

Abstract
Clinical symptoms may vary and not necessarily reflect serum thyroid hormone (TH) levels during acute and chronic hyperthyroidism as well as recovery from hyperthyroidism. We thus examined changes in hepatic gene expression and serum TH/TSH levels in adult male mice treated either with a single T3 (20 μg per 100 g body weight) injection (acute T3) or daily injections for 14 days (chronic T3) followed by 10 days of withdrawal. Gene expression arrays from livers harvested at these time points showed that among positively-regulated target genes, 320 were stimulated acutely and 429 chronically by T3. Surprisingly, only 69 of 680 genes (10.1%) were induced during both periods, suggesting desensitization of the majority of acutely stimulated target genes. About 90% of positively regulated target genes returned to baseline expression levels after 10 days of withdrawal; however, 67 of 680 (9.9%) did not return to baseline despite normalization of serum TH/TSH levels. Similar findings also were observed for negatively regulated target genes. Chromatin immunoprecipitation analysis of representative positively regulated target genes suggested that acetylation of H3K9/K14 was associated with acute stimulation, whereas trimethylation of H3K4 was associated with chronic stimulation. In an in vivo model of chronic intrahepatic hyperthyroidism since birth, adult male monocarboxylate transporter-8 knockout mice also demonstrated desensitization of most acutely stimulated target genes that were examined. In summary, we have identified transcriptional desensitization and incomplete recovery of gene expression during chronic hyperthyroidism and recovery. Our findings may be a potential reason for discordance between clinical symptoms and serum TH levels observed in these conditions.
Hyperthyroidism refers to the clinical condition of hypermetabolism and hyperactivity, resulting from serum elevations in thyroid hormone (TH) levels. Classic symptoms of hyperthyroidism include heat intolerance, palpitations, anxiety, weight loss, and tremor. However, in some patients with Graves' disease, the most frequent cause of hyperthyroidism, initial symptoms can abate despite elevated serum TH levels. Also, some patients with Graves' disease report residual symptoms suggestive of hyperthyroidism, even after normalization of serum TH levels (1). In the liver, previous studies showed that triglyceride accumulation and mitochondrial activity adapted during acute and chronic hyperthyroidism (2, 3). Additionally, SHBG, a hepatic protein secreted by the liver that is highly sensitive to TH, was normal in 7 of 20 of thyrotoxic patients (35%) (4). These findings suggest that the sensitivity of hepatic target genes to TH may change between acute and chronic hyperthyroidism and during the transition from hyperthyroidism to euthyroidism.
TH regulates a wide range of processes including growth, development, and metabolism (5). The effects of TH are mainly mediated by the transcriptional activity of TH receptors (TRs), members of the nuclear hormone receptor superfamily. Circulating free TH enters the cell via plasma membrane transporters, and the intracellular concentration of the more active form of TH, T3, can be increased by converting T4 to T3 via type 2 iodothyronine deiodinases, or decreased by type 3 iodothyronine deiodinases (6). T3 then enters the nucleus, in which it binds to the nuclear TRs mainly as a heterodimer with retinoid X receptor isoforms. In T3-dependent activation of transcription (positive regulation), liganded TRs bind to TH response elements (TREs), which are typically located in the promoter regions of target genes, and recruit coactivator complexes that have histone acetyltransferase activity (5). Histone acetyltransferases (HATs) such as steroid receptor coactivators and p300/cAMP response element-binding protein-associated factor induce histone acetylation, formation of euchromatin, and recruitment of RNA polymerase II, leading to transactivation of T3-regulated target genes (7). In the absence of ligand, TRs also can bind to the promoter regions of target genes and recruit corepressors such as nuclear receptor corepressor 1 or silencing mediator for retinoid and TH receptors (or nuclear receptor corepressor-2), which then recruit histone deacetylase 3 to decrease histone acetylation, resulting in transcriptional repression. This TR/corepressor complex dissociates when TR binds T3.
To better understand transcriptional changes by TH at the genomic level, we previously examined hepatic gene expression profiles in hypothyroid and hyperthyroid mice using gene expression arrays (8). Subsequent studies using different TR-knockout mouse models in various nutritional states have identified both novel and previously described target genes involved in a wide range of cellular functions, including gluconeogenesis, lipogenesis, and cell proliferation (9–12). However, the transcriptional patterns and mechanisms that occur after chronic TH treatment and its withdrawal remain poorly understood. Accordingly, we analyzed the hepatic gene expression patterns and associated epigenetic changes in T3-regulated target genes as well as serum TH/TSH levels after acute and chronic TH treatment as well as during the transition from hyperthyroidism to euthyroidism. We also analyzed the expression of some representative T3-target genes in male mice deficient in monocarboxylate transporter 8 (MCT8), which have chronic intrahepatic hyperthyroidism since birth (13).
Our findings showed that transcriptional desensitization and incomplete recovery of gene expression occur during chronic hyperthyroidism and recovery, which may account for the variable clinical phenotypes seen in patients with these conditions. We also found that transcriptional desensitization of target genes after chronic T3 treatment may be due to specific histone modifications.
Materials and Methods
Animal experimental protocol
C57BL/6 male mice were maintained on a 12-hour light, 12-hour dark cycle and had access to food and water ad libitum. To induce systemic thyrotoxicosis, C57BL/6 male mice between ages 8 and 10 weeks received daily an ip injection of 20 μg liothyronine (L-T3; Sigma-Aldrich) per 100 g body weight for 14 days (Figure 1A). Animal experiments were performed as two subsets, with nontreated mice used to compare acute T3 effects and another batch of nontreated mice used for chronic and withdrawal effects of T3. The corresponding serum levels of total T3, TSH, and total T4 in nontreated mice showed no significant differences between two subsets of animal experiments. As controls, mice were treated with the same volume of saline and killed at 6 hours and on day 10 after the last (14th) injection, which induced no significant changes in serum T3 and TSH concentrations (Supplemental Table 1). Each group and each treatment consisted of four to five mice. Mice appeared healthy during daily L-T3 injections for 14 days, and there were no significant changes in body weight (mean ± SEM) between nontreated (27.5 ± 0.6 g) and chronically treated mice (27.0 ± 0.3 g). The animals were anesthetized before blood was drawn by cardiac puncture, and plasma was separated by centrifugation. Livers were rapidly collected, snap frozen in liquid nitrogen, and stored at −80°C until analysis. To exclude a diurnal pattern of gene expression, L-T3 was injected at the same time of day (ie, 9:00 am), and mice were sacrificed at 3:00 pm (6 h after injection) or 9:00 am (24 h after injection). For microarray and DNA methylation analysis, three mice were randomly picked up for each group. This animal protocol was approved by the Institutional Animal Care and Use Committee of the Biopolis Resource Centre, Agency for Science and Technology (A*STAR), Singapore.
Figure 1.

Design of animal experiments to study thyroid hormone treatment and withdrawal and corresponding thyroid function test measurements during the study. A, Schematic of animal experiments. Adult male mice (8–10 wk old) were treated with 20 μg liothyronine (L-T3) per 100 g body weight for 1 day or 14 days followed by cessation of L-T3 for 10 days. Mice were killed at each of the indicated time points. Arrowheads represent L-T3 injections and arrows represent the time points of the animals were killed and tissue collection. B–D, Measurements of serum thyroid hormone and TSH levels during the study described above. Serum total T3 (B), total T4 (C), and TSH (D) concentrations were determined at indicated T3-treatment periods after the first and the last L-T3 injection. No treatment (blank); acute T3 treatment (black); chronic T3 treatment (hashed); early T3 withdrawal (hashed); T3 withdrawal (striped) are shown. Note that the same data points were used for the chronic T3 treatment and early T3 withdrawal groups (hashed). The limits of detection were 0.25 μg/dL for serum total T4 and 10 mU/L for TSH. Data are presented as means ± SEM (n = 4–5/group). *, P < .05, **, P < .01, ***, P < .001, compared with nontreated mice (0 d) by one-way ANOVA.
Mct8-knockout mice were generated, housed, and genotyped as described previously (14). Experiments were performed on male wild-type (WT) and Mct8-knockout mice between ages 10 and 12 weeks. After mice were anesthetized, collected livers were immediately frozen on dry ice and stored −80°C until analysis. Each group consisted of 12 mice. This animal protocol was approved by the University of Chicago Institutional Animal Care and Use Committee.
Measurement of serum thyroid function tests
Serum total T3, T4, and TSH concentrations were measured by RIAs as detailed elsewhere (13).
RNA isolation
Total RNA was extracted from liver (15–20 mg) with TRIzol reagent (Invitrogen) followed by InviTrap spin cell RNA minikit (Stratec Molecular) according to the manufacturer's instructions.
Microarray analysis
Gene expression microarray profiling was performed using the MouseWG-6 version 2.0 Expression BeadChip kit (Illumina) by hybridizing RNA from liver tissues (n = 3/group). cRNA generation, labeling, and hybridization were performed at Duke-NUS Genome Biology Facility, Duke-NUS Medical School, Singapore. Gene expression signals were quantile normalized using the Partek Genomics Suite (version 6.6) software (Partek), and subsequent gene set enrichment analysis for acute and chronic T3, and T3 withdrawal were conducted (15). After the rank metric score of each gene was converted to a Z-score, differentially expressed genes were defined using a cut of value of 2.58 (Z-score > 2.58; significantly up-regulated gene, Z-score < −2.58; significantly down-regulated gene). In a heat map, expression values are represented in colors, in which the colors red, pink, light blue, and dark blue represent high, moderate, low, and lowest. An additional pathway overrepresentation analysis was conducted using the Database for Annotation, Visualization, and Integrated Discovery Bioinformatics Resources 6.7 (16). Raw microarray data sets have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under accession number GSE68867.
Reverse transcription-quantitative PCR (RT-qPCR)
The concentration of RNA was measured by NanoDrop 8000 (Thermo Scientific), and cDNA was reverse transcribed from 500 ng of total RNA using high-capacity cDNA reverse transcription kits (Applied Biosystems). The quantitative PCRs (qPCRs) were performed using the QuantiFast SYBR Green PCR kit (QIAGEN) on the 7900HT Fast real-time PCR system (Applied Biosystems). Relative mRNA levels were calculated using the 2-δδCt method and normalized to 18S ribosomal RNA (Rn18S), which is regarded as a suitable reference gene following TH exposure (17). The stabilities of Rn18S and β-actin (Actb) were evaluated under the different T3-treated conditions, revealing Rn18S transcripts were less responsive to T3 (data not shown). Data are expressed as the fold change relative to the mean value of nontreated mice. Primer sequences are provided in Supplemental Table 2.
Chromatin immunoprecipitation (ChIP)-qPCR
The ChIP assays were performed on liver tissues (n = 4–5/group) using the LowCell# ChIP kit protein G (kch-maglow-G48; Diagenode) according to the manufacturer's protocol with some modifications. Minced liver (30–35 mg) was fixed with 1% formaldehyde, lysed, and sheared with the bioruptor (Diagenode) for 15 cycles of 30-second intervals. Samples were precleared and incubated with antibodies against histone H3 acetylated at lysines 9 and 14 (H3K9/K14ac; number 06-599; Millipore), histone H4 pan-acetylated at lysines 5, 8, 12, and 16 (Pan-H4ac; number 06-598; Millipore), histone H3 trimethylated at lysine 4 (H3K4me3; number 07-473; Millipore), or normal rabbit IgG (sc2027; Santa Cruz Biotechnology) overnight at 4°C (Table 1). For each immunoprecipitation, 4 μg of antibody and 30–35 mg of tissue were used. Immune complexes were pulled down with magnetic beads, reverse cross-linked, and purified with phenol-chloroform. Immunoprecipitated chromatin was quantified by RT-qPCR on FAST7900HT (Applied Biosystems). Signal of immunoprecipitated DNA relative to 100% input was calculated according to the manufacturer's protocol (kch-maglow-G48; Diagenode).
Table 1.
Antibodies Used for ChIP-qPCR
| Antibody Specificity | Commercial Name | Source | Catalog Number | Dilution Used |
|---|---|---|---|---|
| H3K9/K14ac | Antiacetyl-histone H3 antibody | Millipore | 06-599 | 1:75 |
| Pan-H4ac | Antiacetyl-histone H4 antibody | Millipore | 06-598 | 1:75 |
| H3K4me3 | Antitrimethyl-histone H3 (Lys4) antibody | Millipore | 07-473 | 1:75 |
| IgG | Normal rabbit IgG | Santa Cruz Biotechnology | sc2027 | 1:75 |
The specific primer sequences used in ChIP-qPCR were as follows: Dio1 (−174 to +10), forward, 5′-AGAGAGAGCTCTGTGCCCTGG-3′, reverse, 5′-GAAGTGGCTCTGAGCCTGCAG-3′; Thrsp (−145 to −42), forward, 5′-ACAGACACTGGGGACCAAAC-3′, reverse, 5′-AGGCTTTTGAGCAGACAGCA-3′; Cyp17a1 (−142 to −27), forward, 5′-AGGATCCATAGCGCAGAAGC-3′, reverse, 5′-GAGGGAATTTGGCCTCCACA-3′; Idh3a (−312 to −193), forward, 5′-TGGTCCCCTAATAGGTCGGA-3′, reverse, 5′-CTTTTGATGACGCGCGGAG-3′; Fasn (−225 to −51), forward, 5′-TGACCGGTAGTAACCCCGC-3′, reverse, 5′-GAAACCAATTGGACACCGAGG-3′; Fgf21 (−188 to −78), forward, 5′-CCCATGCCTAGCCCTTTTCA-3′, reverse, 5′-TGACACACTTGGCAGGAACC-3′; Alb (−116 to +7), forward, 5′-CTTTTTGGCAAAGATGGTATG-3′, reverse, 5′-TGTGTGCAGAAAGACTCGCTC-3′. Negative controls using either the IgG antibody or the Alb promoter did not show any detectable changes in ChIP signals (data not shown).
Statistics
All data represent the mean ± SEM. Statistical significance of differences (P < .05) was examined by a one-way ANOVA followed by a Fisher's protected least significant difference using GraphPad PRISM (version 6.0) software (GraphPad Software Inc).
Results
Serum total T3 and TSH concentrations in mice after T3 treatment and its withdrawal
To evaluate hepatic gene expression patterns and associated epigenetic changes during chronic TH treatment and recovery, we treated adult male mice either with a single L-T3 (20 μg per 100 g body weight) injection (acute T3) or multiple injections for 14 days (chronic T3) followed by a 10-day withdrawal period (T3 withdrawal; Figure 1A). Serum thyroid function tests were determined to confirm thyrotoxicosis induced by these treatment protocols (Figure 1, B–D, and Supplemental Table 1). Significant elevations (10- to 30-fold) in serum total T3 concentrations were noted in the early phases (6 h) after both a single L-T3 injection (acute T3) and 14 days of L-T3 injections (chronic T3). In contrast, serum TSH and total T4 concentrations decreased in both the early and late phases (24 h) of L-T3 injections, consistent with the down-regulation of pituitary TSH secretion by exogenous T3. During the withdrawal period, serum T3 and TSH levels returned to near basal levels 10 days after the last L-T3 injection. Interestingly, serum TSH concentrations returned to baseline by day 3 and transiently increased to a maximum level 5 days after the last L-T3 before returning to baseline after 10 days. This rise in TSH likely was driven by the pituitary response to low serum T4 and T3 levels due to the suppression of endogenous TH synthesis by the thyroid gland during the first 3 days after exogenous T3 withdrawal. Taken together, we confirmed that both acute and chronic T3 exhibited similar changes in serum total T3 and TSH concentrations when compared with untreated control mice.
Global hepatic gene expression patterns in mice after T3 treatment and its withdrawal
To characterize the T3-regulated global expression patterns, gene expression microarrays were performed on livers harvested from mice before T3 treatment as well as 6 hours after acute and chronic T3 treatments. The microarray data were validated by comparing the changes in expression of several genes with those obtained by RT-qPCR, which resulted in high concordance (Spearman's rank order correlation coefficient [rs] = 0.916, n = 27, P < .001) (data not shown). A total of 680 positively and 537 negatively regulated genes were identified (Figure 2, A and B) that could be divided according to their characteristic into three general patterns: genes regulated only acutely by T3 (251 positive, 207 negative), only chronically by T3 (360 positive, 265 negative), and by both treatments (69 positive, and 65 negative). Most of positively regulated T3 target genes were transactivated either acutely or chronically, whereas only 69 of 680 genes (10.1%) were regulated both acutely and chronically. Similar findings were noted for negatively-regulated target genes. Several well-described T3-regulated target genes (10, 11, 17–20), such as thyroid hormone-responsive protein (Thrsp, or Spot14), isocitrate dehydrogenase 3α (Idh3a), type 1 iodothyronine deiodinase (Dio1), B-cell lymphoma 3 (Bcl3), fatty acid synthase (Fasn), cytochrome P450 17α-hydroxylase/17,20-lyase (Cyp17a1), and fibroblast growth factor 21 (Fgf21), were among the top 50 up-regulated genes induced by acute and/or chronic T3 treatment (Figure 2C).
Figure 2.

Distinct hepatic target gene expression patterns after acute and chronic T3. Adult male mice (8–10 wk old) were treated with 20 μg liothyronine (L-T3) per 100 g body weight for 14 days. Livers were harvested from mice 6 hours after the first L-T3 injection (acute T3) as well as after the last L-T3 injection (chronic T3). A and B, Venn diagrams showing differentially expressed genes at indicated T3-treatment periods in positively (A) and negatively (B) regulated genes from microarray data as described in Materials and Methods. C, Representative heat map displaying the top 50 up-regulated genes after acute T3 and/or chronic T3, compared with nontreated mice. Genes underlined show genes induced in common with both exposure protocols. Genes in bold show genes analyzed by RT-qPCR. Row-normalized gene expression levels are represented according to the colored bar scale at the bottom of the figure (n = 3/group).
We also examined gene expression 10 days after the last L-T3 injection (T3 withdrawal) and found that about 90% of genes regulated acutely and/or chronically by T3 returned to baseline (Figure 3, A and B). However, there were subsets of genes that remained up- or down-regulated relative to baseline even after serum T3 returned to near basal levels. For positively regulated target genes (Figure 3A), 58 of 680 (8.5%) remained up-regulated and 9 of 680 genes (1.3%) were paradoxically down-regulated. These findings suggest that 67 of 680 genes (9.8%) had not returned back to baseline expression levels despite normalization of serum T3 and TSH levels.
Figure 3.
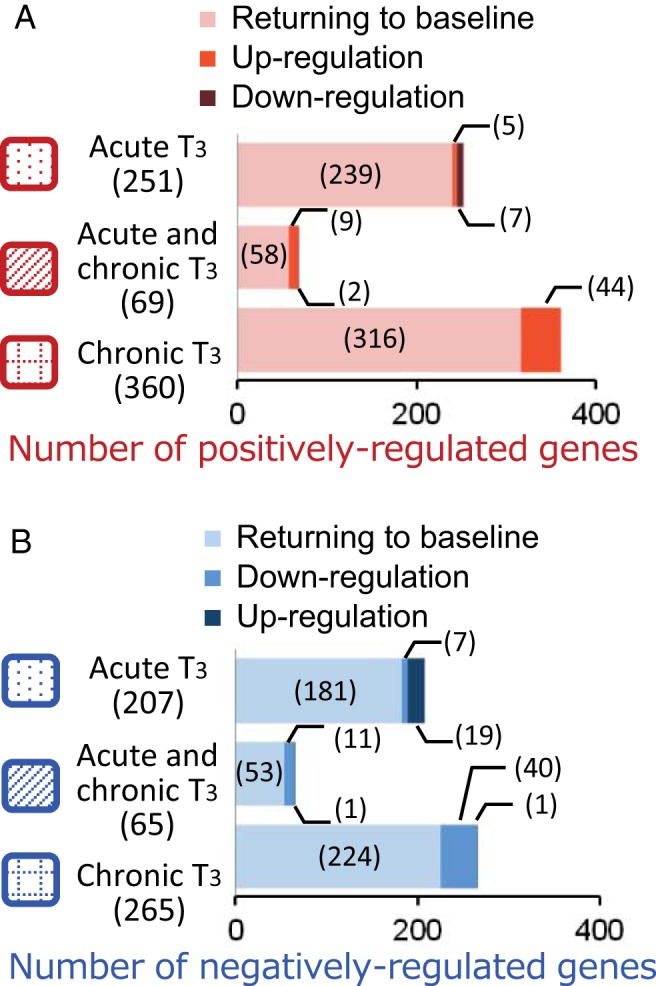
Distinct hepatic target gene expression patterns after T3 withdrawal. Adult male mice (8–10 wk old) were treated with 20 μg liothyronine (L-T3) per 100 g body weight for 14 days. Livers were harvested from mice 6 hours after the first L-T3 injection (acute T3), 6 hours after the last L-T3 injection given on day 14 (chronic T3), and 10 days after the last L-T3 injection (T3 withdrawal). A and B, Bar graph depicting the number of differentially expressed genes during T3 withdrawal and recovery. There were three categories of genes during recovery: returning to baseline, persistent up-regulation, or persistent down-regulation after T3 withdrawal in the indicated response groups for positively (A) and negatively (B) regulated genes described in Figure 2. Numbers in parentheses represent the number of differentially expressed genes.
Subsequent pathway analysis was performed using significantly up- and down-regulated genes, revealing distinct pathways for each condition (Supplemental Table 3). Acute T3 treatment induced substantial numbers of gene sets, including several previously reported T3-regulated pathways such as lipid synthesis or sterol metabolism (8, 10), whereas chronic T3 treatment up-regulated fewer genes, and they were involved in immune response. Interestingly, the steroid metabolism pathway was induced by both acute and chronic T3 treatment. Furthermore, pathways involved in immune and lipid processes remained up-regulated even after T3 withdrawal.
Individual gene expression patterns in T3-treated mice after T3 treatment and its withdrawal
To further characterize the temporal expression patterns of hepatic T3-targeted genes, we measured their mRNA in relation to serum TH and TSH levels at more time points (Figure 1A): 6 and 24 hours after the first L-T3 (acute T3) as well as 6 and 24 hours, and 3, 5, 7, and 10 days after the last L-T3 injection (chronic T3 and T3 withdrawal). For positively regulated target genes, we focused on eight well-characterized genes, reported to have functional TREs: Dio1, Thrsp, Cyp17a1, Fasn, Idh3a, Bcl3, Fgf21, and carnitine palmitoyltransferase 1a (Cpt1a) (10, 11, 17–20). T3 acutely stimulated gene expression of six target genes (except for Cyp17a1 and Fgf21) with their maximum inductions occurring 6 hours after acute T3 (Figure 4, A, B, and D). Similar to our transcriptome analyses (Figure 2C), Dio1 and Idh3a were stimulated after acute and chronic T3 treatment (Figure 4A), whereas the other four genes were stimulated acutely but not chronically (Figure 4, B and D) and thus had become desensitized to T3. Cyp17a1 and Fgf21 were stimulated only after chronic T3 treatment (Figure 4C).
Figure 4.

Differential temporal expression patterns of individual positively regulated hepatic target genes after T3 treatment and its withdrawal. Adult male mice (8–10 wk old) were treated with 20 μg liothyronine (L-T3) per 100 g body weight for 14 days. Relative mRNA expression was determined at indicated T3-treatment periods after the first and the last L-T3 injection. Shown are the temporal expression patters of genes that were both acutely and chronically induced, Dio1 and Idh3a (A,); genes that were only acutely induced, Thrsp and Bcl3 (B); and genes that were only chronically induced (C), Cyp17a1 and Fgf21. D, Genes that were paradoxically up-regulated during T3 withdrawal: Fasn and Cpt1a. No treatment (blank); acute T3 treatment (black); chronic T3 treatment (hashed); early T3 withdrawal (hashed); and T3 withdrawal (striped) are shown. Note that the same data points were used for the chronic T3 treatment and early T3 withdrawal groups (hashed). Data are presented as means ± SEM (n = 4–5/group). *, P < .05, **, P < .01, ***, P < .001, compared with nontreated mice (0 d) by one-way ANOVA.
During the withdrawal period, we observed three distinct temporal expression patterns in positively regulated target genes. First, Dio1 mRNA expression closely followed serum T3 levels by increasing at 6 and 24 hours before dropping significantly below baseline 3 days after the last L-T3 and gradually returning to baseline after 10 days (Figure 4A). Second, Fasn and Cpt1a transcripts showed no significant changes 6 and 24 hours after the last L-T3 dose but paradoxically increased after 5–7 days and remained elevated after 10 days withdrawal despite normal serum T3 concentrations (Figures 1B and 4D). Third, the remaining five genes showed no significant changes during the T3 withdrawal period.
We also examined the temporal patterns after acute and chronic T3 treatment, and T3 withdrawal of several negatively regulated T3 target genes (18, 19): stearoyl-CoA desaturase 1 (Scd1), β-ureidopropionase (Upb1), sterol regulatory element binding transcription factor 1 (Srebf1, or Srebp1c), and choline dehydrogenase (Chdh) (Supplemental Figure 1). Regarding acute vs chronic T3 treatment, Scd1 and Upb1 transcripts were down-regulated acutely and chronically, whereas Srebf1 and Chdh transcripts were down-regulated only after chronic T3 treatment. After T3 withdrawal, Scd1 and Upb1 transcripts were returned to baseline levels by 3–5 days and remained there. In contrast, Srebf1 and Chdh mRNA were initially down-regulated at 6 and 24 hours before their expression returned to baseline and above at 5 and 7 days, respectively. Taken together, different positively and negatively regulated target genes exhibited distinct and variable temporal transcriptional patterns after acute and chronic T3 treatment and during T3 withdrawal.
Transcription of representative hepatic T3-regulated target genes in Mct8-knockout mice
From our foregoing analyses of the transcriptome and individual target genes, we observed that most of the well-characterized positive genes became refractory to T3 stimulation after chronic T3 treatment (Figure 4, B and D). To better understand this desensitization of gene expression in another physiological context, we analyzed T3 target gene expression in WT and Mct8-knockout mouse liver. Several previous studies on adult Mct8-knockout mice showed that T3 levels are elevated within the liver (13, 14, 21). Recently, high concentrations of T3 in the livers were detected, even at postnatal day 0, indicating that adult Mct8-knockout mice have chronic intrahepatic exposure to high T3 levels from birth (14). In agreement with our findings in the mice that underwent acute and chronic T3 treatment (Figure 4, B and D), there was no induction of hepatic Thrsp, Bcl3, Fasn, and Cpt1a mRNA expression in Mct8-knockout mice when compared with WT mice (Figure 5). However, significant increases in Dio1 and Cyp17a1 were observed in Mct8-knockout mice. In the T3-injected mouse model, Dio1 was induced by both acute and chronic T3 treatment, whereas Cyp17a1 was induced by only chronic T3 treatment. Idh3a and Fgf21 mRNA expression was induced by chronic T3 treatment (Figure 4, A and C); however, mRNA levels were similar between Mct8-knockout and WT mice (Figure 5). This desensitization of chronically stimulated target genes may be due to the lifetime exposure to increased intrahepatic TH that occurs in Mct8-knockout mice. Taken together, our results showed desensitization to T3 stimulation after chronic TH exposure in vivo in both Mct8-knockout mice and T3-injected mice.
Figure 5.
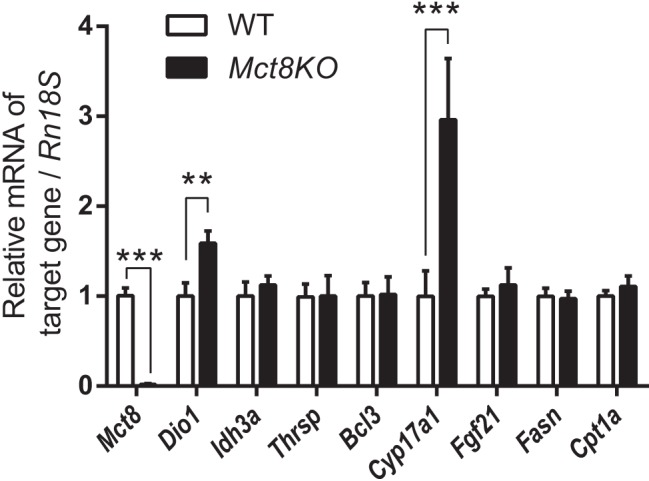
Differential hepatic gene expression due to chronically increased hepatocellular T3 concentration in MCT8 deficiency. Relative mRNA expression of Mct8 and representative positively regulated T3-target genes in WT and Mct8-knockout (Mct8KO) mice (10–12 wk old). Data are presented as means ± SEM (n = 12/group). **, P < .01, ***, P < .001, by two-tailed Student's t test.
Histone modification patterns after T3 treatment and its withdrawal
To determine whether there may be an epigenetic mechanism for this desensitization, we investigated the induction of histone modifications that were reported previously for T3-regulated target genes in Xenopus laevis and rat pituitary cells (22, 23). We initially performed ChIP-qPCR on liver samples from T3-treated mice with an antibody against histone H3 acetylated at lysines 9 and 14 (H3K9/K14ac). Rapid increases in H3 acetylation within the promoter region of Dio1, Thrsp, and Fasn occurred 6 hours after acute T3 treatment (Figure 6, A and B, and Supplemental Figure 2B). However, these increases in histone H3 acetylation returned to baseline levels by 24 hours and remained unchanged after chronic T3. We observed no significant changes in histone H4 acetylation between acute vs chronic T3 treatments when we performed ChIP-qPCR with an antibody against pan-acetylated histone H4 lysines (Pan-H4ac; data not shown).
Figure 6.

Distinct temporal patterns of histone modifications induced by T3 on Dio1, Thrsp, and Cyp17a1 gene promoters. Adult male mice (8–10 wk old) were treated with 20 μg liothyronine (L-T3) per 100 g body weight for 14 days. A–C, Histone modifications in the promoters of these three target genes induced by T3. ChIP-qPCR analyses were performed with anti-H3K9/K14ac or anti-H3K4me3 at indicated T3-treatment periods after the first and the last L-T3 injections. No treatment (blank); acute T3 treatment (black); chronic T3 treatment (hashed); early T3 withdrawal (hashed); and T3 withdrawal (striped) are shown. Note that the same data points were used for the chronic T3 treatment and early T3 withdrawal groups (hashed). Parentheses represent primer location referring to the transcription start site of reference sequencing. Data are presented as means ± SEM (n = 4–5/group). *, P < .05, **, P < .01, ***, P < .001, compared with nontreated mice (0 d) by one-way ANOVA.
The Dio1 promoter showed a transient increase in histone H3 acetylation acutely but did not exhibit any H3 or H4 acetylation after chronic T3 despite a persistent increase in gene expression at 14 days (Figures 4A and 6A). To better understand the reason for this persistent sensitivity to T3 stimulation in Dio1, we examined histone H3 at lysine 4 (H3K4) methylation, an epigenetic modification that is associated with both T3-dependent and general transcription (24, 25). H3K4 trimethylation (H3K4me3) sites were shown previously to interact with the PHD finger regions of HATs such as p300 and cAMP response element-binding protein, to promote HAT activity and transcription at certain target genes (26). In addition, the methylation of histones is thought to be a stable epigenetic mark, making it an attractive candidate as a mechanism for long-term transcriptional activation (25). As shown in Figure 6A (right panel), both acute and chronic T3 increased H3K4me3 on Dio1 promoter; in contrast, no changes in H3K4me3 were observed on the Thrsp and Fasn promoters (Figure 6B and Supplemental Figure 2B). This increase in histone methylation correlated with the sustained increase in Dio1 mRNA levels observed during acute and chronic T3 (Figure 4A). Furthermore, chronic T3, but not acute T3, treatment induced significant increases in H3K4me3 on Cyp17a1 and Fgf21 promoters, genes that were stimulated only chronically (Figure 6C and Supplemental Figure 2C).
We also analyzed histone modifications on Idh3a as another representative gene that showed persistent transactivation (Figure 4A); however, neither H3K9/K14ac nor H3K4me3 showed any significant changes (Supplemental Figure 2A). Although we studied only a limited number of target genes, our data suggest that H3 acetylation may play a role in acute T3 stimulation, whereas H3K4me3 may be associated with chronic T3 stimulation. However, it is possible that these histone modifications may not occur in all target genes (eg, Idh3a) because changes in histone markers still may be gene or gene class specific, given the relatively few genes that we have characterized so far.
Discussion
In this report, we studied global and individual hepatic gene expression patterns as well as possible epigenetic modifications after acute and chronic T3 treatment and withdrawal from sustained exposure to T3. Surprisingly, analyses of gene expression patterns showed that most acutely regulated target genes became insensitive to T3 after chronic T3 treatment, whereas other target genes that were initially unresponsive later became sensitive to T3. Only a small number of genes were regulated both acutely and chronically. Most previous gene expression profiles in cell culture and in vivo models examined only the short-term effects of T3 on target gene expression, although studies of individual target genes have demonstrated variable kinetics of T3 induction (8, 27). Of note, Flores-Morales et al (10) reported differences in global hepatic gene expression patterns between acute and chronic T3 treatment in hyperthyroid mice because they identified early, late, and sustained patterns of transcriptional responses after acute and/or chronic TH treatment. However, their TH regimens and harvest times after TH treatment were different for the acute TH treatment group (2 h after the single injection of combined T3 and T4) and the chronic TH treatment group (24 h after the last injection of only T3). These differences in TH regimens and harvest times could potentially affect mRNA expression levels because the serum half-life of T3 in mice is approximately 4 hours (17); thus, mice from the two groups might be exposed to different serum TH concentrations. In our studies, we measured mRNA expression at the same time points after T3 injections in both the acute and chronic T3 treatment groups. This protocol enabled us to compare mRNA expression levels accurately and determine temporal patterns due to acute vs chronic T3 under the same treatment and harvest conditions. Moreover, we also were able to compare directly the sensitivity of hepatic target genes to T3 between 6 and 24 hours after the first T3 dose with 6 and 24 hours after the last dose at 14 days.
Recent studies have raised the possibility that TH can indirectly regulate transcription of target genes by primary induction of transcription factors that regulate other genes. For example, Ohguchi et al (28) proposed that T3 induction of Krüppel-like transcription factor 9 (KLF9) cooperates with hepatocyte nuclear factor 4α (HNF4A) and GATA binding protein 4 (GATA4) to stimulate Dio1 gene expression in mouse hepatocytes. Although Klf9, Hnf4a, or Gata4 was not among the 320 genes induced acutely by T3 in the present study, several other transcriptional factors, such as D site albumin promoter binding protein (Dbp), Bcl3, or peroxisome proliferative activated receptor γ, coactivator 1β (Ppargc1b) were identified as acutely induced genes (Figure 2C). Thus, it is possible that transcriptional factors that are acutely induced by T3 could be involved in the secondary induction of some target genes after chronic T3 treatment.
It has been reported that TH regulates FGF21 expression (29) and that exogenous FGF21 administration stimulates some hepatic T3-regulated target genes (30). Additionally, a small subset of genes that were commonly regulated by TH and FGF21 had diminished response to T3 in the Fgf21-knockout mice (31). Of note, some of those genes overlapped with our set of chronically induced genes, including Cyp17a1, Dio1, ecto-5′-nucleotidase (Nt5e), and (Kcnk1) (Figures 2C, and 4, A and C). Accordingly, these findings raise the possibility that FGF21 might partially contribute to the delayed induction in subset of T3-target genes. Dio1 transcription is currently thought to be the most sensitive indicator of thyroid status in the mouse liver (17). We also observed both acute and chronic induction of Dio1 mRNA expression by T3 (Figure 4A). Ramadoss et al (18) recently identified two TR binding sites in the first intron of the Dio1 gene by using ChIP-sequencing technology. It is possible that liganded TR can stimulate acute Dio1 mRNA expression, whereas it may require coregulation with other factors such as KLF9 and FGF21 for chronic Dio1 mRNA expression. Consistent with different transcriptional mechanisms for acute and chronic regulation of Dio1 gene expression by T3, we observed H3 acetylation only during acute T3 treatment and trimethylation of H3K4 during both acute and chronic T3 treatment (Figure 6, B and D).
In contrast to the well-characterized mechanism(s) for positive regulation, little is known about the molecular mechanism(s) for negative transcriptional regulation by TH (5–7). After the identification of TREs involved in positive regulation, negative TREs have long been postulated (32). However, a consensus sequence for negative TREs has remained elusive and controversial, despite the localization of inhibitory regions in the promoters of a large number of negatively regulated target genes. Other possible mechanisms for negative regulation that have been proposed include negative coregulators (33), microRNA-mediated negative regulation (34), DNA-independent repression via protein-protein interactions (tethering mechanism) (35), and diminished TR recruitment to TREs in the presence of T3 (18). Interestingly, our microarray analysis of negatively regulated genes revealed similar findings as those observed in positively regulated target genes (Figure 2B and Supplemental Figure 2). In particular, transcriptional sensitivity varied according to three major patterns (acute, chronic, or both), and most genes that were acutely regulated by T3 became desensitized after chronic T3 treatment. Therefore, similar to positively regulated target genes, it is likely that chronic negative transcriptional regulation by TH may involve complex processes other than just TR binding to TREs.
Our microarray studies identified differing patterns of recovery because 23 of 320 of acutely induced genes (7.2%) did not return to baseline expression levels despite renormalization of serum TH and TSH levels (Figure 3A). Of note, subsequent analyses of individual target genes revealed paradoxical increases in Fasn and Cpt1a mRNA during T3 withdrawal (Figure 4D). These findings resembled the overshoot of TSH during recovery that was observed 5 days after T3 withdrawal (Figure 1D). Thus, it is possible that these genes may be more sensitive to T3 during the transition from hypothyroidism to euthyroidism because the mice were slightly hypothyroid 3 days after T3 withdrawal before they were able to return to baseline TH/TSH levels when normal pituitary and thyroid function resumed (Figure 1B). Indeed, our previous microarray analyses identified a subset of hepatic target genes, including Fasn, that were activated preferentially by T3 during the transition from the hypothyroid to euthyroid states (11). In support of this notion, tissue levels of T3 have been shown to decrease disproportionately when compared with serum T3 levels under certain conditions (17). Alternatively, it is possible that elevated TSH during T3 withdrawal may have stimulated Fasn and Cpt1a transcripts because TSH reportedly induced liver triglyceride accumulation via increased expression of lipogenic genes (36). This same group also described a direct effect of TSH on hepatic bile acid homeostasis by repressing Cyp7a1 expression in a manner that was independent of serum TH levels (37).
Although our analysis of chromatin modifications only characterized six representative positive genes, it demonstrated an association between transcriptional response by acute T3 treatment with H3K9/K14ac for Thrsp and Fasn (Figure 6B and Supplemental Figure 2B) and chronic T3 exposure with H3K4me3 for Cyp17a1 and Fgf21 (Figure 6C and Supplemental Figure 2C). We also did not observe any persistent histone modifications in genes that returned back to baseline levels 10 days after T3 withdrawal. On the other hand, no significant changes in histone modifications were noted on Fasn promoter (Supplemental Figure 2B) despite its failure to return to baseline gene expression level 10 days after withdrawal. It is likely that other histone modifications may be playing a predominant role during recovery for some genes such as Fasn. In further support that histone modifications appear to play the major role in regulating acute and chronic transcriptional response to T3 and recovery from chronic hyperthyroidism, DNA methylome analyses during these conditions did not show any significant changes in the methylation status of more than 485 000 methylation sites across the entire genome during acute and chronic treatment and recovery (data not shown). In this connection, Kasai et al (38) also did not identify significant changes in the methylation status on the promoter region of thrb gene in a X laevis cell line or in tadpole tissues after T3 treatment.
To the best of our knowledge, this is the first study to evaluate epigenetic modifications after acute vs chronic T3 treatments and withdrawal after chronic T3 treatment. Importantly, our ChIP-qPCR analyses suggest that increased H3K9/K14ac and H3K4me3 may be important for transactivation during acute and chronic T3 treatments, respectively. In a similar manner, for a negatively regulated target gene, chronic T3 treatment in a rat pituitary cell line caused sustained repression of TRH gene promoter activity that was associated with a prolonged decrease in H3K4me3 and only a transient reduction in H3K9/K14ac (23). Additionally, the H3K4 methyltransferase, SET7, has been shown to increase H3K4 methylation and estrogen receptor α methylation to activate transcription (39). In addition to changes in the level of histone modifying enzymes, it also is possible that decreased expression of TRs and/or coactivators or increased expression of corepressors could contribute to the changes in histone acetylation and methylation of desensitized target genes. The ubiquitin proteasome pathway plays a significant role in the down-regulation of liganded glucocorticoid, TH, and progesterone receptors (40–42) as well as the degradation of coactivators (43). Taken together, our data strongly point to the notion that histone modifications contribute to persistent sensitivity and desensitization of target genes during chronic T3 exposure.
Desensitization due to chronic hormone exposure has been established for several membrane receptors including the adrenergic and insulin receptors (44–46); however, little is known about this phenomenon in nuclear hormone receptors. Recently Jeong et al (47) reported differential temporal responses to progesterone that were gene specific. Similar to our findings, they identified early, late, and sustained patterns of transcriptional responses by uterine genes after acute and chronic progesterone treatment. They also found that most acutely regulated genes became desensitized after chronic progesterone treatment. Thus, it is likely that other nuclear receptors may display desensitization after chronic exposure to their cognate hormone or agonists. We do not know the precise mechanism for the desensitization to chronic T3; however, decreased TR expression (perhaps due to proteasomal degradation) and/or changes in the amount or type of coregulators may contribute to changes in chromatin modifications in target genes after acute and chronic T3 treatment and explain the desensitization after chronic T3 observed in our study. Recently Vella et al (48) proposed that the balance of corepressors and coactivators, especially nuclear receptor corepressor 1 and nuclear coactivator 1 (or steroid receptor coactivator-1), may determine sensitivity to T3 in the liver. Better understanding of these effects in chronic hormone exposure and withdrawal may lead to novel strategies to increase hormone sensitivity of specific genes in conditions of hormone resistance such as aging or to decrease their hormone sensitivity in pathological states such as hormone-dependent cancers.
In conclusion, our data demonstrated that hepatic target genes showed differential gene-specific transcriptional patterns during the acute and chronic phases of T3 treatment and its withdrawal period. In particular, 251 of 320 of acutely regulated positive target genes (78.4%) became desensitized after chronic T3 treatment despite elevated serum T3 levels. Additionally 67 of 680 acutely and chronically induced target genes (9.9%) demonstrated incomplete recovery after T3 withdrawal. These findings suggest that, although circulating TH and TSH levels reflect the integrity and negative feedback of the hypothalamic/pituitary/thyroid axis, they may not portray accurately the T3 responsiveness of specific tissues and their genes during chronic hyperthyroidism as well as the transition from hyperthyroidism to euthyroidism. These findings also may help explain the persistence of metabolic and physiological abnormalities observed clinically in some patients despite renormalization of serum thyroid function tests after therapy (1).
Acknowledgments
We thank Ms Vania Lim, Mr Patrick Lee, Ms Dawn Koh, Dr Madhulika Tripathi, Dr Jin Zhou, Dr Winifred Yau, Mr Sherwin Xie, Ms Andrea Lim, and Dr Benjamin Farah for technical assistance.
This work was supported by Singapore National Medical Research Council Clinician Scientist Award and Clinician Scientist Individual Research Grant 12May0004 (to P.M.Y.), by an Endocrine and Metabolic Society of Singapore grant (to M.K.-S.L.), by the A*STAR intramural funding for the Neuroepigenetics Laboratory under the Integrative Neuroscience Programme, Singapore Institute for Clinical Sciences (to J.C.G.S.), and by Grant R37-DK15070 from the National Institutes of Health (to S.R.).
Disclosure Summary: The authors have nothing to disclose.
For News & Views see page 1326
- ChIP
- chromatin immunoprecipitation
- FGF21
- fibroblast growth factor 21
- GATA4
- GATA binding protein 4
- HAT
- histone acetyltransferase
- H3K9/K14ac
- histone H3 acetylated at lysines 9 and 14
- H3K4me3
- histone H3 trimethylated at lysine 4
- KLF9
- Krüppel-like transcription factor 9
- MCT8
- monocarboxylate transporter 8
- Pan-H4ac
- histone H4 pan-acetylated at lysines 5, 8, 12, and 16
- qPCR
- quantitative PCR
- RT-qPCR
- reverse transcription-quantitative PCR
- TH
- thyroid hormone
- TR
- TH receptor
- TRE
- TH response element
- WT
- wild type.
References
- 1. Fahrenfort JJ, Wilterdink AM, van der Veen EA. Long-term residual complaints and psychosocial sequelae after remission of hyperthyroidism. Psychoneuroendocrinology. 2000;25:201–211. [DOI] [PubMed] [Google Scholar]
- 2. Piatnek-Leunissen DA, Leunissen RL. Liver mitochondrial function in acute vs. chronic hyperthyroidism. Endocrinology. 1969;84:456–461. [DOI] [PubMed] [Google Scholar]
- 3. Varas SM, Oliveros LB, Gimenez MS. Lipids in rat liver submitted to acute and chronic hyperthyroidism. Horm Metab Res. 1999;31:514–518. [DOI] [PubMed] [Google Scholar]
- 4. Sarne DH, Refetoff S, Rosenfield RL, Farriaux JP. Sex hormone-binding globulin in the diagnosis of peripheral tissue resistance to thyroid hormone: the value of changes after short term triiodothyronine administration. J Clin Endocrinol Metab. 1988;66:740–746. [DOI] [PubMed] [Google Scholar]
- 5. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122:3035–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sinha R, Yen PM. Cellular Action of Thyroid Hormone. In: De Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, Koch C, McLachlan R, New M, Rebar R, Singer F, Vinik A, Weickert MO, eds. Endotext. South Dartmouth, MA: MD Text.com, Inc.; 2014. [Google Scholar]
- 8. Feng X, Jiang Y, Meltzer P, Yen PM. Thyroid hormone regulation of hepatic genes in vivo detected by complementary DNA microarray. Mol Endocrinol. 2000;14:947–955. [DOI] [PubMed] [Google Scholar]
- 9. Miller LD, Park KS, Guo QM, et al. Silencing of Wnt signaling and activation of multiple metabolic pathways in response to thyroid hormone-stimulated cell proliferation. Mol Cell Biol. 2001;21:6626–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flores-Morales A, Gullberg H, Fernandez L, et al. Patterns of liver gene expression governed by TRβ. Mol Endocrinol. 2002;16:1257–1268. [DOI] [PubMed] [Google Scholar]
- 11. Yen PM, Feng X, Flamant F, et al. Effects of ligand and thyroid hormone receptor isoforms on hepatic gene expression profiles of thyroid hormone receptor knockout mice. EMBO Rep. 2003;4:581–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yuan C, Lin JZ, Sieglaff DH, et al. Identical gene regulation patterns of T3 and selective thyroid hormone receptor modulator GC-1. Endocrinology. 2012;153:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferrara AM, Liao XH, Gil-Ibanez P, et al. Changes in thyroid status during perinatal development of MCT8-deficient male mice. Endocrinology. 2013;154:2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006;147:4036–4043. [DOI] [PubMed] [Google Scholar]
- 15. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 17. Bianco AC, Anderson G, Forrest D, et al. American Thyroid Association guide to investigating thyroid hormone economy and action in rodent and cell models. Thyroid. 2014;24:88–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ramadoss P, Abraham BJ, Tsai L, et al. Novel mechanism of positive versus negative regulation by thyroid hormone receptor β1 (TRβ1) identified by genome-wide profiling of binding sites in mouse liver. J Biol Chem. 2014;289:1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sinha RA, Singh BK, Yen PM. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol Metab. 2014;25:538–545. [DOI] [PubMed] [Google Scholar]
- 20. Park E, Kim Y, Lee HJ, Lee K. Differential regulation of steroidogenic enzyme genes by TRα signaling in testicular Leydig cells. Mol Endocrinol. 2014;28:822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trajkovic M, Visser TJ, Mittag J, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grimaldi A, Buisine N, Miller T, Shi YB, Sachs LM. Mechanisms of thyroid hormone receptor action during development: lessons from amphibian studies. Biochim Biophys Acta. 2013;1830:3882–3892. [DOI] [PubMed] [Google Scholar]
- 23. Umezawa R, Yamada M, Horiguchi K, et al. Aberrant histone modifications at the thyrotropin-releasing hormone gene in resistance to thyroid hormone: analysis of F455S mutant thyroid hormone receptor. Endocrinology. 2009;150:3425–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. [DOI] [PubMed] [Google Scholar]
- 25. Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taverna SD, Ilin S, Rogers RS, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell. 2006;24:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S. Thyroid hormone action on liver, heart, and energy expenditure in thyroid hormone receptor β-deficient mice. Endocrinology. 1998;139:4945–4952. [DOI] [PubMed] [Google Scholar]
- 28. Ohguchi H, Tanaka T, Uchida A, et al. Hepatocyte nuclear factor 4α contributes to thyroid hormone homeostasis by cooperatively regulating the type 1 iodothyronine deiodinase gene with GATA4 and Kruppel-like transcription factor 9. Mol Cell Biol. 2008;28:3917–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Adams AC, Astapova I, Fisher FM, et al. Thyroid hormone regulates hepatic expression of fibroblast growth factor 21 in a PPARα-dependent manner. J Biol Chem. 2010;285:14078–14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Domouzoglou EM, Fisher FM, Astapova I, et al. Fibroblast growth factor 21 and thyroid hormone show mutual regulatory dependency but have independent actions in vivo. Endocrinology. 2014;155:2031–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang A, Sieglaff DH, York JP, et al. Thyroid hormone receptor regulates most genes independently of fibroblast growth factor 21 in liver. J Endocrinol. 2015;224:289–301. [DOI] [PubMed] [Google Scholar]
- 32. Chin WW, Carr FE, Burnside J, Darling DS. Thyroid hormone regulation of thyrotropin gene expression. Recent Prog Horm Res. 1993;48:393–414. [DOI] [PubMed] [Google Scholar]
- 33. Treuter E, Albrektsen T, Johansson L, Leers J, Gustafsson JA. A regulatory role for RIP140 in nuclear receptor activation. Mol Endocrinol. 1998;12:864–881. [DOI] [PubMed] [Google Scholar]
- 34. Yap CS, Sinha RA, Ota S, Katsuki M, Yen PM. Thyroid hormone negatively regulates CDX2 and SOAT2 mRNA expression via induction of miRNA-181d in hepatic cells. Biochem Biophys Res Commun. 2013;440:635–639. [DOI] [PubMed] [Google Scholar]
- 35. Iwaki H, Sasaki S, Matsushita A, et al. Essential role of TEA domain transcription factors in the negative regulation of the MYH 7 gene by thyroid hormone and its receptors. PLoS One. 2014;9:e88610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan F, Wang Q, Lu M, et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J Hepatol. 2014;61:1358–1364. [DOI] [PubMed] [Google Scholar]
- 37. Song Y, Xu C, Shao S, et al. Thyroid-stimulating hormone regulates hepatic bile acid homeostasis via SREBP-2/HNF-4α/CYP7A1 axis. J Hepatol. 2015;62:1171–1179. [DOI] [PubMed] [Google Scholar]
- 38. Kasai K, Nishiyama N, Izumi Y, Otsuka S, Ishihara A, Yamauchi K. Exposure to 3,3′,5-triiodothyronine affects histone and RNA polymerase II modifications, but not DNA methylation status, in the regulatory region of the Xenopus laevis thyroid hormone receptor βA gene. Biochem Biophys Res Commun. 2015;467:33–38. [DOI] [PubMed] [Google Scholar]
- 39. Zhou Q, Shaw PG, Davidson NE. Epigenetics meets estrogen receptor: regulation of estrogen receptor by direct lysine methylation. Endocr Relat Cancer. 2009;16:319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okret S, Poellinger L, Dong Y, Gustafsson JA. Down-regulation of glucocorticoid receptor mRNA by glucocorticoid hormones and recognition by the receptor of a specific binding sequence within a receptor cDNA clone. Proc Natl Acad Sci USA. 1986;83:5899–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dace A, Zhao L, Park KS, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proc Natl Acad Sci USA. 2000;97:8985–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ismail A, Nawaz Z. Nuclear hormone receptor degradation and gene transcription: an update. IUBMB Life. 2005;57:483–490. [DOI] [PubMed] [Google Scholar]
- 44. Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci. 2004;25:413–422. [DOI] [PubMed] [Google Scholar]
- 45. Kasuga M, Kahn CR, Hedo JA, Van Obberghen E, Yamada KM. Insulin-induced receptor loss in cultured human lymphocytes is due to accelerated receptor degradation. Proc Natl Acad Sci USA. 1981;78:6917–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tozzo E, Desbuquois B. Effects of STZ-induced diabetes and fasting on insulin receptor mRNA expression and insulin receptor gene transcription in rat liver. Diabetes. 1992;41:1609–1616. [DOI] [PubMed] [Google Scholar]
- 47. Jeong JW, Lee KY, Kwak I, et al. Identification of murine uterine genes regulated in a ligand-dependent manner by the progesterone receptor. Endocrinology. 2005;146:3490–3505. [DOI] [PubMed] [Google Scholar]
- 48. Vella KR, Ramadoss P, Costa ESRH, et al. Thyroid hormone signaling in vivo requires a balance between coactivators and corepressors. Mol Cell Biol. 2014;34:1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]