prepared for immunofluorescence detection by addition of mounting medium (90% glycerol in Tris buffer, pH 8.8, + 0.25% DABCO). The fluorescence was analyzed with FITC, TRITC, and DAPI filters on an Olympus Microscope BX61. Pictures were made in triplicates. The fluorescence signal of control cells was subtracted from pictures incubated with primary antibodies. DAPI is a fluorescent stain that binds strongly to A-T-rich regions of DNA. When it is bound to double-stranded DNA it has an absorption maximum at a wavelength of 358 nm (ultraviolet), and its emission maximum is at 461 nm (blue). For fluorescence microscopy, DAPI is excited with ultraviolet light and is detected through a blue/cyan filter. FITC has excitation and emission wavelengths of ∼495 nm and 521 nm. TRITC (tetramethylrhodamine isothiocyanate) has excitation and emission wavelengths of ∼545 and 572 nm.
Effects of 2-ME on tubulin polymerization.
The influence of 2-ME on the dynamics of tubulin polymerization was assayed by immunofluorescence microscopy and as described before (4). Briefly, HASMCs grown to subconfluence in 8-well chamber slides were growth arrested for 48 h with DMEM-F12. Subsequently, the starved/synchronized cells were treated for 24 h with 2-ME (0, 0.1, 1, 3 μmol/l) in the presence of 2.5% FCS. Following the treatment, the cells were washed twice with PBS and fixed at 37°C with 2% paraformaldehyde containing 0.04% Triton X-100. The polymerized tubulin in fixed HASMCs was labeled with primary anti-α-tubulin (1:2,500 dilution) and secondary (goat anti-mouse FITC-conjugated, ICN, 1:50 dilution) antibodies. The staining was analyzed with fluorescence microscopy.
Cell morphology studies.
For the investigation of cell morphology, HASMCs were plated on 6-well plates at a density of 200,000 cells/well. The next day, the cells were starved for 24 h in DMEM-F12 supplemented with 1× antibiotic-antimycotic, 0.1% sodium bicarbonate solution, and 0.4% steroid-free FCS. After treatment with the test agents, the cells were placed under the microscope, and pictures were taken at different time points for 12 h. Computerized image analysis was utilized to assess changes in nuclear size and tubulin polymerization in response to 2-ME.
Live cell division.
Live time lapse microscopy was employed to assess the impact of 2-ME on HASMC cytokinesis. To analyze the effects of the test agents on cytokinesis, HASMCs were plated on 6-well plates at a density of 200,000 cells/well. The cells were than synchronized in S-phase by a thymidine double block before treatment in growth medium supplemented with SMGS. Cells were placed under a microscope in standard tissue-similar conditions (37°C, 5% CO2). Pictures were taken automatically every 6 min for 12 h.
Carotid artery injury studies.
Balloon injury-induced neointima formation was assessed in animals (male Wistar Kyoto rats, 350 to 400 g; RCC, Fullinsdorf, Switzerland), as described previously (24). Osmotic pumps (ALZET minipumps) containing either vehicle or 2-ME were implanted for intravenous delivery of the vehicle or drug at a rate of 350 μg·kg−1·day−1. This dose was selected based on our previously published studies (13, 31, 33). After 14 days, the animals were euthanized and perfused fixed for morphometric analysis as described before (24). The extent of neointimal proliferation was quantified by measuring the area (μm2) of the neointima in hematoxylin and eosin (H&E)-stained cross sections of each left carotid artery. To assess the impact of 2-ME on ROCK1 and tissue factor in vivo, rats (placebo n = 4, treated n = 4) were euthanized on day 8 and the carotid arteries snap-frozen in liquid nitrogen. Subsequently, segments from placebo- or 2-ME-treated animals were homogenized and lysed, and various proteins were analyzed using Western blotting, as described above.
Microarray experiments and analysis.
HASMCs grown to subconfluence were treated with either vehicle or 3 μmol/l 2-ME in the presence of 5% FCS for 30 h. Following treatment with 2-ME, cells were washed with PBS and processed for total RNA isolation. Total RNA was extracted from vehicle-treated and 2-ME-treated samples using an RNeasy Mini kit (Qiagen) according to the manufacturer's instruction. The quality of the isolated RNA was determined with a NanoDrop ND 1000 (NanoDrop Technologies) and a Bioanalyzer 2100 (Agilent, Waldbronn, Germany). Only samples with a 260/280 nm ratio between 1.8–2.1 and a 28S/18S ratio within 1.5–2.0 were further processed. Total RNA samples (2 μg) were reverse-transcribed into double-stranded cDNA, which was subsequently in vitro transcribed in the presence of biotin-labeled nucleotides, using an IVT Labeling Kit (Affymetrix, Santa Clara, CA). The labeled cRNA was purified and quantified using BioRobot Gene Exp - cRNA Target Prep (Qiagen) (16). Biotin-labeled cRNA samples (15 μg) were fragmented to 35–200 bp at 94°C in fragmentation buffer (Affymetrix) and were mixed in 300 μl of hybridization buffer containing a hybridization control cRNA and control Oligo B2 control (Affymetrix), 0.1 mg/ml herring sperm DNA, and 0.5 mg/ml acetylated bovine serum albumin in MES buffer, pH 6.7, before hybridization to GeneChip Human Genome U_133 Plus 2.0 arrays for 16 h at 45°C. Arrays were then washed using an Affymetrix Fluidics Station 450 EukGE-WS2v5_450 protocol. An Affymetrix GeneChip Scanner 3000 was used to measure the fluorescence intensity emitted by the labeled target. Normalization and computation of expression values was performed using the Robust Multichip Average algorithm (16). The expression data were analyzed both at the single gene level and at the pathway level, as previously described (20) and published in the GEO data repository (identification code GSE12261). In a first step, the statistically most relevant differentially regulated transcripts are identified using Significance Analysis of Microarrays (32), a method that provides a ranking of the transcripts found to be differentially regulated between any two given protocols. Statistical significance is measured by the q-value, which is the lowest false discovery rate (= percent of genes that are expected to be identified by chance) at which a gene is described as significantly regulated. In a second step, the association between 2-ME treatment and functionally related group of genes and pathways was studied using Gene Set Enrichment Analysis (GSEA) (30). GSEA is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (“phenotypes”). Results are ranked by their normalized enrichment score (NES): a P value and the false discovery rate (q-value). Pathway information and references are available at The Molecular Signatures Database (http://www.broad.mit.edu/gsea/msigdb), which is a searchable online collection of gene sets used with GSEA (identification code GSE12261). Several pathways related to artherosclerosis and/or ischemic heart disease were manually curated (1, 22, 26).
Statistics.
Microarray experiments were conducted in triplicates, whereas all growth experiments were performed in triplicates or quadruplicates with three to four separate cultures. Statistical analysis of the microarray data was performed according to standard methods. Treatment effects on cross-sectional areas were analyzed using ANOVA or the nonparametric Kruskal-Wallis test. Other statistical analyses were performed using ANOVA, paired Students't-test, or Fisher's least significant difference test as appropriate. A value of P < 0.05 was considered statistically significant. Changes in protein expression following different treatments were normalized to vehicle-treated controls, which was 100%. To ascertain reproducibility and variation in controls within each experiment, the individual values were normalized against the overall mean value of the controls and presented as means ± SE.
RESULTS
Treatment of HASMCs with 2-ME (1–3 μmol/l) for 3 days inhibited PDGF-BB-induced cell proliferation (cell number) from 100 ± 5.5 to 77.5 ± 5.9 and 63.1 ± 2.8% (P < 0.05; Fig. 2A). Similar to the effects on cell number, 2-ME significantly inhibited PDGF-BB-induced DNA synthesis ([3H]thymidine incorporation), collagen synthesis ([3H]proline incorporation), and cell migration (Fig. 2A). Tubulin polymerization is required for mitosis and cytokinesis. Notably, treatment with 2-ME inhibited tubulin polymerization in a concentration-dependent fashion (Fig. 2B). The percentage of tubulin-positive cells was reduced from 100% in controls to 34 ± 5% in HASMCs treated with 3 μmol/l 2-ME. To further assess the impact of 2-ME on cell division, we employed time lapse live imaging microscopy. HASMCs were synchronized in early S-phase by double thymidine blockade and subsequently allowed to grow in complete growth medium in the presence or absence of 2-ME. In control cells, cytokinesis to two daughter cells was accomplished within 2 h (Fig. 2C). However, in cells treated for 11 h with 5 μmol/l 2-ME cytokinesis was blocked by 47 ± 5% (Fig. 2C). The cells attempted cytokinesis without success for 11 h, and subsequently they degraded into many vesicles/vacuoles, indicating apoptosis (Fig. 2C; see white arrows). Since tubulin disruption influences nuclear separation, we assessed the impact of 2-ME on nuclear size in growing HASMCs. As shown in Fig. 2D, treatment with 2-ME increased nuclear size in HASMCs, indicating interference with cytokinesis.
Fig. 2.

Inhibitory effects of 2-ME on PDGF-BB (20 ng/ml) induced effects in HASMCs. A: inhibitory effects of 2-ME (1 or 3 μmol/l) on cell number, DNA synthesis ([3H]thymidine incorporation), collagen synthesis ([3H]proline incorporation), and cell migration in HASMCs treated for 3 days, 24 h, 36 h, and 8 h, respectively. Results are expressed as means ± SE; n = 3 experiments in triplicates. B: representative photomicrographs and bar graph depicting concentration-dependent inhibitory effects of 2-ME on tubulin polymerization in proliferating HASMCs. Tubulin polymerization experiments were conducted in triplicate. C: time lapse phase contrast imaging of effects of 2-ME on mitogen-induced cytokinesis in HASMCs presynchronized in early S-phase by double thymidine blockade. Pictures were taken every 6 min for 24 h or until cell division. In untreated cells, cell division was accomplished within 2 h (white arrow), whereas 2-ME-treated cells failed to divide and underwent apoptotic disintegration (white arrow) after 11 h. D: modulatory effects of 2-ME on nuclear size in proliferating HASMCs treated for 5 days. Nuclear size in DAPI-stained HASMCs was assessed using the Olympus imaging system; changes are depicted in the bar graph. At least 20 cells were counted for each condition. *P < 0.05 vs. vehicle-treated control.
Figure 3 compares time-lapsed photographs of HASMCs undergoing mitosis and cytokinesis with no treatment (Fig. 3A), treatment with 5 μmol/l 2-ME (Fig. 3B), or treatment with 1 μmol/l H-1152 (Fig. 3C), a ROCK1 inhibitor. As with 2-ME, H-1152 caused mitotic cell rounding and inhibition of cytokinesis; however, unlike with 2-ME there was no indication of apoptosis. Phenotypic changes were observed in 53 ± 6% of HASMCs treated for 11 h with 2-ME (5 μmol/l).
Fig. 3.

Photomicrographs showing the process of cell division of HASMCs in growth medium (A), with 5 µmol/l 2-ME (B), and with 1 µmol/l H-1152 (C). Cells were grown in 6-well plates and synchronized in early S-phase by double thymidine block. After the second release, cells were treated and placed under the microscope in cell culture conditions. Images were taken automatically every 6 min for 12 h.
To investigate further the contribution of the RhoA/ROCK1 pathway in mediating the inhibitory effects of 2-ME on cell proliferation, we assessed the concentration-dependent effects of H-1152 on cell proliferaiton. As shown in Fig. 4A, H-1152 inhibited PDGF-BB-induced proliferation of HASMCs in a concentration-dependent fashion.
Fig. 4.

A: effects of ROCK1 inhibitor H-1152 (1–3 μmol/l) on PDGF-BB-induced proliferation (cell number) of HASMCs. Data are means ± SE; n = 3 experiments in triplicates. B: microarray analysis of transcriptional changes in HASMCs treated with 2-ME (3 μmol/l) for 30 h. Expression levels are depicted as color scale from red (upregulation) to blue (downregulation). Gene Set Enrichment Analysis (GSEA) revealed upregulation of key RhoA/ROCK1 pathway-associated transcripts. C: significant inhibitory effects of 2-ME (3 μmol/l) on FCS (5%)-induced expression of GTP-bound RhoABC in HASMCs. D: inhibitory effects of 2-ME on PDGF-BB (20 ng/ml)-induced membrane-bound RhoA (m-RhoA) expression in HASMCs. Data are presented as means ± SE; n = 3. *P < 0.05 vs. vehicle-treated control; §significant inhibition vs. mitogen-treated cells.
Next, we performed a high-density oligonucleotide microarray analysis and evaluated the potential impact of 2-ME on the RhoA/ROCK1 pathway. Employing GSEA, we found that 2-ME significantly inhibited the mRNA expression of genes involved in the RhoA/ROCK1 pathway (Fig. 4B). These findings suggested modulation of the RhoA/ROCK1 pathway by 2-ME as a potential mechanism for 2-ME's antiproliferative actions in HASMCs. To test this, we further assessed the impact of 2-ME on key components of the RhoA/ROCK1 pathway at the protein level. Consistent with the microarray analysis, treatment of HASMCs with 2-ME abrogated serum-induced levels of GTP-activated RhoA + RhoB +Rho C (RhoABC pulldown assay) (Fig. 4C). Furthermore, 2 and 5 μmol/l 2-ME significantly inhibited the expression of membrane-bound (activated) RhoA from 195.3 ± 33.8% (PDGF-BB) to 67.8 ± 2.8 and 22.27 ± 13.2% (all results expressed as %control), respectively (Fig. 4D). Treatment with PDGF-BB alone induced ROCK1 (primary downstream signaling mediator for RhoA) expression in a time-dependent fashion, with a maximal stimulation at 48 h (Fig. 5A); moreover, treatment with 2 and 5 μmol/l 2-ME for 48 h attenuated the PDGF-BB-induced expression of ROCK1 (Fig. 5A). In contrast to 2-ME, H-1152 had no effect on ROCK1 expression (Fig. 5A). This observation is consistent with the fact that H-1152 inhibits ROCK1 activity but not its expression. ROCK1 is known to phosphorylate MYPT [myosin-targeting subunit of myosin light-chain phosphatase (MLCP)]. The phosphorylation of MYPT by ROCK1 at MYPT's inhibitory site (Thr696) blocks the phosphatase activity of MLCP, resulting in an increase in rMLC (regulatory subunit of myosin light chain) phosphorylation at Ser19. Phosphorylation of rMLC at Ser19 activates contraction. Stimulation of HASMCs with PDGF-BB induced MYPT phosphorylation at Thr696 in a time-dependent fashion with a maximal stimulation of 256.5 ± 29.9% (P < 0.05) after 10 min, as shown in Fig. 5B. 2-ME (2 and 5 μmol/l) concentration-dependently inhibited PDGF-BB-induced phosphorylation of MYPT (P < 0.05), and these effects were mimicked by H-1152 (Fig. 5B). Additionally, 2-ME inhibited angiotensin II-induced expression of phosphorylated MYPT (Fig. 5C).
Fig. 5.

A: time-dependent stimulatory effects of PDGF-BB (20 ng/ml) on expression of ROCK1 and inhibitory effects of 2-ME and H-1152 (ROCK1 inhibitor) on PDGF-BB-induced expression of ROCK1 in HASMCs. Data are presented as means ± SE; n = 3. *P < 0.05 vs. vehicle treated control. B: time-dependent stimulatory effects of PDGF-BB (20 ng/ml) on phosphorylation of MYPT and inhibitory effects of 2-ME and H-1152 on PDGF-BB-induced phosphorylation of myosin-targeting subunit of myosin light-chain phosphatase (MYPT) in HASMCs. C: inhibitory effects of 2-ME on angiotensin II-induced phosphorylated MYPT expression in HASMCs. Data are presented as means ± SE; n = 3.
In serum-starved HASMCs, PDGF-BB induced rMLC phosphorylation in a time-dependent fashion with maximum stimulation (≈10-fold; 1124.3 ± 103.5% of control, P < 0.05) at 8 h (Fig. 6A). Treatment with 2-ME abrogated PDGF-BB-induced phosphorylation of rMLC, with maximal inhibition of 46% at 5 μmol/l (Fig. 6A). Similarly, H-1152 decreased rMLC phosphorylation by 35% (P < 0.05; Fig. 6A). Figure 6B shows photomicrographs of rMLC phosphorylation at Ser19 (green FITC signal) as well as total rMLC by immunofluorescence, confirming the inhibitory effects of 2-ME and H-1152 on rMLC phosphorylation.
Fig. 6.

A: time-dependent stimulatory effects of PDGF-BB (20 ng/ml) on phosphorylation of rMLC and inhibitory effects of 2-ME and H-1152 (ROCK1 inhibitor) on PDGF-BB-induced phosphorylation of regulatory subunit of myosin light chain (rMLC) in HASMCs. Data are presented as means ± SE; n = 3. *P < 0.05 vs. vehicle-treated control; §significant inhibition vs. mitogen-treated cells. B: representative fluorescent staining of HASMCs for inhibitory effects of 2-ME or H-1152 on PDGF-BB (20 ng/ml)-induced phosphorylation of rMLC (green fluorescence) and internal control nonphosphorylated rMLC (pink fluorescence). Three separate experiments were conducted, each in triplicate.
Our previous findings (4) show that 2-ME inhibits phosphorylation of ERK1/2 and Akt and consequently reduces the expression of cyclin D1 and cyclin B1. To decipher the involvement of the RhoA/ROCK1 pathway in mediating these effects, we compared the effects of 2-ME and H-1152 on phosphorylation of ERK1/2 and Akt and expression of cyclin D1 and cyclin B1. As shown in Fig. 7, the inhibitory effects of 2-ME on ERK1/2 and Akt phosphorylation and on cyclin D1 and cyclin B1 protein expression were not mimicked by H-1152. These findings indicate that inhibition of the RhoA/ROCK1 pathway by 2-ME represents yet another site of action of 2-ME to inhibit cell proliferation. The inhibitory effects of 2-ME, but not H-1152, on Akt phosphorylation may also explain their differential actions on HASMCs on apoptosis.
Fig. 7.

Western blots and bar graph (changes in optical density; means ± SE) depicting effects of 2-ME on cyclin D1, cyclin B1, phosphorylated (p)-ERK1/2, total ERK1/2, p-Akt and total Akt. Data are presented as means ± SE; n = 3. *P < 0.05 vs. vehicle-treated control; §significant inhibition vs. mitogen-treated cells.
To assess the pathophysiological significance of the inhibitory effects of 2-ME on the RhoA/ROCK1 pathway, we further investigated the effects of 2-ME on tissue factor, a downstream target of RhoA/ROCK1 and an important player in the pathophysiology of injury-induced neointimal thickening. In HASMCs, thrombin (4 U/ml) induced the expression of tissue factor (Figs. 8 and 9A), and this effect was accompanied by increased expression of ROCK1 (Fig. 9A) and increased phosphorylation of rMLC and MYPT (Fig. 9A). All of these effects of thrombin were inhibited by 2-ME (Figs. 8 and 9A). Also, treatment of HASMCs with other mitogens implicated in inflammation and vascular remodeling (PDGF-BB, TNFα, serum) induced tissue factor expression, and these effects were abrogated by 2-ME (Fig. 9B). To assess whether the inhibitory effects of 2-ME on tissue factor and ROCK1 in HASMCs are also seen in vivo following balloon injury, we assessed changes in the expression of these proteins in the neointima of rat carotid arteries. Consistent with our previous observations (4), 2-ME inhibited neointima formation (Fig. 9C); moreover, compared with vehicle, treatment with 2-ME downregulated the expression of tissue factor and ROCK1 (P < 0.05 vs. vehicle; Fig. 9C).
Fig. 8.

Concentration-dependent inhibitory effects of 2-ME on thrombin (4 U/ml)-induced expression of tissue factor (TF) in HASMCs.
Fig. 9.

A: representative Western blot demonstrating inhibitory effects of 2-ME on thrombin (Thr; 4 U/ml)-induced TF, ROCK1, p-rMLC, and p-MYPT expression in HASMCs. Similar effects were observed in 3 separate experiments. B: stimulatory effects of PDGF-BB (20 ng/ml), thrombin (4 U/ml), TNF-α (10 ng/ml), and FCS (5%) on TF expression in cultured HASMCs, and inhibitory effects of 2-ME (1 μmol/l) on PDGF-BB, TNF-α, and FCS-induced expression of TF in cultured HASMCs. C: Inhibitory effects of 2-ME on TF and ROCK1 protein expression in rat carotids in vivo and on neointima formation following balloon injury. Rats were treated with placebo or 2-ME (350 μg·kg−1·day−1 iv via osmotic pumps, n = 4) and were euthanized on day 8. Carotid arteries were snap-frozen in liquid nitrogen, subsequently lysed, and proteins analyzed using Western blotting. Data are presented as means ± SE; n = 4. *P < 0.05 vs. controls.
DISCUSSION
Our high-density oligonucleotide microarray analysis provides strong evidence that 2-ME downregulates genes in the RhoA/ROCK1 pathway. This concept is corroborated by the fact that 2-ME inhibits mitogen-induced expression of activated (GTP-bound) RhoABC as well as membrane-bound RhoA (indicating inhibition of RhoA's activation), events upstream of ROCK1. We also demonstrate that 2-ME inhibits the expression of ROCK1, the major downstream mediator of activated RhoA. Thus, it appears that 2-ME blocks the RhoA/ROCK1 pathway both by inhibiting the activation of RhoA and by reducing the expression of ROCK1. That 2-ME blocks the RhoA/ROCK1 pathway is further supported by our observations that 2-ME blocks signaling downstream of ROCK1, including phosphorylation of MYPT and rMLC and expression of tissue factor. Blockade of the RhoA/ROCK1 pathway by 2-ME likely contributes to 2-ME's antiproliferative effects, because inhibiting the RhoA/ROCK1 pathway with H-1152 attenuates cell proliferation.
ROCKs are serine-threonine protein kinases that contribute to many downstream effects of Rho GTPases (19, 29). There are two isoforms of ROCK (ROCK1 and ROCK2), but ROCK1 appears to be more involved than ROCK2 in vascular remodeling and VSMC proliferation, migration, adhesion molecule expression, contraction, and actin- and tubulin-mediated cytoskeletal reorganization (19, 29). Downstream of ROCK1 are MYPT and MLC. ROCK1 phosphorylates MYPT at Thr696, a process that inhibits MLCP activity and therefore increases the phosphorylation at Ser19 of rMLC (19, 29). Phosphorylation of rMLC at Ser19 regulates the activity of myosin II, which is an essential motor in the contractile process from contractile ring formation through cleavage furrow ingression to the separation of the two daughter cells (2, 19, 29). The present study shows that 2-ME inhibits the phosphorylation of MYPT at Thr696 and rMLC at Ser19, which would explain how 2-ME blocks cytokinesis. Consistent with this concept, the present study also shows that 2-ME 1) blocks tubulin polymerization, 2) hinders separation of daughter cells, and 3) increases nuclear size. Our previous study demonstrated that 2-ME blocks VSMC growth in the G2/M phase of the cell cycle (4). Downregulation of the RhoA/ROCK1 pathway may be responsible for this effect. Taken together, our results provide strong evidence that the inhibition of the RhoA/ROCK1 pathway by 2-ME importantly contributes to its antiproliferative action in VSMCs.
Our findings that the ROCK1 inhibitor H-1152 attenuates PDGF-BB-induced VSMC proliferation as well as the phosphorylation of MYTP and rMLC suggest active involvement of the RhoA/ROCK1 pathway in VSMC proliferation. The fact that 2-ME attenuates the expression of ROCK1, whereas treatment with H-1152 does not attenuate ROCK1 expression, implies that 2-ME and H-1152 inhibit RhoA/ROCK1 pathway by different mechanisms. This is consistent with the mechanism of action of kinase inhibitors like H-1152, which block the ability of ROCK1 to phosphorylate its targets without affecting the expression of ROCK1.
There is an additional distinction between direct ROCK1 inhibition by H-1152 and inhibition of the RhoA/ROCK1 pathway by 2-ME. Untreated cells divide, starting from mitotic rounding to cytokinesis, in a time frame of 2–3 h. Although this time frame is altered by both H-1152 and 2-ME, these compounds have different effects on the process. HASMCs treated with 2-ME first stop in the mitotic rounding state, then attempt cytokinesis without success, and finally disintegrate into vacuole-like vesicles. In contrast to 2-ME, treatment with H-1152 arrests cells in mitotic rounding for over 12 h without progression to cytokinesis. These results are entirely consistent with the studies by Gui and Zheng (15), who were the first to show that 2-ME causes mitotic arrest and apoptosis in HASMCs. Evidence suggests that ROCK1 may play a role in caspase activation (29), explaining the absence of apoptosis-like behavior in HASMCs treated with H-1152. However, since 2-ME downregulates ROCK1, we conclude that the apoptotic effects of 2-ME are mediated independently of its effects on the RhoA/ROCK1 pathway.
Our previous studies have shown that 2-ME arrests HASMCs in both the G0/G1 and G2/M phases of the cell cycle (4). On the basis of these observations, we postulated that the inhibition of the RhoA/ROCK1 pathway is responsible for the blockade in G2/M phase via reduction of MLC phosphorylation and consequent inhibition of cytokinesis; however, 2-ME-mediated blockade of HASMCs in the G0/G1 phase of the cell cycle is likely independent of the RhoA/ROCK1 pathway. In support of this concept, we observed that 2-ME, but not H-1152, inhibits PDGF-induced ERK1/2 and Akt phosphorylation and that 2-ME, but not H-1152, downregulates PDGF-induced cyclin D1 and B1 expression. Consistent with our past (4) and present investigations, studies by Jennings et al. (17) in mice provide additional evidence that endogenous 2-ME inhibits ERK1/2 and Akt phosphorylation. The results by Jennings et al., along with our findings, suggest that 2-ME, but not H-1152, downregulates the phosphorylation of proteins important for G0/G1 progression. Moreover, 2-ME interferes with microtubule organization, which perturbs the correct alignment of chromosomes at metaphase and their correct segregation during anaphase. These actions would activate the spindle assembly checkpoint and arrest the mitotic process (29) (Fig. 1). Taken together, we conclude that 2-ME inhibits proliferation by at least three distinct mechanisms: 1) arresting VSMCs in G0/G1 phase by inhibiting mitotic signaling and key cell cycle regulators, 2) blocking mitosis by disrupting tubulin organization, and 3) blocking cytokinesis by inhibiting the RhoA/ROCK1 (Fig. 1).
How 2-ME blocks the RhoA/ROCK1 pathway is unknown. Studies show that 2-ME also has inhibitory effects on the activity of calmodulin (3), which associates with mitotic spindles to initiate cytokinesis. Therefore, it is conceivable that 2-ME inhibits the RhoA/ROCK1 pathway by blocking calmodulin activity.
Apart from proliferation, another effect of 2-ME that one can observe in cell culture is on cell morphology. The change in cell shape is in part due to 2-ME-induced interference with microtubules (4). Since RhoA proteins also regulate VSMC contraction (29), we investigated the role of the RhoA pathway in the morphological change induced by 2-ME. We found that 1) 2-ME induced a rounding response in HASMCs in a concentration-dependent manner; and 2) H-1152 had no effect on cell morphology. Rounded cells show less extended microtubules, and the inhibition of tubulin polymerization by 2-ME may explain this effect. Taken together, the above findings suggest that the RhoA pathway is not involved in 2-ME-induced change in cell morphology. This is in accord with the findings that the ROCK1-dependent contractile system regulates the organization of the fine central stress fibers, while peripheral stress fibers responsible for cell morphology are dependent on MLCK and the Ca2+ pathway (19, 29).
Tissue factor is another potential target of the RhoA/ROCK1 pathway (18, 19), and the present study shows that treatment of HASMCs with atherogenic/mitogenic agents (thrombin, TNF-α, PDGF-BB, FCS) induces tissue factor expression and that these effects are abrogated by 2-ME. Importantly, 2-ME inhibits not only thrombin-induced tissue factor expression but also the expressions of ROCK1, p-MYPT, and p-rMLC, suggesting that 2-ME attenuates tissue factor expression by inhibiting the RhoA/ROCK1 pathway. Indeed, the RhoA/ROCK1 pathway directly induces tissue factor expression in VSMCs (18, 19).
Importantly, the present study shows that 2-ME reduces neointima formation and inhibits both ROCK1 and tissue factor expression in rat carotid arteries following balloon injury. These findings suggest that the inhibitory effects of 2-ME on the RhoA/ROCK1/tissue factor axis may contribute to its antivasoocclusive and vascular protective actions in vivo. Because our in vitro experiments were conducted in steroid-stripped and phenol red-free medium, we conducted our in vivo experiments in male rats to avoid high levels of endogenous 17β-estradiol and 2-ME. However, this is a limitation of the present study, and additional studies using ovariectomized female rats are required to fully confirm and validate our findings in vivo.
Although our findings suggest that 2-ME inhibits tissue factor expression by inactivating the RhoA/ROCK1 pathway, other direct or indirect mechanisms may also be involved. Our previous studies using microarray analysis showed that 2-ME modulates multiple mechanisms that can affect tissue factor expression. Specifically, 2-ME induces COX2 expression, activates PPARα and -γ, inhibits 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA), blocks squalene epoxidase and mevalonate (diphospho) decarboxylase (which convert mevalonic acid to isopentenyl-PP and squalene), and inhibits NF-κB and phosphatidylinositol 3-kinase (6, 7, 23). Moreover, 2-ME inhibits MAPK, a known activator of tissue factor (21). Irrespective of whether 2-ME inhibits tissue factor expression directly or indirectly via the RhoA/ROCK1 pathway, this downregulation would protect against vascular remodeling associated with cardiovascular disease. The fact that 2-ME inhibits tissue factor expression induced by multiple factors linked to cardiovascular disease further highlights its importance as a vascular protective agent.
Similarly to 2-ME, paclitaxel/taxol inhibits neointima formation (28); however, use of paclitaxel in drug-eluting stents induces hyperconstriction and ROCK expression (27). A potential explanation for the differential effects of 2-ME and paclitaxel on ROCK may be the difference in their mode of microtubule action. Indeed, taxol is a tubulin-stabilizing agent, whereas 2-ME inhibits tubulin polymerization and induces its breakdown (19).
In conclusion, our results highlight the potential use of 2-ME as therapeutic agent against vasoocclusive disorders in cardiovascular disease. Our findings that the protective effects of 2-ME are in part mediated by the RhoA/ROCK1 pathway and tissue factor provide new insights into the mechanism of action of this molecule. The use of ROCK1 and tissue factor inhibitors is known to be vasoprotective (19, 21, 25), and the observation that 2-ME inhibits rMLC and MYPT phosphorylation as well as tissue factor may be of therapeutic and clinical interest. The above notion, together with the fact that 2-ME inhibits growth of multiple other cell types (cardiac fibroblasts, glomerular mesangial cells) (9, 10) where these pathways are active suggests that 2-ME may be effective against multiple proliferative disorders.
GRANTS
This work was supported by the Olten Heart Foundation (to E. Lucchinetti and R. K. Dubey); the Swiss National Science Foundation Grant nos. IZERO-142213/1 and 31003A-138067 (to R. K. Dubey); and National Institutes of Health Grants NS-087978, HL-109002, DK-091190, HL-069846, DK-068575, and DK-079307 (to E. K. Jackson).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.R., F.B.B., J.F., B.I., E.K.J., and R.K.D. conception and design of research; L.R., F.B.B., E.L., M.Z., J.F., M.R., E.K.J., and R.K.D. performed experiments; L.R., F.B.B., M.Z., J.F., M.R., and R.K.D. analyzed data; L.R., F.B.B., E.L., J.F., E.K.J., and R.K.D. interpreted results of experiments; L.R., E.K.J., and R.K.D. prepared figures; L.R., E.K.J., and R.K.D. drafted manuscript; L.R., F.B.B., E.L., M.Z., J.F., M.R., B.I., E.K.J., and R.K.D. edited and revised manuscript; L.R., F.B.B., E.L., M.Z., J.F., M.R., B.I., E.K.J., and R.K.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Marzanna Künzli-Gontarczyk (Functional Genomics Center, Zurich) for help with Affymetrix GeneChip hybridization; and Delbert G. Gillespie (University of Pittsburgh, Pittsburgh, PA) for the HASMC growth assays. Part of this work was presented at AHA Council for HBPR research meeting in Chicago 2009 (Barchiesi et al.) and published as an abstract.
REFERENCES
- 1.Archacki SR, Angheloiu G, Tian XL, Tan FL, DiPaola N, Shen GQ, Moravec C, Ellis S, Topol EJ, Wang Q. Identification of new genes differentially expressed in coronary artery disease by expression profiling. Physiol Genomics 15: 65–74, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Asano S, Hamao K, Hosoya H. Direct evidence for roles of phosphorylated regulatory light chain of myosin II in furrow ingression during cytokinesis in HeLa cells. Genes Cells 14: 555–568, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Attalla H, Makela TP, Adlercreutz H, Andersson LC. 2-Methoxyestradiol arrests cells in mitosis without depolymerizing tubulin. Biochem Biophys Res Commun 228: 467–473, 1996. [DOI] [PubMed] [Google Scholar]
- 4.Barchiesi F, Jackson EK, Fingerle J, Gillespie DG, Odermatt B, Dubey RK. 2-Methoxyestradiol, an estradiol metabolite, inhibits neointima formation and smooth muscle cell growth via double blockade of the cell cycle. Circ Res 99: 266–274, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Barchiesi F, Jackson EK, Gillespie DG, Zacharia LC, Fingerle J, Dubey RK. Methoxyestradiols mediate estradiol-induced antimitogenesis in human aortic SMCs. Hypertension 39: 874–879, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Barchiesi F, Lucchinetti E, Zaugg M, Ogunshola OO, Wright M, Meyer M, Rosselli M, Schaufelberger S, Gillespie DG, Jackson EK, Dubey RK. Candidate genes and mechanisms for 2-methoxyestradiol-mediated vasoprotection. Hypertension 56: 964–972, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Degraeve F, Bolla M, Blaie S, Creminon C, Quere I, Boquet P, Levy-Toledano S, Bertoglio J, Habib A. Modulation of COX-2 expression by statins in human aortic smooth muscle cells. Involvement of geranylgeranylated proteins. J Biol Chem 276: 46849–46855, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Dubey RK, Gillespie DG, Zacharia LC, Rosselli M, Korzekwa KR, Fingerle J, Jackson EK. Methoxyestradiols mediate the antimitogenic effects of estradiol on vascular smooth muscle cells via estrogen receptor-independent mechanisms. Biochem Biophys Res Commun 278: 27–33, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Dubey RK, Imthurn B, Jackson EK. 2-Methoxyestradiol: a potential treatment for multiple proliferative disorders. Endocrinology 148: 4125–4127, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Dubey RK, Jackson EK. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol Metab 20: 374–379, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubey RK, Jackson EK, Gillespie DG, Zacharia LC, Imthurn B, Keller PJ. Clinically used estrogens differentially inhibit human aortic smooth muscle cell growth and mitogen-activated protein kinase activity. Arterioscler Thromb Vasc Biol 20: 964–972, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Dubey RK, Jackson EK, Luscher TF. Nitric oxide inhibits angiotensin II-induced migration of rat aortic smooth muscle cell. Role of cyclic-nucleotides and angiotensin1 receptors. J Clin Invest 96: 141–149, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubey RK, Jackson EK, Rupprecht HD, Sterzel RB. Factors controlling growth and matrix production in vascular smooth muscle and glomerular mesangial cells. Curr Opin Nephrol Hypertens 6: 88–105, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Foe VE, von Dassow G. Stable and dynamic microtubules coordinately shape the myosin activation zone during cytokinetic furrow formation. J Cell Biol 183: 457–470, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gui Y, Zheng XL. 2-Methoxyestradiol induces cell cycle arrest and mitotic cell apoptosis in human vascular smooth muscle cells. Hypertension 47: 271–280, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings BL, George LW, Pingili AK, Khan NS, Estes AM, Fang XR, Gonzalez FJ, Malik KU. Estrogen metabolism by cytochrome P450 1B1 modulates the hypertensive effect of angiotensin II in female mice. Hypertension 64: 134–140, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunieda Y, Nakagawa K, Nishimura H, Kato H, Ukimura N, Yano S, Kawano H, Kimura S, Nakagawa M, Tsuji H. HMG CoA reductase inhibitor suppresses the expression of tissue factor and plasminogen activator inhibitor-1 induced by angiotensin II in cultured rat aortic endothelial cells. Thromb Res 110: 227–234, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 98: 322–334, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Lucchinetti E, Hofer C, Bestmann L, Hersberger M, Feng J, Zhu M, Furrer L, Schaub MC, Tavakoli R, Genoni M, Zollinger A, Zaugg M. Gene regulatory control of myocardial energy metabolism predicts postoperative cardiac function in patients undergoing off-pump coronary artery bypass graft surgery: inhalational versus intravenous anesthetics. Anesthesiology 106: 444–457, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Moons AH, Levi M, Peters RJ. Tissue factor and coronary artery disease. Cardiovasc Res 53: 313–325, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Papaspyridonos M, Smith A, Burnand KG, Taylor P, Padayachee S, Suckling KE, James CH, Greaves DR, Patel L. Novel candidate genes in unstable areas of human atherosclerotic plaques. Arterioscler Thromb Vasc Biol 26: 1837–1844, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Paumelle R, Staels B. Peroxisome proliferator-activated receptors mediate pleiotropic actions of statins. Circ Res 100: 1394–1395, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Powell JS, Clozel JP, Muller RK, Kuhn H, Hefti F, Hosang M, Baumgartner HR. Inhibitors of angiotensin-converting enzyme prevent myointimal proliferation after vascular injury. Science 245: 186–188, 1989. [DOI] [PubMed] [Google Scholar]
- 25.Satoh K, Fukumoto Y, Shimokawa H. Rho-kinase: important new therapeutic target in cardiovascular diseases. Am J Physiol Heart Circ Physiol 301: H278–H296, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Satterthwaite G, Francis SE, Suvarna K, Blakemore S, Ward C, Wallace D, Braddock M, Crossman D. Differential gene expression in coronary arteries from patients presenting with ischemic heart disease: further evidence for the inflammatory basis of atherosclerosis. Am Heart J 150: 488–499, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Shiroto T, Yasuda S, Tsuburaya R, Ito Y, Takahashi J, Ito K, Ishibashi-Ueda H, Shimokawa H. Role of Rho-kinase in the pathogenesis of coronary hyperconstricting responses induced by drug-eluting stents in pigs in vivo. J Am Coll Cardiol 54: 2321–2329, 2009. [DOI] [PubMed] [Google Scholar]
- 28.Sollott SJ, Cheng L, Pauly RR, Jenkins GM, Monticone RE, Kuzuya M, Froehlich JP, Crow MT, Lakatta EG, Rowinsky EK, et al. Taxol inhibits neointimal smooth muscle cell accumulation after angioplasty in the rat. J Clin Invest 95: 1869–1876, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Street CA, Bryan BA. Rho kinase proteins—pleiotropic modulators of cell survival and apoptosis. Anticancer Res 31: 3645–3657, 2011. [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tofovic SP, Salah EM, Dubey RK, Melhem MF, Jackson EK. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic nitric oxide synthase inhibition. J Cardiovasc Pharmacol 46: 25–35, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zacharia LC, Piche CA, Fielding RM, Holland KM, Allison SD, Dubey RK, Jackson EK. 2-hydroxyestradiol is a prodrug of 2-methoxyestradiol. J Pharmacol Exp Ther 309: 1093–1097, 2004. [DOI] [PubMed] [Google Scholar]