

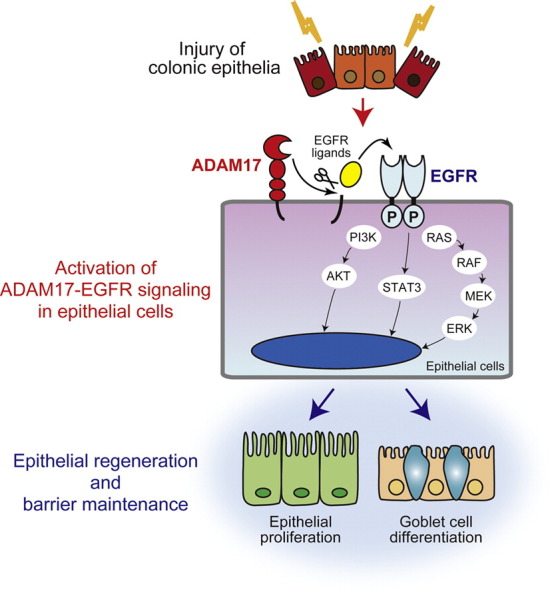
Abstract
Epithelial regeneration is a key process for the recovery from ulcerative colitis (UC). Here we demonstrate that a disintegrin and metalloproteinase-17 (ADAM17), a main sheddase for tumor necrosis factor (TNF)-α, is essential for defensive epithelial properties against UC by promoting epithelial cell growth and goblet cell differentiation in mouse and human. Mice with systemic deletion of Adam17 developed severe dextran sulfate sodium-induced colitis when compared to mice with myeloid cell Adam17 deletion or control littermates. ADAM17 was predominantly expressed by regenerating epithelia in control mice, and its loss or inhibition attenuated epidermal growth factor receptor (EGFR) activation, epithelial proliferation, mucus production and barrier functions. Conversely, ectopic EGFR stimulation promoted epithelial regeneration thereby partially rescuing the severe colitis caused by ADAM17 deficiency. In UC patients, epithelial ADAM17 expression positively correlated with both cell proliferation and goblet cell number. These findings suggest that maintaining ADAM17–EGFR epithelial signaling is necessary for the recovery from UC and would be beneficial to therapeutic strategies targeting ADAM17-mediated TNF-α shedding.
Abbreviations: UC, ulcerative colitis; ADAM, a disintegrin and metalloproteinase; TNF, tumor necrosis factor; EGFR, epidermal growth factor receptor; MMP, matrix metalloproteinase; EGF, epidermal growth factor; TACE, tumor necrosis factor-α converting enzyme; IBD, inflammatory bowel disease; DSS, dextran sulfate sodium; TGF, transforming growth factor; pIpC, polyinosinic–polycytidylic acid; BrdU, bromodeoxyuridine; PCNA, proliferation cell nuclear antigen; pEGFR, phosphorylated EGFR; RT-qPCR, real-time quantitative PCR; MAPK, mitogen activated protein kinase; PI3K, phosphatidylinositol 3-kinase; STAT3, signal transducer and activator of transcription 3; TGM, transglutaminase
Keywords: A disintegrin and metalloproteinase 17 (ADAM17), Ulcerative colitis, Epidermal growth factor receptor (EGFR), Goblet cell, Epithelial barrier
Graphical Abstract
Highlights
-
•
Mice with systemic deletion of ADAM17, but not with its myeloid cell-specific deficiency, are more sensitive to colitis.
-
•
ADAM17-EGFR axis promotes repair processes through epithelial cell proliferation and goblet cell differentiation.
-
•
Epithelial ADAM17 expression correlates with cell growth and mucus production in ulcerative colitis patients.
Epithelial regeneration is a key process for the recovery from ulcerative colitis (UC). We now demonstrate that a disintegrin and metalloproteinase-17 (ADAM17) is essential for defensive epithelial properties against UC by driving repair processes in mouse and human. During colonic inflammation, ADAM17 is up-regulated in regenerating epithelia, and its loss or inhibition attenuated epidermal growth factor receptor (EGFR) activation, epithelial proliferation, mucus production and barrier functions. These findings suggest that maintaining ADAM17–EGFR epithelial signaling is necessary for the recovery from UC and would be beneficial to therapeutic strategies targeting ADAM17-mediated tumor necrosis factor-α shedding.
1. Introduction
ADAMs (a disintegrin and metalloproteinases) are multifunctional proteins involved in ectodomain shedding of transmembrane proteins and thereby regulate cell adhesion, migration, and cell–cell communication. Ectodomain shedding, which is characterized by proteolytic release of extracellular domains of membrane-bound proteins, is a crucial post-translational regulator for the function and availability of membrane-bound proteins. The human genome contains 25 ADAMs including four pseudogenes, and of the 21 ADAM proteins there are 13 proteolytic and 8 nonproteolytic ADAMs (Mochizuki and Okada, 2007, Blobel, 2005, Edwards et al., 2008). Proteolytic ADAMs share the metalloproteinase domain of matrix metalloproteinases (MMPs), and a typical proteolytic ADAM protein is comprised of propeptide, metalloproteinase, disintegrin-like, cysteine-rich, epidermal growth factor (EGF)-like, transmembrane, and cytoplasmic domains (Mochizuki and Okada, 2007, Blobel, 2005, Edwards et al., 2008). Previous studies have shown that several proteolytic ADAM species contribute to inflammatory diseases and cancers by shedding pro-inflammatory cytokines, growth factors and their receptors in addition to the degradation of extracellular proteins including extracellular matrix (ECM) components (Shimoda et al., 2007, Shimoda et al., 2014, Murphy, 2008).
ADAM17, also called tumor necrosis factor (TNF)-α converting enzyme (TACE), was originally identified as a proteinase responsible for the ectodomain shedding of the membrane-bound form of TNF-α (Black et al., 1997). Subsequent studies showed that ADAM17 is also involved in the processing of various membrane-bound molecules including EGF receptor (EGFR) ligands, CD44, Kit ligand and L-selectin (Blobel, 2005). This ectodomain shedding event regulates key functions of these molecules that impact immune responses and cancer development in vivo (Scheller et al., 2011). However, the early lethality of Adam17-deficient mice has hampered the analysis of ADAM17 functions in postnatal development, adult homeostasis and disease (Peschon et al., 1998). To circumvent this issue, we generated conditional Adam17-deficient mice, which exhibit no phenotype in adult animals, enabling us to analyze the roles of ADAM17 in various disease models (Horiuchi et al., 2007, La Marca et al., 2011).
Ulcerative colitis (UC) is an intractable inflammatory bowel disease (IBD), showing diffuse mucosal inflammation that extends proximally from the rectum. Onset of UC typically occurs in the second and third decades of life and the majority of affected individuals deteriorate to chronic disease (Farmer et al., 1993, Ordas et al., 2012). Many factors affecting innate and adaptive immunity and epithelial barrier, which include genetic predisposition, eating habits and changes in intestinal flora, may contribute to the complexity of UC, but the underlying mechanisms are still poorly understood (Ananthakrishnan, 2015). Metalloproteinases including ADAMs are produced by many different cell types and regulate intestinal immune responses as well as wound healing (Khokha et al., 2013, Shimoda and Khokha, 2013). Actually, mucosal ADAM17 activity is reported to increase in UC patients (Brynskov et al., 2002). Transient upregulation of ADAM17 activity is implicated in promotion of neutrophil transepithelial migration and high colitis activity (Cesaro et al., 2009) and ADAM17-dependent shedding of TNF-α from intestinal epithelial cells initiates a pro-inflammatory state and mucosal atrophy in a mouse model of total parenteral nutrition (Feng et al., 2015), whereas mice with reduced ADAM17 levels were shown to exhibit increased sensitivity to colitis (Chalaris et al., 2010, Brandl et al., 2010). Therefore, the functions of ADAM17 and the origin of ADAM17-producing cells in UC remain unclear. Previous studies have shown that anti-TNF-α therapy is useful for human inflammatory diseases with excess TNF-α production (Abraham and Cho, 2009), but at present, most of emerging selective ADAM17 inhibitors show various side-effects and are limited to phase I and II trials (Duffy et al., 2011). It is still an open question whether ADAM17 is a promising target for UC treatments.
In this study, we examined the role of ADAM17 in colitis by developing dextran sulfate sodium (DSS)-induced colitis using two different conditional Adam17-deficient mice, i.e. mice with systemic deletion by the inducible Mx1-Cre gene of Adam17 (Adam17flox/floxMx1-Cre+) or those with its deletion in myeloid cells by the lysozyme M promoter (Adam17flox/floxLysM-Cre+). We also studied the significance of the ADAM17-EGFR pathway in cell proliferation and goblet cell maintenance by utilizing colonic epithelial cell lines, and finally investigated the relevance of our findings to human UC tissues. Our data demonstrate a protective function of epithelial ADAM17 as a gatekeeper molecule against intestinal inflammation.
2. Materials and Methods
2.1. Study Approval
All animal experiments were conducted following ARRIVE standard in accordance with protocols approved by the Institutional Animal Care and Use Committee of the Keio University School of Medicine (Protocol No. 071120, No. 10236-2). For experiments using human samples, informed consent for the study was obtained from the patients in accordance with the Declaration of Helsinki and IRB approval of the Keio University School of Medicine (Protocol No. 2012-377-2).
2.2. Generation of Conditional ADAM17-Deficient Mice
The following mouse strains were used: Adam17flox/flox, Mx1-Cre and LysM-Cre mice (Horiuchi et al., 2007). Adamflox/flox mice and Adamflox/floxMx1-Cre knock-in mice were generated as previously described (Horiuchi et al., 2007, Kuhn et al., 1995). To allow temporal systemic deletion of ADAM17, six-week-old Adam17flox/floxMx1-Cre mice were injected i.p. with 250 μg of pIpC (polyinosinic–polycytidylic acid) (Sigma-Aldrich) three times at 2-day intervals (Horiuchi et al., 2007). Recombination of the Adam17 gene in Adam17flox/floxMx1-Cre mice was confirmed by PCR of genomic DNAs isolated from colon and liver tissues (Horiuchi et al., 2007). As for a control, Adam17flox/flox mice were treated with pIpC three times. Adamflox/floxLysM-Cre mice, which delete the ADAM17 gene in myeloid cells, were also generated as described previously (Horiuchi et al., 2007, Clausen et al., 1999). All the mice were maintained under a 12 hour light–dark cycle with ad libitum access to regular food and water in a specific-pathogen free environment. In all experiments with Adam17flox/floxCre mice, age- and sex-matched littermates were served as controls.
2.3. DSS-Induced-Colitis
Age- and sex-matched male mice (8-week-old) were administered with 3% DSS (molecular weight: 36,000–50,000 Da; MP Biomedicals) in their drinking water for 8 days and thereafter they were provided with regular water for 4 days (Wirtz et al., 2007). They were observed and weighed every day for determination of percent weight, which was calculated as follows: (weight at day X / weight at day 0) × 100. For histological and gene expression analyses, the mice were sacrificed at days 0, 4, 8 or 12 after the initiation of DSS treatment. For recovery experiments, recombinant transforming growth factor (TGF)-α (8 μg per injection) or PBS was intraperitoneally injected into the mice at days 0, 2, 4 and 6 after DSS administration.
Protocols for all other procedures are provided in the Supplementary Materials and Methods.
3. Results
3.1. Systemic Deletion of ADAM17 Develops Severe Inflammation in Response to DSS
To investigate the effects of ADAM17 on the pathogenesis of colitis, we used pIpC-treated Adam17flox/floxMx1-Cre+ mice, which show systemic deletion of Adam17 (Horiuchi et al., 2007). The mice exhibited a normal phenotype without any evident histological defects in the colon mucosa or spontaneous colitis up to 6 weeks after pIpC injection (data not shown). DSS-induced colitis, which is a rapid and reproducible model of colitis and mimics human UC, was developed by the administration of 3% DSS in drinking water for 8 days to age- and sex-matched Adam17flox/floxMx1-Cre+ mice and their control littermates, followed by 4 days of regular drinking water. Both Adam17flox/floxMx1-Cre+ mice and control mice developed signs of colitis 4 days after DSS administration. However, colitis was significantly worse in Adam17flox/floxMx1-Cre+ mice, as evidenced by severe weight loss (p < 0.05 and p < 0.01 at days 7, 8, 9 and 10) and a high mortality rate (p < 0.05 at day 12) compared to controls (Fig. 1a). Adam17flox/floxMx1-Cre+ mice showed significantly shorter colons than controls after treatment (p < 0.05 and p < 0.01 at days 4 and 8) (Fig. 1b). Microscopically, ulcer lesions appeared to increase in Adam17flox/floxMx1-Cre+ mice compared to control mice (Fig. 1c). According to the methods by Egger et al. (1997), we determined the histological scores (Supplementary Fig. 1), and found that mean areas of Grade III lesion and Crypt damage scores at days 8 and 12 are significantly increased in Adam17flox/floxMx1-Cre+ mice compared to control littermates (Grade III lesion, p < 0.05 and p < 0.01; Crypt damage score, p < 0.01) (Fig. 1d and e). These data indicate a protective role of ADAM17 against DSS-induced colitis.
Fig. 1.

Systemic deletion of Adam17 but not its deletion in myeloid cells develops severe inflammation in response to DSS. a. Percent weight change (left panel) and survival (right panel) of 8-week-old control (Control) (n = 10 mice) and Adam17flox/floxMx1-Cre+ (ΔAdam17_Mx1) mice (n = 10 mice) after DSS administration. Bars, mean ± s.d.; *, p < 0.05; **, p < 0.01. b. Macroscopic images of representative colons from control and ΔAdam17_Mx1 mice at day 8 after DSS challenge (left panel). Scale bar, 1 cm. Right panel shows the ratio of length/initial weight of the colon from these mice at days 0, 4, 8 and 12. Bars, mean ± s.d.; n = 3–5 colon tissues; **, p < 0.01. c. Representative HE-stained colon sections from control (left panels) and ΔAdam17_Mx1 mice (right panels). Upper panels, whole colon sections from ascending colon (A) to rectum (R); lower panels, representative high-power view. Ulcer lesions are indicated by red lines. Scale bars, 50 μm. d. Grade III lesions in control and ΔAdam17_Mx1 mice at days 0, 4, 8 and 12 after DSS administration. Bars, mean ± s.d.; n = 3–6 colon tissues; *, p < 0.05; **, p < 0.01. e. Crypt damage score in control and ΔAdam17_Mx1 mice at days 0, 4, 8 and 12 after DSS administration. Bars, mean ± s.d.; n = 3–6 colon tissues; **, p < 0.01. f. Percent weight change (left panel) and survival (right panel) of 8-week-old control (Control) (n = 8 mice) and Adam17flox/floxLysM-Cre+ (ΔAdam17_LysM) mice (n = 9 mice) after DSS administration. Bars, mean ± s.d. g. Macroscopic images of representative colon tissues from control and ΔAdam17_LysM mice at day 8 of DSS challenge (left panel). Scale bar, 1 cm. Right panel shows the ratio of length/initial weight of the colon from these mice at days 0, 4, 8 and 12. Bars, mean ± s.d.; n = 3–5 colon tissues. h. Crypt damage score in control and ΔAdam17_LysM mice at days 0, 4, 8 and 12 after DSS administration. Bars, mean ± s.d.; n = 3–5 colon tissues. i. Infiltrating leukocytes in colon sections from control and Adam17flox/floxMx1-Cre+ (ΔAdam_Mx1) mice after DSS administration. Quantification of immunostained cells per high-power field is shown. Bars, mean ± s.d.; n = 3–6 colon tissues. Results between the two independent groups were determined by Student's t-test. For survival analysis, Kaplan–Meier test was used. P values smaller than 0.05 are indicated on respective plots.
3.2. Loss of Myeloid Cell-Derived ADAM17 Has No Effect on Development of Colitis
ADAM17 is known as the major TNF-α sheddase in myeloid cells (Horiuchi et al., 2007). We therefore examined the possible involvement of myeloid cell-derived ADAM17 in DSS-induced colitis by using Adam17flox/floxLysM-Cre+ mice, which lack ADAM17 in the myeloid compartment (Horiuchi et al., 2007). These mice showed no evident histological abnormalities in the colon (data not shown). Unexpectedly, DSS-induced colitis in Adam17flox/floxLysM-Cre+ mice was less severe compared to the colitis in Adam17flox/floxMx1-Cre+ mice, showing similar severity to control mice, based on the observations of weight loss and survival (Fig. 1f). Length of the colon and histology of the colitis at different time points were comparable between Adam17flox/floxLysM-Cre+ mice and control littermates (Fig. 1g and h). Immunohistochemical analyses of individual infiltrated leukocytes demonstrated no significant differences in the number of neutrophils, macrophages or T-lymphocytes at various time points post-DSS administration between Adam17flox/floxMx1-Cre+ and the control (Fig. 1i and Supplementary Fig. 2). These data indicate that myeloid cell-derived ADAM17 has no impact on DSS-induced colitis model.
3.3. ADAM17 is Expressed in Regenerating Epithelial Cells and Involved in Epithelial Barrier Functions
The pIpC-induced recombination in Adam17flox/floxMx1-Cre mice is known to occur in various organs with different efficiency, leading to almost complete recombination in the bone marrow, liver and spleen (Horiuchi et al., 2007). As shown in Fig. 2a, Adam17flox/floxMx1-Cre+ mice showed effective gene recombination in the distal colon after pIpC injection at a similar level to the liver, as indicated by detection of increased null gene and decreased floxed ADAM17. Protein expression of ADAM17 was negligible in colon tissues from untreated Adam17flox/floxMx1-Cre+ and control mice, but at day 8 after DSS administration, ADAM17 was expressed in control mice, but was still absent in Adam17flox/floxMx1-Cre+ colon tissue (Fig. 2b). Immunohistochemical analysis demonstrated that ADAM17 is expressed predominantly by epithelial cells of the DSS-induced inflamed colon in control mice, although negligible expression of ADAM17 was detected in the crypts of Adam17flox/floxMx1-Cre+ mice (Fig. 2c). Further analysis indicated that bromodeoxyuridine (BrdU)-positive or proliferation cell nuclear antigen (PCNA)-positive intestinal epithelial cells are decreased in Adam17flox/floxMx1-Cre+ mice compared to the control at day 8 after DSS administration (Fig. 2d, p < 0.05 each and Supplementary Fig. 3). In addition, the number of Alcian-Blue-positive goblet cells was significantly decreased in Adam17flox/floxMx1-Cre+ mice at day 8 compared to control mice (Fig. 2e, p < 0.01). Importantly, gastrointestinal permeability, as determined by serum FITC-dextran intensity 4 h after gavage, was significantly increased in Adam17flox/floxMx1-Cre+ mice at day 8 compared to control mice (Fig. 2f, p < 0.01). Therefore, both regeneration and barrier integrity are reduced in the absence of epithelial ADAM17.
Fig. 2.

Adam17flox/floxMx1-Cre+ mice exhibit decreases in epithelial cell proliferation, number of goblet cells and barrier function. a. Efficiency of Cre-induced Adam17 excision in the colon (distal, proximal and cecum) and liver evaluated by RT-PCR. b. Immunoblot of ADAM17 in the distal colon from control and Adam17flox/floxMx1-Cre+ (ΔAdam17_Mx1) mice at days 0 and 8 after DSS challenge. β-Actin was used as a loading control. c. Representative ADAM17-immunostained colon sections from control and ΔAdam17_Mx1 mice at days 0 and 8 after DSS challenge. The highlighted areas are magnified to the right. Scale bars, 100 μm. d. Quantification of number of BrdU-positive epithelial cells per gland and percentage of PCNA-positive epithelial cells is shown. Bars, mean ± s.d.; n = 3–4 colon tissues; *, p < 0.05. e. Images and quantification of colonic goblet cells in control and ΔAdam17_Mx1 mice. Representative Alcian-Blue-stained sections from control and ΔAdam17_Mx1 mice at days 0 and 8 after DSS challenge are indicated in the left panel. Scale bars, 100 μm. Quantification of number of Alcian-Blue-positive goblet cells per 100 epithelial cells is shown in the right panel. Bars, mean ± s.d.; n = 4 colon tissues; **, p < 0.01. f. In vivo permeability assay performed by measuring the translocation of FITC-dextran, given by gavage, into mouse serum after 4 h. Bars, mean ± s.d.; n = 4–6 mice; **, p < 0.01. Results between the two independent groups were determined by Student's t-test. P values smaller than 0.05 are indicated on respective plots.
3.4. Loss of ADAM17 Greatly Inhibits EGFR Activation and Mucus Production in Colonic Epithelia
Considering the decreased proliferation observed in the colon tissues of DSS-treated Adam17flox/floxMx1-Cre+ mice, we examined the downstream impact on EGFR signaling by measuring the expression of total and phosphorylated EGFR (pEGFR) in colon tissues from Adam17flox/floxMx1-Cre+ mice and their control littermates at day 0 (untreated) and day 8 after treatment with DSS. As shown in Fig. 3a, increased expression of pEGFR was detected in the distal colon tissues of littermate controls at day 8 after DSS challenge, but only weak or negligible expression was seen in Adam17flox/floxMx1-Cre+ mice. Immunohistochemical expression of pEGFR was clearly observed in the colonic epithelia of control mice at day 8, but negligible staining was present in those of Adam17flox/floxMx1-Cre+ mice (Fig. 3b). Colonic epithelia of both Adam17flox/floxMx1-Cre+ and control mice expressed EGFR and TGF-α, a major EGFR ligand (Fig. 3b). To further study the morphological changes and pEGFR expression in colonic epithelia, we isolated colonic crypts from the distal colon. The morphology of isolated colonic crypts was almost identical in control and Adam17flox/floxMx1-Cre+ mice under untreated conditions, but the crypts from DSS-treated mice at day 8 showed different morphology: large regenerative crypts were present among small fragmented crypts in the control, whereas most crypts from Adam17flox/floxMx1-Cre+ mice were fragmented (Fig. 3c). Similar to the in vivo setting, the crypts isolated from Adam17flox/floxMx1-Cre+ mice showed lower levels of ADAM17 and pEGFR expression compared to control mice (Fig. 3d). In addition, by real-time quantitative PCR (RT-qPCR) (Fig. 3e) and dot blotting using anti-MUC2 antibody (Fig. 3f), the crypts from DSS-treated Adam17flox/floxMx1-Cre+ mice showed less expression of MUC2, a mucin polypeptide specific for goblet cells, compared to control crypts. Since the secretion of mucins into the intestinal lumen by goblet cells creates the first line of defense against microbial encroachment (Peterson and Artis, 2014), ADAM17 appears to protect defective epithelial barriers of the colon against inflammation and damage through goblet cell differentiation or maintenance.
Fig. 3.

Loss of ADAM17 abrogates EGFR signaling in colonic epithelia. a. Immunoblot of EGFR and pEGFR in the distal colon from control and Adam17flox/floxMx1-Cre+ (ΔAdam17_Mx1) mice at days 0 and 8 after DSS administration. β-Actin was used as a loading control. b. Phosphorylated EGFR (pEGFR), EGFR and TGF-α immunostaining of representative colon sections from control and ΔAdam17_Mx1 mice at day 8 after DSS administration. NI, non-immune IgG (Control for anti-pEGFR and anti-EGFR antibodies). Scale bars, 100 μm. c. Morphology of isolated colonic crypts from control and ΔAdam17_Mx1 mice at days 0 and 8 after DSS administration. Arrows indicate regenerative large crypts. Scale bars, 50 μm. d. Immunoblot of ADAM17, pEGFR and EGFR in isolated colonic crypts from control and ΔAdam17_Mx1 mice at day 8 after DSS administration. β-Actin was used as a loading control. e. Relative gene expression of MUC2 by RT-qPCR in isolated colonic crypts from control and ΔAdam17_Mx1 mice at day 8 after DSS administration. Bars, mean ± s.d.; n = 4 independently isolated colonic crypts; **, p < 0.01. f. Dot immunoblot of MUC2 in isolated colonic crypts from control and ΔAdam17_Mx1 mice at day 8 after DSS administration. Two μg of tissue lysates were loaded per dot and GAPDH was used as a loading control. Results between the two independent groups were determined by Student's t-test. P values smaller than 0.05 are indicated on respective plots.
3.5. ADAM17–EGFR Signaling is Responsible for Cell Proliferation and Goblet Cell Differentiation
To address the link between the ADAM17–EGFR signaling and the epithelial cell proliferation and goblet cell differentiation, we utilized two human colonic epithelial cell lines, Caco-2 and LS174T cells, both of which express ADAM17, EGFR and its ligand TGF-α (Supplementary Fig. 4). As shown in Fig. 4a, pEGFR expression was suppressed by treatment with ADAM17-selective inhibitor S-44029 (Supplementary Fig. 5) or an EGFR inhibitor AG1478 in both Caco-2 and LS174T cells. S-44029 inhibits LPS-induced TNF-α release in THP-1 cells with an IC50 of 5.9 μM (a gift from Kaken Pharmaceutical Co., LTD.; Patent No. WO2003/022801), and has 10-fold more selectivity for the ADAM17 over ADAM10 and > 37-fold selectivity over MMP-1, -2, -3, -8, -9, -13, -14 or -17 in in vitro enzyme assays (Kawasaki et al., 2006). Importantly, these inhibitors significantly reduced cell growth as well (Fig. 4b, p < 0.01). LS174T cells are endowed with characteristics of goblet cells such as mucus granules (Van Klinken et al., 1996), and here we found that the size and number of mucus granules, stained by Alcian-Blue, are significantly decreased by treatment with S-44029 or AG1478 (Fig. 4c, p < 0.05 or p < 0.01). These inhibitors also significantly reduced the mRNA expression level of the goblet cell marker MUC2 in LS174T cells (Fig. 4d, p < 0.05 or p < 0.01). These data suggest that the ADAM17–EGFR signaling is involved in maintaining or promoting the goblet cell differentiation in addition to cell proliferation.
Fig. 4.

ADAM17–EGFR signaling is responsible for cell proliferation and goblet cell phenotype. a. Effect of ADAM17-selective (20 μM, S-44029) or EGFR-specific (20 μM, AG1478) inhibitor on pEGFR and EGFR expression in Caco-2 and LS174T cells. β-Actin was used as a loading control. b. Time-lapse cell growth assay showing the effect of S-44029 or AG1478 inhibitor on Caco-2 and LS174T cell growth. Bars, mean ± s.d.; n = 3 wells; **, p < 0.01. c. Effect of S-44029 or AG1478 inhibitor on mucus production in LS174T cells. Representative Alcian-Blue (AB)-stained LS174T cells in the presence of S-44029 or AG1478 inhibitor are indicated in the left panel. Scale bars, 20 μm. The ratio of AB-positive area in the total cell area was also determined (right panel). Bars, mean ± s.d.; n = 4 dishes; *, p < 0.05; **, p < 0.01. d. Effect of S-44029 or AG1478 inhibitor on MUC2 expression in LS174T cells. Relative gene expression of MUC2 was examined by RT-qPCR. Bars, mean ± s.d.; n = 4 dishes; *, p < 0.05; **, p < 0.01. e. Time-lapse cell growth assay showing the effect of MEK-specific (10 μM, U0126), PI3K-specific (20 μM, LY294002) or STAT3-specific inhibitor (100 μM, S3I) on Caco-2 and LS174T cell growth. Bars, mean ± s.d.; n = 3 wells; *, p < 0.05; **, p < 0.01. U0124 (10 μM) was used as a control for U0126. f. Effect of MEK-specific (U0126), PI3K-specific (LY294002) or STAT3-specific inhibitor (S3I) on MUC2 expression in LS174T cells. The expression of MUC2 was evaluated by dot immunoblot (upper panel) and RT-qPCR (lower panel). Bars, mean ± s.d.; n = 3 dishes; **, p < 0.01. U0124 was used as a control for U0126. GAPDH was used as a loading control for dot immunoblot analysis. Results between the two independent groups were determined by Student's t-test, and comparisons among three or more groups were determined by one-way ANOVA followed by Bonferroni's post-hoc testing. P values smaller than 0.05 are indicated on respective plots.
We then examined the impact of the EGFR downstream pathways on colonic epithelial cell proliferation and goblet cell differentiation by treating LS174T cells with inhibitors for the mitogen activated protein kinase (MAPK), the phosphatidylinositol 3-kinase- (PI3K-), or signal transducer and activator of transcription 3 (STAT3) signaling, all of which are pathways downstream of EGFR. As shown in Fig. 4e, time-lapse cell growth analysis indicated that cell growth of Caco-2 and LS174T cells is significantly reduced by treatment with MEK inhibitor (U0126), PI3K inhibitor (LY294002) or STAT3 inhibitor (S3I) compared to controls or those treated with a negative control agent (U0124). In addition, U0126 or S3I also significantly down-regulated the expression of MUC2 in LS174T cells, while LY294002 showed no effects on MUC2 expression (Fig. 4f, p < 0.01).
3.6. ADAM17 Knockdown Reduces EGFR Signaling in Colonic Epithelial Cells
We asked whether specific knockdown of ADAM17 expression by siRNAs affects the cell growth and goblet cell phenotype in colonic epithelial cell lines. When Caco-2 and LS174T cells were transfected with siRNAs for ADAM17 (siADAM17#1 and siADAM17#2) or control non-silencing siRNA, ADAM17-targeting siRNAs effectively reduced the level of ADAM17 and pEGFR when compared to non-silencing control (Fig. 5a). Importantly, ADAM17-knockdown cells demonstrated significantly reduced cell growth (Fig. 5b, p < 0.01), decreased Alcian-Blue-positive mucin production, and MUC2 expression in LS174T cells (Fig. 5c and d, p < 0.01).
Fig. 5.

ADAM17 knockdown inactivates EGFR signaling and reduces cell proliferation and goblet cell phenotype. a. Immunoblot of ADAM17, pEGFR and EGFR in Caco-2 and LS174T cells transfected with ADAM17-specific siRNAs #1 and #2 or non-silencing siRNA. β-Actin was used as a loading control. b. Time-lapse cell growth assay showing the effect of ADAM17 knockdown by siRNAs on Caco-2 and LS174T cell growth. Bars, mean ± s.d.; n = 3 wells; **, p < 0.01. c. Effect of ADAM17 knockdown by siRNAs on mucus production in LS174T cells. Mucus production was evaluated by Alcian-Blue (AB) staining (left panel). Scale bars, 20 μm. The ratio of AB-positive area in the total cell area is determined (right panel). Bars, mean ± s.d.; n = 4 dishes; **, p < 0.01. d. Effect of ADAM17 knockdown by siRNAs on MUC2 expression in LS174T cells. Relative gene expression of MUC2 was examined by RT-qPCR. Bars, mean ± s.d.; n = 3 dishes; **, p < 0.01. Results between the two independent groups were determined by Student's t-test, and comparisons among three or more groups were determined by one-way ANOVA followed by Bonferroni's post-hoc testing. P values smaller than 0.05 are indicated on respective plots.
3.7. Ectopic EGFR Stimulation Partially Rescues Severe Colitis in Adam17flox/floxMx1-Cre+ Mice
We further asked whether injection of recombinant TGF-α would rescue the severity of intestinal inflammation in DSS-treated Adam17flox/floxMx1-Cre+ mice. As shown in Fig. 6a, Adam17flox/floxMx1-Cre+ mice treated with recombinant TGF-α showed less weight loss than those treated with PBS (p < 0.01). Histological analysis demonstrated that mean areas of Grade III lesion are significantly decreased in Adam17flox/floxMx1-Cre+ mice upon injection of recombinant TGF-α compared to PBS-injected mice (Fig. 6b, p < 0.01). Concomitantly, TGF-α injection restored Alcian-Blue-positive mucus production by epithelial cells to a degree, which was markedly decreased in PBS-treated Adam17flox/floxMx1-Cre+ mice after DSS administration (Fig. 6c, p < 0.05), suggesting that adequate EGFR stimulation could be a potential avenue of therapeutic intervention.
Fig. 6.

Ectopic EGFR stimulation reduces severe colitis in Adam17flox/floxMx1-Cre+ mice. a. Percent weight change of PBS-treated Adam17flox/floxMx1-Cre+ (ΔAdam17_Mx1) (n = 6 mice) and TGF-α-treated ΔAdam17_Mx1 mice (n = 6 mice) after DSS administration. ΔAdam17_Mx1 mice were intraperitoneally injected with recombinant TGF-α (8 μg per injection) or with PBS at days 0, 2, 4 and 6 after DSS administration. Bars, mean ± s.d.; **, p < 0.01. b. Grade III lesions in PBS-treated ΔAdam17_Mx1 (n = 3 colon tissues) and TGF-α-treated ΔAdam17_Mx1 mice (n = 3 colon tissues) at day 8 after DSS administration. Bars, mean ± s.d.; **, p < 0.01. c. Images and quantification of colonic goblet cells in PBS-treated control and TGF-α-treated ΔAdam17_Mx1 mice at day 8 after DSS administration. Representative HE- and Alcian-Blue (AB)-stained colon sections from control and TGF-α-treated ΔAdam17_Mx1 mice at day 8 after DSS challenge are indicated in the left panel. Note that AB-positive mucus production is restored in ΔAdam17_Mx1 mice by TGF-α administration. Scale bars, 50 μm. Quantification of number of AB-positive goblet cells per 100 epithelial cells is shown in the right panel. Bars, mean ± s.d.; n = 3 colon tissues; *, p < 0.05. Results between the two independent groups were determined by Student's t-test. P values smaller than 0.05 are indicated on respective plots.
3.8. ADAM17 is Upregulated in Colonic Epithelia of UC Patients
Finally, to address the relevance of our findings to human UC, we investigated by immunohistochemistry whether ADAM17 is upregulated by colonic epithelia in UC as compared to normal epithelia of colon mucosae, which were obtained from the colon remote from colon cancer. ADAM17 was strongly immunostained in cytokeratin AE1/AE3 positive-epithelial cells of the colon in UC patients, while only negligible staining was seen in the colonic mucosa from control subjects (Fig. 7a and data not shown for AE1/AE3 staining). Similar expression pattern was observed for pEGFR, while expression of total EGFR was comparable in both UC and normal colonic mucosae. When the immunohistochemical staining for ADAM17 was evaluated by the scoring system described in the Methods, the mean score of epithelial ADAM17 expression was significantly higher in the UC patients than in the control (Fig. 7b, p < 0.01) and this score trended lower in UC patients under remission, although not significantly different. We also analyzed correlations of Ki-67-positive cell index and number of goblet cells between ADAM17-low and ADAM17-high groups of active UC patients. As shown in Fig. 7c, ADAM17-high expression group showed higher Ki-67-positive cell index (p < 0.01) and an increased number of goblet cells (p < 0.05) compared to the ADAM17-low expression group. All these immunohistochemical data are in accord with the findings obtained in our mouse model, and support our hypothesis on the protective actions of ADAM17 in colitis.
Fig. 7.

ADAM17 is upregulated in colonic epithelia of UC patients. a. Representative colon sections from control normal subjects or UC patients which are stained with HE or immunostained for ADAM17, pEGFR and EGFR. NI, non-immune IgG. The highlighted areas are magnified to the lower. Scale bars, 100 μm. b. ADAM17 expression score in colonic epithelia from control subjects (n = 30 patients), the patients with active UC (n = 25 patients) or under remission (n = 4 patients). Bars, mean ± s.d.; **, p < 0.01. c. Correlations between epithelial ADAM17 expression levels (ADAM17-low (n = 11 patients) and -high groups (n = 14 patients) of active UC patients) and Ki-67-positive cell index (left panel) or number of goblet cells per 100 epithelial cells (right panel). Bars, mean ± s.d.; *, p < 0.05; **, p < 0.01. Results between the two independent groups were determined by Student's t-test, and comparisons among three or more groups were determined by one-way ANOVA followed by Bonferroni's post-hoc testing. P values smaller than 0.05 are indicated on respective plots.
4. Discussion
In the present study, we have developed DSS-induced colitis in two different conditional Adam17-deficient mice, and demonstrated that ADAM17 derived from epithelial cells, but not myeloid cells, confers resistance to colitis by driving repair processes through epithelial cell proliferation and goblet cell differentiation. We propose that EGFR activation and its signaling pathways initiated by ADAM17 contribute to the repair of damaged epithelial cells and maintenance of the epithelial barrier in human UC (Supplementary Fig. 6).
Chalaris et al. developed hypomorphic ADAM17 mice (Adam17ex/ex mice) that resulted in aggravation of DSS-induced colitis. This study suggested that the impaired phosphorylation of STAT3 via EGFR activation in epithelial cells is implicated for the increased sensitivity to DSS-induced colitis (Chalaris et al., 2010). However, the mice had abnormalities in the eyes, heart and hair follicles during their development, and more importantly, they developed spontaneous inflammation of the skin. Since DSS-induced colitis is known to generate systemic responses in mice (Dong et al., 2013), unexpected responses to DSS due to the innate inflammatory features are likely to be superimposed on top of the colitis induced by this model. Adam17wavedX/wavedX mice generated by Brandl et al. also showed severe weight loss during DSS administration (Brandl et al., 2010), and the authors demonstrated that ADAM17-mediated EGFR signaling in non-hematopoietic cells plays a protective role against DSS-induced colitis. Although the study provided no information about which cell types in the non-hematopoietic compartment are responsible for the accelerated colitis, we have disclosed that ADAM17 derived from colonic epithelial cells is essential for these processes, and further demonstrated the relevance of the experimental findings to human UC tissues.
Epithelial regeneration is a key process for the recovery from IBDs, and is one of the most significant prognostic factors for long-term remission (Lichtenstein and Rutgeerts, 2010). Epithelial cell proliferation during regeneration is stimulated by several growth factors produced by local epithelial and mesenchymal cells in crypts near the damaged mucosal area. EGFR ligands are one major group of growth factors involved in rapid expansion of crypt epithelial cells, and they are secreted mainly from epithelial cells surrounding the damaged epithelial region (Okamoto and Watanabe, 2005). Previous studies showing that defective EGFR signaling in mice deficient for TGF-α leads to severe inflammation upon DSS challenge (Egger et al., 1997) and overexpression of TGF-α reduces susceptibility of DSS-induced colitis (Egger et al., 1998) have suggested that TGF-α acts as a principal growth factor for protection from colitis. In the present study, TGF-α was overexpressed by the colonic epithelial cells in both DSS-administered control and Adam17flox/floxMx1-Cre+ mice, but aggravation of the colitis was observed only in the latter mouse group. The severity of colitis in DSS-treated Adam17flox/floxMx1-Cre+ mice was partially rescued by treatment with recombinant TGF-α. Since ADAM17 is the major sheddase for membrane-type TGF-α and its shedding is suppressed in our Adam17flox/floxMx1-Cre+ mice (Horiuchi et al., 2007), exacerbation of the colitis in Adam17flox/floxMx1-Cre+ mice could be explained by the inability of the colonic epithelia to activate TGF-α.
One of our most intriguing findings in the present study is that decreased goblet cell differentiation and mucus production were associated with severe inflammation in response to DSS in Adam17flox/floxMx1-Cre+ mice. Complete knockout of the Adam17 gene displays multiple pathological changes in several organs, resulting in disturbed development of the eyes, lung, heart, immune system, hair and skin (Peschon et al., 1998). The phenotype is ascribed to result from disturbance of cell differentiation and tissue development, which may be caused by failure of ADAM17 to activate EGFR. This ADAM17-EGFR axis has been reported to regulate terminal differentiation of keratinocytes (Franzke et al., 2012) and chondrocytes (Hall et al., 2013) and oligodendrogenesis during postnatal myelination (Palazuelos et al., 2014). Our data indicate that this axis also enhances goblet cell differentiation of colon epithelial cells during inflammation.
Repair of the damaged intestinal epithelium is carried out by two continuous steps, i.e. restitution by proliferated epithelial cells and subsequent maturation into functionally differentiated epithelial cells (Okamoto and Watanabe, 2005). We have shown that the ADAM17-EGFR axis commands both the processes through different EGFR-downstream signaling pathways. All the MAPK, STAT3 and PI3K pathways were required for epithelial cell proliferation, but the PI3K pathway was dispensable for goblet cell differentiation. ADAM17 is implicated in the shedding of five of the seven EGFR ligands (TGF-α, epiregulin, epigen, amphiregulin and HB-EGF) (Sahin and Blobel, 2007, Sahin et al., 2004). Therefore, it might be possible that the shedding of diverse EGFR ligands by ADAM17 may sequentially control epithelial cell proliferation and differentiation. Further in vivo work is needed to gain deeper mechanistic insight.
The integrity of the epithelial barrier is maintained mainly by tight junctions (Laukoetter et al., 2008). However, the barrier is reinforced by several factors, which are dependent on multiple types of epithelia. In colon mucosa, mucin plays a key barrier function by coating the epithelium and preventing entry of microbes (Laukoetter et al., 2008). Genome-wide association studies showed that genetic mutations of MUC2, the most abundant protective mucin, are associated with UC (Abraham and Cho, 2009). Our data on the goblet cell differentiation and MUC2 expression have indicated that ADAM17 supports mucosal epithelial barrier functions mostly by promoting mucus production. In the skin, keratin layer formed by keratinocytes is known to have a supportive role in the epidermal barrier. Recent experimental studies provided mechanistic evidence that keratinocyte-derived ADAM17 strengthens the epidermal barrier function by increasing keratin cross-linking through the activation of transglutaminase 1 (TGM1) (Franzke et al., 2012), and promotes resistance to bacterial infection (Brooke et al., 2014). Although no direct evidence is available for the relationship between TGM1 expression and intestinal barrier function, TGM1 is known to be downregulated in the colon of UC patients (D'argenio et al., 2005). Interestingly, patients with ulcerative colitis are reported to have a higher risk for skin disorders (Huang et al., 2012). Altogether, these data suggest the possibility that ADAM17 plays an important role in maintaining the barrier function of both colon and skin. In this context, the recently reported autosomal recessive disease linked to ADAM17 deletion (Blaydon et al., 2011), in which patients exhibit severe inflammatory skin lesions and symptoms of bowel disease such as diarrhea, seems to support this hypothesis, since these symptoms may be triggered by challenges to defective epithelial barriers of the skin and intestine.
A potential limitation of this study was the use of the DSS-induced colitis model as a clinically relevant model. Although DSS-induced colitis is a disease model resembling human UC, T and B cells are not involved in development of the colitis, indicating that adaptive immunity is not required for this model (Chassaing et al., 2014). Nevertheless, DSS-induced colitis model may be useful for studying the contribution of the epithelial barrier function and innate immune system to the initiation of colitis, and our results suggest that epithelial cell-derived ADAM17 is essential for repair processes through epithelial cell proliferation and goblet cell differentiation. Regulation mechanisms for the ADAM17 expression by colonic epithelial cells during colitis also remain unclear in this study. Previous studies suggested that TNF-α and wild-type p53 up-regulate ADAM17 expression (Guinea-Viniegra et al., 2012, Charbonneau et al., 2007), but further studies are needed to determine regulators of ADAM17 in intestinal inflammation.
Treatments against TNF-α exhibit a remarkable clinical response in some UC patients (Danese et al., 2013), but the majority of emerging selective ADAM17 inhibitors which target TNF-α shedding in human inflammatory diseases are not successful (Duffy et al., 2011). The information in the present study that epithelial cell-derived ADAM17 is required for colonic epithelial regeneration and barrier function indicates that remedies targeting specifically stromal cells, but not epithelial cells, would be necessary to avoid negative consequences during clinical trials of ADAM17 inhibitors in UC patients. In addition, this work provides important mechanistic insight for goblet cell- and mucin-based therapy to protect the epithelial barrier and compensate for impaired epithelial regenerative activity when ADAM17 inhibitors are introduced.
Funding Sources
This work was supported by Grant-in-Aid for Scientific Research (A) (24249022) (to Y.O.) and Grant-in-Aid for Young Scientists (B) (20790291) (to M.S.) from Japan Society for the Promotion of Science, Keio Gijuku Academic Development Funds (to M.S.) and Nagao Memorial Fund (041) (to M.S.).
Conflict of Interest Statement
We declare that this study does not have financial or other relationships that might lead to a conflict of interest.
Author Contributions
Study concept and design, M.S. and Y.O.; generation and maintenance of Adam17-deficient mice, K.H., M.S. and A.S.; acquisition, analysis and interpretation of data, M.S., A.S. and Y.O.; writing the manuscript, M.S. and Y.O.; contribution to analysis on human samples, M.S., A.S., K.O., H.H. and Y.K.; technical or material support, K.H. and T.T.
Acknowledgments
We thank Ms. Aya Shiraishi and Ms. Yuko Hashimoto (Department of Pathology, Keio University School of Medicine) for their technical assistance. We are also grateful to Dr. Hartland W. Jackson (Institute for Molecular Life Sciences, University of Zürich) for reviewing the manuscript.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2016.02.007.
Contributor Information
Masayuki Shimoda, Email: shimoda@a2.keio.jp.
Yasunori Okada, Email: okada@z6.keio.jp, ya-okada@juntendo.ac.jp.
Appendix A. Supplementary Data
Supplementary material
References
- Abraham C., Cho J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015;12:205–217. doi: 10.1038/nrgastro.2015.34. [DOI] [PubMed] [Google Scholar]
- Black R.A., Rauch C.T., Kozlosky C.J., Peschon J.J., Slack J.L., Wolfson M.F., Castner B.J., Stocking K.L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K.A., Gerhart M., Davis R., Fitzner J.N., Johnson R.S., Paxton R.J., March C.J., Cerretti D.P. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Blaydon D.C., Biancheri P., Di W.L., Plagnol V., Cabral R.M., Brooke M.A., Van Heel D.A., Ruschendorf F., Toynbee M., Walne A., O'toole E.A., Martin J.E., Lindley K., Vulliamy T., Abrams D.J., Macdonald T.T., Harper J.I., Kelsell D.P. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011;365:1502–1508. doi: 10.1056/NEJMoa1100721. [DOI] [PubMed] [Google Scholar]
- Blobel C.P. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- Brandl K., Sun L., Neppl C., Siggs O.M., Le Gall S.M., Tomisato W., Li X., Du X., Maennel D.N., Blobel C.P., Beutler B. MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. U. S. A. 2010;107:19967–19972. doi: 10.1073/pnas.1014669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke M.A., Etheridge S.L., Kaplan N., Simpson C., O'toole E.A., Ishida-Yamamoto A., Marches O., Getsios S., Kelsell D.P. iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Hum. Mol. Genet. 2014;23:4064–4076. doi: 10.1093/hmg/ddu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brynskov J., Foegh P., Pedersen G., Ellervik C., Kirkegaard T., Bingham A., Saermark T. Tumour necrosis factor alpha converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut. 2002;51:37–43. doi: 10.1136/gut.51.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesaro A., Abakar-Mahamat A., Brest P., Lassalle S., Selva E., Filippi J., Hebuterne X., Hugot J.P., Doglio A., Galland F., Naquet P., Vouret-Craviari V., Mograbi B., Hofman P.M. Differential expression and regulation of ADAM17 and TIMP3 in acute inflamed intestinal epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2009;296:G1332–G1343. doi: 10.1152/ajpgi.90641.2008. [DOI] [PubMed] [Google Scholar]
- Chalaris A., Adam N., Sina C., Rosenstiel P., Lehmann-Koch J., Schirmacher P., Hartmann D., Cichy J., Gavrilova O., Schreiber S., Jostock T., Matthews V., Hasler R., Becker C., Neurath M.F., Reiss K., Saftig P., Scheller J., Rose-John S. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010;207:1617–1624. doi: 10.1084/jem.20092366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau M., Harper K., Grondin F., Pelmus M., Mcdonald P.P., Dubois C.M. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J. Biol. Chem. 2007;282:33714–33724. doi: 10.1074/jbc.M704041200. [DOI] [PubMed] [Google Scholar]
- Chassaing B., Aitken J.D., Malleshappa M., Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014;104 doi: 10.1002/0471142735.im1525s104. (Unit 15 25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen B.E., Burkhardt C., Reith W., Renkawitz R., Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Danese S., Colombel J.F., Peyrin-Biroulet L., Rutgeerts P., Reinisch W. Review article: the role of anti-TNF in the management of ulcerative colitis — past, present and future. Aliment. Pharmacol. Ther. 2013;37:855–866. doi: 10.1111/apt.12284. [DOI] [PubMed] [Google Scholar]
- D'argenio G., Calvani M., Della Valle N., Cosenza V., Di Matteo G., Giorgio P., Margarucci S., Petillo O., Jori F.P., Galderisi U., Peluso G. Differential expression of multiple transglutaminases in human colon: impaired keratinocyte transglutaminase expression in ulcerative colitis. Gut. 2005;54:496–502. doi: 10.1136/gut.2004.049411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F., Zhang L., Hao F., Tang H., Wang Y. Systemic responses of mice to dextran sulfate sodium-induced acute ulcerative colitis using 1H NMR spectroscopy. J. Proteome Res. 2013;12:2958–2966. doi: 10.1021/pr4002383. [DOI] [PubMed] [Google Scholar]
- Duffy M.J., Mullooly M., O'donovan N., Sukor S., Crown J., Pierce A., Mcgowan P.M. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin. Proteomics. 2011;8:9. doi: 10.1186/1559-0275-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D.R., Handsley M.M., Pennington C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008;29:258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger B., Procaccino F., Lakshmanan J., Reinshagen M., Hoffmann P., Patel A., Reuben W., Gnanakkan S., Liu L., Barajas L., Eysselein V.E. Mice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitis. Gastroenterology. 1997;113:825–832. doi: 10.1016/s0016-5085(97)70177-x. [DOI] [PubMed] [Google Scholar]
- Egger B., Carey H.V., Procaccino F., Chai N.N., Sandgren E.P., Lakshmanan J., Buslon V.S., French S.W., Buchler M.W., Eysselein V.E. Reduced susceptibility of mice overexpressing transforming growth factor alpha to dextran sodium sulphate induced colitis. Gut. 1998;43:64–70. doi: 10.1136/gut.43.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer R.G., Easley K.A., Rankin G.B. Clinical patterns, natural history, and progression of ulcerative colitis. A long-term follow-up of 1116 patients. Dig. Dis. Sci. 1993;38:1137–1146. doi: 10.1007/BF01295733. [DOI] [PubMed] [Google Scholar]
- Feng Y., Tsai Y.H., Xiao W., Ralls M.W., Stoeck A., Wilson C.L., Raines E.W., Teitelbaum D.H., Dempsey P.J. Loss of ADAM17-mediated tumor necrosis factor alpha signaling in intestinal cells attenuates mucosal atrophy in a mouse model of parenteral nutrition. Mol. Cell. Biol. 2015;35:3604–3621. doi: 10.1128/MCB.00143-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzke C.W., Cobzaru C., Triantafyllopoulou A., Loffek S., Horiuchi K., Threadgill D.W., Kurz T., Van Rooijen N., Bruckner-Tuderman L., Blobel C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 2012;209:1105–1119. doi: 10.1084/jem.20112258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinea-Viniegra J., Zenz R., Scheuch H., Jimenez M., Bakiri L., Petzelbauer P., Wagner E.F. Differentiation-induced skin cancer suppression by FOS, p53, and TACE/ADAM17. J. Clin. Invest. 2012;122:2898–2910. doi: 10.1172/JCI63103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall K.C., Hill D., Otero M., Plumb D.A., Froemel D., Dragomir C.L., Maretzky T., Boskey A., Crawford H.C., Selleri L., Goldring M.B., Blobel C.P. ADAM17 controls endochondral ossification by regulating terminal differentiation of chondrocytes. Mol. Cell. Biol. 2013;33:3077–3090. doi: 10.1128/MCB.00291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K., Kimura T., Miyamoto T., Takaishi H., Okada Y., Toyama Y., Blobel C.P. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 2007;179:2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- Huang B.L., Chandra S., Shih D.Q. Skin manifestations of inflammatory bowel disease. Front. Physiol. 2012;3:1–13. doi: 10.3389/fphys.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T., Matsuzaki K., Horiuchi K., Toyama Y., Takaishi H. A novel ADAM inhibitor, S-44029, supresses osteoclastogenesis in vitro. J. Bone Miner. Res. 2006;21:S394. [Google Scholar]
- Khokha R., Murthy A., Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013;13:649–665. doi: 10.1038/nri3499. [DOI] [PubMed] [Google Scholar]
- Kuhn R., Schwenk F., Aguet M., Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- La Marca R., Cerri F., Horiuchi K., Bachi A., Feltri M.L., Wrabetz L., Blobel C.P., Quattrini A., Salzer J.L., Taveggia C. TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci. 2011;14:857–865. doi: 10.1038/nn.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoetter M.G., Nava P., Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2008;14:401–407. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein G.R., Rutgeerts P. Importance of mucosal healing in ulcerative colitis. Inflamm. Bowel Dis. 2010;16:338–346. doi: 10.1002/ibd.20997. [DOI] [PubMed] [Google Scholar]
- Mochizuki S., Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007;98:621–628. doi: 10.1111/j.1349-7006.2007.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer. 2008;8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- Okamoto R., Watanabe M. Cellular and molecular mechanisms of the epithelial repair in IBD. Dig. Dis. Sci. 2005;50(Suppl. 1):S34–S38. doi: 10.1007/s10620-005-2804-5. [DOI] [PubMed] [Google Scholar]
- Ordas I., Eckmann L., Talamini M., Baumgart D.C., Sandborn W.J. Ulcerative colitis. Lancet. 2012;380:1606–1619. doi: 10.1016/S0140-6736(12)60150-0. [DOI] [PubMed] [Google Scholar]
- Palazuelos J., Crawford H.C., Klingener M., Sun B., Karelis J., Raines E.W., Aguirre A. TACE/ADAM17 is essential for oligodendrocyte development and CNS myelination. J. Neurosci. 2014;34:11884–11896. doi: 10.1523/JNEUROSCI.1220-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon J.J., Slack J.L., Reddy P., Stocking K.L., Sunnarborg S.W., Lee D.C., Russell W.E., Castner B.J., Johnson R.S., Fitzner J.N., Boyce R.W., Nelson N., Kozlosky C.J., Wolfson M.F., Rauch C.T., Cerretti D.P., Paxton R.J., March C.J., Black R.A. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- Peterson L.W., Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- Sahin U., Blobel C.P. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett. 2007;581:41–44. doi: 10.1016/j.febslet.2006.11.074. [DOI] [PubMed] [Google Scholar]
- Sahin U., Weskamp G., Kelly K., Zhou H.M., Higashiyama S., Peschon J., Hartmann D., Saftig P., Blobel C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller J., Chalaris A., Garbers C., Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011;32:380–387. doi: 10.1016/j.it.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Shimoda M., Khokha R. Proteolytic factors in exosomes. Proteomics. 2013;13:1624–1636. doi: 10.1002/pmic.201200458. [DOI] [PubMed] [Google Scholar]
- Shimoda M., Hashimoto G., Mochizuki S., Ikeda E., Nagai N., Ishida S., Okada Y. Binding of ADAM28 to P-selectin glycoprotein ligand-1 enhances P-selectin-mediated leukocyte adhesion to endothelial cells. J. Biol. Chem. 2007;282:25864–25874. doi: 10.1074/jbc.M702414200. [DOI] [PubMed] [Google Scholar]
- Shimoda M., Principe S., Jackson H.W., Luga V., Fang H., Molyneux S.D., Shao Y.W., Aiken A., Waterhouse P.D., Karamboulas C., Hess F.M., Ohtsuka T., Okada Y., Ailles L., Ludwig A., Wrana J.L., Kislinger T., Khokha R. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat. Cell Biol. 2014;16:889–901. doi: 10.1038/ncb3021. [DOI] [PubMed] [Google Scholar]
- Van Klinken B.J., Oussoren E., Weenink J.J., Strous G.J., Buller H.A., Dekker J., Einerhand A.W. The human intestinal cell lines Caco-2 and LS174T as models to study cell-type specific mucin expression. Glycoconj. J. 1996;13:757–768. doi: 10.1007/BF00702340. [DOI] [PubMed] [Google Scholar]
- Wirtz S., Neufert C., Weigmann B., Neurath M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material