

Abstract
The NRF2 (also known as NFE2L2) transcription factor is a critical regulator of genes involved in defense against oxidative stress. Previous studies suggest that Nrf2 plays a role in adipogenesis in vitro, and deletion of the Nrf2 gene protects against diet-induced obesity in mice. Here, we demonstrate that resistance to diet-induced obesity in Nrf2−/− mice is associated with a 20–30% increase in energy expenditure. Analysis of bioenergetics revealed that Nrf2−/− white adipose tissues exhibit greater oxygen consumption. White adipose tissue showed a >2-fold increase in Ucp1 gene expression. Oxygen consumption is also increased nearly 2.5-fold in Nrf2-deficient fibroblasts. Oxidative stress induced by glucose oxidase resulted in increased Ucp1 expression. Conversely, antioxidant chemicals (such as N-acetylcysteine and Mn(III)tetrakis(4-benzoic acid)porphyrin chloride) and SB203580 (a known suppressor of Ucp1 expression) decreased Ucp1 and oxygen consumption in Nrf2-deficient fibroblasts. These findings suggest that increasing oxidative stress by limiting Nrf2 function in white adipocytes may be a novel means to modulate energy balance as a treatment of obesity and related clinical disorders.
Keywords: adipose tissue, antioxidant, oxidative stress, reactive oxygen species (ROS), uncoupling protein
Introduction
Obesity is characterized by excess adipose tissue mass, and it is associated with various chronic diseases, including type 2 diabetes, hypertension, atherosclerosis, and cancer. Obesity is currently one of the leading causes of morbidity and mortality globally. There are two different types of adipose tissues in mammals: white adipose tissue and brown adipose tissue. White adipose tissue (WAT)2 serves an important role in maintaining energy homeostasis in the body by storing surplus energy as triglyceride and releasing free fatty acids to meet the needs of other organs when energy intake is less than energy expenditure. White adipocytes contain few mitochondria and are characterized by a single, large lipid droplet. Brown adipose tissue (BAT) is specialized in energy consumption through heat production in response to cold or excess calories. This response, known as adaptive thermogenesis, protects against cold exposure and plays a beneficial role in regulating energy balance in mice that have a variable diet (1, 2). Brown adipocytes contain multiple small lipid droplets and are highly enriched in mitochondria. In humans, BAT is abundant in newborns, and it was originally thought to be nonexistent or nonfunctional in adults. However, recent studies have shown metabolically active BAT in adult humans through the use of PET/CT scans (positron emission tomography associated with computed tomography) (3–5). These BAT depots are observed in the neck and in other locations, including the suprarenal, supraclavicular, and paravertebral region of the body (6–11).
The thermogenic function of BAT is dependent on UCP1, a protein expressed in the inner mitochondrial membrane of brown fat cells that functions to dissipate the proton gradient generated during oxidative phosphorylation. This is achieved by increasing the permeability of the inner mitochondrial membrane, allowing protons that have been pumped into the intermembrane space to return to the mitochondrial matrix. This proton leak, termed mitochondrial uncoupling, is physiologically relevant, accounting for 20–25% of the basal metabolic rate (12–14). Studies have shown that mice lacking Ucp1 are more susceptible to diet-induced obesity (15). Conversely, transgenic mice with increased Ucp1 expression in WAT are resistant to high fat diet-induced obesity (16). In addition, transgenic mice expressing Ucp1 in skeletal muscle showed improved glucose tolerance after being fed a high fat diet when compared with wild type mice (17). In humans, it has been estimated that as little as 50 g of BAT (<0.1% of body weight) could utilize up to 20% of basal caloric needs if maximally stimulated (2). Because of the enormous thermogenic capacity of BAT, there has been intense interest in the determinants of BAT function and the potential for treating obesity by manipulating BAT function. In addition to brown adipose tissue, several studies have shown that brown-like adipocytes (often referred to as “brite” or “beige” adipocytes) occur in some classical white adipose depots as well as skeletal muscle and that these serve as extremely promising targets for treatment of obesity (18–21).
NRF2 (nuclear factor-erythroid 2-related factor 2, also known as NFE2L2) is a member of the Cap-N-Collar subfamily of bZIP transcription factors that also includes NRF1, p45NFE2, and NRF3 (22–24). Under basal conditions, NRF2 is maintained at low levels through the ubiquitin proteasome proteolytic pathway mediated by KEAP1 (Kelch-like ECH-associated protein 1) (24, 25). In response to oxidative stress, NRF2 escapes KEAP1-mediated negative regulation, resulting in nuclear accumulation of NRF2 (26). NRF2 is able to form heterodimers with other transcription factors, including sMAF (small Maf proteins), JUND, and ATF4 (activation transcription factor 4), to bind and activate gene transcription via cis-acting antioxidant response elements in the promoters of cellular defense genes (27–30). These genes include a wide variety of enzymes involved in detoxification, such as NAD(P)H quinone oxidoreductase 1 and heme oxygenase 1 (31, 32). Nrf2 is also involved in driving the expression of key antioxidant enzymes, such as the catalytic and regulatory subunits of γ-glutamate cysteine ligase, and phase 2 enzymes, such as glutathione transferases (33). Mice lacking the Nrf2 gene demonstrate no obvious outward defects; however, they have an increased susceptibility to toxicant and oxidative stress-induced diseases, and they are also more prone to developing autoimmune disorders as they age (34–38). Compared with wild type mice, Nrf2 mutant mice have also been shown to be more sensitive to carcinogens in the stomach, suggesting an important role of Nrf2 and antioxidant response in the protection against cancer (39).
Recent findings suggest that Nrf2 plays a role in regulating the expression of key genes involved in the process of adipogenesis (40, 41). In addition, mice lacking Nrf2 demonstrate a resistance to high fat diet-induced obesity, but the underlying mechanism is not clear (41). In this study, we explored the role of Nrf2 in mitochondrial function. Specifically, we assessed energy expenditure, changes in adipose tissue mass, morphology, and gene expression in Nrf2−/− mice fed both a normal and high fat diet. In addition, Nrf2−/− cells were assessed for mitochondrial function and gene expression. Our studies are consistent with the hypothesis that oxidative stress modulates mitochondrial function to control fat storage and energy expenditure in Nrf2 knock-outs.
Experimental Procedures
Reagents
Dulbecco's modified Eagle's medium (DMEM), α-minimum essential medium, fetal bovine serum (FBS), l-glutamine, penicillin, streptomycin, CM-H2DCFDA, MitoTracker Green FM, nanoyl-acridine orange, and Superscript III reverse transcriptase were purchased from Invitrogen. Glucose oxidase, butylated hydroxyanisole (BHA), and anti-β-actin antibody (A1978) were purchased from Sigma. UCP1 (ab23841) antibody was purchased from Abcam (Cambridge, MA). α-Tubulin antibody (ab3873) was purchased from Cell Signaling (Beverly, MA). TRIzol RNA reagent, 2× FastStart SYBR Green Master mix, was purchased from Roche Applied Science. The RNeasy MinElute cleanup kit was from Qiagen (Valencia, CA). The enhanced chemiluminescence substrate kit was from Pierce. Mn(III)tetrakis(4-benzoic acid)porphyrin chloride (MnTBAP) was purchased from EMD Millipore (Billerica, MA).
Cells
Mouse embryonic fibroblasts (MEFs) were generated as described previously (41) and cultured in DMEM supplemented with 10% FBS, 8.3 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Oxidative stress was induced in cells by incubating with 25 milliunits/ml glucose oxidase for 6 h at 37 °C, and antioxidant treatment was accomplished by incubation with 40 μm BHA or with 10 μm MnTBAP.
Mice
All experiments were carried out following guidelines of the University of California Irvine institutional animal care and use committee. Nrf2 knock-out mice have been described previously (42) and were back-crossed 10 generations to C57BL/6J background to produce nearly isogenic lines. Studies were done using age-matched 12–16-week-old Nrf2−/− and wild type male mice. Mice were housed on 12-h light and dark cycles under controlled environmental settings (23 ± 1 °C), with free access to food and water. Age-matched wild type and Nrf2−/− mice were fed either a standard global rodent diet (Harland Teklad 2020x) or a high fat diet chow containing 60% kcal from fat (Harlan Teklad TD.06414). The fatty acid profile of the high fat diet as a percentage of total fat consists of 37% saturated, 47% monounsaturated, of 16% polyunsaturated fat.
Metabolic and Indirect Calorimetry Studies
Indirect calorimetry to measure oxygen (O2) consumption, carbon dioxide (CO2) production, activity, and food intake was done using the Comprehensive Laboratory Animal Monitoring System (Columbus Instruments, Columbus, OH). Mice were individually housed in metabolic chambers maintained at 20–22 °C in a 12-h light/12-h dark cycle. After the mice had adapted to the environment of the metabolic chamber for 72 h, metabolic parameters were assessed. Activity was monitored by the number of times a mouse moved through infrared beams crossing the cages. Body fat and lean mass were measured using an EchoMRI-100 (Echo Medical Systems, Houston, TX). Glucose tolerance tests were conducted in animals that were fasted overnight. Glucose was administered by intraperitoneal injection at a dose of 1 mg/g body weight. Blood samples were collected from the tail vein at time 0 (prior to the glucose injection) and 30, 60, and 120 min after glucose loading for blood glucose. Blood glucose concentrations were measured using a TRUE-Test blood glucose meter (Nipro Diagnostics, Fort Lauderdale, FL). Fasting serum triglyceride levels were analyzed with a serum triglyceride kit according to the manufacturer's instructions (Sigma), and serum concentrations of insulin were determined using the Ultra Sensitive Mouse Insulin ELISA Kit from Crystal Chem (Downers Grove, IL).
RNA Isolation and Quantitative Real-time PCR
RNA was extracted using TRIzol RNA reagent and further purified using the RNeasy MinElute Cleanup Kit. cDNA synthesis was generated with a Superscript III first-strand synthesis kit according to the manufacturer's recommendation. Amplification of cDNA occurred in a Step One Plus PCR machine (ABI) using the FastStart SYBR Green reagent in duplicate 10-μl reactions. The ribosomal phosphoprotein large P0 (RPLP0) gene was used as a reference control, and relative expression was calculated using the expression, 2(Ct test gene − CtRPLP0). -Fold change in expression was determined from the difference between the averaged expression levels relative to control. Primers used are listed in Table 1.
TABLE 1.
List of primers
| Gene | Forward | Reverse |
|---|---|---|
| Pgc1α | CGGAAATCATATCCAACCAG | TGAGGACCGCTAGCAAGTTTG |
| NRF1 | CGCAGCACCTTTGGAGAA | CCCGACCTGTGGAATACTTG |
| Dio2 | AAGCACGTGGCGTTGCTTCTG | TGGAGACGTGCACCACACTGG |
| Ucp1 | ACTGCCACACCTCCAGTCATT | CTTTGCCTCACTCAGGATTGG |
| Cox7a1 | GGGCGAAGAGGGGAGGTGAC | TTGTCCATTCCCCCGCCTTT |
| Cox8b | GAACCATGAAGCCAACGACT | GCGAAGTTCACAGTGGTTCC |
| Sirt3 | TGGCGTTGTGAAACCCGACA | AACGAACGGCCCCACCAAGT |
| CideA | TGCTCTTCTGTATCGCCAGT | GCCGTGTTAAGGAATCTGCTG |
| Tfam | GGAATGTGGAGCGTGCTAAAA | TGCTGGAAAACACTTCGGAATA |
| Rplpo | CCCCTTCTCCTTCGGGCTGAT | GTGTGGCACCGAGGCAACAGT |
Western Blotting
Protein lysates were prepared using Nonidet P-40 lysis buffer, and protein concentrations were determined using Bio-Rad protein assay reagent with BSA as the protein standard. Samples were electrophoresed on SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Membranes were then blocked in 5% milk at room temperature for 1 h and incubated with primary antibody overnight at 4 °C. Subsequently, blots were washed, and peroxidase-conjugated secondary antibody was added and incubated for 1 h. Blots were visualized using a chemiluminescent detection system.
Histology and Immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin and imbedded in paraffin. Tissue sections were stained with H&E and visualized using a Nikon microscope equipped with a CCD camera.
RNA Sequencing (RNA-seq) Analysis
From WAT of mice fed a high fat diet, total RNA was isolated using TRIzol RNA reagent and further purified using RNeasy MinElute Cleanup Kit. Three independent replicates were used for both wild type and Nrf2−/− mice. Integrity and purity of the samples were assessed using an Agilent Bioanalyzer, and libraries for RNA-Seq were generated using the Illumina Truseq kit version 2 per the manufacturer's instructions. RNA-seq libraries were sequenced on an Illumina HiSeq 2500 next generation sequencer using single read 50-bp (SR50) chemistry with a read depth of >10 million reads/sample. Reads are processed and mapped to a reference genome (mm10, built name NCBIM37) using ELAND. Transcript abundance quantification was assigned using normalized read counts, such as FPKM, and statistical testing for differential expression was performed using Cufflinks. Differentially expressed genes were further analyzed using Ingenuity Pathways Analysis.
Flow Cytometry
For intracellular reactive oxygen species measurements, cells were incubated with 10 μm CM-H2DCFDA in PBS for 15 min at 37 °C. Fluorescence was analyzed by flow cytometry. Oxidative stress was induced in cells by incubating with 25 milliunits/ml glucose oxidase for 6 h at 37 °C prior to CM-H2DCFDA staining. Antioxidant treatment was accomplished by incubation with 100 μm BHA for 6 h at 37 °C.
Measurement of Oxygen Consumption in Adipocytes and MEF Cells Using the XF24 Extracellular Flux Analyzer
The Seahorse XF24 Extracellular Flux Analyzer was utilized for measuring the oxygen consumption of adherent MEF cells as well as extracted adipose tissue explants. The protocol for acquisition of intra-abdominal WAT, preparation of tissue, and usage of XF24 was performed as described previously (43), with minor changes. The sensor cartridge was calibrated with calibration buffer (Seahorse Bioscience, North Billerica, MA) at 37 °C the day before oxygen consumption rate (OCR) measurements. Intra-abdominal WAT was dissected into small pieces (∼5 mg) and washed extensively with XF-Basal Medium containing 25 mm HEPES and 25 mm glucose. Two pieces of isolated WAT weighing a total of 10 mg were placed into each well of a XF24 Islet Capture Microplate (Seahorse Bioscience), and each well was covered with a capture screen to hold the tissues in place while allowing for free perfusion. XF Basal Medium supplemented with 25 mm glucose, 1 mm sodium pyruvate, and 2 mm l-glutamine (450 μl) was then added, and the OCR was measured in a XF24 extracellular flux analyzer (Seahorse Bioscience) according to the manufacturer's protocol. OCR readings were taken over time under basal conditions and after the addition of mitochondrial inhibitors, oligomycin (8 μg/ml), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 8 μm), and a mixture of rotenone (3 μm) and antimycin-A (12 μm), in succession. A minimum of five wells were utilized per condition in any given experiment. Plates were saved, and tissue protein concentrations were measured by a Bradford assay. OCR was determined by Seahorse XF-24 software as a function of time and normalized by protein concentration (pmol/min/μg), respectively. Bioenergetic parameters assessed were basal, ATP-linked respiration, proton leak, maximal, and reserve capacity. The basal respiration rate was calculated by subtracting the residual OCR determined after the addition of rotenone and antimycin-A. ATP-linked respiration was determined from the difference between basal OCR and OCR following oligomycin addition. The difference in OCR between rotenone and oligomycin represented the amount of oxygen consumed that is due to proton leak. Maximal OCR was determined by subtracting the OCR after rotenone addition from the OCR induced by FCCP. Last, the reserve capacity was calculated by the difference between maximal and basal respiration. All data are expressed as a percentage of the basal OCR to allow comparison between the different genotypes. For MEF cells, 50,000 cells were plated in XF 24-well plates 1 day before analysis. This allowed for 70–90% confluence on the day of measurement. The sensor cartridge was hydrated 24 h before the run as indicated by the manufacturer's protocol. On the day of the run, cells were washed two times with Seahorse Basal Medium containing 25 mm glucose, 1 mm sodium pyruvate, and 4 mm l-glutamine and then covered with 450 μl of the same basal medium. The OCR was measured in a XF24 extracellular flux analyzer (Seahorse Bioscience) according to the manufacturer's protocol. OCR readings were taken over time under basal conditions and after the addition of mitochondrial inhibitors, oligomycin (1 μm), FCCP (1 μm), and a mixture of rotenone (1 μm) and antimycin-A (1 μm), in succession. A minimum of five wells were utilized per condition in any given experiment. Plates were saved, and cellular protein concentrations were measured by a Bradford assay. OCR was determined by Seahorse XF-24 software as a function of time and normalized by protein concentration (pmol/min/μg), respectively.
Measurement of Oxygen Consumption Utilizing the BD Oxygen Biosensor Plate
Fluorometric measurements of oxygen consumption in adherent mouse cells were done using BD Oxygen Biosensor plates (BD Biosciences) (44). Triplicate wells were seeded with 2 × 105 cells suspended in 25 mm glucose DMEM without phenol red. Plates were scanned every 5 min for 120 min in a temperature-controlled (37 °C) plate reader (Thermo, Waltham, MA) with an excitation wavelength of 485 nm and an emission wavelength of 630 nm. Fluorescence values for each well were normalized to their initial value and then to the values of no-cell controls at each time point.
GSH and GSSG Assay
Measurements of GSH and GSSG levels were determined using the Glutathione Assay Kit (Cayman Chemical, Ann Arbor, MI) following the manufacturer's protocol. Tissues were homogenized in 5–10 ml of cold 50 mm MES buffer/g of tissue. Homogenates were spun at 10,000 × g for 15 min, and supernatant fractions were collected. Supernatant fractions were assayed for total reduced (GSH) and oxidized (GSSG) glutathione by the standard enzymatic recycling method after deproteination of samples using 5% metaphosphoric acid and the addition of triethanolamine. Samples for GSSG determination were first incubated at room temperature with 2 μl of 4-vinylpyridine/100-μl sample for 1 h after vigorous vortexing in order to derivatize endogenous GSH and allow for measurement of GSSG specifically. The GSSG was then subtracted from the total GSH to determine actual GSH level and GSH/GSSG ratio, which is used as an indicator for oxidative stress (45).
Statistical Analysis
Data are expressed as means ± S.E., and statistical analysis was done using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). For simple comparisons of two groups, a two-tailed Student's t test for unpaired samples was used. For comparisons of wild type and knock-out groups concerning the influence of high fat diet, a two-way ANOVA analysis was used, followed by a post hoc Bonferroni analysis. p values of ≤0.05 were considered statistically significant and are indicated.
Results
Nrf2−/− Mice Are Resistant to High Fat Diet-induced Obesity
The body weights of wild type and Nrf2−/− male mice were indistinguishable at 12 weeks of age when fed a standard chow diet (Fig. 1A). This observation in mice derived from a C57BL/6 background is consistent with our previous results in mice derived from a mixed C57BL/6 × Sv129 genetic background (41). After exposure to a high fat diet for 6 weeks, the mean body weight of Nrf2−/− mice was 25% less than that of wild type mice (Fig. 1A). Wild type mice nearly doubled their starting weight, whereas Nrf2−/− mice only gained 50% of their starting weight (Fig. 1A). A comparison of adipose tissue mass normalized to lean body mass measured by EchoMRI showed that Nrf2−/− mice exposed to a high fat diet had 50% less whole-body fat compared with wild type mice. Importantly, there was no significant change in the lean body mass of Nrf2−/− mice compared with wild type mice (Fig. 1B). The white and brown adipose tissue weights normalized to body weight were 30% lower in Nrf2−/− mice compared with wild type mice. Relative weights of liver, heart, and kidney were not different between wild type and Nrf2−/− mice (Fig. 1C) (data not shown). To rule out possible lipid deposition in other tissues, sections of various tissues were analyzed. Ectopic accumulation of lipids was not observed in heart or skeletal tissues of both wild type and Nrf2−/− mice placed on a high fat diet (Fig. 1D). However, wild type mice demonstrated significant liver steatosis compared with Nrf2−/− mice (Fig. 1D). These results indicate that lower fat mass in Nrf2−/− mice is not associated with increased lipid accumulation in non-adipose tissues. Along with the reduction in fat mass, intra-abdominal WAT depots of Nrf2−/− mice showed smaller unilocular adipocytes compared with wild type mice (Fig. 1E). The frequency of smaller fat cells, quantitated from H&E images, was significantly higher in Nrf2−/− WAT (Fig. 1F). In addition, BAT from Nrf2−/− mice showed smaller multilocular lipid droplets (Fig. 1G). However, strain differences in subcutaneous adipocytes were not observed (data not shown).
FIGURE 1.

Nrf2−/− mice are resistant to high fat diet-induced obesity. A, comparison of body weights of wild type and Nrf2−/− mice on a regular diet (RD) and after 6 weeks of high fat diet (HFD). B, echo MRI analysis of body compositions of wild type and Nrf2−/− mice. C, comparison of WAT, BAT, and liver and heart weights, normalized to body weights, of wild type and Nrf2−/− mice after 6 weeks of high fat diet. Shown are representative histological sections of skeletal muscle, heart, liver (D), and peritoneal fat from wild type and Nrf2−/− mice (E). F, histogram depicting the size distribution of the measured white adipocytes in wild type and Nrf2−/− mice. G, representative interscapular brown fat from wild type and Nrf2−/− mice. All tissues are taken from mice after 6 weeks of high fat diet feeding. Error bars, S.E.
Nrf2−/− Mice on a High Fat Diet Showed Improved Metabolic Profiles
Nrf2−/− mice on a high fat diet exhibited decreased fasting glucose levels compared with wild type controls (Fig. 2A). Glucose tolerance tests showed significantly lower blood glucose levels in Nrf2−/− mice at the 90 and 120 min time points (Fig. 2B). The area under the curve (AUC)0–120 min of blood glucose was also significantly lower in Nrf2−/− mice than that in wild type controls (Fig. 2C). The improvement in glucose tolerance in Nrf2−/− mice is not due to increased insulin secretion because plasma insulin levels were lower in Nrf2−/− mice compared with wild type controls (Fig. 2D). In addition, serum triglyceride levels were also lower in Nrf2−/− mice (Fig. 2E). Taken together, these findings suggest that Nrf2 deficiency provides protection against the deleterious effects of a high fat diet on plasma levels of glucose and triglycerides.
FIGURE 2.

Nrf2−/− mice on a high fat diet showed improved metabolic profiles. A, fasting glucose levels in wild type and Nrf2−/− mice. B, glucose tolerance test. Mice starved for 12 h were given an intraperitoneal injection of glucose (1 mg/g body weight). Blood samples were taken at the times indicated from the tail vein of the same animals. Glucose was measured as indicated under “Experimental Procedures.” C, area under the curve (AUC) of blood glucose levels during intraperitoneal glucose tolerance test. Shown are plasma insulin (D) and triglyceride levels (E) of wild type and Nrf2−/− mice. Experiments were performed after 6 weeks of high fat diet feeding in all groups. Data are expressed as means ± S.E. (error bars) (n = 8 mice/group), and significance was determined by Student's t test (*, p < 0.05).
Nrf2−/− Mice on High Fat Diet Demonstrate Increased Energy Expenditure
Potential mechanisms, including reduced food intake or increased energy expenditure, could contribute to reduced adiposity in Nrf2−/− mice. To investigate this possibility, we monitored the food consumption and metabolic activities using the Comprehensive Laboratory Animal Monitoring System. Food intake per body weight in Nrf2−/− mice was comparable with that of wild type mice fed the same diet (Fig. 3A). The activity of mice was also measured for 72 h under a 12-h light-dark cycle. No significant difference was observed in either horizontal activity or rearing behavior between Nrf2−/− and wild type mice (Fig. 3B). Although energy expenditure levels in wild type and Nrf2−/− mice fed a regular diet were similar (data not shown), O2 consumption and CO2 generation were significantly higher in Nrf2−/− mice compared with wild type controls under high fat feeding (Fig. 3, C and D). Respiratory quotient (RER) levels were identical in Nrf2−/− and wild type mice, indicating that the utilization of carbohydrate and fat was similar for both groups (Fig. 3E). This suggests that despite the increase in metabolic activity, Nrf2−/− mice have not shifted their energy source dramatically. These findings suggest that resistance to obesity of Nrf2−/− mice is due to increased energy expenditure by increasing metabolic rate.
FIGURE 3.

Nrf2−/− mice on high fat diet demonstrate increased energy expenditure. A, daily food intake of wild type and Nrf2−/− mice over a 6-day period. B, physical activity of wild type and Nrf2−/− mice over a 3-day period measured by horizontal activity and rearing light beam breaks using the Comprehensive Laboratory Animal Monitoring System. Counts of horizontal activity and rearing are shown for both dark and light cycles. Wild type and Nrf2−/− mice were analyzed for oxygen consumption (VO2) (C), energy expenditure (D), and respiratory exchange ratio (E) determined by the Comprehensive Laboratory Animal Monitoring System over a 3-day period. Values from dark and light cycles are shown. Experiments were performed after 6 weeks of high fat diet feeding in all groups. Data are expressed as means ± S.E. (error bars) (n = 6 mice/group), and significance was determined by Student's t test (*, p < 0.05).
Nrf2-deficient WAT Show Increased Oxygen Consumption
To further substantiate these findings, we investigated whether loss of Nrf2 affects mitochondrial function by comparing OCRs of adipose tissues from wild type and Nrf2−/− mice. WAT isolated from Nrf2−/− mice fed either a normal or high fat diet showed an increase in basal OCR in comparison with wild type (Fig. 4A). Proton leak-dependent respiration measured in the presence of the ATP synthase inhibitor, oligomycin, was significantly higher in WAT of Nrf2−/− mice fed a normal diet and was further increased in Nrf2−/− mice fed a high fat diet (Fig. 4B), suggesting that mitochondrial coupling was diminished. High fat diet feeding led to an increase in maximal and reserve mitochondrial respiratory capacities in both wild type and Nrf2−/− mice, but no difference was seen between the genotypes (Fig. 4, C and D). Although WAT of Nrf2−/− mice showed a decrease in reserve capacity, this difference did not reach statistical significance (Fig. 4D). Taken together, these results suggest that mitochondrial activity is increased in Nrf2−/− adipocytes and that this increase in activity is potentially caused by increased proton leak across the mitochondrial inner membrane.
FIGURE 4.

Nrf2-deficient WAT show increased oxygen consumption. A–D, quantitation of basal OCR (A), proton leak (oligomycin OCR − rotenone/antimycin OCR) (B), maximal rate (FCCP OCR − rotenone/antimycin OCR) (C), and reserve capacity (rotenone/antimycin OCR) (D) in WAT of wild type and Nrf2−/− mice. Data were normalized to total protein and represented as means ± S.E. (error bars) (n = 3/group). Significance was determined by two-way ANOVA and Bonferroni post hoc comparison. HFD, high fat diet; RD, regular diet.
Nrf2-deficient MEF Cells Show Increased Oxygen Consumption
To determine whether increased cellular respiration is intrinsic to Nrf2 deficiency, we measured OCR in MEFs utilizing the XF24 extracellular flux analyzer. Compared with wild type MEF cells, basal oxygen consumption rates in Nrf2−/− MEF cells were increased 2.5-fold (Fig. 5A). To confirm these results, oxygen utilization of wild type and Nrf2−/− MEFs was also compared using oxygen biosensor plates in which the fluorescent signal from oxygen-sensitive dye is quenched by O2. In this assay, Nrf2−/− MEF cells showed a similar increase in oxygen consumption compared with wild type MEFs (Fig. 5B). To demonstrate that this effect is specific to Nrf2 deficiency, we asked if re-expression of Nrf2 would lower the oxygen consumption rates in Nrf2−/− MEF cells. As shown in Fig. 5C, retrovirus-mediated expression of Nrf2 in Nrf2−/− MEF cells decreased oxygen consumption by >50%.
FIGURE 5.
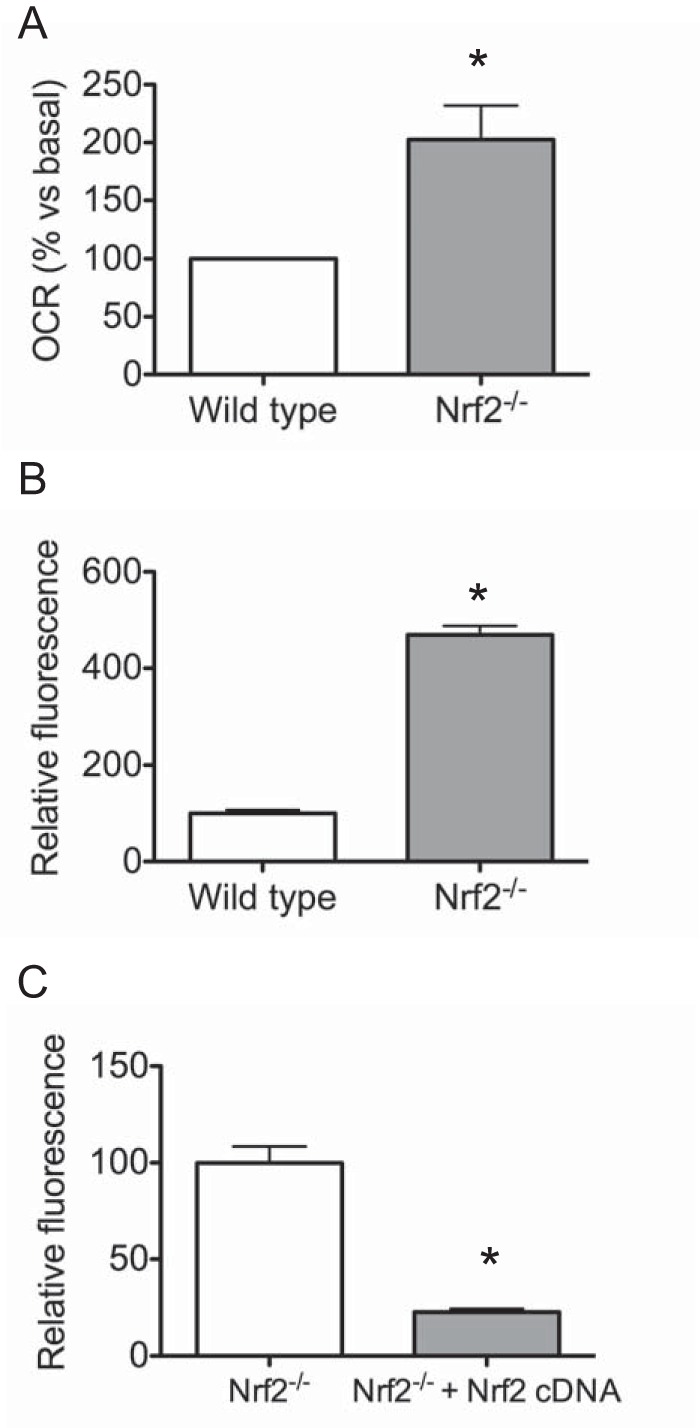
Nrf2-deficient MEF cells show increased oxygen consumption. A, Seahorse X-24 analysis of basal OCR normalized to total protein in wild type and Nrf2−/− MEF cells. Data represent means ± S.E. (n = 3/group), and significance was determined by Student's t test (*, p < 0.05). B, oxygen consumption of wild type and Nrf2−/− MEF cells; C, Nrf2−/− and Nrf2−/− MEF cells stably expressing Nrf2 cDNA. Quantitative changes in respiratory rate were calculated from oxygen consumption curves measured using BD Oxygen Biosensor plates. Data represent means of maximum fluorescence intensity normalized to cell number ± S.E. (error bars) of 3–4 independent experiments.
Brown Fat Markers Are Up-regulated in White Adipose Tissues of Nrf2−/− Mice
To determine the molecular basis of the lean phenotype of Nrf2−/− mice, we performed gene expression analysis by high throughput RNA-seq of adipocytes derived from intra-abdominal fat pads of wild type and Nrf2 mice fed a high fat diet. Genes that were differentially expressed were analyzed with Ingenuity Pathway Analysis software. The top biological functions and disease networks identified by IPA in Nrf2−/− included cell signaling, small molecule biochemistry, lipid metabolism, cancer, and cell death (Fig. 6A). Overall, the top ranked networks were significantly enriched for pathways involved in lipid metabolism. Of the genes identified in these pathways, Nrf2−/− white adipose tissue showed increased expression of Ucp1 and other brown adipocyte markers. UCP1 functions as an uncoupler of mitochondrial ATP production, and its up-regulation might account for the change in energetics in Nrf2−/− mice. Hence, we focused our attention on these genes to validate the RNA-seq data. Quantitative RT-PCR analysis showed significant up-regulation of Ucp1, Cox7a1, Cox8b, Sirt3, CideA, Pgc1a, Tfam, and Nrf1 (nuclear respiratory factor 1) in WAT of Nrf2−/− mice fed a high fat diet (Fig. 6B). Interestingly, expression of some of these genes associated with brown fat was also up-regulated in Nrf2−/− WAT under regular diet conditions (Fig. 6B). Western blotting analysis showed increased UCP1 protein expression in Nrf2−/− WAT that was further enhanced by high fat diet (Fig. 6, C–E). Unlike WAT, expression of Ucp1 and other brown fat markers was not altered significantly in Nrf2−/− brown adipose tissues under regular diet or high fat diet conditions (data not shown). Taken together, these data suggest that WAT in Nrf2−/− mice gained some BAT-like features.
FIGURE 6.

Brown fat markers are up-regulated in white adipose tissues of Nrf2−/− mice. A, Ingenuity Pathway Analysis of differentially expressed genes in wild type and Nrf2−/− white adipose tissues. Genes that were significantly different between wild type and Nrf2−/− white adipose tissues were used as input for pathway analysis. The top 10 overrepresented canonical pathways of up-regulated genes found in Nrf2−/− white adipose tissues compared with controls are shown. Category names are presented on the y axis. The x axis indicates the −log(p) value of the overrepresentation analysis. B, quantitative RT-PCR analysis of brown fat markers in abdominal white adipose tissues of wild type and Nrf2−/− mice fed a high fat diet (HFD) or regular diet (RD). Expression levels were quantitated relative to Rplpo levels as an internal reference and calculated as 2(Ct test gene − CtRPLP0). Bar graphs depict mean values ± S.E. (error bars) of 4–6 different samples of each genotype, and significance was assessed by two-way ANOVA and post-hoc Bonferroni analysis. *, significance between wild type and Nrf2−/− regular diet samples; #, significance between regular diet and high fat diet samples. C–E, Western blotting analysis of UCP1 protein levels in abdominal white adipose tissues of wild type and Nrf2−/− mice fed a high fat or regular diet, respectively. β-Actin levels were used as a loading control. E, bar graph shows quantitation of protein levels; significance was assessed by two-way ANOVA and post hoc Bonferroni analysis.
Oxidative Stress Modulates Oxygen Consumption and UCP1 Expression
Nrf2 plays a pivotal role in oxidative stress response. Hence, we sought to determine whether elevated oxygen consumption rates in Nrf2-deficient cells are potentially linked to oxidative stress. To confirm oxidative stress in our model, we assessed glutathione status in adipose tissues of wild type and Nrf2−/− mice. WAT from Nrf2−/− mice fed a high fat diet showed a decrease in GSH with a corresponding increase in GSSG, leading to a reduction in GSH/GSSG ratio (Fig. 7, A–C). A decreased GSH/GSSG ratio is indicative of oxidative stress. To further test the effects of oxidative stress on cellular respiration, we compared oxygen consumption rates in MEF cells derived from wild type and Nrf2-deficient mice. Oxygen consumption rates were 2-fold higher in Nrf2−/− cells compared with wild type (Fig. 7D). Wild type cells treated with glucose oxidase to induce oxidative stress also showed a 2-fold increase in oxygen consumption (Fig. 7D). However, glucose oxidase treatment did not lead to further enhancement of oxygen consumption in Nrf2−/− cells. As expected, elevated intracellular reactive oxygen species levels were detected in Nrf2−/− cells and wild type cells treated with glucose oxidase (Fig. 7E). We then examined the effects of BHA, a free radical scavenging antioxidant, on oxygen consumption in Nrf2−/− cells. As shown in Fig. 7D, oxygen consumption in Nrf2−/− cells was reduced by BHA treatment. BHA treatment had no effect in wild type cells. Next, we examined the effects of oxidative stress on UCP1 expression. Wild type MEF cells treated with glucose oxidase led to a marked induction of Ucp1 mRNA and protein (Fig. 7, F and G). Consistent with the effects of oxidative stress, Nrf2−/− MEF cells showed increased Ucp1 mRNA and protein compared with wild type cells (Fig. 7, H and I), and treatment with antioxidants (N-acetylcysteine, a free radical scavenging compound, or MnTBAP, a cell-permeable superoxide dismutase mimetic) led to a decrease in Ucp1 in Nrf2−/− MEF cells (Fig. 7J). To test the involvement of Ucp1 in cellular oxygen consumption, we used SB203580, a p38 MAPK inhibitor known to suppress Ucp1 expression (46). SB203580 treatment resulted in a dose-dependent decrease in OCR in Nrf2−/− MEF cells (Fig. 7K). Taken together, these data suggest that Nrf2 deficiency in MEFs results in oxidative stress, which in turn up-regulates Ucp1 expression and cellular respiration.
FIGURE 7.

Oxidative stress modulates oxygen consumption and UCP1 expression. A–C, GSH (A), GSSG (B), and GSH/GSSG levels (C) of WAT from wild type and Nrf2−/− mice after 6 weeks of high fat diet feeding. Data represent means (n = 3/group) ± S.E. (error bars); *, p < 0.05. D, Seahorse X-24 analysis of basal oxygen consumption rate in wild type and Nrf2−/− MEF cells with and without treatment with glucose oxidase or BHA. Data were normalized to total protein and are represented as means ± S.E. (n = 6/group). Significance was assessed by two-way ANOVA and post hoc Bonferroni analysis. *, p < 0.05; **, p < 0.001. E, dichlorodihydrofluorescein fluorescence levels in wild type, Nrf2−/−, and wild type cells treated with glucose oxidase. Data represent means (n = 3/4/group) ± S.E.; *, p <0.05. F, expression of Ucp1 mRNA in wild type MEF cells treated with glucose oxidase to induce oxidative stress for the indicated times. G, Western blotting analysis of UCP1 protein in wild type MEF cells treated with glucose oxidase for the indicated times. Densitometric quantitations of band intensities are shown. H, basal expression of Ucp1 mRNA levels in wild type and Nrf2−/− MEF cells. Results represent means (n = 3/group) ± S.E.; *, p < 0.05. I, Western blotting analysis of basal UCP1 protein in wild type and Nrf2−/− MEF cells. J, expression of Ucp1 mRNA levels in Nrf2−/− MEF cells treated with N-acetylcysteine (NAC) or MnTBAP. Bar graphs represent mean values ± S.E. (n = 3); *, p < 0.05. K, Seahorse X-24 analysis of basal OCR in Nrf2−/− MEF cells treated with SB203580. Data represent means ± S.E. (n = 3/group), and significance was determined by Student's t test (*, p < 0.05).
Discussion
Obesity and its related disorders are leading causes of morbidity and mortality worldwide. Various treatment modalities for obesity have been met with limited success. Hence, identification of novel targets that can potentially be employed for prevention and treatment of obesity is of great interest. In this study, we have demonstrated that loss of Nrf2 protects mice from diet-induced obesity. Nrf2−/− mice exhibit increased energy expenditure and brown adipose-like changes in white adipose depots. Loss of Nrf2 in cells results in increased oxygen consumption and expression of UCP1, and we have shown that oxidative stress can modulate UCP1 expression and oxygen consumption in cells. Taken together, these results suggest that increased uncoupled respiration due to increased oxidative stress brought on by the loss of Nrf2 helps to contribute to the lean phenotype of Nrf2−/− animals fed a high fat diet.
Nrf2 plays an important role in oxidative stress response by regulating the basal and inducible expression of a large battery of antioxidant genes. It is interesting to note that a deficiency in various Nrf2 target genes leads to a similar decrease in adiposity and improved metabolic parameters. Knock-outs of Gclm and Gpx1, which are genes involved in antioxidant defense and glutathione synthesis, result in mice with decreased adipose tissue mass and with protection from high fat diet-induced obesity. In addition, these mice show increased energy expenditure, decreased serum insulin levels, and increased insulin sensitivity (47, 48). Conversely, it has been shown that transgenic mice overexpressing Gpx1 are insulin-resistant and obese (49). Another pathway of potential interest involves thioredoxins, which are cellular antioxidants that protect against oxidative stress by promoting the reduction of other proteins via a cysteine thiol-disulfide exchange reaction. Thioredoxins are kept in the reduced state via TXNRD1 (thioredoxin reductase 1). TXNIP (thioredoxin-interacting protein), on the other hand, can increase reactive oxygen species by inhibiting TXNRD1. It has been demonstrated that Nrf2 suppresses Txnip gene expression. Thus, loss of Nrf2 results in increased Txnip expression and a subsequent increase in oxidative stress (50). Interestingly, Txnip knock-out mice also demonstrate increased adiposity when exposed to high fat diet feeding (51). Whereas findings suggest that the lean phenotype in Nrf2−/− mice is linked to antioxidant dysfunction and oxidative stress, Nrf2 affects the expression of many genes besides just those involved in oxidative stress, and it is possible that multiple mechanisms are contributing to the ability of Nrf2 knock-out to attenuate diet-induced weight gain and accumulation of body fat. Additionally, our model represents a whole animal knock-out of Nrf2. The possibility that Nrf2 deficiency in non-adipose tissue may contribute to the observed phenotype will require further experimentation using adipose tissue-specific knock-out models of Nrf2 deficiency.
UCP1 uncouples the proton motive force generated in the mitochondria from ATP production to produce heat, and UCP1 has been shown to play an important role in energy homeostasis and obesity. Mice deficient in Ucp1 are shown to be susceptible to weight gain, whereas overexpression of Ucp1 provides protection against diet-induced obesity (16, 17). UCP2 and UCP3, which are homologs of UCP1, also have uncoupling function in the mitochondria and serve as potential targets for increased metabolic capacity. Ucp2 is ubiquitously expressed at low levels throughout the body, whereas Ucp3 is limited to skeletal and cardiac muscle (52, 53). Mice overexpressing Ucp3 in muscle are lean and have increased energy metabolism as well as decreased levels of serum glucose (54). Additionally, polymorphisms in the Ucp2 gene have been associated with obesity and decreased metabolic rates in humans (55–57). In this study, we found that Nrf2−/− mice fed a high fat diet have an elevated metabolic rate in both whole animals and isolated white adipose tissue that is associated with an up-regulation of Ucp1 in their abdominal white adipose depots. Similarly, the basal oxygen consumption rate in Nrf2-deficient cells is increased, and Ucp1 expression is induced. However, up-regulation of Ucp2 and Ucp3 expression was not observed in Nrf2-deficient fat tissues (data not shown). Additionally, we observed decreased levels of Ucp3 in muscle tissues of Nrf2−/− mice (data not shown) that are consistent with data demonstrating direct regulation of Ucp3 by Nrf2 (58). Thus, UCP2 and UCP3 do not appear to provide additional mitochondrial uncoupling in the adipose tissue of Nrf2 knock-out animals. These findings suggest that UCP1-dependent thermogenesis is a mechanism by which energy expenditure in Nrf2−/− mice is up-regulated.
Whereas a wide range of stimuli, including cold exposure, diet, and exercise, controls Ucp1 expression (15, 59), these factors are not likely to play a role here because mice in this study were housed and maintained in a controlled environment, and both wild type and Nrf2−/− mice showed similar food intake and physical activity. In addition, expression of Rip140 and Lxr, both of which are negative regulators of Ucp1 expression (60–62), was not altered in Nrf2−/− adipose tissues (data not shown). Expression of Ucp1 is activated by oxidative stress (46, 63, 64). Consistent with this, we show that glucose oxidase-induced oxidative stress in MEF cells up-regulates Ucp1 expression, and antioxidant treatment down-regulates Ucp1 expression in Nrf2−/− MEF cells. We also found that Pgc1a expression is increased in WAT of Nrf2−/− mice fed a high fat diet, which is known to exacerbate oxidative stress in fat cells (65). It has been shown that oxidative stress increases the expression of Pgc1a, and PGC1a interacts with other nuclear hormone receptors in white adipose cells to enhance mitochondrial biogenesis and the expression of Ucp1 (64, 66, 67). Hence, we surmise that acquisition of brown fat features by WAT depots in Nrf2−/− mice is driven in part by oxidative stress induction of Pgc1a expression.
Data obtained previously showed that Nrf2−/− mice of C57/129 mixed genetic background are resistant to high fat diet-induced obesity (41). In agreement with this, our results also show that Nrf2−/− mice derived on a C57/BL/6 background are resistant to diet-induced obesity. Here we demonstrated that Nrf2−/− mice have increased energy expenditure and acquisition of brown fat-like gene expression in their WAT depots. There is growing evidence that the acquisition of brown-like fat cells in WAT protects against both genetic and diet-induced obesity through increased metabolic activity, and this “browning” phenomenon has become a subject of considerable interest for possible treatment of obesity and metabolic syndrome (68–70). Browning of WAT has been shown to occur in mouse models of prolactin receptor deficiency, IkB kinase ϵ deficiency, COXII overexpression, and increased prostaglandin synthesis (71–74). In these instances, mice displayed a protection against diet-induced obesity as well as increased oxygen consumption. Additionally, FGF21 has been shown to exert anti-obesogenic effects and ameliorates insulin and leptin resistance in ob/ob mice (75). It has been shown that mice lacking Nrf2 demonstrate protection against diet-induced obesity, maintain insulin sensitivity, and demonstrate increased circulating levels of FGF21 and expression of Fgf21 in liver and WAT (76). However, no difference in Fgf21 expression was found between wild type and Nrf2-deficient white adipose tissue or liver in our experiments (data not shown). The basis of this discrepant result is not known currently; however, our high fat diet duration of 6 weeks was significantly shorter than the 180 days tested in the above study. We propose that the metabolic and gene expression changes observed in our Nrf2−/− model are due in part to increased levels of oxidative stress, caused by a combination of decreased antioxidant capacity through the loss of Nrf2 function and the additional oxidative stress provided through exposure to a high fat diet. Currently, we cannot rule out the possibility that increased UCP1 expression in Nrf2−/− mice is through modulation of downstream target genes, independent of oxidative stress effects. Previous data have shown that deficiency of Nrf2 results in down-regulation of Pparγ (75). Pparγ knock-out mice demonstrate decreased adipose tissue mass, protection against diet-induced obesity, and increased expression of Ucp1 in white adipose tissue (77, 78). It is thought that decreased PPARγ activity drives increased expression of prostaglandin E synthase, which, in concert with COXII, drives the formation of prostaglandin, which is hypothesized to be important in the formation of brown adipose tissue within white adipose tissue depots (77). It is likely that the phenotype observed in Nrf2−/− mice is due to a complex interplay of a variety of pathways.
Further supporting this complex role of Nrf2, recent studies have shown that overexpression of Nrf2 through the use of graded knockdown of Keap1 actually provides protection against the onset of diabetes in genetic models of diabetes in mice (79). This suggests a beneficial role for Nrf2 in the protection against the development of diabetes with increased expression of Nrf2 resulting in decreased blood glucose and protection against obesity in a db/db mouse background. However, it is worth noting that in addition to the protective effects of Nrf2, the same group also found that mice lacking Nrf2 demonstrate decreased serum glucose levels compared with control mice, which correlates with data from our model of Nrf2 deficiency. In mice doubly deficient in Keap1 and leptin, there was a resistance to high fat diet-induced obesity and a decrease in adipose tissue weight that was also associated with impaired insulin signaling, hyperglycemia, and glucose intolerance (80). Additional studies utilizing the ob/ob genetic model of obesity have yielded varied results. Nrf2 knock-out animals crossed into the ob/ob mouse background, using either a whole animal or fat-specific Nrf2 knock-out, demonstrated that loss of Nrf2 resulted in decreased adipose tissue mass yet aggravated insulin resistance, hyperglycemia, and hypertriglyceridemia (81). This seems to demonstrate that in genetic models of obesity and diabetes, the results of Nrf2 modulation can vary drastically, depending on the specific conditions of the model. In order to most closely compare results with our own, it is important to look at systems similar to ours, in which the experiments are performed looking at strictly diet-induced models of obesity rather than genetic models of obesity and diabetes. For example, Keap1 knockdown mice in the C57BL/6 background without crossing to any genetic models of obesity or diabetes gained significantly more weight than wild type animals as well as developed glucose intolerance, increased inflammation, and hepatic steatosis when placed on a high fat diet (82). As mentioned previously, other groups have also shown that Nrf2 knock-out mice were found to be resistant to long term high fat diet-induced obesity (68). These results coincide with our data and seem to suggest that increased Nrf2 levels lead to increased obesity and that Nrf2 deficiency may provide protective benefits in a diet-induced obesity model. These varied results in the literature suggest a complex interaction between Nrf2, obesity, and diabetes, and further research will probably provide insight.
In summary, Nrf2-deficient mice are protected from diet-induced obesity and exhibit increased energy expenditure associated with the up-regulation of brown adipose tissue-specific genes in white adipose depots. Cell-based experiments demonstrated that increased UCP1 expression is associated with increased cellular oxidative stress. These observations suggest a promising model for increasing energy expenditure in white adipose tissue through Nrf2 and the possibility of pharmacological modulation of Nrf2 or regulation of oxidative stress as treatments for obesity and its related disorders.
Author Contributions
J. Y. C. and T. W. K. conceived and wrote the paper. K. S. coordinated the studies and performed the studies in Figs. 1–7 and also wrote the paper. M. V. analyzed the experiments shown in Fig. 6A. B. G. helped to design and provided technical assistance in experiments shown in Figs. 4 and 5. J. N., J. V., and B. G. provided technical assistance and contributed to the preparation of figures. All authors reviewed the results and approved the final version of the manuscript.
This work was supported in part by National Institute of Health Research Grant CA091907 to J. Y. C. The authors declare that they have no conflicts of interest with the contents of this article.
- WAT
- white adipose tissue(s)
- BAT
- brown adipose tissue
- CM-H2DCFDA
- chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- BHA
- butylated hydroxyanisole
- MaTBAP
- Mn(III)tetrakis(4-benzoic acid)porphyrin chloride
- MEF
- mouse embryo fibroblast
- OCR
- oxygen consumption rate
- FCCP
- carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- ANOVA
- analysis of variance
- RNA-seq
- RNA sequencing.
References
- 1. Lowell B. B., and Spiegelman B. M. (2000) Towards a molecular understanding of adaptive thermogenesis. Nature 404, 652–660 [DOI] [PubMed] [Google Scholar]
- 2. Tseng Y. H., Cypess A. M., and Kahn C. R. (2010) Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 9, 465–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohade C., Osman M., Pannu H. K., and Wahl R. L. (2003) Uptake in supraclavicular area fat (“U.S.A.-Fat”): description on 18F-FDG PET/CT. J. Nucl. Med. 44, 170–176 [PubMed] [Google Scholar]
- 4. Enerbäck S. (2010) Human brown adipose tissue. Cell metabolism 11, 248–252 [DOI] [PubMed] [Google Scholar]
- 5. Ravussin E., and Galgani J. E. (2011) The implication of brown adipose tissue for humans. Annu. Rev. Nutr. 31, 33–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., Kolodny G. M., and Kahn C. R. (2009) Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nedergaard J., Bengtsson T., and Cannon B. (2007) Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 293, E444–E452 [DOI] [PubMed] [Google Scholar]
- 8. Saito M., Okamatsu-Ogura Y., Matsushita M., Watanabe K., Yoneshiro T., Nio-Kobayashi J., Iwanaga T., Miyagawa M., Kameya T., Nakada K., Kawai Y., and Tsujisaki M. (2009) High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58, 1526–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., and Teule G. J. (2009) Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508 [DOI] [PubMed] [Google Scholar]
- 10. Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T., Taittonen M., Laine J., Savisto N. J., Enerbäck S., and Nuutila P. (2009) Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525 [DOI] [PubMed] [Google Scholar]
- 11. Zingaretti M. C., Crosta F., Vitali A., Guerrieri M., Frontini A., Cannon B., Nedergaard J., and Cinti S. (2009) The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 23, 3113–3120 [DOI] [PubMed] [Google Scholar]
- 12. Porter R. K., and Brand M. D. (1995) Causes of differences in respiration rate of hepatocytes from mammals of different body mass. Am. J. Physiol. 269, R1213–1224 [DOI] [PubMed] [Google Scholar]
- 13. Rolfe D. F., Newman J. M., Buckingham J. A., Clark M. G., and Brand M. D. (1999) Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. Am. J. Physiol. 276, C692–C699 [DOI] [PubMed] [Google Scholar]
- 14. Rothwell N. J., and Stock M. J. (1983) Luxuskonsumption, diet-induced thermogenesis and brown fat: the case in favour. Clin. Sci. 64, 19–23 [DOI] [PubMed] [Google Scholar]
- 15. Feldmann H. M., Golozoubova V., Cannon B., and Nedergaard J. (2009) UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209 [DOI] [PubMed] [Google Scholar]
- 16. Kopecky J., Clarke G., Enerbäck S., Spiegelman B., and Kozak L. P. (1995) Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J. Clin. Invest. 96, 2914–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li B., Nolte L. A., Ju J. S., Han D. H., Coleman T., Holloszy J. O., and Semenkovich C. F. (2000) Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat. Med. 6, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 18. Barbatelli G., Murano I., Madsen L., Hao Q., Jimenez M., Kristiansen K., Giacobino J. P., De Matteis R., and Cinti S. (2010) The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am. J. Physiol. Endocrinol. Metab. 298, E1244–E1253 [DOI] [PubMed] [Google Scholar]
- 19. Bartelt A., and Heeren J. (2014) Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 10, 24–36 [DOI] [PubMed] [Google Scholar]
- 20. Kozak L. P. (2011) The genetics of brown adipocyte induction in white fat depots. Front. Endocrinol. 2, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petrovic N., Walden T. B., Shabalina I. G., Timmons J. A., Cannon B., and Nedergaard J. (2010) Chronic peroxisome proliferator-activated receptor gamma (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 285, 7153–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Biswas M., and Chan J. Y. (2010) Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 244, 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kobayashi A., Ito E., Toki T., Kogame K., Takahashi S., Igarashi K., Hayashi N., and Yamamoto M. (1999) Molecular cloning and functional characterization of a new Cap 'n' collar family transcription factor Nrf3. J. Biol. Chem. 274, 6443–6452 [DOI] [PubMed] [Google Scholar]
- 24. Motohashi H., and Yamamoto M. (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549–557 [DOI] [PubMed] [Google Scholar]
- 25. Itoh K., Wakabayashi N., Katoh Y., Ishii T., O'Connor T., and Yamamoto M. (2003) Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8, 379–391 [DOI] [PubMed] [Google Scholar]
- 26. Wakabayashi N., Dinkova-Kostova A. T., Holtzclaw W. D., Kang M. I., Kobayashi A., Yamamoto M., Kensler T. W., and Talalay P. (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. U.S.A. 101, 2040–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giudice A., and Montella M. (2006) Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays 28, 169–181 [DOI] [PubMed] [Google Scholar]
- 28. Kwak M. K., Wakabayashi N., Itoh K., Motohashi H., Yamamoto M., and Kensler T. W. (2003) Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 278, 8135–8145 [DOI] [PubMed] [Google Scholar]
- 29. Nguyen T., Sherratt P. J., and Pickett C. B. (2003) Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43, 233–260 [DOI] [PubMed] [Google Scholar]
- 30. Osburn W. O., and Kensler T. W. (2008) Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat. Res. 659, 31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kobayashi M., and Yamamoto M. (2006) Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 46, 113–140 [DOI] [PubMed] [Google Scholar]
- 32. Wang L., He X., Szklarz G. D., Bi Y., Rojanasakul Y., and Ma Q. (2013) The aryl hydrocarbon receptor interacts with nuclear factor erythroid 2-related factor 2 to mediate induction of NAD(P)H:quinoneoxidoreductase 1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Biochem. Biophys. 537, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu S. C. (2009) Regulation of glutathione synthesis. Mol. Aspects Med. 30, 42–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan K., Han X. D., and Kan Y. W. (2001) An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 98, 4611–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He X., Chen M. G., and Ma Q. (2008) Activation of Nrf2 in defense against cadmium-induced oxidative stress. Chem. Res. Toxicol. 21, 1375–1383 [DOI] [PubMed] [Google Scholar]
- 36. He X., Lin G. X., Chen M. G., Zhang J. X., and Ma Q. (2007) Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2. Recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol. Sci. 98, 298–309 [DOI] [PubMed] [Google Scholar]
- 37. Ma Q., Battelli L., and Hubbs A. F. (2006) Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 168, 1960–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramos-Gomez M., Kwak M. K., Dolan P. M., Itoh K., Yamamoto M., Talalay P., and Kensler T. W. (2001) Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 3410–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee J. M., Li J., Johnson D. A., Stein T. D., Kraft A. D., Calkins M. J., Jakel R. J., and Johnson J. A. (2005) Nrf2, a multi-organ protector? FASEB J. 19, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 40. Hou Y., Xue P., Bai Y., Liu D., Woods C. G., Yarborough K., Fu J., Zhang Q., Sun G., Collins S., Chan J. Y., Yamamoto M., Andersen M. E., and Pi J. (2012) Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein β during adipogenesis. Free Radic. Biol. Med. 52, 462–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pi J., Leung L., Xue P., Wang W., Hou Y., Liu D., Yehuda-Shnaidman E., Lee C., Lau J., Kurtz T. W., and Chan J. Y. (2010) Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 285, 9292–9300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan K., Lu R., Chang J. C., and Kan Y. W. (1996) NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. U.S.A. 93, 13943–13948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dunham-Snary K. J., Sandel M. W., Westbrook D. G., and Ballinger S. W. (2014) A method for assessing mitochondrial bioenergetics in whole white adipose tissues. Redox Biol. 2, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rolo A. P., Palmeira C. M., and Cortopassi G. A. (2009) Biosensor plates detect mitochondrial physiological regulators and mutations in vivo. Anal. Biochem. 385, 176–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kemp M., Go Y. M., and Jones D. P. (2008) Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic. Biol. Med. 44, 921–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ro S. H., Nam M., Jang I., Park H. W., Park H., Semple I. A., Kim M., Kim J. S., Park H., Einat P., Damari G., Golikov M., Feinstein E., and Lee J. H. (2014) Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 111, 7849–7854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kendig E. L., Chen Y., Krishan M., Johansson E., Schneider S. N., Genter M. B., Nebert D. W., and Shertzer H. G. (2011) Lipid metabolism and body composition in Gclm(−/−) mice. Toxicol. Appl. Pharmacol. 257, 338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B. J., Skiba B., Ooms L. M., Stepto N., Wu B., Mitchell C. A., Tonks N. K., Watt M. J., et al. (2009) Reactive oxygen species enhance insulin sensitivity. Cell Metab. 10, 260–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McClung J. P., Roneker C. A., Mu W., Lisk D. J., Langlais P., Liu F., and Lei X. G. (2004) Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. U.S.A. 101, 8852–8857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He X., and Ma Q. (2012) Redox regulation by nuclear factor erythroid 2-related factor 2: gatekeeping for the basal and diabetes-induced expression of thioredoxin-interacting protein. Mol. Pharmacol. 82, 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chutkow W. A., Birkenfeld A. L., Brown J. D., Lee H. Y., Frederick D. W., Yoshioka J., Patwari P., Kursawe R., Cushman S. W., Plutzky J., Shulman G. I., Samuel V. T., and Lee R. T. (2010) Deletion of the α-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity. Diabetes 59, 1424–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boss O., Samec S., Paoloni-Giacobino A., Rossier C., Dulloo A., Seydoux J., Muzzin P., and Giacobino J. P. (1997) Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 408, 39–42 [DOI] [PubMed] [Google Scholar]
- 53. Fleury C., Neverova M., Collins S., Raimbault S., Champigny O., Levi-Meyrueis C., Bouillaud F., Seldin M. F., Surwit R. S., Ricquier D., and Warden C. H. (1997) Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 15, 269–272 [DOI] [PubMed] [Google Scholar]
- 54. Clapham J. C., Arch J. R., Chapman H., Haynes A., Lister C., Moore G. B., Piercy V., Carter S. A., Lehner I., Smith S. A., Beeley L. J., Godden R. J., Herrity N., Skehel M., Changani K. K., et al. (2000) Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature 406, 415–418 [DOI] [PubMed] [Google Scholar]
- 55. Mahadik S. R., Lele R. D., Saranath D., Seth A., and Parikh V. (2012) Uncoupling protein-2 (UCP2) gene expression in subcutaneous and omental adipose tissue of Asian Indians: relationship to adiponectin and parameters of metabolic syndrome. Adipocyte 1, 101–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mexitalia M., Yamauchi T., Utari A., Sjarif D. R., Subagio H. W., Soemantri A., and Ishida T. (2013) The role of uncoupling protein 2 and 3 genes polymorphism and energy expenditure in obese Indonesian children. J. Pediatr. Endocrinol. Metab. 26, 441–447 [DOI] [PubMed] [Google Scholar]
- 57. Qian L., Xu K., Xu X., Gu R., Liu X., Shan S., and Yang T. (2013) UCP2–866G/A, Ala55Val and UCP3–55C/T polymorphisms in association with obesity susceptibility: a meta-analysis study. PLoS One 8, e58939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Anedda A., López-Bernardo E., Acosta-Iborra B., Saadeh Suleiman M., Landázuri M. O., and Cadenas S. (2013) The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 61, 395–407 [DOI] [PubMed] [Google Scholar]
- 59. Ringholm S., Grunnet Knudsen J., Leick L., Lundgaard A., Munk Nielsen M., and Pilegaard H. (2013) PGC-1α is required for exercise- and exercise training-induced UCP1 up-regulation in mouse white adipose tissue. PLoS One 8, e64123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Christian M., Kiskinis E., Debevec D., Leonardsson G., White R., and Parker M. G. (2005) RIP140-targeted repression of gene expression in adipocytes. Mol. Cell. Biol. 25, 9383–9391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Leonardsson G., Steel J. H., Christian M., Pocock V., Milligan S., Bell J., So P. W., Medina-Gomez G., Vidal-Puig A., White R., and Parker M. G. (2004) Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc. Natl. Acad. Sci. U.S.A. 101, 8437–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang H., Zhang Y., Yehuda-Shnaidman E., Medvedev A. V., Kumar N., Daniel K. W., Robidoux J., Czech M. P., Mangelsdorf D. J., and Collins S. (2008) Liver X receptor α is a transcriptional repressor of the uncoupling protein 1 gene and the brown fat phenotype. Mol. Cell. Biol. 28, 2187–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Echtay K. S., Roussel D., St-Pierre J., Jekabsons M. B., Cadenas S., Stuart J. A., Harper J. A., Roebuck S. J., Morrison A., Pickering S., Clapham J. C., and Brand M. D. (2002) Superoxide activates mitochondrial uncoupling proteins. Nature 415, 96–99 [DOI] [PubMed] [Google Scholar]
- 64. Fernandez-Marcos P. J., and Auwerx J. (2011) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 93, 884S–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Matsuzawa-Nagata N., Takamura T., Ando H., Nakamura S., Kurita S., Misu H., Ota T., Yokoyama M., Honda M., Miyamoto K., and Kaneko S. (2008) Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 57, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 66. St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D. K., Bachoo R., and Spiegelman B. M. (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 [DOI] [PubMed] [Google Scholar]
- 67. Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., and Spiegelman B. M. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 [DOI] [PubMed] [Google Scholar]
- 68. Lee P., Werner C. D., Kebebew E., and Celi F. S. (2013) Functional thermogenic beige adipogenesis is inducible in human neck fat. Int. J. Obes. 38, 170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Whittle A., Relat-Pardo J., and Vidal-Puig A. (2013) Pharmacological strategies for targeting BAT thermogenesis. Trends Pharmacol. Sci. 34, 347–355 [DOI] [PubMed] [Google Scholar]
- 70. Zafrir B. (2013) Brown adipose tissue: research milestones of a potential player in human energy balance and obesity. Horm. Metab. Res. 45, 774–785 [DOI] [PubMed] [Google Scholar]
- 71. Auffret J., Viengchareun S., Carré N., Denis R. G., Magnan C., Marie P. Y., Muscat A., Fève B., Lombès M., and Binart N. (2012) Beige differentiation of adipose depots in mice lacking prolactin receptor protects against high-fat-diet-induced obesity. FASEB J. 26, 3728–3737 [DOI] [PubMed] [Google Scholar]
- 72. Chiang S. H., Bazuine M., Lumeng C. N., Geletka L. M., Mowers J., White N. M., Ma J. T., Zhou J., Qi N., Westcott D., Delproposto J. B., Blackwell T. S., Yull F. E., and Saltiel A. R. (2009) The protein kinase IKKϵ regulates energy balance in obese mice. Cell 138, 961–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fisher F. M., Kleiner S., Douris N., Fox E. C., Mepani R. J., Verdeguer F., Wu J., Kharitonenkov A., Flier J. S., Maratos-Flier E., and Spiegelman B. M. (2012) FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vegiopoulos A., Müller-Decker K., Strzoda D., Schmitt I., Chichelnitskiy E., Ostertag A., Berriel Diaz M., Rozman J., Hrabe de Angelis M., Nüsing R. M., Meyer C. W., Wahli W., Klingenspor M., and Herzig S. (2010) Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 328, 1158–1161 [DOI] [PubMed] [Google Scholar]
- 75. Kharitonenkov A., Shiyanova T. L., Koester A., Ford A. M., Micanovic R., Galbreath E. J., Sandusky G. E., Hammond L. J., Moyers J. S., Owens R. A., Gromada J., Brozinick J. T., Hawkins E. D., Wroblewski V. J., Li D. S., et al. (2005) FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chartoumpekis D. V., Ziros P. G., Psyrogiannis A. I., Papavassiliou A. G., Kyriazopoulou V. E., Sykiotis G. P., and Habeos I. G. (2011) Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 60, 2465–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. García-Alonso V., López-Vicario C., Titos E., Morán-Salvador E., González-Périz A., Rius B., Párrizas M., Werz O., Arroyo V., and Clària J. (2013) Coordinate functional regulation between microsomal prostaglandin E synthase-1 (mPGES-1) and peroxisome proliferator-activated receptor γ (PPARγ) in the conversion of white-to-brown adipocytes. J. Biol. Chem. 288, 28230–28242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jones J. R., Barrick C., Kim K. A., Lindner J., Blondeau B., Fujimoto Y., Shiota M., Kesterson R. A., Kahn B. B., and Magnuson M. A. (2005) Deletion of PPARγ in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 102, 6207–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Uruno A., Furusawa Y., Yagishita Y., Fukutomi T., Muramatsu H., Negishi T., Sugawara A., Kensler T. W., and Yamamoto M. (2013) The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol. Cell. Biol. 33, 2996–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xu J., Kulkarni S. R., Donepudi A. C., More V. R., and Slitt A. L. (2012) Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 61, 3208–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xue P., Hou Y., Chen Y., Yang B., Fu J., Zheng H., Yarborough K., Woods C. G., Liu D., Yamamoto M., Zhang Q., Andersen M. E., and Pi J. (2013) Adipose deficiency of Nrf2 in ob/ob mice results in severe metabolic syndrome. Diabetes 62, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. More V. R., Xu J., Shimpi P. C., Belgrave C., Luyendyk J. P., Yamamoto M., and Slitt A. L. (2013) Keap1 knockdown increases markers of metabolic syndrome after long-term high fat diet feeding. Free Radic. Biol. Med. 61, 85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]