

Abstract
Liver phenylalanine hydroxylase (PheH) is an allosteric enzyme that requires activation by phenylalanine for full activity. The location of the allosteric site for phenylalanine has not been established. NMR spectroscopy of the isolated regulatory domain (RDPheH(25–117) is the regulatory domain of PheH lacking residues 1–24) of the rat enzyme in the presence of phenylalanine is consistent with formation of a side-by-side ACT dimer. Six residues in RDPheH(25–117) were identified as being in the phenylalanine-binding site on the basis of intermolecular NOEs between unlabeled phenylalanine and isotopically labeled protein. The location of these residues is consistent with two allosteric sites per dimer, with each site containing residues from both monomers. Site-specific variants of five of the residues (E44Q, A47G, L48V, L62V, and H64N) decreased the affinity of RDPheH(25–117) for phenylalanine based on the ability to stabilize the dimer. Incorporation of the A47G, L48V, and H64N mutations into the intact protein increased the concentration of phenylalanine required for activation. The results identify the location of the allosteric site as the interface of the regulatory domain dimer formed in activated PheH.
Keywords: allosteric regulation, enzyme kinetics, nuclear magnetic resonance (NMR), site-directed mutagenesis, tetrahydrobiopterin (BH4), binding site, phenylalanine hydroxylase
Introduction
Phenylalanine hydroxylase (PheH)2 catalyzes a key step in phenylalanine catabolism, the hydroxylation of phenylalanine to tyrosine in the liver using tetrahydropterin (BH4) and oxygen. A deficiency in human PheH increases the level of phenylalanine in the blood, resulting in the inherited disease phenylketonuria (PKU) (1). Thus, the activity of PheH must be tightly controlled to maintain appropriate phenylalanine levels. PheH is activated by phenylalanine and inhibited by BH4 (2, 3). In addition, phosphorylation of PheH at Ser-16 is reported to decrease the concentration of phenylalanine required to activate PheH (4).
Mammalian PheH is a homotetramer, and each monomer contains an N-terminal regulatory domain, a central catalytic domain, and a C-terminal tetramerization domain. The other two aromatic amino acid hydroxylases, tyrosine hydroxylase (TyrH) and tryptophan hydroxylase, have similar architectures. The crystal structures of the catalytic domains of all three enzymes show very similar folds and active sites (5–7), consistent with these enzymes sharing a common catalytic mechanism (8). The structures of the regulatory domains of PheH and TyrH show that both contain ACT domains (5, 9, 10), although the two enzymes are regulated differently (2, 11). To date, there is no published structure of a full-length mammalian PheH, or indeed of any eukaryotic aromatic amino acid hydroxylase, in that the available structures are of proteins lacking the N-terminal regulatory domain, much of the C-terminal tetramerization domain, or both. The structure of a dimeric form of rat PheH containing both the catalytic and regulatory domains but lacking the C-terminal 24 residues required for tetramer formation (5) has provided the present model for the structural basis for activation by phenylalanine. In this structure the N-terminal ∼30 residues lie across the active site, likely preventing substrate binding. Removal of these residues results in an active enzyme that does not require activation by phenylalanine (12). Based on these results, activation of PheH by phenylalanine is proposed to involve a conformational change in which the N-terminal residues move away from the active site (12). Activation of PheH by phenylalanine is well established to cause a significant conformational change in the protein (13), readily detectable as an increase in the fluorescence of the enzyme (14) and exposure of a hydrophobic surface (15).
In the absence of structures of the intact protein containing an amino acid ligand, there are no structural data to identify the site at which phenylalanine binds to activate PheH. Shiman et al. (16) originally proposed that the activating site was separate from the active site but was unable to propose a specific location in the absence of any structure. When the structure of the combined regulatory and catalytic domains was first solved, Kobe et al. (5) proposed that the phenylalanine allosteric binding site is located near the interface between the regulatory and catalytic domains of PheH, specifically at residues 42–47 in the loop between helix α1 and strand β1, based on the similar structures of the regulatory domain of PheH and the ACT domain of 3-phosphoglycerate dehydrogenase. A recent computational study came to a similar conclusion (17). In contrast, Jaffe et al. (18) has proposed that the regulatory domains of PheH dimerize and form a phenylalanine-binding site similar to those in ACT domains that are regulated by amino acids. Flydal et al. (19) found that phenylalanine binds to the regulatory domain of Caenorhabditis elegans PheH; however, they concluded that this site is blocked in the human enzyme and that activation of the latter involves phenylalanine binding in the active site (20). The regulatory domain of TyrH also contains an ACT domain but it does not bind tyrosine (9), so the presence of an ACT domain in PheH does not in itself establish that the regulatory domain contains a site for phenylalanine. In addition, there is significant diversity in the sites at which ACT domains bind amino acids (21), making it difficult to predict the binding site on an ACT domain without a structure. A recent structure of a bacterial PheH, which is homologous to the catalytic domain of the eukaryotic enzymes, showed phenylalanine bound at a site 15.7 Å from the active site (22), raising the possibility of a separate allosteric site within the catalytic domain. Recent studies of the isolated regulatory domain of rat PheH (RDPheH) have established that this domain indeed binds phenylalanine (23–25). Moreover, elimination of phenylalanine binding in the active site does not prevent the conformational change associated with phenylalanine activation, consistent with an allosteric site separate from the active site (26).
Identification of the site at which phenylalanine binds to activate PheH is critical to understanding the molecular basis for regulation of the enzyme. Here, we report that the isolated regulatory domain of PheH (RDPheH) forms a side-by-side ACT domain dimer, with helix α1, strand β2, and the loops connected to strand β2 forming the dimer interface and that phenylalanine binds across the two edges of the dimer interface. Mutagenesis of residues in the proposed binding site results in a significant decrease in the ability of phenylalanine to activate PheH. The results provide further support for a regulatory mechanism in which phenylalanine binding to the regulatory domain of PheH is linked to regulatory domain dimerization and that this is the conformational change associated with activation.
Experimental Procedures
Materials
2H8-l-Phenylalanine, 15NH4Cl, 13C6-d-glucose, d-glucose-1,2,3,4,5,6,6-d7, and deuterium oxide were from Cambridge Isotope Laboratories, Inc. (Andover, MA). BH4 was purchased from Schircks Laboratories (Jona, Switzerland). Dithiothreitol was from Inalco, S.p.A. (Milan, Italy). Leupeptin and pepstatin A were from Peptide Institute, Inc. (Osaka, Japan).
Protein Expression and Purification
Mutagenesis of RDPheH(25–117) and PheH was performed using the QuikChange mutagenesis protocol (Agilent Technologies). The mutations were verified by DNA sequencing (Genscript, Piscataway, NJ). The expression and purification of wild-type and variant versions of rat RDPheH(25–117) and PheH were performed as described previously for the wild-type proteins (23, 24). For 15N-labeled or 15N/13C-labeled RDPheH(25–117), the expression was the same as that for the unlabeled proteins except that the cells were grown in M9 minimal media with 15NH4Cl (1 g/liter) in the absence or presence of 13C6-glucose (4 g/liter), respectively (27). For completely deuterated 15N-labeled RDPheH(25–117), the cells were grown in M9 minimal media in 99.99% D2O with 15NH4Cl (1 g/liter) and d-glucose-1,2,3,4,5,6,6-d7 (4 g/liter) as the sole nitrogen and carbon sources, respectively (9). The purities of all protein preparations were greater than 95% based on PAGE in the presence of SDS.
NMR Spectroscopy
NMR experiments were carried out at 298 K on a Brüker Avance 700 MHz spectrometer using cryogenically cooled probes equipped with 13C and 15N decoupling and pulsed-field gradient coils. All NMR samples were prepared in 50 mm phosphate, 100 mm NaCl, 1 μm leupeptin, 1 μm pepstatin A, and 5% D2O, pH 8. Three-dimensional HNCACB, HN(CO)CACB, HNCA, and HN(CO)CA (28) spectra were collected for backbone assignments of RDPheH(25–117); the samples were 0.5–1 mm 15N,13C-labeled RDPheH(25–117) plus 2–5 mm phenylalanine. To identify the dimer interface and phenylalanine-binding sites on RDPheH, 15N-edited NOESY HSQC spectra were collected with a mixing time of 120 ms on three NMR samples as follows: 1 mm 15N,2H-labeled RDPheH(25–117) in the presence of 5 mm phenylalanine; 1 mm 15N,2H-labeled RDPheH(25–117) in the presence of 5 mm 2H8-phenylalanine; and isotopic heterodimers of RDPheH prepared by mixing equal amounts of unlabeled and 15N,2H-labeled RDPheH(25–117) (1 mm each) and subsequently adding 5 mm phenylalanine. All spectra were processed using NMRPipe (29) and analyzed using NMRView (30). The chemical shift assignments for RDPheH(25–117) have been deposited in the Biological Magnetic Resonance Bank with accession number 26703.
Analytical Ultracentrifugation
Sedimentation velocity analytical ultracentrifugation (AUC) was carried out as described previously (24) using ∼15 μm total monomer and 50 μm to 1 mm phenylalanine, with detection at 230 nm. All AUC samples were prepared in 50 mm phosphate, 100 mm NaCl, pH 8.0. The standard c(s) model of SEDFIT (31) version 14.1 was used to generate c(s) distributions. The values for the weighted-average sedimentation coefficient (sw) were determined by integration of the c(s) distribution between 0.8 and 3 S. The data were fitted to Equation 1,
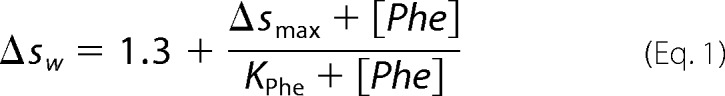 |
where Δsw is difference in the value from the sw value in the absence of phenylalanine; Δsmax is the change in the sw value in the presence of saturating phenylalanine, and Kphe is the dissociation constant for phenylalanine. The value of 1.3 is the sw value determined for protein samples without phenylalanine added. All data fitting was done using KaleidaGraph (Synergy Software).
Stopped-flow Spectroscopy
The binding of phenylalanine to different PheH variants was monitored using fluorescence spectroscopy as described previously (25). Enzyme (10 μm in 0.2 m HEPES, pH 7.5) in one syringe of an Applied Photophysics (Leatherhead, Surrey, UK) SX18 stopped-flow spectrofluorometer was mixed with an equal volume of 0.25–10 mm phenylalanine in the same buffer from the other syringe at 25 °C. The intrinsic tryptophan fluorescence was monitored using excitation at 295 nm and an emission cutoff filter of 340 nm until a stable reading was obtained, usually in 3–5 min. The total fluorescence change as a function of the concentration of phenylalanine was fit to Equation 2,
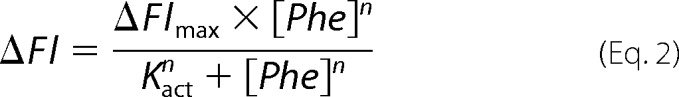 |
where ΔFl is the observed fluorescence change; ΔFlmax is the fluorescence change at saturating concentrations of phenylalanine; n is the Hill coefficient, and Kact is the concentration of phenylalanine at which one-half of the maximum fluorescence change occurs.
Enzyme Assays
The effects of preincubation with phenylalanine on the activities of different PheH mutants were determined as described previously for wild-type PheH (25). Enzyme (25 μm in 200 mm HEPES, pH 7.0) was preincubated with 0 or 250 μm phenylalanine in the same buffer at 23 °C for 10 min. A 5-μl aliquot was then added to a 0.5-ml assay mix containing 200 μm BH4, 1 mm phenylalanine, 50 μg/ml catalase, 1 mm dithiothreitol, 5 μm ferrous ammonium sulfate, 80 mm HEPES, pH 7.0, at 23 °C The reaction was quenched after 30 s, and the amount of tyrosine formed was determined by HPLC using a Gemini-NX C18 150 × 2.0-mm column in 0.1% acetic acid. Tyrosine was detected by fluorescence with the excitation wavelength set at 275 nm and the emission wavelength set at 303 nm. A standard curve of 0–800 μm tyrosine was used to quantify the amount of tyrosine produced. To determine the kinetic parameters of the PheH mutants, the amount of phenylalanine in the preincubation was increased (5 mm for A47G PheH and 10 mm for L48V and H64N PheH), and the tyrosine formed at 0, 30, and 60 s was determined using 0.15 to 10.1 mm phenylalanine. The reaction of each PheH mutant was linear during this time period. Steady-state kinetic parameters were determined by fitting the rates as a function of phenylalanine concentration to Equation 3,
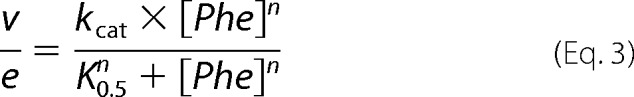 |
where n is the Hill coefficient; e is the enzyme concentration, and K0.5 is the concentration of phenylalanine at which the rate is one-half of kcat.
Results
Solution Structure of the Regulatory Domain of Phenylalanine Hydroxylase
NMR spectroscopy was used to study the solution structure of the dimer formed by the PheH regulatory domain using a protein (RDPheH(25–117)) that lacked the N-terminal 24 residues. X-ray crystallography (5) and NMR spectroscopy (25) have both established that the N-terminal ∼24 residues of PheH are disordered. Removal of the N-terminal 24 residues of RDPheH to generate RDPheH(25–117) results in a significant increase in the stability and solubility of the protein at the concentrations required for NMR spectroscopy, but it has no significant effect on dimerization or phenylalanine binding (24, 25). Previous NMR studies have established that the backbone structure of RDPheH dimer is the same whether the dimer is formed at high (>1 mm) protein concentrations (Kd = 45 μm) in the absence of phenylalanine or at lower concentrations in the presence of saturating phenylalanine (24, 25). To ensure that the protein was overwhelmingly in the dimer form, excess phenylalanine (2–5 mm) was added to NMR samples. Fig. 1A shows a two-dimensional 1H-15N HSQC spectrum of 480 μm RDPheH(25–117) plus 2 mm phenylalanine at pH 8. The presence of nearly 80 cross-peaks compared with the 91 nonproline residues of RDPheH(25–117) suggests that RDPheH(25–117) forms a symmetrical dimer, since an asymmetrical dimer would be expected to exhibit more cross-peaks. It was possible to assign 69 cross-peaks using standard NMR methods (Fig. 1A). No cross-peaks corresponding to residues 27–30, 84–90, 110–112, or Ser-93 could be identified. The backbone NH signals of six nonproline residues (Gln-31, Phe-39, Leu-91, Ile-94, Glu-108, and Lys-116) were also absent from spectra. The absence of cross-peaks for these residue signals likely resulted from intermediate time scale conformational exchange or exchange with water at the high pH required to maintain the protein in solution. Most of the unassigned residues are either at the N terminus or on helix α2, which is close to both termini in space.
FIGURE 1.

Structural analysis of RDPheH(25–117) by NMR spectroscopy. A, two-dimensional 1H-15N HSQC spectrum of 0.48 mm RDPheH(25–117) plus 2 mm phenylalanine showing the assignments of the individual residues. Conditions are as follows: 50 mm sodium phosphate, 100 mm NaCl, 1 μm leupeptin, 1 μm pepstatin A, and 5% D2O, pH 8.0, at 298 K at a magnetic field strength of 14.1 tesla (600 MHz 1H). B, secondary structure prediction for RDPheH(25–117) in the presence of phenylalanine using PECAN (32). The probabilities of α-helices and β-strands are given as positive and negative values, respectively. C, α-helices (red) and β-strands (green) predicted in B mapped to the crystal structure of the regulatory domain of PheH (Protein Data Bank code 2PHM, residues 25–117).
The available backbone chemical shifts for RDPheH(25–117) were analyzed using the program PECAN (32), which provides the secondary structure probabilities on a residue-by-residue basis (Fig. 1B). The predicted secondary structure is in good agreement with the crystal structure of the regulatory domain of PheH in the presence of the catalytic domain (Fig. 1C), establishing that the isolated regulatory domain of PheH in solution folds essentially the same as in the full-length protein. The major difference is in the lengths of helix α2, strand β1, and strand β4. The probabilities suggest that strand β2 continues into residues 61–64 in the RDPheH(25–117) dimer, although these residues form a loop in the crystal structure. The analysis would also appear to predict that helix α2 is shorter in solution than in the crystal; this apparent discrepancy can be attributed to the lack of assignments for residues 84–90. Finally, the C-terminal residues His-107 to Ser-110 do not show a strong probability to form strand β4; this is likely due to the absence of the catalytic domain.
To identify the dimer interface in the RDPheH(25–117) dimer, three-dimensional 15N NOESY HSQC spectra for two NMR samples were collected and compared. The first sample was a mixed dimer of RDPheH(25–117) formed by mixing 1 mm 15N-labeled, perdeuterated RDPheH(25–117) with 1 mm unlabeled RDPheH(25–117) and allowing the subunits to exchange before adding 5 mm phenylalanine. For this sample, cross-peaks between 15N-labeled amide protons and aliphatic protons should be due to intermonomer contacts. The other sample was a homodimer of perdeuterated, 15N-labeled protein, also in the presence of 5 mm phenylalanine; this sample was a control to avoid misassignments of cross-peaks arising from residual protons in the same monomer. The cross-peaks in the 15N NOESY spectrum from the mixed dimer that showed a significant increase in intensity compared with the corresponding cross-peaks from the spectrum of the perdeuterated sample can be identified as residues at the dimer interface. Thirteen cross-peaks were identified in this fashion (Fig. 2). The locations of these residues in the structure of the regulatory domain of PheH are shown in Fig. 3A. Five of the residues (Arg-49, Lys-50, Val-51, Arg-52, and Leu-53) are located on helix α1, three (Ile-65, Glu-66, and Ser-67) are on strand β2, and five (Asn-58, Asp-59, Ile-60, Asn-61, and Leu-72) are on the two loops connected to strand β2. The location of these residues suggests that the dimer interface of RDPheH dimer is between helix α1 and strand β2. This is the same arrangement as is seen in the stable dimer formed by the isolated regulatory domain of TyrH (RDTyrH) (9). The structure of RDPheH(25–117) dimer was modeled by replacing each monomer of the core structure of the RDTyrH dimer with one RDPheH(25–117) monomer (Fig. 3B), because the core structures of the two domains are nearly the same. All 13 residues predicted to be on the dimer interface are located on the dimer interface in the model of the RDPheH dimer, supporting the structure.
FIGURE 2.

Identification of the PheH regulatory domain dimer interface. Selected 15N slices of three-dimensional NOESY-HSQC spectra of the RDPheH(25–117) dimer with excess phenylalanine are shown. For each pair of spectra, the left one is the spectrum of 1 mm 15N,2H-labeled RDPheH(25–117) with 5 mm phenylalanine, whereas the right one is the same region in the spectrum of the mixed dimer of 1 mm unlabeled RDPheH(25–117) and 1 mm 15N,2H-labeled RDPheH(25–117) with 5 mm phenylalanine. Amide proton assignments are provided at the top of the left slice.
FIGURE 3.
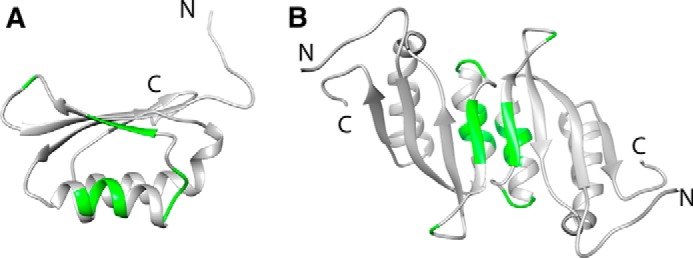
Structure of the RDPheH(25–117) dimer. The dimer was generated by replacing each monomer of the RDTyrH dimer (Protein Data Bank code 2MDA) with one RDPheH(25–117) monomer (Protein Data Bank code 2PHM) using Chimera (36). The dimer interface residues are indicated in green in the RDPheH(25–117) monomer (A) and the RDPheH(25–117) dimer (B).
Identification of the Phenylalanine-binding Site on RDPheH(25–117)
The residues in the allosteric site for phenylalanine were identified using an approach similar to that used to identify the RDPheH dimer interface. Phenylalanine or [2H8]phenylalanine was added at a concentration of 5 mm to 1 mm 15N-labeled, perdeuterated RDPheH(25–117). Comparison of the 15N NOESY spectra of the two samples showed that six residues in the spectrum in the presence of phenylalanine exhibited cross-peaks that were not seen with deuterated phenylalanine (Fig. 4). Glu-44, Ala-47, and Leu-48 showed additional signals at 3.219, 3.602, and 3.983 ppm, close to the proton chemical shifts of the aliphatic protons of phenylalanine in solution. Leu-62, His-64, and Ile-65 showed additional signals at 6.236, 6.906, and 7.385 ppm, close to the proton chemical shifts of the aromatic protons of phenylalanine. These results suggest that phenylalanine binds to the allosteric site of PheH with its Cα and Cβ carbons near Glu-44, Ala-47, and Leu-48, and its aromatic ring near Leu-62, His-64, and Ile-65. The locations of these residues in the structure of RDPheH, shown in Fig. 5, are consistent with two identical phenylalanine-binding sites at the dimer interface, with each site formed by residues from both monomers. The presence of two phenylalanine-binding sites in the RDPheH dimer is consistent with our earlier studies of phenylalanine binding to RDPheH (24).
FIGURE 4.
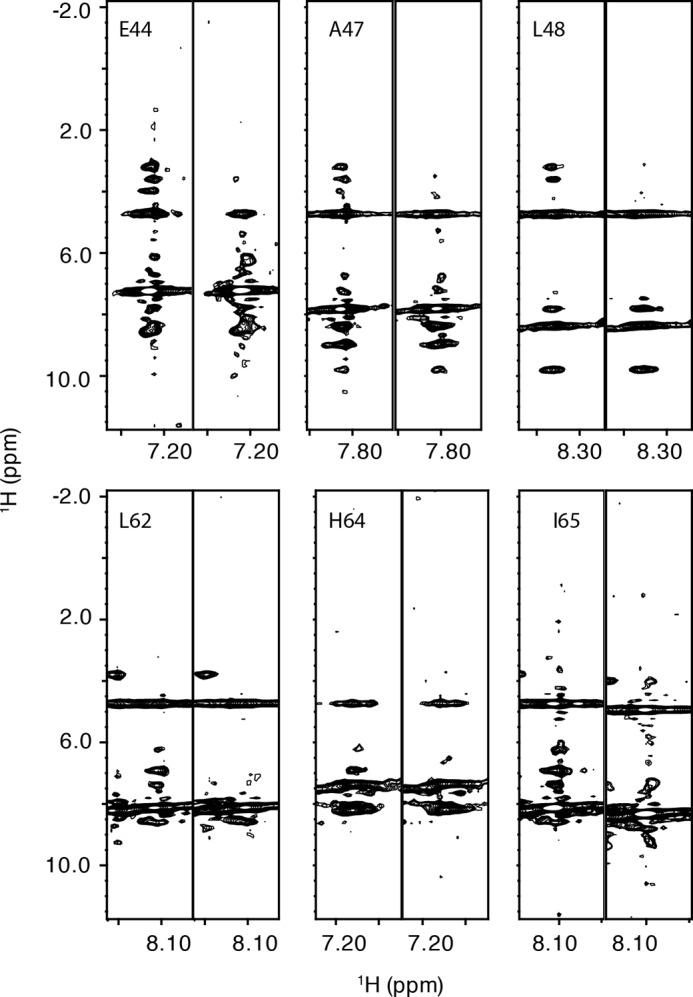
Identification of the phenylalanine-binding site in RDPheH(25–117). Selected 15N slices of three-dimensional NOESY-HSQC spectra of RDPheH(25–117) are shown. For each pair of spectra, the left one is the spectrum of 1 mm 15N,2H-labeled RDPheH(25–117) with 5 mm phenylalanine, whereas the right one is the same region in the spectrum of 1 mm 15N,2H-labeled RDPheH(25–117) with 2H8− phenylalanine. Amide proton assignments are provided at the top of the left slice.
FIGURE 5.
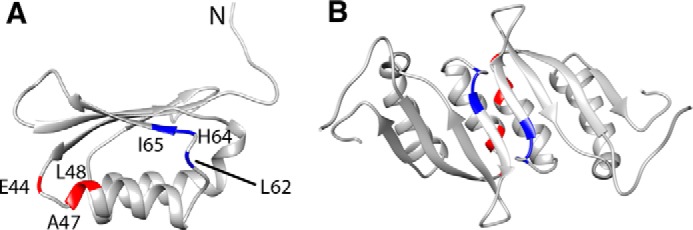
Phenylalanine-binding residues in the RDPheH(25–117) monomer (A) and dimer (B). The aromatic ring of phenylalanine binds to the three blue residues, and the aliphatic region binds to the three red residues.
The binding of phenylalanine to RDPheH is coupled to dimerization of RDPheH (24). To determine whether these six residues were indeed in the phenylalanine-binding site in the regulatory domain, each was first mutated in RDPheH(25–117) to determine whether the phenylalanine concentration dependence for the monomer-dimer equilibrium was affected. Altogether, nine mutant proteins were constructed, including four that incorporated PKU-causing mutations (A47V, L48S, H64N, and I65T). Two of the PKU-related variants (L48S and I65T) were rapidly lost to proteolysis during purification (data not shown), precluding studies of their solution properties. Two variants, E44A and the PKU-related A47V, aggregated to higher oligomers in the presence of excess phenylalanine (data not shown) and were also not studied further. Altogether, mutations could be identified for five of the six residues that yielded versions of RDPheH(25–117) well behaved enough for sedimentation velocity ultracentrifugation experiments (E44Q, A47G, L48V, L62V, and H64N). For each of these five variants, the effect of phenylalanine on the weight-average sedimentation coefficient (sw) value was determined. The results are shown in Fig. 6A. All five mutations yield regulatory domains that require higher concentrations of phenylalanine to form dimers (Table 1), consistent with all five residues being involved in binding phenylalanine. The smallest effects were seen with the L62V and E44Q mutations, whereas the L48V mutation had the largest effect on phenylalanine binding.
FIGURE 6.

Phenylalanine binding and activation of PheH variants. A, effect of the concentration of phenylalanine on the weight-average sedimentation coefficient (sw) of wild type (▵), L62V (○), E44Q (●), A47G (▴), H64N (□), and L48V (■) RDPheH(25–117). The lines are from fits of the data to sw = sm + Δsw × [Phe]/(KPhe + [Phe]). The value of sm was set to 1.3 based on the sw values in the absence of phenylalanine. For H64N and L48V RDPheH(25–117), the sw value of the dimer was assumed to be identical to that for wild-type RDPheH(25–117). The data for wild-type RDPheH(25–117) are from Ref. 25. B, representative fluorescence changes upon binding of phenylalanine to wild-type PheH (▵), A47G PheH (▴), H64N PheH (□), and L48V PheH (■) in 0.2 m HEPES, pH 7.5, at 25 °C. The lines are from fits of the data to Δfluorescence = ΔFlmax x× [Phe]n/(Kactn + [Phe]n). C, activation of PheH variants by phenylalanine. Each enzyme (25 μm) was incubated with 250 μm phenylalanine at 23 °C for 10 min before being diluted 100-fold into assay mix containing 1 mm phenylalanine and all other assay components, and the initial rate of tyrosine formation was determined.
TABLE 1.
Kinetic parameters for PheH variants
| Variant | Kdimera | Kactb | K0.5c | kcatc |
|---|---|---|---|---|
| mm | mm | mm | min−1 | |
| Wild type | 0.016 ± 0.001 | 0.054 ± 0.03d (2.4 ± 0.3)e | 0.30 ± 0.01 (2.0 ± 0.1) | 580 ± 10 |
| A47G | 0.23 ± 0.04 | 0.17 ± 0.01 (2.2 ± 0.5) | 0.63 ± 0.04 (1.9 ± 0.2) | 350 ± 10 |
| E44Q | 0.060 ± 0.010 | NDf | ND | ND |
| L62V | 0.063 ± 0.017 | ND | ND | ND |
| H64N | 1.7 ± 0.1 | 0.79 ± 0.08 (2.0 ± 0.3) | 4.1 ± 0.4 (2.2 ± 0.4) | 390 ± 30 |
| L48V | 4.1 ± 0.5 | 1.6 ± 0.1 (2.5 ± 0.2) | 1.8 ± 0.4 (1.4 ± 0.2) | 130 ± 20 |
a Kd value for phenylalanine binding to RDPheH25–117 determined by AUC.
b Kd value for phenylalanine binding to PheH based on the fluorescence change in the presence of phenylalanine. The values are averages of at least three individual measurements.
c Steady-state kinetic parameters with phenylalanine as substrate, after activation by 5–10 mm phenylalanine.
d Data are from Ref. 25.
e This is the Hill coefficient.
f ND means not determined.
The three mutations causing the greatest decrease in the affinity of RDPheH(25–117) for phenylalanine, A47G, L48V, and H64N, were incorporated into the intact protein to determine directly their effects on activation by phenylalanine. The conformational change in the enzyme that accompanies activation results in a shift in the fluorescence emission to longer wavelengths (14). The effects of the concentration of phenylalanine on this fluorescence change for each of the three mutant proteins and for the wild-type enzyme are shown in Fig. 6B. The relative effects of the mutations on the concentration of phenylalanine required for activation agree with the relative effects on the concentration of phenylalanine required to form the regulatory domain dimer (Table 1), with the L48V enzyme showing the greatest change, providing further evidence that these residues are involved in binding of phenylalanine in the allosteric site. For all three mutant proteins, the effect of the phenylalanine concentration on the protein fluorescence was fit significantly better by the Hill equation than by an equation for noncooperative binding, as is the case for the wild-type enzyme (14), with Hill coefficients of ∼2 (Table 1).
Finally, the effects of these three mutations on phenylalanine activation of PheH were examined directly in tyrosine formation assays. Each enzyme was incubated with or without 250 μm phenylalanine for 10 min before diluting the enzyme into the assay mix containing BH4 and 1 mm phenylalanine. Reactions were stopped after 30 s to minimize any activation during the assay. The results are shown in Fig. 6C. The data are consistent with the effects on dimerization of the isolated regulatory domain and on the fluorescence of the intact protein. A47G PheH showed an activation of ∼4-fold, compared with ∼10-fold activation of WT PheH. Neither L48V PheH nor H64N PheH showed significant activation at this concentration of phenylalanine.
The steady-state kinetic parameters for these three mutants were also determined with phenylalanine as the substrate to determine whether the mutations altered the properties of the activated enzyme. For these analyses, the enzymes were treated with high concentrations of phenylalanine (5 mm phenylalanine for A47G PheH and 10 mm for L48V and H64N PheH) to convert each to the fully activated form. Under these conditions, the fluorescence emission spectra of the mutant proteins were identical to that of the wild-type enzyme (data not shown). The kinetic data were better fit by the Hill equation, as has been observed for the activated wild-type enzyme (3). The kcat values for the A47G and H64N variants were about 60% of the wild-type enzyme, whereas that for the L48V variant was ∼5-fold lower than the wild-type value (Table 1). For all three, the Km values for phenylalanine increased severalfold.
Discussion
Although the allosteric behavior of liver PheH is well established, and the initial structure of a dimeric mutant containing the catalytic and regulatory domains provides a plausible structural basis for the lack of activity of the resting enzyme, the location and even the existence of an allosteric site have been controversial (2, 3). The data presented here strongly support the conclusion that the allosteric site is located at the interface of the regulatory domain dimer that is present in the activated enzyme. The involvement of residues from both monomers in a single allosteric site also provides an obvious structural basis for the stabilization of the regulatory domain dimer by phenylalanine.
There is growing evidence that the conformational change accompanying allosteric activation of PheH involves dimerization of the regulatory domains (24, 25). The present results establish that the regulatory domains form a side-by-side dimer similar in structure to those formed by other ACT domains (21), including that of TyrH (9). The participation of residues on helix α1 and strand β2 in the dimer interface of RDPheH is similar to what is seen in other side-by-side ACT domain dimers (10, 33). The NMR data suggest that five additional residues on loops connected to strand β2 are in the dimer interface (Fig. 3). Four (Asn-58, Asp-59, Ile-60, and Asn-61) are on loop 2 between helix α1 and strand β2, whereas Leu-72 is on loop 3 between strands β2 and β3. In the structural model for the RDPheH(25–117) dimer in Fig. 3B, Leu-72 is not positioned appropriately to be involved in the dimer interface. This suggests that loop 3 has a different conformation in the activated protein.
The NMR data (Fig. 4) establish that the amide nitrogens of six residues (Glu-44, Ala-47, Leu-48, Leu-62, His-64, and Ile-65) on the regulatory domain of PheH are close to phenylalanine when the amino acid is bound to the regulatory domain, and thus they are likely to be in the allosteric site. Conservative mutations of these residues in RDPheH(25–117) decrease the affinity for phenylalanine, confirming that these residues are involved in binding phenylalanine. Although we were unable to obtain a well behaved variant of Ile-65 for AUC analysis, the I65T mutation has been reported to completely abolish phenylalanine binding of maltose-binding protein fusion of PheH (34). The effect on activation by phenylalanine parallels the effect on dimerization for the three variants of the intact protein that were examined, providing further evidence that these residues are part of the allosteric site. The similar effects on dimerization of the isolated domain and on activation are consistent with a model in which activation of PheH by phenylalanine is linked to the dimerization of the regulatory domains.
The two sets of residues identified here as binding in the allosteric site for phenylalanine are far away from each other in one RDPheH(25–117) monomer (Fig. 5A). However, the residues from opposing monomers are closer in space in the dimer (Fig. 5B), suggesting that phenylalanine binds across the interface of the RDPheH dimer, with Glu-44, Ala-47, and Leu-48, at the N terminus of helix α1 in one monomer, interacting with the aliphatic region of phenylalanine, and Leu-62, His-64, Ile-65, at the N terminus of β2 on the other monomer, interacting with the aromatic ring of phenylalanine. This observation supports the orientation of the individual monomers in Fig. 3 based on the structure of the RDTyrH dimer. Several crystal structures of ACT domains that bind small ligands have been reported. The binding site for PheH is most similar to that of bacterial prephenate dehydratase (35); only three of the six residues identified here are conserved in that protein.
Although A47G, H64N, and L48V PheH variants exhibit lower levels of activation at physiological levels of phenylalanine, all three have kcat values ≥20% of the wild-type enzyme when treated with very high levels of phenylalanine. In addition, the K0.5 values are within an order of magnitude of the wild-type values, and the activated variants still exhibit cooperativity. These changes are significantly less than the effects of the same mutations on binding to the allosteric site, establishing that the effects of the mutations are not generalized effects on the protein structure. We also cannot rule out the possibility that the steady-state kinetic parameters in Table 1 underestimate the activity of the activated mutant proteins due to their much weaker affinities for phenylalanine to the allosteric site.
Overall, the present results establish that the regulatory domain of PheH can form a side-by-side ACT domain dimer stabilized by phenylalanine and identify the allosteric site for phenylalanine binding. The data also provide further evidence that the activation of PheH by phenylalanine is linked to phenylalanine-stabilized dimerization of the regulatory domains.
Author Contributions
S. Z. and P. F. F. designed the research and wrote the paper. S. Z performed the experiments.
Acknowledgments
We thank Drs. Andrew P. Hinck and Dmitri Ivanov for helpful suggestions during the course of this work. The assistance of Virgil Schirf with the analytical ultracentrifugation and Dr. Kristin Cano-McCue with the NMR spectroscopy is gratefully acknowledged. Support for the Center for Macromolecular Interactions and the Center for NMR Spectroscopy from the Office of the Vice President for Research at University of Texas Health Science Center at San Antonio is gratefully acknowledged.
This work was supported by National Institutes of Health Grant GM047291 and The Welch Foundation Grant AQ-1245. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PheH
- phenylalanine hydroxylase
- BH4
- tetrahydrobiopterin
- TyrH
- tyrosine hydroxylase
- RDPheH
- the regulatory domain of PheH, residues 2–117
- RDPheH(25–117)
- RDPheH residues 25–117
- HSQC
- heteronuclear single-quantum coherence
- PKU
- phenylketonuria.
References
- 1. Eisensmith R. C., and Woo S. L. (1991) Phenylketonuria and the phenylalanine hydroxylase gene. Mol. Biol. Med. 8, 3–18 [PubMed] [Google Scholar]
- 2. Fitzpatrick P. F. (2012) Allosteric regulation of phenylalanine hydroxylase. Arch. Biochem. Biophys. 519, 194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flydal M. I., and Martinez A. (2013) Phenylalanine hydroxylase: function, structure, and regulation. IUBMB Life 65, 341–349 [DOI] [PubMed] [Google Scholar]
- 4. Døskeland A. P., Martinez A., Knappskog P. M., and Flatmark T. (1996) Phosphorylation of recombinant human phenylalanine hydroxylase: effect on catalytic activity, substrate activation and protection against non-specific cleavage of the fusion protein by restriction protease. Biochem. J. 313, 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kobe B., Jennings I. G., House C. M., Michell B. J., Goodwill K. E., Santarsiero B. D., Stevens R. C., Cotton R. G., and Kemp B. E. (1999) Structural basis of autoregulation of phenylalanine hydroxylase. Nat. Struct. Biol. 6, 442–448 [DOI] [PubMed] [Google Scholar]
- 6. Goodwill K. E., Sabatier C., Marks C., Raag R., Fitzpatrick P. F., and Stevens R. C. (1997) Crystal structure of tyrosine hydroxylase at 2.3 Å and its implications for inherited neurodegenerative diseases. Nat. Struct. Biol. 4, 578–585 [DOI] [PubMed] [Google Scholar]
- 7. Wang L., Erlandsen H., Haavik J., Knappskog P. M., and Stevens R. C. (2002) Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochemistry 41, 12569–12574 [DOI] [PubMed] [Google Scholar]
- 8. Fitzpatrick P. F. (2003) Mechanism of aromatic amino acid hydroxylation. Biochemistry 42, 14083–14091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang S., Huang T., Ilangovan U., Hinck A. P., and Fitzpatrick P. F. (2014) The solution structure of the regulatory domain of tyrosine hydroxylase. J. Mol. Biol. 426, 1483–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grant G. A. (2006) The ACT domain: a small molecule binding domain and its role as a common regulatory element. J. Biol. Chem. 281, 33825–33829 [DOI] [PubMed] [Google Scholar]
- 11. Daubner S. C., Le T., and Wang S. (2011) Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 508, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jennings I. G., Teh T., and Kobe B. (2001) Essential role of the N-terminal autoregulatory sequence in the regulation of phenylalanine hydroxylase. FEBS Lett. 488, 196–200 [DOI] [PubMed] [Google Scholar]
- 13. Li J., Dangott L. J., and Fitzpatrick P. F. (2010) Regulation of phenylalanine hydroxylase: conformational changes upon phenylalanine binding detected by hydrogen/deuterium exchange and mass spectrometry. Biochemistry 49, 3327–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Phillips R. S., Parniak M. A., and Kaufman S. (1984) Spectroscopic investigation of ligand interaction with hepatic phenylalanine hydroxylase: evidence for a conformational change associated with activation. Biochemistry 23, 3836–3842 [DOI] [PubMed] [Google Scholar]
- 15. Shiman R., Gray D. W., and Pater A. (1979) A simple purification of phenylalanine hydroxylase by substrate-induced hydrophobic chromatography. J. Biol. Chem. 254, 11300–11306 [PubMed] [Google Scholar]
- 16. Shiman R., Xia T., Hill M. A., and Gray D. W. (1994) Regulation of rat liver phenylalanine hydroxylase. II. Substrate binding and the role of activation in the control of enzymatic activity. J. Biol. Chem. 269, 24647–24656 [PubMed] [Google Scholar]
- 17. Carluccio C., Fraternali F., Salvatore F., Fornili A., and Zagari A. (2015) Towards the identification of the allosteric Phe-binding site in phenylalanine hydroxylase. J. Biomol. Struct. Dyn. 10.1080/07391102.2015.1052016 [DOI] [PubMed] [Google Scholar]
- 18. Jaffe E. K., Stith L., Lawrence S. H., Andrake M., and Dunbrack R. L. Jr. (2013) A new model for allosteric regulation of phenylalanine hydroxylase: Implications for disease and therapeutics. Arch. Biochem. Biophys. 530, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flydal M. I., Mohn T. C., Pey A. L., Siltberg-Liberles J., Teigen K., and Martinez A. (2010) Superstoichiometric binding of l-Phe to phenylalanine hydroxylase from Caenorhabditis elegans: evolutionary implications. Amino Acids 39, 1463–1475 [DOI] [PubMed] [Google Scholar]
- 20. Thórólfsson M., Ibarra-Molero B., Fojan P., Petersen S. B., Sanchez-Ruiz J. M., and Martínez A. (2002) l-Phenylalanine binding and domain organization in human phenylalanine hydroxylase: a differential scanning calorimetry study. Biochemistry 41, 7573–7585 [DOI] [PubMed] [Google Scholar]
- 21. Lang E. J., Cross P. J., Mittelstadt G., Jameson G. B., and Parker E. J. (2014) Allosteric ACTion: the varied ACT domains regulating enzymes of amino-acid metabolism. Curr. Opin. Struct. Biol. 29, 102–111 [DOI] [PubMed] [Google Scholar]
- 22. Ronau J. A., Paul L. N., Fuchs J. E., Corn I. R., Wagner K. T., Liedl K. R., Abu-Omar M. M., and Das C. (2013) An additional substrate binding site in a bacterial phenylalanine hydroxylase. Eur. Biophys. J. 42, 691–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J., Ilangovan U., Daubner S. C., Hinck A. P., and Fitzpatrick P. F. (2011) Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase. Arch. Biochem. Biophys. 505, 250–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang S., Roberts K. M., and Fitzpatrick P. F. (2014) Phenylalanine binding is linked to dimerization of the regulatory domain of phenylalanine hydroxylase. Biochemistry 53, 6625–6627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang S., Hinck A. P., and Fitzpatrick P. F. (2015) The amino acid specificity for activation of phenylalanine hydroxylase matches the specificity for stabilization of regulatory domain dimers. Biochemistry 54, 5167–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roberts K. M., Khan C. A., Hinck C. S., and Fitzpatrick P. F. (2014) Activation of phenylalanine hydroxylase by phenylalanine does not require binding in the active site. Biochemistry 53, 7846–7853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marley J., Lu M., and Bracken C. (2001) A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 28. Bax A. (1994) Multidimensional nuclear magnetic resonance methods for protein studies. Curr. Opin. Struct. Biol. 4, 738–744 [Google Scholar]
- 29. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 30. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 31. Gabrielson J. P., Randolph T. W., Kendrick B. S., and Stoner M. R. (2007) Sedimentation velocity analytical ultracentrifugation and SEDFIT/c(s): limits of quantitation for a monoclonal antibody system. Anal. Biochem. 361, 24–30 [DOI] [PubMed] [Google Scholar]
- 32. Eghbalnia H. R., Wang L., Bahrami A., Assadi A., and Markley J. L. (2005) Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J. Biomol. NMR 32, 71–81 [DOI] [PubMed] [Google Scholar]
- 33. Schuller D. J., Grant G. A., and Banaszak L. J. (1995) The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nat. Struct. Biol. 2, 69–76 [DOI] [PubMed] [Google Scholar]
- 34. Gjetting T., Petersen M., Guldberg P., and Güttler F. (2001) Missense mutations in the N-terminal domain of human phenylalanine hydroxylase interfere with binding of regulatory phenylalanine. Am. J. Hum. Genet. 68, 1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tan K., Li H., Zhang R., Gu M., Clancy S. T., and Joachimiak A. (2008) Structures of open (R) and close (T) states of prephenate dehydratase (PDT)–implication of allosteric regulation by l-phenylalanine. J. Struct. Biol. 162, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]