

Abstract
Hypothyroidism is the most frequent and earliest endocrine complication in cystinosis, a multisystemic lysosomal storage disease caused by defective transmembrane cystine transporter, cystinosin (CTNS gene). We recently demonstrated in Ctns−/− mice that altered thyroglobulin biosynthesis associated with endoplasmic reticulum stress, combined with defective lysosomal processing, caused hypothyroidism. In Ctns−/− kidney, hematopoietic stem cell (HSC) transplantation provides long-term functional and structural protection. Tissue repair involves transfer of cystinosin-bearing lysosomes from HSCs differentiated as F4/80 macrophages into deficient kidney tubular cells, via tunneling nanotubes that cross basement laminae. Here we evaluated the benefit of HSC transplantation for cystinotic thyroid and investigated the underlying mechanisms. HSC engraftment in Ctns−/− thyroid drastically decreased cystine accumulation, normalized the TSH level, and corrected the structure of a large fraction of thyrocytes. In the thyroid microenvironment, HSCs differentiated into a distinct, mixed macrophage/dendritic cell lineage expressing CD45 and major histocompatibility complex II but low CD11b and F4/80. Grafted HSCs closely apposed to follicles and produced tunneling nanotube-like extensions that crossed follicular basement laminae. HSCs themselves further squeezed into follicles, allowing extensive contact with thyrocytes, but did not transdifferentiate into Nkx2.1-expressing cells. Our observations revealed significant differences of basement lamina porosity between the thyroid and kidney and/or intrinsic macrophage invasive properties once in the thyroid microenvironment. The contrast between extensive thyrocyte protection and low HSC abundance at steady state suggests multiple sequential encounters and/or remanent impact. This is the first report demonstrating the potential of HSC transplantation to correct thyroid disease and supports a major multisystemic benefit of stem cell therapy for cystinosis.
Infantile cystinosis is an inherited multisystemic lysosomal storage disease, in which cystine, an obligatory degradation product of disulphide bearing proteins, fails to exit lysosome due to genetic inactivation of its transmembrane transporter, cystinosin (CTNS gene) (1). Additional roles of cystinosin, such as mammalian target of rapamycin complex 1 regulation, have recently been demonstrated (2). Cystinosis typically manifests itself before 1 year of age as renal Fanconi syndrome, ie, generalized proximal tubular cell (PTC) dysfunction, and inevitably evolves to renal insufficiency. Primary (TSH compensated) hypothyroidism is the earliest and almost obligatory endocrine dysfunction in cystinotic children (3, 4).
Cystinosin-deficient mice in a strict congenic C57/BL6 background (Ctns−/− mice) mimic human cystinosis (5) and proved informative to unravel early pathogenic and adaptive mechanisms in kidneys and thyroid (6–9). In Ctns−/− kidneys, impaired apical receptor-mediated endocytosis, endolysosomal trafficking, and lysosomal proteolysis are associated to PTC dedifferentiation (6, 7). In Ctns−/− thyroid, altered thyroglobulin biosynthesis with endoplasmic reticulum (ER) stress, combined with defective lysosomal processing of iodothyroglobulin, lead to impaired thyroid hormone production, resulting in subclinical hypothyroidism (increased TSH), thyrocyte hyperplasia/hypertrophy, and accelerated cell turnover (8).
Lysosomal cystine accumulation can be corrected by substrate depletion therapy with cysteamine, the only current approved drug. Cysteamine reacts with lysosomal cystine to form mixed cysteamine/cysteine disulphide that exits lysosome by the lysine transporter system (10). However, cysteamine therapy is very demanding and even early implementation in compliant patients fails to prevent renal insufficiency and thyroid dysfunction (11, 12). Alternative therapies are thus needed. Recent reports demonstrated a major benefit of hematopoietic stem cell (HSC) transplantation to prevent kidney disease progression and corneal defects in Ctns−/− mice, as evidenced by decreased cystine level and long-term functional protection (13–16). Engrafted HSC-derived cells mostly expressed the common macrophage markers F4/80 and major histocompatibility complex II (MHCII) and did not fuse with kidney epithelial cells nor keratocytes (15–17). In coculture experiments, bone marrow-derived macrophages generated tunneling nanotubes (TNTs) whereby cystinosin bearing-lysosomes were transported into Ctns-deficient fibroblasts (17). Conversely, lysosomes of Ctns-deficient fibroblasts used the same physical connection toward wild-type (WT) macrophages, to fuse with the competent lysosomal pool (17). In vivo, HSCs grafted in Ctns−/− kidney also generated TNT-like structures that crossed basement laminae and connected with epithelial cells, which acquired cystinosin (17). These data provided the first demonstration of cross-correction of a genetic lysosomal storage disease. The same mechanism was recently documented in the cornea (16). We here document the remarkable benefits of HSC transplantation in Ctns−/− thyroid and define structural protective mechanisms.
Materials and Methods
Mice
WT Ctns+/+, enhanced green fluorescent protein (eGFP)-transgenic mice (C57BL/6-Tg[ACTB-EGFP]1Osb/J) were from Jackson Laboratory. Cross-breeding of DsRed-transgenic mice (B6.Cg-Tg[CAG-DsRed*MST]1Nagy/J; Jackson Laboratory) with C57BL/6 Ctns−/− mice generated by Dr C. Antignac (5) produced transgenic DsRed Ctns−/− mice, ubiquitously expressing the DsRed fluorescent protein (15). Protocols were approved by University of California, San Diego, Animal Care and Use Committee. Mice were fed ad libitum with pellets containing 3 mg/kg iodine (Harlan Laboratories).
HSCs isolation and transplantation
Bone marrow cells were collected from WT eGFP-transgenic mice. Sca1+ HSCs selected by immunomagnetic separation (Miltenyi Biotec) were transplanted iv into 2-month-old DsRed Ctns−/− mice, irradiated (8 Gy) on the previous day (17). Because histological changes appeared in Ctns−/− thyroid around 6 months and became prominent 2–3 months thereafter (8), Ctns−/− mice transplanted with WT-eGFP HSCs (grafted Ctns−/− mice) were analyzed at 8 months of age/6 months after transplantation and compared with age-matched WT and Ctns−/− mice. Older mice were occasionally examined. We studied 10 WT (four males; six females), 15 Ctns−/− mice (four males; 11 females), and 16 grafted Ctns−/− mice (six males; 10 females).
Biochemical assays
Thyroid cystine and plasma TSH, T3, and T4 were measured as described (14, 18).
Microscopy
Perfusion-fixed thyroids were dissected and postfixed overnight with 4% neutral-buffered formaldehyde (7). Thyroid lobes were either equilibrated in 20% sucrose, embedded in Tissue-Tek/optimum cutting temperature, and snap frozen in isopentane or processed for paraffin embedding. For histological analysis, 4-μm-thick paraffin sections were stained with hematoxylin/eosin. Immunofluorescence was performed as described, using antibodies listed in Table 1 (7). HSCs were identified by immunofluorescence instead of intrinsic GFP fluorescence because of its inactivation during tissue paraffin processing and rapid photobleaching in frozen sections and to amplify detection sensitivity in cytoplasmic extensions. For detection of inflammatory cell markers, blocking solution with 10% normal goat serum and 2% milk was used. Alexa-fluor antibodies were selected, at the exclusion of red fluorophores for frozen sections, to avoid confusion with DsRed endogenous emission. Antigen retrieval in citrate buffer (pH 6.0) was performed for E-cadherin, lysosome-associated membrane protein-1 (LAMP-1), the tetra-amino acid sequence for ER retention (KDEL), zonula occludens-1 (ZO-1), F4/80, and NKx2.1. Sections were imaged with a spinning disk confocal microscope using EC Plan-NeoFluar ×40/1.3 or Plan Apochromat ×100/1.4 Oil DIC objectives (cell observer spinning disk; Zeiss), and pseudocolors were assigned afterward using AxioVision Rel. 4.8 software. Electron microscopy was performed as described (8).
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence | Name of Antibody | Manufacturer, Catalog # | Species Raised in; Monoclonal or Polyclonal | Dilution |
|---|---|---|---|---|---|
| E-cadherin | Anti-E-cadherin | DB Bioscience, 610 182 | Mouse monoclonal (clone 36) | 0.25 μg/ml | |
| Ki-67 | Anti-Ki-67 | DB Pharmingen, 556 003 | Mouse monoclonal (clone B56) | 2 μg/ml | |
| Nkx2–1 | Anti-Thyroid Transcription Factor-1 | Dako, M3575 | Mouse monoclonal (clone 8G7G3/1) | 1/500 | |
| GFP | Anti-GFP | Cell signaling, 2956 | Rabbit monoclonal | 1/200 | |
| GFP | aa1-246 | Anti-GFP | Abcam, ab6673 | Goat polyclonal | 1/250 |
| ZO-1 | aa463-1109 | Anti-ZO-1 | Invitrogen, 61-7300 | Rabbit polyclonal | 2.5 μg/ml |
| Laminin | Anti-laminin | Sigma, L9393 | Rabbit polyclonal | 5 μg/ml | |
| F4/80 | Anti-F4/80 | Abcam, ab6640 | Rat monoclonal (clone CI:A3-1) | 3.4 μg/ml | |
| F4/80 | Anti-F4/80 | eBioscience, 14-4801 | Rat monoclonal (clone BM8) | 5 μg/ml | |
| MHCII | Anti-MHC class II (I-A/I-E) | Millipore, MABF33 | Rat monoclonal (clone M5/114) | 10 μg/ml | |
| CD11b | Anti-CD11b | Biolegend, 101 202 | Rat monoclonal (clone M1/70) | 5 μg/ml | |
| CD11c | Anti-CD11c | Abcam, ab11029 | Rat monoclonal (clone 3.9) | 10 μg/ml | |
| CD45 | Anti-CD45 | Biolegend, 103 102 | Rat monoclonal (clone 30-F11) | 5 μg/ml | |
| Ly6C | Anti-Ly6C | Thermo Scientific, MA1-81 899 | Rat monoclonal (ER-MP20) | 10 μg/ml | |
| KDEL | Anti-KDEL | Abcam, ab50601 | Rat monoclonal (MAC 256) | 1/300 | |
| LAMP-1 | Anti-LAMP-1 1D4B | Hybridoma Bank, #1D4B | Rat monoclonal (clone 1D4B) | 1/75 |
Statistical analysis
Statistical significance was tested using the Mann-Whitney test. Differences were considered significant for P < .05.
Results
HSC transplantation into Ctns−/− mice normalizes thyroid function and protects follicular structures
We recently reported that Ctns−/− mice develop between 6 and 9 months subclinical hypothyroidism with multifocal thyrocyte hyperplasia/hypertrophy and colloid exhaustion as cystinotic patients. Increased Ctns−/− thyrocyte apoptosis and proliferative repair revealed accelerated cell turnover (8). Clinical benefit of early WT-eGFP-HSC transplantation was first assessed by measuring thyroid cystine and plasma hormone levels. Grafting of WT-eGFP-HSCs decreased by approximately 75% cystine accumulation in 8-month-old Ctns−/− thyroid (Figure 1A), half of them showing normal plasma T4 and TSH levels (Figure 1B). By conventional histopathology, grafted Ctns−/− thyroids showed striking overall improvement over nongrafted controls, as evidenced by predominantly normal thyrocyte height and homogeneous colloid filling (Figure 1Cc). Thyrocyte proliferation (Ki-67 immunolabeling), was decreased by approximately 60% upon transplantation, in agreement with hyperplasia correction (Figure 1D; Supplemental Figure 1). This analysis also revealed the proliferation of WT-eGFP-HSCs, indicating local graft expansion in cystinotic thyroid (Figure 1Dc and insert). Altogether these data demonstrated a remarkable benefit of early engraftment of WT-eGFP-HSCs to correct thyroid disease progression in Ctns−/− mice.
Figure 1.

WT HSC transplantation into Ctns−/− mice can normalize thyroid function and prevent thyrocyte hyperplasia and hypertrophy. Eight-month-old/6-month posttransplant Ctns−/− mice were compared with age-matched WT and Ctns−/− mice. Because there was no significant difference between males and females, combined values are presented. A, Normalization of cystine accumulation. Cystine levels were assayed in thyroid lysates of WT, Ctns−/−, and grafted Ctns−/− mice and were normalized to protein concentration. *, P < .05; **, P < .01. NS, nonsignificant. B, Protection against primary hypothyroidism. TSH and T4 plasma concentrations were measured in WT, Ctns−/−, and grafted Ctns−/− mice. In about half of the treated Ctns−/− mice, TSH and T4 plasma concentrations are within the normal range. C, Prevention of thyrocyte hyperplasia and hypertrophy. Hematoxylin and eosin staining of thyroid paraffin sections from WT (a), Ctns−/− (b), and grafted Ctns−/− mice (c). a, WT thyroid is made of uniform follicles filled with homogenous colloid and mostly delineated by flat thyrocytes. b, In the Ctns−/− thyroid, the sustained TSH response causes thyrocyte hypertrophy and hyperplasia associated with colloid exhaustion (insert). #, A rare normal follicle. c, In grafted Ctns−/− mice, follicular activation is suppressed, except at the gland center (asterisks). D, Normalization of thyrocyte proliferation. Triple immunofluorescence for E-cadherin (white, thyrocyte basolateral membrane), Ki-67 (red, proliferation marker), and green fluorescent protein (GFP; green, HSCs) in WT (a), Ctns−/− (b), and grafted Ctns−/− mice thyroid (c). Notice in the nongrafted Ctns−/− numerous proliferating cells, mostly thyrocytes (arrows), as compared with the WT and grafted Ctns−/− mice. For quantification, see Supplemental Figure 1. In the right panels, high-magnification views from grafted Ctns−/− mice show individual channels and then merge images of grafted HSCs (GFP) immunolabeled for Ki-67 (red), demonstrating ongoing local graft expansion.
WT-eGFP-HSCs transplantation improves biosynthetic and lysosomal overload in Ctns−/− thyroid
We thus looked for protection by HSC transplantation against subcellular alterations induced by cystinosis (8). Upon grafting, we found a major decrease of ER expansion in most Ctns−/− thyrocytes, as monitored by KDEL immunolabeling, suggesting relief of ER stress (Figure 2A). Endolysosomal status of grafted Ctns−/− thyroids, monitored by LAMP-1 immunolabeling (Figure 2B) and electron microscopy (Supplemental Figure 2), was very heterogeneous: regions apparently normal upon grafting, elsewhere very much altered, but no cystine crystal could be detected, in contrast to nongrafted Ctns−/− thyroids (Supplemental Figure 2). Of note, neither cystinosis (data not shown) nor HSC engraftment and follicular infiltration (see below) disrupted thyrocyte tight junction integrity, thus epithelial barrier (Supplemental Figure 2).
Figure 2.

Improvement by HSC transplantation of biosynthetic and lysosomal overload in Ctns−/− thyroid. Eight-month-old/6-month posttransplant Ctns−/− mice were compared with age-matched WT and Ctns−/− mice. Samples were analyzed by double immunofluorescence for E-cadherin (red, thyrocyte structure) and either (green) KDEL, as generic marker of endoplasmic reticulum residents (A), or LAMP-1, as lysosomal membrane marker (B). A, ER expansion. In WT follicles (a), thyrocytes are flat or cuboidal, and the ER mostly occupies the basal cytoplasm. In nongrafted Ctns−/− mice (b), thyrocytes are columnar (hypertrophic, reaching ∼20 μm in height), and KDEL labeling fills the lumen of dilated ER (arrows), which is spread over the entire cytoplasm. Upon grafting (c), Ctns−/− thyrocyte height is generally decreased and the ER is overall reduced as compared with panel b. However, KDEL antibodies occasionally delineate larger structures devoid of luminal signal (arrow). B, Lysosome abnormalities. In WT thyrocytes (a), LAMP-1-labeled structures are dotty and sparse along the cytoplasm. In nongrafted Ctns−/− thyrocytes (b), many lysosomes are dilated and concentrated at the apical pole (arrows and enlarged in insert). LAMP-1 also labels cell remnants in the follicular lumen. Panels c and d show two distinct patterns of grafted Ctns−/− thyrocytes. In panel c, the aspect is comparable with WT (ie, preserved). In panel d, enlarged lysosomes are obvious, even frequently larger than in panel b. For electron microscopy, see Supplemental Figure 2 (crystals were detected only in nongrafted Ctns−/− thyrocytes).
Thyroid-grafted WT-eGFP-HSCs emit tunneling nanotube-like expansions able to cross follicle basement lamina
We next addressed the mechanism(s) of HSC-mediated tissue protection in a bifluorescent mouse model (17), which allows to discriminate the fate of green WT-eGFP-HSCs transplanted into DsRed mice (here identifying Ctns−/− thyrocytes). No grafted WT-eGFP-HSCs simultaneously expressed DsRed (Figure 3A, no yellow signal), thereby excluding cell fusion as protective mechanism. WT-eGFP-HSCs with dendritic-like shape frequently apposed onto follicular basement laminae (Figure 3, Aa, Ab, and Ba), which they further crossed by long cytoplasmic extensions, either very thin (stricto sensu tunneling nanotubes) or much thicker with budding tips (Figure 3Ba–c). Furthermore, several WT-eGFP-HSCs became completely inserted inside follicles, intertwined with thyrocytes (arrowheads in Figure 3Aa, Ab, and Ba and Supplemental Figure 3). Inserted WT-eGFP-HSCs thus enjoyed extensive close contact with adjacent Ctns−/− thyrocytes, which was never found in Ctns−/− kidney PTCs (see below). Lateral extensions of individually inserted HSCs could further contact several thyrocytes (Supplemental Figure 3). Follicular basement lamina porosity to invading cells could be due to tissue-specific difference between thyroid and kidneys, combined with cystinosis-related alterations (Supplemental Figure 4). Indeed, follicular basement lamina in nongrafted Ctns−/− mice lost normal circularity (indicating lesser tensile strength) and disclosed large discontinuities, which were not seen in grafted congeners.
Figure 3.

Grafted WT-eGFP-HSCs produce tunneling nanotubes that perforate follicular basement lamina and squeeze into the thyrocyte monolayer. Note here the different use of red in panel A (Ctns−/− tissue) vs panel B (laminin). A, General view of spatial relation between WT-eGFP-HSCs and DsRed+-Ctns−/− thyrocytes. Double-fluorescence confocal imaging in cryostat sections of 6- (a) and 11-month (b) posttransplant Ctns−/− mice (endogenous emission from DsRed [red, thyrocytes] and GFP [green, HSCs]). a, Low magnification shows that HSCs either preferentially appose onto follicles (arrows; yellow dotted line at b′ indicating basement lamina in panel b) or appear colinear with, ie, integrate within, thyrocytes (arrowheads; inside follicular space at b″). For further views of HSCs fully incorporated into the follicle, see Supplemental Figures 3 and 5. Colloid space is indicated by asterisks. Notice that intrafollicular HSCs do not fuse with thyrocytes (no yellow signal). B, Extrafollicular HSCs produce tunneling nanotubes. Three-dimensional reconstruction of z-stack series images (0.23 μm increment, 5 μm total thickness) was obtained after double immunolabeling for green fluorescent protein (green, HSCs) and laminin (red, basement lamina) in frozen sections of grafted mice thyroid at 6 months after transplantation. a, Typical extrafollicular HSCs (#), closely apposed to the follicle basement lamina, produce slender cytoplasmic extensions, some of which bend perpendicular to (yellow arrowhead) or cross the basement lamina (white arrowhead). Extensions can be very thin (yellow arrow in panel b) or irregular and varicose (white arrow in panel a). Notice the local absence of laminin indicating basement lamina discontinuities allowing TNT passage (better seen in enlargement in panel c) and the overall mottled aspect of follicular basement lamina. Asterisk indicates another, intrafollicular HSC. For rotating view, see Supplemental Video 1. For comparison of laminin structure between WT and Ctns−/− vs grafted Ctns−/−, see Supplemental Figure 4.
Despite extensive contiguity, inserted WT-eGFP-HSCs do not transdifferentiate into thyrocytes
Grafted WT-eGFP-HSCs did not simultaneously expressed DsRed, thus excluding cell fusion as protective mechanism. However, in response to tissue injury, engrafted HSCs may transdifferentiate into unexpected lineages, including epithelial cells (19). We thus investigated whether follicle-inserted WT-eGFP-HSCs could instead transdifferentiate into thyrocytes. Although inserted WT-eGFP-HSCs were apparently circumscribed by the thyrocyte basolateral membrane marker, E-cadherin (Supplemental Figure 5A), they never engaged in tight junction formation with neighboring thyrocytes (Supplemental Figure 5B). This indicated that HSCs did not transdifferentiate into polarized thyrocytes and suggested that circumscribing E-cadherin signal originated from a single, adjacent epithelial cell, thus arguing against transdifferentiation. Furthermore, none of the inserted WT-eGFP-HSC nuclei were labeled for the thyrocyte-specific transcription factor, Nkx2-1, excluding transdifferentiation into thyrocytes (Supplemental Figure 5C). No WT-eGFP-HSCs expressed calcitonin (not shown), thereby excluding transdifferentiation into epithelial C cells.
Distinct phenotype of thyroid-engrafted WT-eGFP-HSCs
Macrophages form a plastic population (20) yet poorly characterized in thyroid disease. In Ctns−/− liver, brain, and kidneys, HSC-derived cells mainly differentiate into F4/80high macrophages (13–15, 17). We thus looked at macrophage abundance and differentiation markers in WT, Ctns−/−, and grafted Ctns−/− thyroid. In the WT thyroid, F4/80+ macrophages were extremely rare but became massively recruited in Ctns−/− mice between 6 and 9 months of age, concomitant with histoarchitectural alterations (Supplemental Figure 6). Most such endogenous macrophages further inserted into Ctns−/− follicles, like WT-eGFP-HSCs described above (Figure 4Ab and Supplemental Figure 6F). In the grafted Ctns−/− thyroid, much fewer endogenous F4/80+ macrophages were observed, probably due to considerable correction of tissue injury (for full thyroid section, see Supplemental Figure 7). We next attempted to better define the status of engrafted WT-eGFP-HSC-derived cells. CD45 confirmed hematopoietic cell lineage origin (Figure 4B), but F4/80 was rarely detected, arguing against conventional macrophages (Figure 4, Ac and B). This differed from other cystinotic tissues such as the kidneys, indicating a unique role of the thyroid microenvironment (Supplemental Figure 8). Most engrafted WT-eGFP-HSC-derived cells expressed MHCII, consistent with healing differentiation (16). The signal for the CD11b macrophage marker was weak, and neither Ly6c nor CD11c was detected (Figure 4B). These data indicated that HSC-derived cells grafted into Ctns−/− thyroid apparently replaced typical endogenous F4/80+ macrophages recruited in nongrafted congeners and acquired in this particular tissue microenvironment a distinct combination of macrophage/dendritic differentiation markers. Of note, the steady-state abundance of grafted, particularly follicle-apposed and inserted HSCs, was very low as compared with the high prevalence of apparently protected thyrocytes (Supplemental Figure 7).
Figure 4.
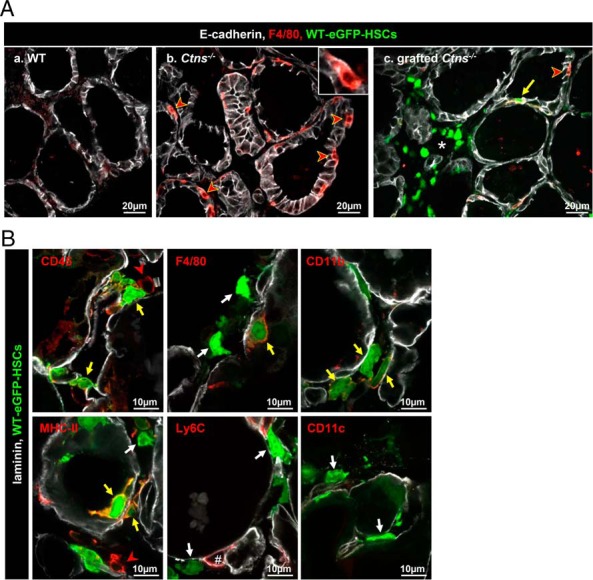
Grafted WT-HSCs replace endogenous Ctns−/− macrophages and differentiate into macrophage/dendritic cell lineages. A, WT-eGFP-HSCs grafted into Ctns−/− mice thyroid replace endogenous macrophages recruitment. Triple immunofluorescence for E-cadherin (white), F4/80 (red, common macrophage marker), and green fluorescent protein (GFP; green, HSCs) on thyroids of WT (a), Ctns−/− (b), and grafted Ctns−/− mice (c) is shown. a, In WT mice, macrophages are rare. b, In Ctns−/− mice, macrophages are abundant around and within follicles (red arrowheads and insert). c, In grafted Ctns−/− mice, much fewer endogenous F4/80+ macrophages (red arrowhead) are seen. Whereas some grafted HSCs also express the detectable F4/80 marker (yellow arrow), most do not (interstitial cluster around the asterisk). B, Phenotype of grafted HSCs. Multiplex immunofluorescence for laminin (white), GFP (green, HSCs), and specific markers of macrophages/dendritic cells (red): the common leukocyte marker (CD45); two common macrophages makers (F4/80 or CD11b); a dendritic cell and macrophage marker (MHCII); a monocyte and endothelial cell marker (Ly6C); and a macrophage and dendritic cell marker (CD11c). Most grafted HSCs are labeled for CD45 and some also express F4/80, CD11b, and MHCII markers (yellow arrow). In our conditions, Ly6C antibodies did not label HSCs but endothelium (#). HSCs that do not express a specific marker tested are indicated by white arrows. Endogenous inflammatory cells expressing CD45 and MHCII are indicated by a red arrowheads.
Discussion
We here report that HSC transplantation can protect thyroid structure and normalize function in a mouse model of cystinosis (Ctns−/−), a lysosomal storage disease due to defective membrane transporter. Thyroid-engrafted HSCs generate extensions that cross follicular basal lamina and fully squeeze into the thyrocyte monolayer, thus acquiring extensive close contact with target cells. HSCs neither fuse with, nor transdifferentiate into, thyrocytes but mostly differentiate into MHCII-positive macrophage/dendritic lineage instead of homogenous F4/80 labeling found in engrafted kidneys and eyes, pointing to a unique role of thyroid microenvironment in macrophage differentiation.
Stem cell transplantation is now a therapeutic option for a broad range of congenital or acquired disorders (21) but has never been validated for thyroid disorders. On the contrary, case reports suggested that HSCs may trigger autoimmune thyroiditis and favor thyroid cancer, although indirect effects of myeloablation are unclear (22, 23). However, in a mouse model of Hashimoto's disease, mesenchymal stem cells efficiently reduced inflammation and improved clinical outcome (24, 25). We here demonstrate that bone marrow-derived HSCs can partially protect Ctns−/− thyroid structure and normalize function. These data further support a major multisystemic benefit of stem cells therapy for cystinosis, in particular thyroid protection, and suggest it may represent a generic treatment for membrane protein defects. A clinical trial is currently being developed for human cystinosis based on autologous transplantation of ex vivo lentivirus-transduced HSCs combined with myeloablation by chemotherapy.
In Ctns−/− kidneys and corneas, tissue repair by engrafted HSCs involves TNT formation and transfer into deficient cells of cystinosin-bearing lysosomes as correcting factor, ie, cross-correction (16, 17). HSC recruitment thus cannot be primarily related to the altered thyroid state but to the general disease (13, 16). However, the thyroid seems so far unique because HSCs not only emit TNT-like structures able to cross (damaged?) follicular basement lamina, but also fully squeeze into the epithelial monolayer and simultaneously contact several thyrocytes. Thus, inserted HSCs adopt optimal membrane disposition to interact closely and promiscuously with diseased thyrocytes. In the nongrafted Ctns−/− thyroid, infiltrating endogenous (cystinosin deficient, F4/80+) macrophages can also squeeze across follicular basement lamina but are obviously inefficient to prevent disease progression because they are unable to provide a gene- or protein-associated correcting factor. Dendritic cells, commonly infiltrating follicles in autoimmune thyroiditis, Graves disease, and iododeficiency goiter (26, 27) also strongly adhere to the basement membrane, extend cytoplasmic extensions in between thyrocytes, and even cross apical tight junctions (26). Furthermore, mutual interactions between invading lymphocytes and thyrocytes, rendering possible transfer of material, have been evidenced in Hashimoto's thyroiditis (28). Altogether these observations suggest that the thyroid is more prone to full HSC infiltration across basement lamina as compared with the kidney in which HSC bodies are restricted around tubules (13, 14, 17) and that HSCs, once fully infiltrated within the epithelial monolayer, enjoy close vicinity with cystinotic thyrocytes, which should favor efficient correction as testified by the normalization of thyroid function in most Ctns−/− mice.
A striking discrepancy was further noted between the low abundance of thyrocyte-contacting HSCs at steady state and the remarkable extent of tissue protection. This discrepancy favors the view of multiple cross-corrective interactions, ie, the ability of individual dendritic-shaped HSCs to simultaneously interact with several adjacent disease cells combined with HSC mobility, allowing iterative exchange of correcting organelles at each new location over time.
Overall histological and functional protection was, however, not matched by general subcellular preservation. Tissue heterogeneity, between and within follicles, is not really surprising if protection depends on local physical connections. A deleterious effect of irradiation also can not be excluded, although not supported by previous investigations (13). Considering the thyroid functional reserve whereby independent follicles contribute in parallel to total thyroid hormone production, incomplete structural protection may still support adequate functional correction.
In the Ctns−/− thyroid, engrafted WT HSCs alleviate the afflux of endogenous cystinotic F4/80+ macrophages, as also observed in the kidney (14), probably due to the correction of tissue injury. Most WT HSC-derived cells in the cystinotic thyroid expressed CD45 and MHCII as opposed to conventional F4/80+ macrophages, suggesting a distinct differentiation in Ctns−/− thyroid vs kidneys and corneas, likely due to the cystinotic thyroid microenvironment. Remarkably, although proinflammatory cytokines are known modifiers of thyroid epithelial barrier function (eg, reference 29), the inflammatory component of either cystinosis itself, or linked to HSC engraftment and infiltration, was apparently not sufficient to disrupt thyrocyte tight junction integrity, thus the epithelial barrier. Whether cytokines specify the tissue-specific cystinotic landscape and whether macrophage specification influences their ability to fully cross basement laminae deserve to be studied.
Acknowledgments
We gratefully acknowledge Dr Corinne Antignac (INSERM Unité 983, Paris, France) for providing the original Ctns−/− mice. We thank Dr A. Cominelli for comments as well as Y. Abid and T. Lac for their help with experiments.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
H.G.C. is postdoctoral researcher, and C.E.P. is a research associate at Belgian Fonds de la Recherche Scientifique (Belgium).
This work was supported mainly by the Cystinosis Research Foundation, Belgian Science Policy Office-Interuniversity Attraction Poles Program Grant IAP P7/43-BeMGI, Belgian Fonds de la Recherche Scientifique and Actions de Recherche Concertées (to P.J.C. and C.E.P.), and National Institutes of Health Grants RO1-DK090058, RO1-DK099338, and R21-NS090066 (to S.C.). This work was also supported in part by Grant R37-DK15070 from the National Institutes of Health (to S.R.). The Platform for Imaging Cells and Tissues was financed by National Lottery, Région Bruxelloise, Région Wallonne, Université Catholique de Louvain, and de Duve Institute (to P.C.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- eGFP
- enhanced green fluorescent protein
- ER
- endoplasmic reticulum
- HSC
- hematopoietic stem cell
- KDEL
- tetra-amino acid sequence for ER retention
- LAMP-1
- lysosome-associated membrane protein-1
- MHCII
- major histocompatibility complex II
- PTC
- proximal tubular cell
- TNT
- tunneling nanotube
- WT
- wild type
- ZO-1
- zonula occludens-1.
References
- 1. Gahl WA, Thoene J, Schneider JA. Cystinosis: a disorder of lysosomal membrane transport. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Basis of Inherited Disease. Vol 3, 8th ed McGraw-Hill Companies, Inc, New York; 2001:5085–5108. Online update; 2013. [Google Scholar]
- 2. Andrzejewska Z, Nevo N, Thomas L, et al. Cystinosin is a component of the v-ATPase-Ragulator-Rag complex controlling mTORC1. J Am Soc Nephrol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan AM, Lynch MJ, Bailey JD, Ezrin C, Fraser D. Hypothyroidism in cystinosis. A clinical, endocrinologic and histologic study involving sixteen patients with cystinosis. Am J Med. 1970;48(6):678–692. [DOI] [PubMed] [Google Scholar]
- 4. Lucky AW, Howley PM, Megyesi K, Spielberg SP, Schulman JD. Endocrine studies in cystinosis: compensated primary hypothyroidism. J Pediatr. 1977;91(2):204–210. [DOI] [PubMed] [Google Scholar]
- 5. Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dialysis Transplant. 2010;25(4):1059–1066. [DOI] [PubMed] [Google Scholar]
- 6. Raggi C, Luciani A, Nevo N, Antignac C, Terryn S, Devuyst O. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum Mol Genet. 2014;23(9):2266–2278. [DOI] [PubMed] [Google Scholar]
- 7. Gaide Chevronnay HP, Janssens V, et al. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. J Am Soc Nephrol. 2014;25(6):1256–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gaide Chevronnay HP, Janssens V, Van Der Smissen P, et al. A mouse model suggests two mechanisms for thyroid alterations in infantile cystinosis: decreased thyroglobulin synthesis due to endoplasmic reticulum stress/unfolded protein response and impaired lysosomal processing. Endocrinology. 2015;156(6):2349–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ivanova EA, De Leo MG, Van Den Heuvel L, et al. Endo-lysosomal dysfunction in human proximal tubular epithelial cells deficient for lysosomal cystine transporter cystinosin. PloS One. 2015;10(3):e0120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jezegou A, Llinares E, Anne C, et al. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc Natl Acad Sci USA. 2012;109(50):E3434–E3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brodin-Sartorius A, Tete MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012;81(2):179–189. [DOI] [PubMed] [Google Scholar]
- 12. Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dialysis Transplant. 2014;29(suppl 4):iv87–iv94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Syres K, Harrison F, Tadlock M, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009;114(12):2542–2552. [DOI] [PubMed] [Google Scholar]
- 14. Yeagy BA, Harrison F, Gubler MC, Koziol JA, Salomon DR, Cherqui S. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011;79(11):1198–1206. [DOI] [PubMed] [Google Scholar]
- 15. Harrison F, Yeagy BA, Rocca CJ, Kohn DB, Salomon DR, Cherqui S. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol Ther. 2013;21(2):433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rocca CJ, Kreymerman A, Ur SN, et al. Treatment of inherited eye defects by systemic hematopoietic stem cell transplantation. Invest Ophthalmol Vis Sci. 2015;56(12):7214–7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naphade S, Sharma J, Gaide Chevronnay HP, et al. Brief reports: lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells. 2015;33(1):301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pohlenz J, Maqueem A, Cua K, Weiss RE, Van Sande J, Refetoff S. Improved radioimmunoassay for measurement of mouse thyrotropin in serum: strain differences in thyrotropin concentration and thyrotroph sensitivity to thyroid hormone. Thyroid. 1999;9(12):1265–1271. [DOI] [PubMed] [Google Scholar]
- 19. Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6(11):1229–1234. [DOI] [PubMed] [Google Scholar]
- 20. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gratwohl A, Baldomero H, Gratwohl M, et al. Quantitative and qualitative differences in use and trends of hematopoietic stem cell transplantation: a global observational study. Haematologica. 2013;98(8):1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Milenkovic T, Vujic D, Vukovic R, et al. Subclinical hypothyroidism in children and adolescents after hematopoietic stem cells transplantation without irradiation. Vojnosanit Pregl. 2014;71(12):1123–1127. [DOI] [PubMed] [Google Scholar]
- 23. Vantyghem MC, Cornillon J, Decanter C, et al. Management of endocrino-metabolic dysfunctions after allogeneic hematopoietic stem cell transplantation. Orphanet J Rare Dis. 2014;9:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi EW, Shin IS, Lee HW, et al. Transplantation of CTLA4Ig gene-transduced adipose tissue-derived mesenchymal stem cells reduces inflammatory immune response and improves Th1/Th2 balance in experimental autoimmune thyroiditis. J Gene Med. 2011;13(1):3–16. [DOI] [PubMed] [Google Scholar]
- 25. Choi EW, Shin IS, Park SY, et al. Characteristics of mouse adipose tissue-derived stem cells and therapeutic comparisons between syngeneic and allogeneic adipose tissue-derived stem cell transplantation in experimental autoimmune thyroiditis. Cell Transplant. 2014;23(7):873–887. [DOI] [PubMed] [Google Scholar]
- 26. Molne J, Jansson S, Ericson LE, Nilsson M. Adherence of RFD-1 positive dendritic cells to the basal surface of thyroid follicular cells in Graves' disease. Autoimmunity. 1994;17(1):59–71. [DOI] [PubMed] [Google Scholar]
- 27. Wilders-Truschnig MM, Kabel PJ, Drexhage HA, et al. Intrathyroidal dendritic cells, epitheloid cells, and giant cells in iodine deficient goiter. Am J Pathol. 1989;135(1):219–225. [PMC free article] [PubMed] [Google Scholar]
- 28. Irvine WJ, Muir AR. An electron microscopic study of Hashimoto thyroiditis. Q J Exp Physiol Cogn Med Sci. 1963;48:13–26. [DOI] [PubMed] [Google Scholar]
- 29. Tedelind S, Ericson LE, Karlsson JO, Nilsson M. Interferon-gamma down-regulates claudin-1 and impairs the epithelial barrier function in primary cultured human thyrocytes. Eur J Endocrinol. 2003;149(3):215–221. [DOI] [PubMed] [Google Scholar]