

ABSTRACT
We show that Streptomyces biogeography in soils across North America is influenced by the regional diversification of microorganisms due to dispersal limitation and genetic drift. Streptomyces spp. form desiccation-resistant spores, which can be dispersed on the wind, allowing for a strong test of whether dispersal limitation governs patterns of terrestrial microbial diversity. We employed an approach that has high sensitivity for determining the effects of genetic drift. Specifically, we examined the genetic diversity and phylogeography of physiologically similar Streptomyces strains isolated from geographically distributed yet ecologically similar habitats. We found that Streptomyces beta diversity scales with geographic distance and both beta diversity and phylogenetic diversity manifest in a latitudinal diversity gradient. This pattern of Streptomyces biogeography resembles patterns seen for diverse species of plants and animals, and we therefore evaluated these data in the context of ecological and evolutionary hypotheses proposed to explain latitudinal diversity gradients. The data are consistent with the hypothesis that niche conservatism limits dispersal, and historical patterns of glaciation have limited the time for speciation in higher-latitude sites. Most notably, higher-latitude sites have lower phylogenetic diversity, higher phylogenetic clustering, and evidence of range expansion from lower latitudes. In addition, patterns of beta diversity partition with respect to the glacial history of sites. Hence, the data support the hypothesis that extant patterns of Streptomyces biogeography have been driven by historical patterns of glaciation and are the result of demographic range expansion, dispersal limitation, and regional diversification due to drift.
IMPORTANCE
Biogeographic patterns provide insight into the evolutionary and ecological processes that govern biodiversity. However, the evolutionary and ecological processes that govern terrestrial microbial diversity remain poorly characterized. We evaluated the biogeography of the genus Streptomyces to show that the diversity of terrestrial bacteria is governed by many of the same processes that govern the diversity of many plant and animal species. While bacteria of the genus Streptomyces are a preeminent source of antibiotics, their evolutionary history, biogeography, and biodiversity remain poorly characterized. The observations we describe provide insight into the drivers of Streptomyces biodiversity and the processes that underlie microbial diversification in terrestrial habitats.
INTRODUCTION
Patterns of microbial biogeography remain ill described because tools for measuring microbial diversity have emerged only recently and are evolving rapidly. It is still unclear to what degree patterns of microbial diversity can be explained by the same ecological and evolutionary forces that govern the diversity of plants and animals. Evolutionary ecology seeks to explain the forces that govern evolutionary diversification in an ecological context. If the same forces govern the diversification of both macro- and microorganisms, then it will be possible to bring the full theoretical framework which underpins evolutionary ecology to bear in evaluating the causes and consequences of microbial diversity.
Microorganisms, due to their small size, large populations, ability to survive dormancy, and potential for wind dissemination (1), may support rates of dispersal that far exceed rates of diversification (2–4). If dispersal rates greatly exceed diversification rates, then microbial diversity is controlled by selection acting on global scales (5). There is now abundant evidence that environmental gradients explain patterns of microbial beta diversity on local and regional scales (for reviews, see references 5, 6, and 7). It remains unclear, however, to what degree these biogeographic patterns are the result of recruitment from globally distributed species pools or from dispersal limitation and evolutionary diversification acting on regional scales.
Microorganisms lack morphological features to enable identification, and definitions for microbial species remain controversial (8). Patterns of microbial biogeography are often inferred from the analysis of small-subunit (SSU) rRNA genes, with operational taxonomic units (OTU) defined using a 3% nucleotide identity cutoff (9). The SSU rRNA gene, however, has an exceptionally low substitution rate, and it is estimated that approximately 50 million years are required to achieve 1% divergence in SSU rRNA sequences (10). As a point of comparison, the family Leguminosae first appeared approximately 60 million years ago (11), and hence all legumes would represent a single OTU if plant diversity was defined in the manner of microbial diversity. Biogeographic patterns detected through the analysis of SSU rRNA genes likely result from physiological traits that map deeply in the tree of life. Indeed, some of the best-documented patterns of microbial biogeography have been made at the phylum level. For example, Betaproteobacteria are common in freshwater environments but uncommon in saltwater; Actinobacteria and Firmicutes are ubiquitous in soils but rare in oceans, and Actinobacteria decrease in abundance in acidic soils while Acidobacteria increase. These patterns are likely due to characteristics, such as cell envelope structure, that are shared by all members of a phylum and which convey selective fitness advantages in certain habitats. However, units of diversity defined by SSU rRNA gene sequences are insensitive to diversification resulting from dispersal limitation (5).
The examination of dispersal limitation is facilitated by analyzing discrete lineages at sufficient genetic resolution to resolve recent evolutionary divergence due to genetic drift (5, 12). Power to detect dispersal limitation is further increased by examining taxa from a single type of habitat that can be found distributed across large spatial scales (5). This level of focus is readily achieved through analysis of microbial strains that can be cultivated in isolation (13). For example, multilocus sequence analysis (MLSA) of Sulfolobus species indicates dispersal limitation and allopatric diversification on local and regional scales (14–16). In addition, genomic analysis of the soilborne pathogen Burkholderia pseudomallei revealed genetic diversification driven by vicariance along Wallace’s line between Southeast Asia and Australia (17). Also, analysis of single nucleotide polymorphisms in Bacillus anthracis indicated introduction of this soilborne pathogen to North America as a consequence of animal migrations during the late Pleistocene (18).
We chose the genus Streptomyces (phylum Actinobacteria) as a model system for exploring bacterial biogeography in soil. There are currently 615 described species of Streptomyces belonging to 130 clades (19). Streptomyces organisms are ubiquitous in soils, where they play an important role in the carbon cycle, particularly in the degradation of insoluble polymers, such as cellulose and chitin (20). These bacteria are also a major source for the discovery of clinically useful antibiotics and secondary metabolites (21). They produce spores that are resistant to starvation, UV light, and desiccation (22), and so they have the potential for widespread dispersal. Furthermore, analysis of the genetic diversity of individual species revealed that Streptomyces species can be distributed across large geographic regions (on scales of hundreds to thousands of kilometers [23, 24]).
Despite the prevalence and importance of Streptomyces species in soil, studies of their biogeography and evolutionary history are limited. Analysis of Streptomyces diversity on local spatial scales suggests that their diversity is influenced by environmental gradients (25, 26). Furthermore, analysis of Streptomyces diversity with respect to antibiotic production and resistance indicates that these phenotypes exhibit regional endemism, suggesting dispersal limitation and regional adaptation (27). We characterized Streptomyces strains from soils across the United States to determine whether their diversity scales with geographic distance and to examine the ecological and evolutionary factors that govern their biogeography. Our focus on a widespread spore-forming organism provides a strong test for the hypothesis of panmixia.
RESULTS AND DISCUSSION
A total of 924 Streptomyces strains were isolated and characterized from 15 sites spanning the United States (see Table S1 in the supplemental material). Sites were selected to represent a narrow range of ecological characteristics, including meadow, pasture, or native grasslands dominated by perennial grasses and having moderately acidic soil (pH 6.0 ± 1.0 [average ± standard deviation, or SD]). Strains were isolated under uniform conditions (see Materials and Methods), which were used to select for strains having similar physiological characteristics. The analysis of physiologically similar strains from ecologically similar sites improves our ability to detect biogeographical patterns that result from drift by minimizing the importance of selection (reviewed in reference 5).
The isolated strains encompassed 208 unique rpoB sequences, which were classified into 107 OTUs with clusters defined at a patristic distance of 0.01 (OTUrpoB) (see Fig. S1 in the supplemental material). This distance has previously been observed to roughly correlate with species boundaries for Streptomyces (28). Good’s coverage was 0.88 for unique rpoB sequences and 0.95 for OTUrpoB, indicating high coverage of Streptomyces taxonomic diversity as captured under our isolation conditions (see Fig. S2 in the supplemental material). An average of 15.5 ± 8.5 OTUrpoB was observed in each site. None of the OTUrpoB had cosmopolitan distribution, and each OTUrpoB occurred in an average of 1.7 ± 1.3 sites, with the most widespread taxon found in 8 sites. Identical rpoB sequences were observed in sites separated by more than 5,000 km (sites MS and AK2), indicating the potential for long-range dispersal.
Despite the potential for long-range dispersal, Streptomyces beta diversity varied in relation to geographic distance on spatial scales of 1,000 to 7,000 km (Fig. 1). The null model of random taxon assortment between sites was rejected, indicating that taxon composition differed significantly between sites (UniFrac analysis; P < 0.01). Both phylogenetic differentiation and Bray-Curtis dissimilarity (BCD) increased in relation to geographic distance (Fig. 1) (Unifrac distance, Mantel R2 = 0.32, P = 0.018; BCD for OTUrpoB, Mantel R2 = 0.45, P = 0.002; BCD for unique rpoB, Mantel R2 = 0.51, P = 0.002); these relationships remained significant if the Alaska (AK) sites were excluded from the analysis. In addition, we observed significant phylogenetic clustering in all sites except for the California, Mississippi, Texas, North Carolina, and Wisconsin sites (see Table S1 in the supplemental material). Phylogenetic clustering indicated that taxa within regions are more closely related to each other than they are to taxa from distant regions (29). These results agreed with our previous observations that Streptomyces populations can have large geographic ranges (23, 24) and with other observations that Streptomyces and other soil microorganisms exhibit dispersal limitation and regional endemism (27, 30–32).
FIG 1 .
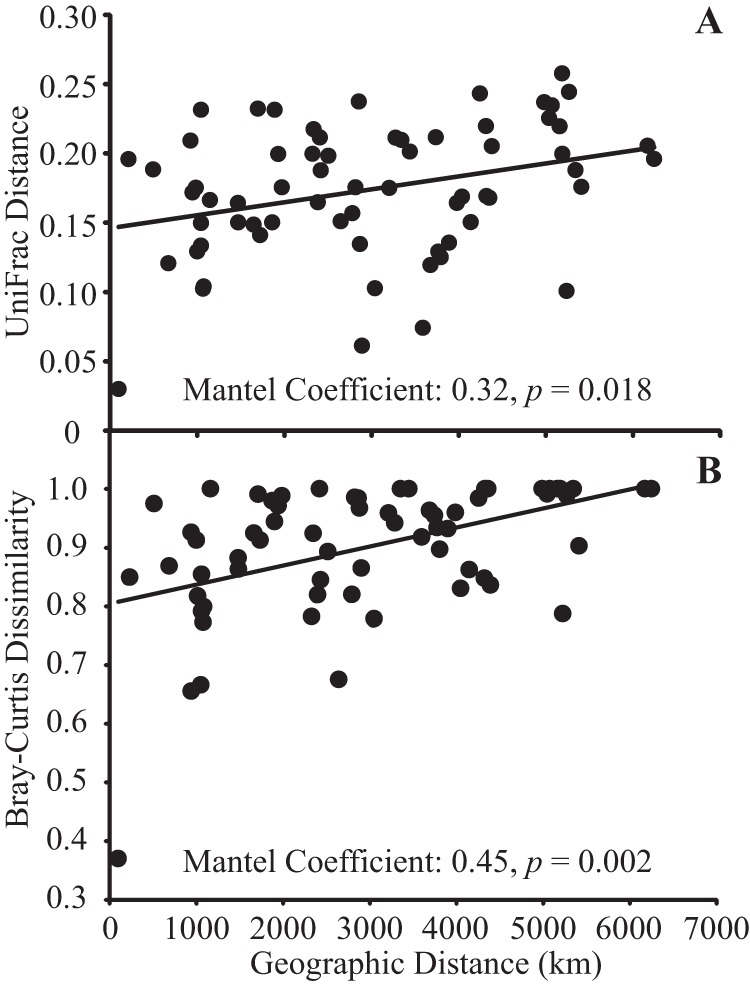
Phylogenetic (A) and taxonomic (B) dissimilarities of Streptomyces increase as a function of geographic distance between sites. The Mantel coefficient is provided along with the linear regression line.
While dispersal limitation is a parsimonious explanation for these results, an alternative hypothesis is panmixia and habitat filtering. This is better known to microbiologists as the Baas Becking hypothesis: “Everything is everywhere, but, the environment selects” (3). Habitat filtering of panmictic species would cause beta diversity to correspond strongly with environmental variables at spatial scales that fall within the Darwinian-Hutchinsonian zone (33), in which dispersal occurs more rapidly than diversification. If environmental characteristics are correlated with geographic distance within the Darwinian-Hutchinsonian zone, then habitat filtering could produce strong relationships between beta diversity and geographic distance. However, our sampling design reduced habitat variation and physiological variation of strains, and this should have likewise reduced the potential for habitat filtering to explain our results. In addition, observation of phylogenetic clustering across scales of thousands of kilometers is indicative of biogeographic rather than ecological processes, as dispersal limitation and regional diversification cause the species within regions to be more related to each other than they are to species from other regions (29, 33). Talbot et al., who likewise sampled soils associated with a single vegetation type (Pinaceae), also found little impact of environmental variation on large spatial scales when they examined fungal biogeography across North America (32).
To determine whether environmental characteristics could explain the patterns of beta diversity we observed, environmental variables were used to calculate the environmental distance between sites. Bray-Curtis distance varied in relation to the environmental distance between sites (Mantel R2 = 0.275, P = 0.008), but this result was not significant if the Alaska sites were excluded (Mantel R2 = 0.157, P = 0.146). Canonical correspondence analysis found no significant correlation between beta diversity and environmental characteristics whether the Alaska sites were included (P = 0.73) or excluded (P = 0.14). With respect to discrete environmental variables, Streptomyces beta diversity varied in relation to latitude, temperature, and soil pH, but not in relation to soil organic matter or annual precipitation (Table 1). Support for a relationship between beta diversity and temperature declined when the Alaska sites were excluded, though other results were largely unaffected by removing the Alaska sites (see Table S2 in the supplemental material). These results indicated that environmental variables have a minor though significant impact on Streptomyces beta diversity, with the greatest amount of variation due to latitude and soil pH. Soil pH is well known to impact the beta diversity of Actinobateria in soil (34, 35), and certain Streptomyces species are known to have habitat constraints which are circumscribed by soil pH (36).
TABLE 1 .
Relationships between environmental factors and Streptomyces phylogenetic (UniFrac distance) and taxonomic (Bray-Curtis dissimilarity) valuesa
| Analysis type | Sequence inclusion | Correlation (R2 value) withb: |
||||
|---|---|---|---|---|---|---|
| Latitude | Soil pH | Temp | SOM | PPT | ||
| UniFrac | Weighted | 0.52** | 0.35 | 0.50** | 0.26 | 0.28 |
| Unweighted | 0.48*** | 0.41* | 0.44** | 0.32 | 0.34 | |
| Bray-Curtis | Weighted | 0.46*** | 0.40* | 0.44** | 0.21 | 0.33 |
| Unweighted | 0.46*** | 0.44** | 0.44** | 0.25 | 0.31 | |
Analyzed by using the adonis program (permutational multivariate analysis of variance) within the R package.
Bold values indicate statistically significant correlations (*, P < 0.05; **, P < 0.01; ***, P < 0.001). The analyses were performed by either including all rpoB sequences (weighted) or excluding duplicate sequences for each OTU (unweighted). Abbreviations: Temp, average annual temperature; SOM, soil organic matter content; PPT, annual average precipitation.
Streptomyces phylogenetic diversity was negatively correlated with latitude (Table 2; Fig. 2). A latitudinal gradient was observed in relation to Faith’s phylogenetic diversity (PD) (Fig. 2A) (r = −0.70, P = 0.012), the net relatedness index (NRI) (Fig. 2B) (r = 0.72, P = 0.008), and mean root distance (MRD) (r = 0.61, P = 0.035). The correlation between Streptomyces phylogenetic diversity and latitude corresponded with the slope of the latitudinal diversity gradient observed for a wide range of taxa (average slope, −0.73), as determined in the meta-analysis reported by Hillebrand (37). As expected, Streptomyces diversity was also observed to correlate with temperature (which correlates strongly with latitude), but a significant correlation was not observed between phylogenetic diversity (as defined by PD, NRI, or MRD) and soil pH, or between soil pH and latitude (Table 2). Hence, while soil pH does explain some variation in beta diversity, it does not underlie the latitudinal diversity gradient we observed.
TABLE 2 .
Correlation coefficients for relationships between Streptomyces diversity and environmental characteristics across sitesa
| Characteristic | Correlation (R2 value) based on: |
|||||||
|---|---|---|---|---|---|---|---|---|
| PD | NRI | MRD | Lat. | Long. | Temp | SOM | PPT | |
| NRI | −0.86** | |||||||
| MRD | −0.70* | 0.67* | ||||||
| Lat. | −0.70* | 0.72** | 0.61* | |||||
| Long. | 0.62* | −0.51 | −0.54 | −0.75** | ||||
| Temp | 0.61* | −0.64* | −0.47 | −0.97** | 0.60* | |||
| SOM | 0.45 | −0.24 | −0.14 | −0.16 | −0.06 | 0.27 | ||
| PPT | 0.42 | −0.24 | 0.01 | −0.40 | 0.54 | 0.38 | 0.25 | |
| pH | 0.16 | −0.39 | −0.42 | −0.45 | 0.11 | 0.43 | 0.09 | −0.41 |
Bold values indicate statistically significant correlations (*, P < 0.05; **, P < 0.01). Abbreviations: Lat., latitude; Long., longitude; Temp, average annual temperature; SOM, soil organic matter content; PPT, annual average precipitation; pH, soil pH.
FIG 2 .
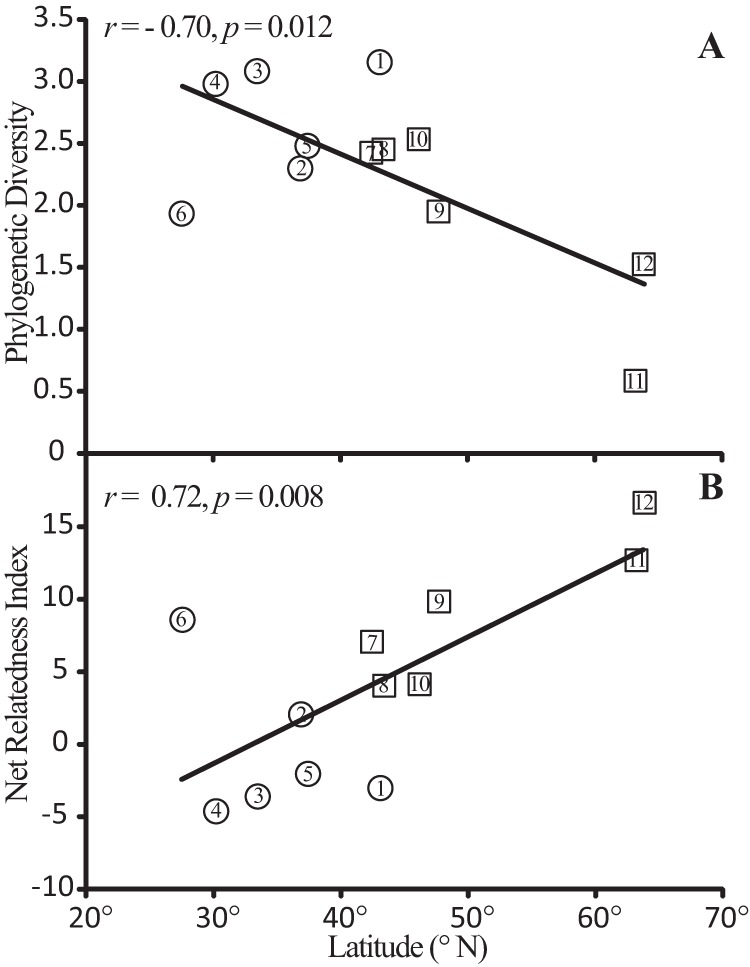
Latitude correlates with the phylogenetic diversity of Streptomyces as measured by both Faith’s phylogenetic diversity (A) and the net relatedness index (B). The Pearson correlation coefficient is provided along with the linear regression line. Symbols indicate the presence (□) or absence (○) of glaciation during the late Pleistocene. Numbers rank sites by time available for colonization, as follows: 1, WI; 2, NC; 3, MS; 4, TX; 5, CA; 6, FL; 7, NY; 8, ME; 9, WA; 10, OR; 11, AK1; 12, AK2. The FL site was below sea level at the beginning of the late Pleistocene, and sites in southern Wisconsin bound the Driftless Area, which escaped glaciation and has remained above sea level since the late Paleozoic.
The latitude diversity gradient is a fundamental pattern in ecology, has been well-documented since the days of Wallace (38), and is consistent across a wide taxonomic range of plant and animal species (37). Far fewer studies have been able to document a latitude diversity gradient in bacteria. The most likely explanation for this absence of evidence is the low taxonomic resolution at which microbial diversity is typically measured (5). Latitudinal patterns of diversity are typically observed at the population to class level in plants and animals (38). And yet, while the domain Bacteria is far older and far more diverse than the domain Eukarya, it is common practice to quantify patterns of microbial diversity for all Bacteria as if this domain represented a single ecologically coherent unit. However, the ecological and evolutionary factors that cause patterns of diversity are best understood when we quantify diversity at the level of taxonomic resolution that is sufficient to describe processes of diversification (5).
Taxon-specific, culture-dependent approaches to describing microbial diversity allow us to evaluate the processes that contribute to microbial diversification. The notable limitations of cultivation-dependent approaches are the potential for bias against certain physiological traits and the lack of sensitivity to taxa present at very low relative abundance. For example, the cultivation method we used would be unable to detect strains present at less than 1.25 × 105 cells per g of soil. The biogeographic sampling of plants and animals is likewise subject to both veil line effects that obscure the presence of rare taxa and to sampling artifacts that may bias samples against certain physiological types (for example, nocturnal species are rarely collected during the day). However, a distinct advantage of a taxon-specific approach is that it allows for the physiological and genotypic characterization of discrete microbial taxa. Such data are highly valuable, because hypotheses that describe patterns of diversification often make predictions that can be tested with physiological and genomic data. Hence, taxon-specific, culture-dependent approaches provide evidence suitable for evaluating hypotheses derived from the broader field of evolutionary ecology.
The evolutionary forces that generate latitudinal diversity gradients remain under debate, and several hypotheses have been proposed to explain this biogeographical pattern (reviewed in references 39 and 40). Ecological hypotheses explain differences in species richness as a function of ecological factors, such as carrying capacity, productivity, and niche availability, which vary across climate gradients (41). Evolutionary tempo hypotheses invoke the relationship between higher temperatures and increased kinetics of metabolism to predict that evolutionary rates and cladogenesis vary across temperature gradients (42). Hypotheses based on historical contingency propose that diversity gradients result from historical geologic, ecological, or demographic events that influenced dispersal and diversification (40, 43, 44).
The niche conservatism hypothesis explains the latitude diversity gradient as a function of historical climate change (45–47). Specifically, the hypothesis posits that climate oscillations and patterns of glaciation have constrained time for speciation at higher latitudes and that species found at higher latitudes are derived as a result of demographic range expansion from species occupying lower latitudes. Niche conservatism is facilitated by high-density blocking, whereby early colonists subsequently impose density-dependent barriers to colonization by late dispersing individuals (48). Furthermore, derived populations at higher latitudes are expected to evolve adaptations to their new habitats, thereby imposing further barriers to latitudinal dispersal across climate regimes (48). Glacial retreat at the end of the Pleistocene caused demographic range expansions in diverse plant and animal species, and the legacy of these events is readily observed in extant patterns of genetic diversity (49–53). In addition, dispersal limitation and strong priority effects suggest that historical contingency has played an important role in determining the diversity of soil fungal communities (54, 55), and this may explain why the diversity of soil fungi varies with respect to the biogeographic provinces described for North America (32).
The Wisconsin Glacier occurred between 30,000 and 10,000 years BP (maximum extent, approximately 15,000 to 16,000 years BP) and covered nearly all of Canada and the northern part of the United States except for a small region in southwestern Wisconsin known as the Driftless Area (56, 57). The legacy of Pleistocene glaciation in North America can often be observed as discontinuity in genetic diversity, which manifests at roughly 40°N latitude (58). In several instances, the genetic diversity of terrestrial species of Bacteria has been shaped by glacial dynamics during the late Pleistocene (18, 59), including evidence for discontinuity in the genetic diversity of Actinobacteria along a chronosequence formed by recession of Wisconsin glaciation (60).
The latitudinal gradient of diversity we observed for Streptomyces is broadly consistent with the niche conservatism hypothesis. The niche conservatism hypothesis predicts that latitude will correlate negatively with phylogenetic diversity and positively with phylogenetic clustering and mean root distance (61, 62), and these predictions are consistent with our observations for the Streptomyces diversity gradient (Table 2; Fig. 2). As predicted by the niche conservatism hypothesis, PD is lower (glaciated, 1.9 ± 0.8; nonglaciated, 2.7 ± 0.5; P = 0.071), and both NRI phylogenetic clustering (glaciated, 10.0 ± 4.9; nonglaciated, −0.5 ± 5.0; P = 0.008) and MRD (glaciated, 26.1 ± 3.0; nonglaciated, 22.1 ± 2.7; P = 0.037) are higher in sites subjected to glaciation than in nonglaciated sites. In addition, we observed a correlation between diversity and time available for colonization with respect to phylogenetic diversity (Spearman’s r = 0.64, P = 0.026), net relatedness index (Spearman’s r = −0.82, P = 0.001), and mean root distance (Spearman’s r = 0.70, P = 0.012), and this is also a prediction of niche conservatism (see Table S1 in the supplemental material).
The niche conservatism hypothesis predicts that sites at higher latitudes were colonized following glacial retreat as a result of demographic range expansion from species at lower latitudes. Network analysis of OTUrpoB sharing between sites indicated a strong latitudinal delineation in the OTUrpoB compositions of sites (Fig. 3). The network showed that 95% (21/22) of the shared OTUs observed in glaciated sites were also found in a nonglaciated site, and most of these OTUs (18/22) were common to Wisconsin. The WI site also stood out in relationships between NRI, PD, and latitude (Fig. 2), having lower NRI and PD than sites of comparable latitude (NY and ME sites) and being the only northern latitude site to lack significant phylogenetic clustering (see Table S1 in the supplemental material). We note that the Driftless Area in Wisconsin has been proposed as a refugium for certain species of plants (63), and the unusual diversity of Streptomyces in this region may be due to the fact that much of Wisconsin was never glaciated and thus strains from this region had greater opportunity than lower-latitude strains to disperse into habitat exposed by glacial retreat. In addition, niche conservatism posits that time for speciation limits diversity, and hence it is interesting that the FL site was below sea level during the last interglacial period, as recently as 118,000 years ago (64). The FL site, despite being at the lowest latitude, also had the lowest phylogenetic diversity of any nonglacial site and was the only nonglacial site to demonstrate significant phylogenetic clustering (Fig. 2; see also Table S1).
FIG 3 .
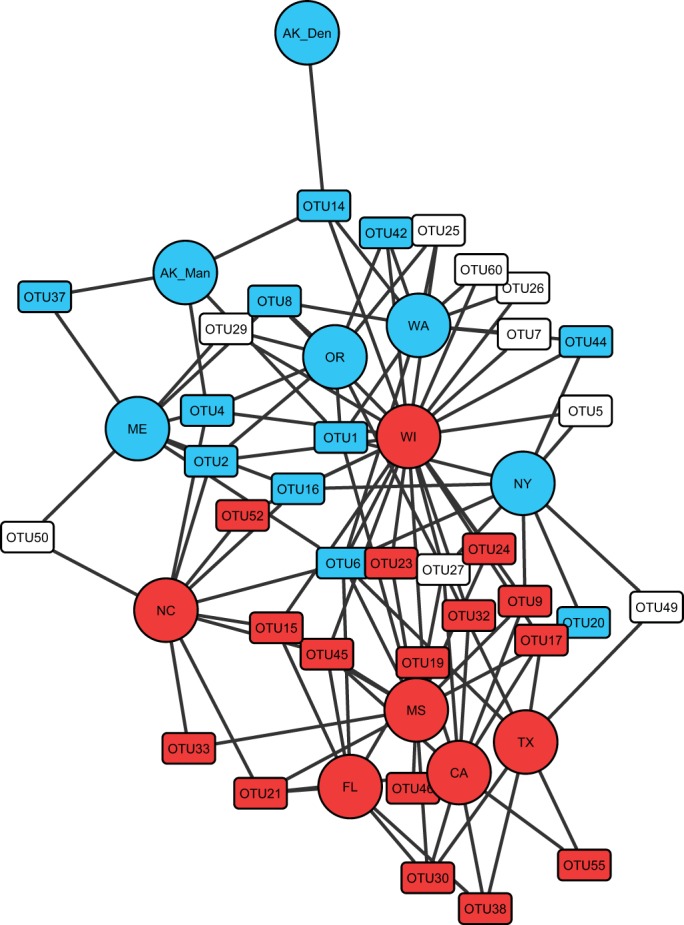
Network analysis illustrating OTUrpoB sharing across sample sites, indicating a latitudinal gradient of beta diversity consistent with glaciation history. Sample sites (circles) are colored by history of glaciation (blue) or nonglaciation (red) during the late Pleistocene. OTUs (rectangles) are colored if both >90% of sequences were recovered from glacial (blue) or nonglacial (red) sites and the null hypothesis of random assortment was rejected (Fisher exact test, P < 0.05). The WI site was the most highly connected site in the network and has previously been proposed as a glacial refugium for certain plant species, as discussed in the text. OTUs found in only one site were excluded from the analysis.
Finally, network analyses of haplotype phylogeography provided evidence for the regional diversification of Streptomyces clades as a consequence of population expansion and isolation by distance (see Fig. S3 and Table S3 in the supplemental material). The largest observed rpoB haplotype network represented 156 strains. Nested clade analysis revealed that the pattern of ancestry in this network was consistent with divergence due to population expansion (representing 82% of the strains in the network) and subsequent isolation of clades by distance (representing 95% of the strains in the prior subset) (see Fig. S3 and Table S3). Overall, 421 strains belonged to clades whose pattern of ancestry was consistent with dispersal limitation, and these represented 91% of the strains in networks that supported evolutionary inference (see Fig. S3 and Table S3). These findings are consistent with the hypothesis that historical demographic processes explain patterns of Streptomyces biogeography.
Strong correlations between latitude, temperature, and climate make it difficult to determine the ultimate mechanisms that generate latitudinal diversity gradients. For example, the kinetic effects of temperature have been previously shown to impact latitudinal diversity gradients for marine bacteria (65). In addition, the diversity gradient we observed could be due to an ecological correlation between Streptomyces diversity and unmeasured differences in biotic or abiotic variables at our sites. However, the ability of temperature to increase evolutionary tempo would not explain the low diversity in our FL site or the high diversity in Wisconsin. Likewise, neither the effects of temperature nor the effects of biotic or abiotic variables would readily predict discontinuity in phylogenetic clustering or mean root depth with respect to historically glaciated and nonglaciated sites; nor would these hypotheses explain well the inference of demographic expansion provided by analysis of haplotype networks. Finally, observations of the genetic diversity within Streptomyces populations indicate that, despite evidence for dispersal limitation, multiple species are in linkage equilibrium across large latitudinal gradients (23, 24). The most parsimonious explanation of these apparently conflicting results is that these populations have experienced an evolutionarily recent demographic range expansion from low to high latitudes. While the results we describe are broadly consistent with niche conservatism, they do not rigorously exclude other hypotheses. Ultimately, it is likely that latitudinal diversity gradients can result from a combination of ecological, evolutionary, and historical processes and that the relative importance of these different mechanisms varies between different taxa.
In summary, these data indicate that Streptomyces diversity varies in relation to geographic distance and manifests in a latitudinal diversity gradient. Furthermore, the data suggest that these patterns result from dispersal limitation and the regional diversification of clades. Habitat filtering is often invoked to explain microbial biogeography, but while habitat filtering could produce patterns of beta diversity that vary with latitude, this hypothesis does not predict latitudinal gradients of PD, NRI, and MRD. While soil pH was shown to influence beta diversity (Table 1), soil pH did not correlate with latitude across our sites, and soil pH did not correlate with patterns of PD, NRI, and MRD (Table 2). Hence, we conclude that soil pH is unlikely to underlie the latitudinal diversity gradient. The hypothesis that is most parsimonious given the data we have described is that historical demographic events underlie the Streptomyces latitudinal diversity gradient, though these data do not exclude completely either evolutionary or ecological hypotheses. The hypothesis of niche conservatism leads to specific predictions about the genetic consequences of demographic expansion and the phylogenetic conservation of phenotypic traits associated with different climate regimes (45–47). Physiological and genomic analyses of Streptomyces strains from our culture collection should make it possible to further test specific predictions of niche conservatism.
MATERIALS AND METHODS
Sampling and strain collection.
Streptomyces strains (n = 924) were isolated from 15 sites across the United States (see Table S1 in the supplemental material). Soils were collected exclusively from sites dominated by perennial grasses with neutral to acidic pH (lawn, meadow, and pasture). Soils were sampled at a 0- to 5-cm depth and air dried at room temperature. Soil organic matter content was measured by loss on ignition, and soil pH was determined for a 1:2 (wt/vol) dilution of soil in 0.01 M CaCl2 (66). Precipitation and temperature data were obtained from the U.S. National Centers for Environmental Information (http://www.ncdc.noaa.gov/) and represent 30-year climate normal data unless otherwise described. Glacial extent and chronology were determined based on the methods of Peltier (67).
For Streptomyces isolation, 50 mg of soil was diluted 1:100 (wt/vol) in phosphate-buffered saline and mixed vigorously (1 to 2 min), after which 25 to 50 µl was spread onto glycerol-arginine plates containing 300 mg/liter cycloheximide and 30 mg/liter Rose bengal (68, 69), with the pH adjusted to 8.7 as previously described (23). Colonies developed after 5 to 7 days of incubation at room temperature, and strains were isolated by repeated streaking. DNA was extracted from purified cultures, which were grown with shaking at 30°C in liquid yeast extract-malt extract medium (YEME) containing 0.5% glycine (22), using a standard phenol-chloroform-isoamyl alcohol protocol (70). The resulting DNA was resuspended in 150 µl of Tris-EDTA buffer.
Sequence analysis.
PCR amplification and sequencing of rpoB was performed as described elsewhere (71). Briefly, PCR was performed in 25-µl volumes containing 1× AmpliTaq gold buffer (Applied Biosystems, Foster City, CA), 3 mM MgCl2, 2.5 mM each deoxynucleoside triphosphate (Promega, Madison, WI), 0.4 µM rpoB forward primer (5′-GAGCGCATGACCACCCAGGACGTCGAGGC-3′), 0.4 µM rpoB reverse primer (5′-CCTCGTAGTTGTGACCCTCCCACGGCATGA-3′), 10% dimethyl sulfoxide, 1.25 U AmpliTaq Gold (Applied Biosystems, Foster City, CA), and 50 to 200 ng DNA. The following reaction conditions were used: 95°C for 10 min for initial denaturation, followed by 35 cycles of 95°C for 20 s, 65°C for 30 s, and 72°C for 45 s, and final extension at 72°C for 10 min. Sequencing of PCR products was performed at the Cornell Life Sciences Core Laboratories Center. Sequences were assembled manually, and trace files were inspected visually and uniformly trimmed to achieve a final length of 377 bp.
Sequences used in phylogenetic analyses were aligned using MUSCLE (72). Maximum likelihood trees were constructed with the general time-reversible model of nucleotide substitution (73), incorporating an estimated proportion of invariant sites and discrete gamma distribution (GTR+I+G) within the RAxML program (74). Trees were rooted using Mycobacterium smegmatis as the outgroup.
Analyses of Streptomyces diversity and phylogeography.
Operational taxonomic units based on rpoB sequences were defined at a 0.01 nucleotide dissimilarity cutoff using patristic distances as implemented in RAMI (75). This dissimilarity cutoff roughly delineates the genetic divergence between characterized Streptomyces species (28). In the case of the Wisconsin and North Carolina sites, sequences were aggregated across two or three soil samples, respectively, to ensure that >30 strains were available to represent each region. The decision to aggregate was justified by similarities in climate, geography, and soil characteristics across the aggregated samples. In addition, previous analyses had indicated that Streptomyces species are broadly distributed at regional scales (>1,000 km) (23, 24). This provided 12 sites with 77 ± 30 (mean ± SD) strains characterized per site. Beta diversity was evaluated through hierarchical clustering implemented within UniFrac (76). Mantel correlations between matrices of geographic distance and either UniFrac or Bray-Curtis distances were performed with the R package ecodist (77) and the Pearson correlation method with 1,000 permutations. Patterns of OTUrpoB sharing were visualized using Cytoscape 2.8 and the y-files organic layout (78).
Values for the NRI, nearest taxon index (NTI), and Faiths PD were calculated using Phylocom v.4.2 (79). For both the NRI and NTI, positive values indicate phylogenetic clustering (i.e., closely related taxa cooccur more than expected by chance), negative values indicate overdispersal or phylogenetic evenness, and values close to zero suggest a phylogenetically random assembly of species. Significance was determined by permutation (n = 999) in comparison to a null model where taxa are assigned to each site by random draw without replacement from the list of all taxa. The MRD was calculated as the average number of nodes separating the species in a site from the root of their phylogenetic tree (80).
Haplotype networks were created using a statistical parsimony procedure (81, 82) as implemented in TCS v1.18 (83). Closed loops representing network ambiguities were resolved using the nesting rules proposed by Templeton et al. (82). The final nested clade information was used as input in the program GeoDis v2.2 (84). GeoDis analyzes the nested haplotype network to make inferences on the processes that could have produced the association of the haplotype distribution and geography. Both TCS v1.18 and GeoDis v2.2 were performed with the ANeCA platform (85).
Nucleotide sequence accession numbers.
rpoB gene sequences determined in this study are available from GenBank under accession numbers KU238378:KU238472 and KU956103:KU956931.
SUPPLEMENTAL MATERIAL
Streptomyces strains isolated and characterized from the series of 15 sites described in the text (In the cases of Wisconsin and North Carolina, samples were aggregated across two or three soil samples, respectively, to represent each region. “Time,” the time available for colonization; mya, millions of years ago; SOM, soil organic matter. Asterisks identify NRI values that showed clustering [positive] or overdispersion [negative] that are unlikely to result from chance [P < 0.01].)
Results from Table 2 alongside results obtained when sites from Alaska were excluded (The column reporting “sites” indicates results obtained when Alaska sites were included [“+AK” rows are the same as those depicted in Table 2] or excluded [−AK]. Results that reveal different outcomes between the +AK and –AK analyses are indicated with shading. Relationships between environmental factors and Streptomyces phylogenetic [UniFrac distance] and taxonomic [Bray-Curtis dissimilarity] differences were analyzed by using adonis [permutational multivariate analysis of variance]. Values indicate R2 values, and results that are unlikely due to chance are indicated with asterisks [*, P < 0.05; **, P < 0.01; ***, P < 0.001]. The analyses were performed by either including all rpoB sequences [weighted] or excluding duplicate sequences for each OTU [unweighted].)
Significant inferences from nested clade analysis of Streptomyces rpoB haplotypes (see Fig. S3).
Maximum likelihood tree based on rpoB sequences of Streptomyces strains. A total of 924 Streptomyces strains were isolated from 15 sites in North America and classified into 107 OTUs on the basis of rpoB sequence dissimilarity. The inner colored ring includes sequential color blocks to indicate the 107 OTUrpob identified in this study. The outer colored ring identifies the history of the site from which each strain was isolated, as indicated in the color scale (FL and WI are indicated as well, due to their distinctive geologic history). The site from which each strain was isolated can be determined by the strain name, as described in Table S1. Download
The rarefied collectors curve indicates that OTUrpoB were well sampled from the sampling sites (with respect to the constraints imposed by strain collection). Good’s coverage was 0.88 for unique rpoB sequences and 0.95 for OTUrpoB. Download
Nested clade analysis of Streptomyces rpoB haplotype networks provides evidence for contiguous range expansion and dispersal limitation. Circles represent rpoB haplotypes, with radii proportional to the number of strains that belong to the haplotype. Haplotypes are shaded to represent strain source, with the fraction of strains isolated from previously glaciated or nonglaciated sites indicated in blue and red, respectively. Each line symbolizes one mutational step, with dots indicating inferred haplotypes not sampled. The complete set of haplotype networks is shown (Table S2 shows the evolutionary inferences for each clade). Download
ACKNOWLEDGMENTS
We are extremely grateful to Cindy Barton, Julie Brown, Steve Campbell, Sherry Dollhopf, Matthew Elverson, Heather Fullerton, Robin Judge, Ginger Knussmann, Peter McCarthy, Molly Meserve, Tyrrell Nelsen, Armanda Roco, Jacob Ross, Nathan Ross, Jonathon Rudd, Allen Sharpe, Kevin Smith, and Beth Tyson for their invaluable help in collecting soil samples.
This material is based upon work supported by the National Science Foundation under grants DEB-1050475 and DEB-1456821.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Funding Statement
This material is based upon work supported by the National Science Foundation under Grants No. DEB-1050475 and DEB-1456821. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Andam CP, Doroghazi JR, Campbell AN, Kelly PJ, Choudoir MJ, Buckley DH. 2016. A latitudinal diversity gradient in terrestrial bacteria of the genus Streptomyces. mBio 7(2):e02200-15. doi:10.1128/mBio.02200-15.
REFERENCES
- 1.Kellogg CA, Griffin DW. 2006. Aerobiology and the global transport of desert dust. Trends Ecol Evol 21:638–644. doi: 10.1016/j.tree.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Finlay BJ. 2002. Global dispersal of free-living microbial eukaryote species. Science 296:1061–1063. doi: 10.1126/science.1070710. [DOI] [PubMed] [Google Scholar]
- 3.Baas Becking LGM. 1934. Geobiologie of inleiding tot de milieukunde. W.P. van Stockum and Zoon, The Hague, The Netherlands. [Google Scholar]
- 4.Finlay BJ, Fenchel T. 2004. Cosmopolitan metapopulations of free-living microbial eukaryotes. Protist 155:237–244. doi: 10.1078/143446104774199619. [DOI] [PubMed] [Google Scholar]
- 5.Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JB. 2012. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10:497–506. doi: 10.1038/nrmicro2795. [DOI] [PubMed] [Google Scholar]
- 6.Nemergut DR, Costello EK, Hamady M, Lozupone C, Jiang L, Schmidt SK, Fierer N, Townsend AR, Cleveland CC, Stanish L, Knight R. 2011. Global patterns in the biogeography of bacterial taxa. Environ Microbiol 13:135–144. doi: 10.1111/j.1462-2920.2010.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martiny JB, Bohannan BJ, Brown JH, Colwell RK, Fuhrman JA, Green JL, Horner-Devine MC, Kane M, Krumins JA, Kuske CR, Morin PJ, Naeem S, Øvreås L, Reysenbach AL, Smith VH, Staley JT. 2006. Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4:102–112. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 8.Achtman M, Wagner M. 2008. Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6:431–440. doi: 10.1038/nrmicro1872. [DOI] [PubMed] [Google Scholar]
- 9.Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 67:4399–4406. doi: 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ochman H, Elwyn S, Moran NA. 1999. Calibrating bacterial evolution. Proc Natl Acad Sci U S A 96:12638–12643. doi: 10.1073/pnas.96.22.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle JJ, Luckow MA. 2003. The rest of the iceberg. Legume diversity and evolution in a phylogenetic context. Plant Physiol 131:900–910. doi: 10.1104/pp.102.018150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green J, Bohannan BJ. 2006. Spatial scaling of microbial biodiversity. Trends Ecol Evol 21:501–507. doi: 10.1016/j.tree.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, Polz MF, Alm EJ. 2012. Population genomics of early events in the ecological differentiation of Bacteria. Science 336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cadillo-Quiroz H, Didelot X, Held NL, Herrera A, Darling A, Reno ML, Krause DJ, Whitaker RJ. 2012. Patterns of gene flow define species of thermophilic Archaea. PLoS Biol 10:e1001265. doi: 10.1371/journal.pbio.1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitaker RJ, Grogan DW, Taylor JW. 2005. Recombination shapes the natural population structure of the hyperthermophilic archaeon Sulfolobus islandicus. Mol Biol Evol 22:2354–2361. doi: 10.1093/molbev/msi233. [DOI] [PubMed] [Google Scholar]
- 16.Whitaker RJ, Grogan DW, Taylor JW. 2003. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301:976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- 17.Pearson T, Giffard P, Beckstrom-Sternberg S, Auerbach R, Hornstra H, Tuanyok A, Price EP, Glass MB, Leadem B, Beckstrom-Sternberg JS, Allan GJ, Foster JT, Wagner DM, Okinaka RT, Sim SH, Pearson O, Wu Z, Chang J, Kaul R, Hoffmaster AR, Brettin TS, Robison RA, Mayo M, Gee JE, Tan P, Currie BJ, Keim P. 2009. Phylogeographic reconstruction of a bacterial species with high levels of lateral gene transfer. BMC Biol 7:78. doi: 10.1186/1741-7007-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kenefic LJ, Pearson T, Okinaka RT, Schupp JM, Wagner DM, Ravel J, Hoffmaster AR, Trim CP, Chung WK, Beaudry JA, Foster JT, Mead JI, Ravel J, Keim P. 2009. Pre-Columbian origins for North American anthrax. PLoS One 4:e4813. doi: 10.1371/journal.pone.0004813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labeda DP, Goodfellow M, Brown R, Ward AC, Lanoot B, Vanncanneyt M, Swings J, Kim SB, Liu Z, Chun J, Tamura T, Oguchi A, Kikuchi T, Kikuchi H, Nishii T, Tsuji K, Yamaguchi Y, Tase A, Takahashi M, Sakane T, Suzuki KI, Hatano K. 2012. Phylogenetic study of the species within the family Streptomycetaceae. Antonie Van Leeuwenhoek 101:73–104. doi: 10.1007/s10482-011-9656-0. [DOI] [PubMed] [Google Scholar]
- 20.Schrempf H. 2001. Recognition and degradation of chitin by Streptomycetes. Antonie Van Leeuwenhoek 79:285–289. doi: 10.1023/A:1012058205158. [DOI] [PubMed] [Google Scholar]
- 21.Watve MG, Tickoo R, Jog MM, Bhole BD. 2001. How many antibiotics are produced by the genus Streptomyces? Arch Microbiol 176:386–390. doi: 10.1007/s002030100345. [DOI] [PubMed] [Google Scholar]
- 22.Kieser T, Bibb M, Buttner M, Charter K, Hopwood D. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom. [Google Scholar]
- 23.Doroghazi JR, Buckley DH. 2010. Widespread homologous recombination within and between Streptomyces species. ISME J 4:1136–1143. doi: 10.1038/ismej.2010.45. [DOI] [PubMed] [Google Scholar]
- 24.Choudoir M, Doroghazi J, Buckley D 1 March 2016. Latitude delineates patterns of biogeography in terrestrial Streptomyces. bioRxiv doi: 10.1101/032169. [DOI] [PubMed] [Google Scholar]
- 25.Davelos AL, Xiao K, Samac DA, Martin AP, Kinkel LL. 2004. Spatial variation in Streptomyces genetic composition and diversity in a prairie soil. Microb Ecol 48:601–612. doi: 10.1007/s00248-004-0031-9. [DOI] [PubMed] [Google Scholar]
- 26.Antony-Babu S, Stach JE, Goodfellow M. 2008. Genetic and phenotypic evidence for Streptomyces griseus ecovars isolated from a beach and dune sand system. Antonie Van Leeuwenhoek 94:63–74. doi: 10.1007/s10482-008-9246-y. [DOI] [PubMed] [Google Scholar]
- 27.Kinkel LL, Schlatter DC, Xiao K, Baines AD. 2014. Sympatric inhibition and niche differentiation suggest alternative coevolutionary trajectories among Streptomycetes. ISME J 8:249–256. doi: 10.1038/ismej.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rong X, Huang Y. 2012. Taxonomic evaluation of the Streptomyces hygroscopicus clade using multilocus sequence analysis and DNA-DNA hybridization, validating the MLSA scheme for systematics of the whole genus. Syst Appl Microbiol 35:7–18. doi: 10.1016/j.syapm.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Webb CO, Pitman NC. 2002. Phylogenetic balance and ecological evenness. Syst Biol 51:898–907. doi: 10.1080/10635150290102609. [DOI] [PubMed] [Google Scholar]
- 30.Wawrik B, Kutliev D, Abdivasievna UA, Kukor JJ, Zylstra GJ, Kerkhof L. 2007. Biogeography of actinomycete communities and type II polyketide synthase genes in soils collected in New Jersey and Central Asia. Appl Environ Microbiol 73:2982–2989. doi: 10.1128/AEM.02611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho JC, Tiedje JM. 2000. Biogeography and degree of endemicity of fluorescent pseudomonas strains in soil. Appl Environ Microbiol 66:5448–5456. doi: 10.1128/AEM.66.12.5448-5456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talbot JM, Bruns TD, Taylor JW, Smith DP, Branco S, Glassman SI, Erlandson S, Vilgalys R, Liao H-L, Smith ME, Peay KG. 2014. Endemism and functional convergence across the North American soil mycobiome. Proc Natl Acad Sci U S A 111:6341–6346. doi: 10.1073/pnas.1402584111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vamosi SM, Heard SB, Vamosi JC, Webb CO. 2009. Emerging patterns in the comparative analysis of phylogenetic community structure. Mol Ecol 18:572–592. doi: 10.1111/j.1365-294X.2008.04001.x. [DOI] [PubMed] [Google Scholar]
- 34.Fierer N, Jackson RB. 2006. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kampfer P. 2006. The family Streptomycetaceae. Part I: taxonomy, p 538–604. In Dworkin M, Falkow S, Rosenberg E, Schleifer K, Stackebrandt E (ed), The prokaryotes, vol. 3 Springer, New York, NY. [Google Scholar]
- 37.Hillebrand H. 2004. On the generality of the latitudinal diversity gradient. Am Nat 163:192–211. doi: 10.1086/381004. [DOI] [PubMed] [Google Scholar]
- 38.Wallace AR. 1878. Tropical nature and other essays. MacMillan, New York, NY. [Google Scholar]
- 39.Mittelbach GG, Schemske DW, Cornell HV, Allen AP, Brown JM, Bush MB, Harrison SP, Hurlbert AH, Knowlton N, Lessios HA, McCain CM, McCune AR, McDade LA, McPeek MA, Near TJ, Price TD, Ricklefs RE, Roy K, Sax DF, Schluter D, Sobel JM, Turelli M. 2007. Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecol Lett 10:315–331. doi: 10.1111/j.1461-0248.2007.01020.x. [DOI] [PubMed] [Google Scholar]
- 40.Wiens JJ, Donoghue MJ. 2004. Historical biogeography, ecology and species richness. Trends Ecol Evol 19:639–644. doi: 10.1016/j.tree.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 41.Currie DJ, Mittelbach GG, Cornell HV, Field R, Guegan J, Hawkins BA, Kaufman DM, Kerr JT, Oberdorff T, O’Brien E, Turner JRG. 2004. Predictions and tests of climate-based hypotheses of broad-scale variation in taxonomic richness. Ecol Lett 7:1121–1134. doi: 10.1111/j.1461-0248.2004.00671.x. [DOI] [Google Scholar]
- 42.Allen AP, Brown JH, Gillooly JF. 2002. Global biodiversity, biochemical kinetics, and the energetic-equivalence rule. Science 297:1545–1548. doi: 10.1126/science.1072380. [DOI] [PubMed] [Google Scholar]
- 43.Wiens JJ, Ackerly DD, Allen AP, Anacker BL, Buckley LB, Cornell HV, Damschen EI, Jonathan Davies T, Grytnes J-A, Harrison SP, Hawkins BA, Holt RD, McCain CM, Stephens PR. 2010. Niche conservatism as an emerging principle in ecology and conservation biology. Ecol Lett 13:1310–1324. doi: 10.1111/j.1461-0248.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 44.Stevens RD. 2006. Historical processes enhance patterns of diversity along latitudinal gradients. Proc Biol Sci 273:2283–2289. doi: 10.1098/rspb.2006.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hewitt GM. 2004. Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc Lond Soc B Biol Sci 359:183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hewitt G. 2000. The genetic legacy of the quaternary ice ages. Nature 405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- 47.Hewitt GM. 1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. [Google Scholar]
- 48.Waters JM, Fraser CI, Hewitt GM. 2013. Founder takes all: density-dependent processes structure biodversity. Trends Ecol Evol 28:78–85. doi: 10.1016/j.tree.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 49.Soltis DE, Gitzendanner MA, Strenge DD, Soltis PS. 1997. Chloroplast DNA intraspecific phylogeography of plants from the Pacific Northwest of North America. Plant Syst Evol 206:353–373. doi: 10.1007/BF00987957. [DOI] [Google Scholar]
- 50.Milá B, Smith TB, Wayne RK. 2006. Postglacial population expansion drives the evolution of long-distance migration in a songbird. Evolution 60:2403–2409. doi: 10.1111/j.0014-3820.2006.tb01875.x. [DOI] [PubMed] [Google Scholar]
- 51.Wilson AB, Eigenmann Veraguth I. 2010. The impact of Pleistocene glaciation across the range of a widespread European coastal species. Mol Ecol 19:4535–4553. doi: 10.1111/j.1365-294X.2010.04811.x. [DOI] [PubMed] [Google Scholar]
- 52.Bernatchez L, Wilson CC. 1998. Comparative phylogeography of Nearctic and Palearctic fishes. Mol Ecol 7:431–452. doi: 10.1046/j.1365-294x.1998.00319.x. [DOI] [Google Scholar]
- 53.Conroy CJ, Cook JA. 2000. Phylogeography of a post-glacial colonizer: Microtus longicaudus (Rodentia: Muridae). Mol Ecol 9:165–175. doi: 10.1046/j.1365-294x.2000.00846.x. [DOI] [PubMed] [Google Scholar]
- 54.Kennedy P. 2010. Ectomycorrhizal fungi and interspecific competition: species interactions, community structure, coexistence mechanisms, and future research directions. New Phytol 187:895–910. doi: 10.1111/j.1469-8137.2010.03399.x. [DOI] [PubMed] [Google Scholar]
- 55.Fukami T, Dickie IA, Paula Wilkie J, Paulus BC, Park D, Roberts A, Buchanan PK, Allen RB. 2010. Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol Lett 13:675–684. doi: 10.1111/j.1461-0248.2010.01465.x. [DOI] [PubMed] [Google Scholar]
- 56.Blaise B, Clague JJ, Mathewes RW. 1990. Time of maximum late Wisconsin glaciation, west-coast of Canada. Quat Res 34:282–295. doi: 10.1016/0033-5894(90)90041-I. [DOI] [Google Scholar]
- 57.Mickelson DM, Clayton L, Baker RW, Mode WN, Schneider AF. 1984. Pleistocene stratigraphic units of Wisconsin. Wisconsin Geological and Natural History Survey, Madison, WI. [Google Scholar]
- 58.Williams D, Dunkerley D, DeDeckker P, Kershaw P, Chappell M. 1998. Quarternary environments. Arnold, London, England. [Google Scholar]
- 59.Mikheyev AS, Vo T, Mueller UG. 2008. Phylogeography of post-Pleistocene population expansion in a fungus-gardening ant and its microbial mutualists. Mol Ecol 17:4480–4488. doi: 10.1111/j.1365-294X.2008.03940.x. [DOI] [PubMed] [Google Scholar]
- 60.Eisenlord SD, Zak DR, Upchurch RA. 2012. Dispersal limitation and the assembly of soil Actinobacteria communities in a long-term chronosequence. Ecol Evol 2:538–549. doi: 10.1002/ece3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ricklefs RE. 2006. Evolutionary diversification and the origin of the diversity-environment relationship. Ecology 87:S3–S13. doi: 10.1890/0012-9658(2006)87[3:EDATOO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 62.Algar AC, Kerr JT, Currie DJ. 2009. Evolutionary constraints on regional faunas: whom, but not how many. Ecol Lett 12:57–65. doi: 10.1111/j.1461-0248.2008.01260.x. [DOI] [PubMed] [Google Scholar]
- 63.Li P, Li M, Shi Y, Zhao Y, Wan Y, Fu C, Cameron KM. 2013. Phylogeography of North American Herbaceous smilax (Smilacaceae): combined AFLP and cpDNA data support a northern refugium in the Driftless Area. Am J Bot 100:801–814. doi: 10.3732/ajb.1200250. [DOI] [PubMed] [Google Scholar]
- 64.Muhs DR, Simmons KR, Schumann RR, Halley RB. 2011. Sea-level history of the past two interglacial periods: new evidence from U-series dating of reef corals from south Florida. Quat Sci Rev 30:570–590. doi: 10.1016/j.quascirev.2010.12.019. [DOI] [Google Scholar]
- 65.Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL, Brown JH. 2008. A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci U S A 105:7774–7778. doi: 10.1073/pnas.0803070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Staff SS. 2004. Soil survey laboratory methods manual. Service NRC, Lincoln, NE. [Google Scholar]
- 67.Peltier W. 1993. Time dependent topography through glacial cycle. IGBP Pages/World Data Center-A for Paleoclimatology Data Contribution, series no. 93-015. NOAA/NGDC Paleoclimatology Program, Boulder, CO. [Google Scholar]
- 68.Ottow JCG. 1972. Rose-Bengal as a selective aid in isolation of fungi and actinomycetes—from natural sources. Mycologia 64:304. doi: 10.2307/3757834. [DOI] [PubMed] [Google Scholar]
- 69.El-nakeeb MA, Lecheval HA. 1963. Selective isolation of aerobic actinomycetes. Appl Microbiol 11:75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roberts MA, Crawford DL. 2000. Use of randomly amplified polymorphic DNA as a means of developing genus- and strain-specific Streptomyces DNA probes. Appl Environ Microbiol 66:2555–2564. doi: 10.1128/AEM.66.6.2555-2564.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo Y, Zheng W, Rong X, Huang Y. 2008. A multilocus phylogeny of the Streptomyces griseus 16S rRNA gene clade: use of multilocus sequence analysis for streptomycete systematics. Int J Syst Evol Microbiol 58:149–159. doi: 10.1099/ijs.0.65224-0. [DOI] [PubMed] [Google Scholar]
- 72.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lanave C, Preparata G, Saccone C, Serio G. 1984. A new method for calculating evolutionary substitution rates. J Mol Evol 20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- 74.Stamatakis A. 2006. RAxML-VI HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 75.Pommier T, Canbäck B, Lundberg P, Hagström A, Tunlid A. 2009. Rami: a tool for identification and characterization of phylogenetic clusters in microbial communities. Bioinformatics 25:736–742. doi: 10.1093/bioinformatics/btp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lozupone C, Hamady M, Knight R. 2006. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goslee SC, Urban DL. 2007. The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19. [Google Scholar]
- 78.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Webb CO, Ackerly DD, Kembel SW. 2008. Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 24:2098–2100. doi: 10.1093/bioinformatics/btn358. [DOI] [PubMed] [Google Scholar]
- 80.Kerr JT, Currie DJ. 1999. The relative importance of evolutionary and environmental controls on broad-scale patterns of species richness in North America. Ecoscience 6:329–337. [Google Scholar]
- 81.Templeton AR, Boerwinkle E, Sing CF. 1987. A cladistic-analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping 0.1. Basic theory and an analysis of alcohol-dehydrogenase activity in Drosophila. Genetics 117:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Templeton AR, Routman E, Phillips CA. 1995. Separating population structure from population history—a cladistic analysis of the geographical distribution of mitochondrial-DNA haplotypes in the tiger salamander, Ambystoma tigrinum. Genetics 140:767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 84.Posada D, Crandall KA, Templeton AR. 2000. GeoDis: a program for the cladistic nested analysis of the geographical distribution of genetic haplotypes. Mol Ecol 9:487–488. doi: 10.1046/j.1365-294x.2000.00887.x. [DOI] [PubMed] [Google Scholar]
- 85.Panchal M, Beaumont MA. 2007. The automation and evaluation of nested clade phylogeographic analysis. Evolution 61:1466–1480. doi: 10.1111/j.1558-5646.2007.00124.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Streptomyces strains isolated and characterized from the series of 15 sites described in the text (In the cases of Wisconsin and North Carolina, samples were aggregated across two or three soil samples, respectively, to represent each region. “Time,” the time available for colonization; mya, millions of years ago; SOM, soil organic matter. Asterisks identify NRI values that showed clustering [positive] or overdispersion [negative] that are unlikely to result from chance [P < 0.01].)
Results from Table 2 alongside results obtained when sites from Alaska were excluded (The column reporting “sites” indicates results obtained when Alaska sites were included [“+AK” rows are the same as those depicted in Table 2] or excluded [−AK]. Results that reveal different outcomes between the +AK and –AK analyses are indicated with shading. Relationships between environmental factors and Streptomyces phylogenetic [UniFrac distance] and taxonomic [Bray-Curtis dissimilarity] differences were analyzed by using adonis [permutational multivariate analysis of variance]. Values indicate R2 values, and results that are unlikely due to chance are indicated with asterisks [*, P < 0.05; **, P < 0.01; ***, P < 0.001]. The analyses were performed by either including all rpoB sequences [weighted] or excluding duplicate sequences for each OTU [unweighted].)
Significant inferences from nested clade analysis of Streptomyces rpoB haplotypes (see Fig. S3).
Maximum likelihood tree based on rpoB sequences of Streptomyces strains. A total of 924 Streptomyces strains were isolated from 15 sites in North America and classified into 107 OTUs on the basis of rpoB sequence dissimilarity. The inner colored ring includes sequential color blocks to indicate the 107 OTUrpob identified in this study. The outer colored ring identifies the history of the site from which each strain was isolated, as indicated in the color scale (FL and WI are indicated as well, due to their distinctive geologic history). The site from which each strain was isolated can be determined by the strain name, as described in Table S1. Download
The rarefied collectors curve indicates that OTUrpoB were well sampled from the sampling sites (with respect to the constraints imposed by strain collection). Good’s coverage was 0.88 for unique rpoB sequences and 0.95 for OTUrpoB. Download
Nested clade analysis of Streptomyces rpoB haplotype networks provides evidence for contiguous range expansion and dispersal limitation. Circles represent rpoB haplotypes, with radii proportional to the number of strains that belong to the haplotype. Haplotypes are shaded to represent strain source, with the fraction of strains isolated from previously glaciated or nonglaciated sites indicated in blue and red, respectively. Each line symbolizes one mutational step, with dots indicating inferred haplotypes not sampled. The complete set of haplotype networks is shown (Table S2 shows the evolutionary inferences for each clade). Download