

Abstract
Background/Aim:
Despite the significant number of studies on H. pylori pathogenesis, not much data has been published concerning its ability to form biofilm in the host stomach. This study aims to evaluate the potential of clinical isolates of H. pylori to form biofilm in C57BL/6J mice model.
Materials and Methods:
Two strains of H. pylori were selected from a collection of clinical isolates; one (19B), an efficient biofilm producer and the other (4B), with weak biofilm-forming ability. Mice infected through gastric avages were examined after one and two weeks. Colonization was determined by CFU and urease activity; the anti-H. pylori IgA was measured by ELISA, and chronic infections were evaluated by histopathology. Bacterial communities within mucosal sections were studied by immunofluorescence and scanning electron microscopy (SEM).
Results:
Successful infection was obtained by both test strains. Strain 19B with higher ability to form biofilm in vitro also showed a higher colonization rate in the mice stomach one week after infection. Difference (P < 0.05) in IgA titers was observed between the infected mice and the controls as well as between 19B and 4B infected mice, two weeks after the last challenge. Immunofluorescence and SEM results showed tightly colonizing H. pylori in stomach mucosal sections and in squamous and glandular epithelium.
Conclusion:
H. pylori is able to form biofilm in the mouse stomach and induce IgA production, reflecting the same potential as in humans. Firm attachment of coccoid form bacteria to host cells suggests the importance of this state in biofilm formation by H. pylori. Occurrence of biofilm in squamous and glandular epithelium of the mouse stomach proposes that H. pylori can all parts of the upper gastrointestinal tract.
Keywords: Biofilm, C57BL/6J mice, chronic infection, coccoid form, Helicobacter pylori
Helicobacter pylori colonize human stomach in approximately half of the world's population. Many of the pathogenic effects of H. pylori infection are related to chronic active inflammation, which persist lifelong and cause peptic ulcer disease (PUD) or gastric cancer in the absence of appropriate treatment.[1,2]
The relationship between persistence of pathogenic bacteria in host and the ability of biofilm formation, has been studied for numerous bacteria. The National Institute of Health and Center for Disease Control and Prevention has estimated that over 80% of all bacterial infections involve a biofilm stage in their disease period.[3,4,5]
The common characteristics of the biofilm formation ability are the capability to attach to a substratum by embedding into a matrix of extracellular polymeric substances.[3,4,5] This property allows more resistance to host defense and antibiotics by reducing their access to the bacterial population residing within the biofilms. Furthermore, microbial populations within the biofilm can better tolerate nutrient starvation, pH changes, and toxic compounds such as oxygen radicals.[6] Resistance of biofilms to multiple factors may be due to specific characteristics such as slower growth rate and physiological heterogeneity of the inhabitants, as well as its matrix, which is predominantly composed of exopolysaccharides.[7] Furthermore, bacteria growing within a biofilm can secrete substances that may play a role in signaling of gene expression, resulting in phenotypic heterogeneity within the biofilm.[7] The features of biofilm formation have been studied for several pathogenic bacteria.[8] However, despite the significant number of studies published on the pathogenesis of H. pylori, not much data is available concerning its biofilm-formation behavior in the host stomach. The investigators have proposed that H. pylori has the ability to form biofilms on various surfaces in aquatic environments.[9] Carron et al. and Coticchia et al. proposed that urease-positive organisms formed thick biofilms in the host stomach compared with urease-negative isolates.[10,11] Yonezawa et al. isolated a strong biofilm-forming strain of H. pylori and suggested that the growth rate of biofilm-forming populations is a principal factor in the biofilm development in vitro.[12] However, the effects of these complex phenomena and their consequence in the pathogenesis of H. pylori infection in vivo remain to be determined.
Establishing in vivo models of biofilm formation may enable us to understand the conditions, which play important roles in H. pylori pathogenesis.
This study aims to evaluate the biofilm formation ability of H. pylori clinical isolates associated with chronic infection in a C57BL/6J mouse model.
MATERIALS AND METHODS
Bacterial strains and growth condition
A collection of 30 clinical H. pylori isolates from children and adult patients with chronic gastritis were employed for this study. The isolates were grown on modified Campy blood agar plates containing brucella agar base (Merck, Germany) with defibrinated sheep blood (5%), and antibiotics. The plates were incubated at 37°C under microaerophilic atmosphere for 3 days. The grown colonies were identified by Gram staining, standard biochemical tests, and polymerase chain reaction using H. pylori-specific primers for 16SrRNA and ureC.[13]
In vitro screening of the isolates for biofilm formation ability
The isolates were screened for biofilm formation as previously described with some modifications.[14] Colonies from culture plates were inoculated into brucella broth (Biolife, Italy) supplemented with fetal calf serum (2%) and glucose (0.3%) (Merck, Germany). Bacterial suspensions were incubated at 37°C under microaerophilic atmosphere, centrifuged at 100 rpm to an optical density of 0.2 at 600 nm (A600) equivalent to 5-8 x 103 CFU/mL approximately, at the beginning of the exponential phase. Portions (250 μL) of the cultures were inoculated into the wells of 96-well flat-bottomed tissue culture plates (BIOFIL, Jet Bio-Filtration Products Co., China) and were incubated at 37°C under microaerophilic conditions for 6 days. Three independent experiments with eight replicates for each strain were performed and in each test the brucella broth without bacteria was used as the negative control. The wells were vigorously washed three times with sterile phosphate-buffered saline (PBS), in order to remove nonadherent bacteria. The bacteria attached tightly to the wells were fixed with 99% ethanol (200 μL per well) for 20 min, and air-dried. Plates were then stained with 1% crystal violet (200 μL per well) for 5 min and the excess of stain was rinsed by running tap water. To dissolve the dye attached to the adherent cells, the dried plates were treated with 33% glacial acetic acid (160 μL per well) and the optical density (OD) of the wells was measured at 505 nm using an ELISA reader (SCO GmbH, Thu, Dingelstadt, Germany). Culture medium without bacterial cells was used as negative control. Quantitative evaluation of the biofilms was reported as the mean ± standard deviation of three independent experiments.
Animals
All experiments with animals were in accordance with the Institutional Animal Ethical Committee guidelines. Five groups of 10 mice (C57BL/6J, Razi (Razi Institute, Iran), aging between 6 and 8 weeks were employed. Mice (two groups for each strains) were infected (3×) through gastric gavage with 108 of 19B and 4B strains, respectively. Negative controls (also 10 mice) received PBS. One week and two weeks after the last challenge, blood samples were collected and the mice were humanely killed. The stomachs were then removed and divided into four portions for determining the CFU, measurement of urease activity, histopathologic and microscopic examination.
Determination of urease activity and CFU
Urease activity in the homogenized gastric tissue was performed according to the previously described method.[15] For enumeration of colonized bacteria, stomach samples were homogenized with sterile PBS; cultured on the brucella agar plates and incubated at 37°C under microaerobic conditions. Colony counts were performed after 3–5 days of incubation.
Histopathology
Portions of mice stomach were processed for histologic examination according to the standard protocols.[16] Briefly, the segments containing all parts of the stomach including the antrum and corpus were fixed at 10% neutral buffered formalin, embedded in paraffin, sectioned by standard methods, and stained with hematoxylin and eosin (H and E) as well as Giemsa stain.[16] Density of polymorphonuclear leukocytes, and the degree of lymphocytes and plasma cell infiltration in the gastric mucosa were evaluated on the sections stained by H and E. Colonization and bacterial density were evaluated by Geimsa staining. The degree of cell infiltrations was graded according to the protocol previously adopted by Chen et al.[17] The stained biopsies were scored by observing chronic inflammatory cell densities (lymphocytes, plasma cells) as follows: 0, none; 1: Less than 10 in each high power field; 2: >10 cells/high power field; 3: Some areas with heavy inflammatory cell infiltration; 4: Diffuse and dense cell infiltration; 5: Presence of dense chronic inflammatory cells nearly in all parts of the whole mucous such that they separate the gastric glands; 6: Entire mucosa contains a dense chronic inflammatory cell infiltrate.
Measurement of the anti- H. pylori IgA
To measure the amount of IgAs in mouse serum, a method described by O'Riordan et al. and Akhiani et al.[15,18] was employed. Their method is based on comparison of optical density (OD) obtained for increasing dilution of mice antiserums. As increasing dilution of the antiserums was made until reaching to the background level (the level of OD for negative control), statistical analysis of OD values may be of a high-quality criteria. Therefore, the titers of IgAs for each mouse would be the reciprocal value of the dilution without unite.
Ninety-six-well microtiter plates (Nunc, Kamstrup, Denmark) were coated with 108 CFU/mL of the H. pylori in carbonate buffer (0.1 M sodium carbonate, pH 9.6). The plates were washed with Bovine serum albumin (BSA) (1% in PBS) and incubated for 1 h at room temperature (RT), after which time they were washed with PBS containing 0.05%Tween-20 (PBST). The wells were then inoculated with 100 µL of serially diluted mouse serum (1:100 to 1:12000 in PBST) and incubated at 37°C for 1 h. The plates were washed with PBS (6×) and diluted (5000×) goat peroxidase-coated anti-mouse-IgA (Sigma, St. Louis, MO, USA) was added to each well before incubation at RT for 1 h. The wells were then washed (6×), TMB peroxidase substrate (Sigma, Deisenhofer, Germany) was added and incubated for 10 min. The reaction was stopped by hydrochloric acid (1N) and absorbance was measured at 405 nm using an ELISA reader (SCO GmbH, Germany).
Immunofluorescence
Specimens were fixed in 4% paraformaldehyde at 4°C overnight, washed (30 min) with PBS (3×), dehydrated using 50% and 70% ethanol (ETOH), respectively. The fixed tissues were embedded in paraffin, placed in xylene for 5 min (3×), followed by ETOH (100%) for 5 min (2×) and air-dried. Slides were washed in PBST for 5 min, blocked with BSA–Tween-20 (3% in PBS) for 1 h and washed in PBST for 5 min. The slides were then incubated with a rabbit polyclonal anti-H. pylori antibody (1:1000 in PBST containing 1% BSA) raised against H. pylori outer membrane proteins.[19] After overnight incubation, the slides were washed with PBST (3×) for 15 min. For immunoflourescence examination, the slides were treated with the goat antirabbit IgG (0.01 µg/mL in PBST with 1% BSA) conjugated to Rhodamine (Sigma, SAB3700877), incubated in the dark for 1.5 h and washed with PBST (3×) for 15 min. Nuclear staining of the cells was performed according to a previously described method.[20] Briefly, the slides were placed in DAPI (4', 6-diamidino-2-phenylindole, Roche, Mannheim, Germany) or PI (propidium iodide, Molecular Probes) diluted with PBST (1000×) for 5 min, rinsed once with PBS for 5 min and air dried.
Scanning electron microscopy
All histological samples were fixed in 2.5% glutaraldehyde for 3 h followed by dehydration with increasing concentrations of ETOH: 25% (15 min), 30% (15 min), 50% (15 min), 70% (15 min), 90% (15 min), and 100% (15 min, 2×). Samples were stored in desiccators until coated with gold–palladium sputter for two 200-s intervals (Nano Structured Coating Co. Iran). Scanning electron Microscopy (SEM) was performed using TESCAN VEGA3S electron microscope, at 30 KV.[11]
Statistical analysis
Statistical analysis was carried out using analysis of variance (ANOVA) one-way test with Minitab 16 statistical software, and probability levels of <0·05 were considered as statistically significant.
Significant difference between the numbers of bacteria colonizing the mice stomachs (counted by CFU method), was assessed by ANOVA one-way, Dunnett's simultaneous tests. Also, significant differences between the IgA titers, (comparing their ODs) was assessed by ANOVA one-way, Dunnett's simultaneous tests.
RESULTS
Screening of clinical strains for biofilm formation
Two biofilm-forming strains were selected: A strong biofilm producer (strain 19B) and a weak biofilm producer (strain 4B). Figure 1 displays the results of in vitro biofilm formation by 19B and 4B strains studied by SEM. H. pylori strain 19B showed the highest levels of biofilm formation and strain 4B the lowest amount of biofilm. The OD at 505 nm was 0.136 ± 0.0152 for strain 19B, 0.0692 ± 0.0084 for strain 4B and 0.0306 ± 0.00517 for control.
Figure 1.
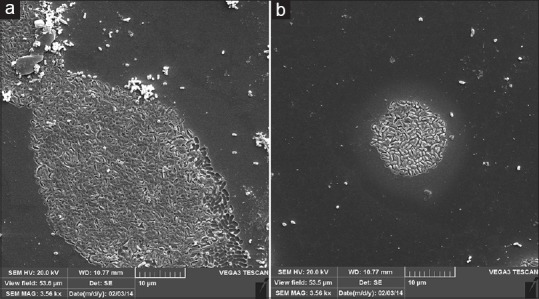
Representative scanning electron microscope images of mature H. pylori biofilms on cover slip. (a) H. pylori strain 19B (b) H. pylori strain 4B
Colonization of the mice stomach
Colonization of the gastric mucosa by 19B and 4B H. pylori strains was evaluated by bacterial enumeration and urease test. Both H. pylori strains were able to colonize the mouse stomach and produce a positive urease reaction. Bacterial enumeration, revealed a higher number for 19B strain colonization, one week after infection. However, no significant difference was observed between the two strains two weeks after infection [Table 1].
Table 1.
Results of bacterial enumeration, urease, and production of anti-H. pylori IgA
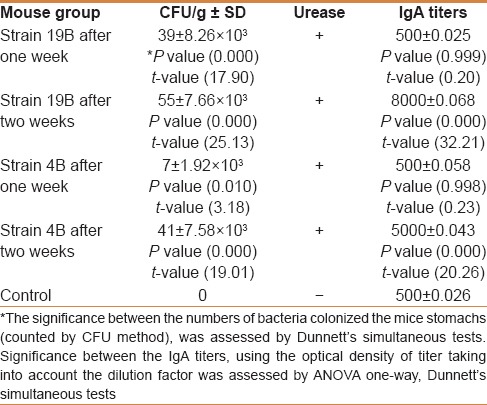
Serum IgA
There was no difference in serum IgA titers between the two mice groups (19B and 4B) as well as the control mice, one week after infection [Table 1]. Both strains were able to induce humoral immune response two weeks after infection. However, the higher biofilm-producing strain, 19B induced a significantly higher IgA response compared with 4B strain, two weeks after infection (P < 0.05).
Histopathology
One week after challenge, no significant cell infiltration was seen in the infected mice stomachs by any of the strains. However, after two weeks, a significant cell infiltration was observed by both strains in comparison with the controls. However, no significant difference was observed between two strains (Score of chronic inflammatory infiltrates in the gastric mucosa were 2.66 ± 0.33 and 2.33 ± 0.33 for 19B and 4B strain, respectively). Microscopic examination of the stained stomach sections by H and E revealed significant cell infiltration and tightly attached organisms with the coccoid H. pylori morphology to the gastric glands compared with the control group [Figure 2].
Figure 2.

Mice stomach sections, stained by H and E. (a) mice challenged by H. pylori strain 19B displaying moderate cell infiltration after two weeks; (b and c) mice challenged by H. pylori strain 19B after one week where no cell infiltration was observed; (d and e) mice challenged by H. pylori strain 4B after two weeks where no cell infiltration was seen; (f) mice challenged by H. pylori strain 4B after one week where no cell infiltration was seen; and (g) mice control group (Scale bar, 10 μm)
No bacterial cell was observed in the gastric glands of the controls sections stained either by H and E or by Giemsa. In addition, the bacteria embedded in the amorphous matrix, were also observed in the squamous and glandular epithelium of the stomach in the infected mice groups, two weeks after challenge [Figures 3 and 4].
Figure 3.

Representative light microscopic image demonstrating biofilm formation by H. pylori in sections stained by Geimsa. (a and b) biofilm formation by 19B strain after one week in the squamous area of the mice stomach; (c and d) biofilm formation by 19B strain after two weeks in the squamous and antral area of the mice stomach respectively; (e and f) biofilm formation by 4B strain after one week in the antral and squamous area of the mice stomach respectively; (g and h) biofilm formation by 4B strain after two weeks in the squamous area of the mice stomach; and (i) squamous area of the mice stomach in control group. (Scale bar, 10 μm)
Figure 4.
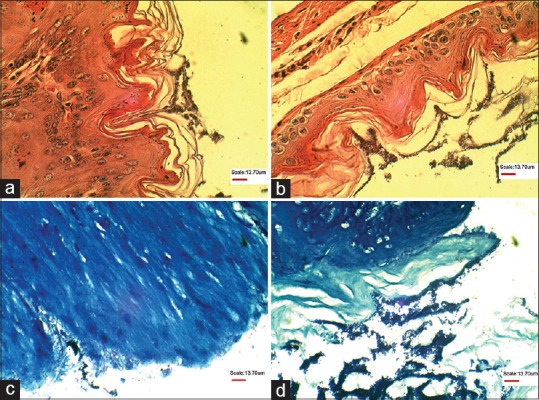
Representative light microscopic image demonstrating biofilm formation by 19B strain in the squamous area of the mice stomach after two weeks in sections stained by Hematoxylin and Eosin (a and b), and Geimsa (c and d)
Visualization of biofilm by immunofluorescence and SEM
Evaluation of the sections by immunofluorescence using anti-H. pylori-specific antibodies demonstrated the dense H. pylori communities in the glandular epithelium and squamous of the stomach [Figures 5 and 6]. Similarly, bacterial presence was visualized by SEM images in both mice groups [Figure 7]. SEM images showed higher densities for strain 19B compared with 4B to some extent [Figure 7].
Figure 5.
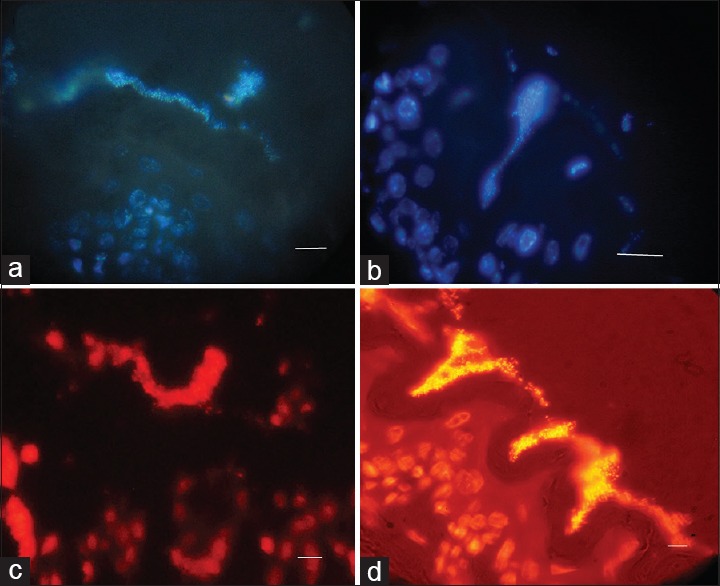
Representative immunofluorescence image of a mature biofilm formed by 19B formed in the antral and squamous areas of mice stomach respectively after two weeks. Fluorescent bacteria are caught near the gastric mucosa. (a and b) Nuclear staining with DAPI visualized by UV filter (c and d) Nuclear staining with PI visualized by red filter. (Scale bar, 10 μm)
Figure 6.
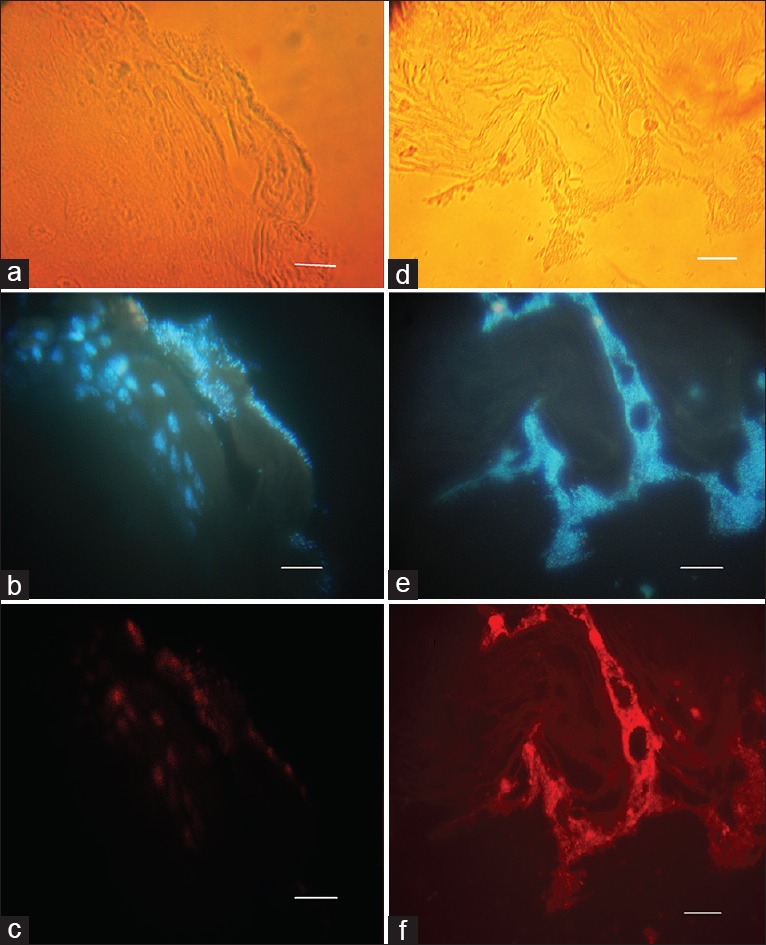
The representative immunofluorescence image of a mature H. pylori biofilm corresponding to 19B and 4B strains, respectively, after two weeks, respectively, formed in the antral and squamous areas of mice stomach. (a and d) visualized by visible light; (b and e) Nuclear staining with DAPI visualized by UV filter; (c and f) Immunofluorescence staining with rhodamin conjugated to antirabbit antibody visualized by red filter. Fluorescent bacteria are seen near the gastric mucosa (Scale bar, 10 μm)
Figure 7.

Representative scanning electron microscope images of a mature H. pylori biofilm formed in mice stomach after two weeks. (a) mice control group; (b and c) mice challenged by H. pylori strain 19B where the microcolonies of bacteria are seen near the gastric mucosa; and (d–f) mice challenged by H. pylori strain 4B
DISCUSSION
In this study, the higher ability of 19B strain to form biofilm in vitro and higher colonization rate in the mouse stomach only one week after infection is consistent with the conclusion of Yang et al., that formation of H. pylori biofilm is time dependent in vitro and may also be time dependent in vivo.[21] However, no difference was observed between the ability of the two strains to colonize or form biofilm in the stomach of the experimental mice after two weeks. Furthermore, induction of mucosal immunity in mice occurred by both strains of H. pylori two weeks after challenge [Table 1], a significantly higher titer of IgA was observed in response to 19B strain, which could be related to its rapid ability to produce a successful infection/biofilm in vivo. In contrast, absence of significant IgA titers one week after challenge suggests that mucosal immune response initiates at least two weeks after colonization. Overall, enumeration of bacterial cells in the infected mice stomach, measurement of IgA titer, and images of light microscopy confirmed that the rate of colonization and and subsequent biofilm formation, was not similar for the two selected strains. The formation of less strain 4B biofilm in vitro may be due to its slower growth rate. However, in vitro it is not practically possible to find a strain that produces no biomass (null) since the biofilms formed by various strains may only be quantitatively different in a shorter time. Costerton et al. have previously suggested that growth rates of the isolates growing in rich media in batch cultures may differ profoundly for the same species.[22] They also showed that the organization of fine structures named biofilms in vivo are different from those formed by pure cultures growing in nutrient-rich media in vitro.
The presence of inflammation in infected mice characterized by observation of chronic inflammatory cell infiltration [Figure 2] confirmed occurrence of chronic infection in the mouse model infected with H. pylori, somehowmimicking the situation observed in humans.
As described by O'Toole et al., biofilms are communities of microorganisms, surrounded by amorphous matrices.[23] Images of light microscopy [Figures 3 and 4], SEM, and immunofluorescence [Figures 5–7] obtained for two H. pylori strains exhibited thick communities of coccoid form bacteria in gastric mucosa suggesting formation of typical biofilms in our mouse model.
In the present work, the coccoid form of H. pylori was dominant in gastric mucosa; these bacterial cells were not only embedded in amorphous matrix, but were also seen in squamous and glandular epithelium suggesting that H. pylori is able to form a biofilm in all parts of the stomach in C57BL/6J mice [Figures 6 and 7]. Presence of H. pylori strains in the coccoid form might play an important role in resistance and transmission of infection.[24] Studies have shown that H. pylori in its coccoid form viable and can decrease its DNA and RNA levels.[25] In humans, H. pylori is commonly colonizes the antrum but colonization in the esophagus has not been proved yet. This may be due to the difference between the gastric anatomy of mice and humans. The stomachs of rodents are divided into glandular and nonglandular portions. The nonglandular part is generally thin walled and lined by keratinized stratified squamous epithelium used for food storage and digestion. The glandular part is thick walled and covered by columnar epithelia. Furthermore, the squamocolumnar junction in mice is not entirely near the gastroesophageal junction as seen in normal human anatomy.[26,27] Thus, H. pylori may be able to form biofilm in all parts of the mouse stomach. This would be interesting since there are a few reports indicating that H. pylori can also colonize other parts of the gastrointestinal tract in humans.[28] For example, Jabbari Moghadam et al. observed that H. pylori can colonize children's tonsillar tissues and may have a possible role as reservoir for H. pylori in children. In addition, Aslan et al. observed that despite no visible colonization, H. pylori may contribute to chronic tonsillitis, especially at the mucosal layers.[29] Furthermore, Gutierrez et al. showed that H. pylori can colonize heterotypic gastric mucosa in the upper esophagus (inlet patch) and provide a reservoir for oral–oral transmission or a niche for H. pylori to avoidantibiotics.[30] Biofilm formation on the medical implants have also been considered as real problem with serious consequences in recent years.[31] Although the significance of biofilm formation has not been well evaluated in pathogenesis of H. pylori infection, many researchers have suggested that it may contribute to the failure in eradication of H. pylori infection. One reason may be survival of microorganisms in the focal sites and further possibility of re-infection, which can help to increase resistance of H. pylori to currently used antimicrobials.[1,32] Consistent with this, Yonezawa et al. demonstrated that expression of efflux pump genes and resistance to clarithromycin significantly increased in the biofilm-forming cells compared with the planktonic populations.[33] Cammarota et al. reported that targeting H. pylori biofilm infections with molecules such as N-acetylcysteine prior to antibiotic treatment, increased the eradication rate compared with that of nontreated group.[34]
CONCLUSION
An important finding of the present study was showing the ability of H. pylori to form typical biofilms in our mouse model during chronic infection, which may reflect the same potential in the human stomach. Furthermore, biofilm formation in squamous and glandular epithelium of the mouse stomach suggests that H. pylori can colonize the upper gastrointestinal tract, such as esophagus, which can also act as a reservoir for H. pylori. Although this study was performed in a mouse model, presence of firmly attached coccoid cells, suggests that this state may also occur in humans. This may be shown by microbiological examinations of biopsies obtained from chronically infected human subjects.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest
Acknowledgment
We cordially thank Dr. Fatemeh Mahjoub from Department of Pathology, Tehran University of Medical Sciences, Tehran, Iran, for help in performing histology and Dr. Mahboubi from Alzahra University, Tehran, Iran, for help with animal models. We especially thank Dr. Fereshteh Eftekhar from the Department of Microbiology, School of Biology, Shahid Beheshti University for critical reading of manuscript and English editing.
REFERENCES
- 1.Bińkowska A, Biernat M, Duś I, Gościniak G. The role of biofilm formation in pathogenesis of Helicobacter pylori infections. Prz Gastroenterol. 2013;8:27–30. [Google Scholar]
- 2.Inoue I, Kato J, Tamai H, Iguchi M, Maekita T, Yoshimura N, et al. Helicobacter pylori-related chronic gastritis as a risk factor for colonic neoplasms. World J Gastroenterol. 2014;20:1485–92. doi: 10.3748/wjg.v20.i6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costerton JW, Lewandowski Z, DeBeer D, Caldwell D, Korber D, James G. Biofilms, the customized microniche. J Bacteriol. 1994;176:2137–42. doi: 10.1128/jb.176.8.2137-2142.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kishen A, Haapasalo M. Biofilm models and methods of biofilm assessment. Endod Topics. 2010;22:58–78. [Google Scholar]
- 5.Goodman SD, Obergfell KP, Jurcisek JA, Novotny LA, Downey JS, Ayala EA, et al. Biofilms can be dispersed by focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 2011;4:625–37. doi: 10.1038/mi.2011.27. [DOI] [PubMed] [Google Scholar]
- 6.Yonezawa H, Osaki T, Kurata S, Zaman C, Hanawa T, Kamiya S. Assessment of in vitro biofilm formation by Helicobacter pylori. J Gastroenterol Hepatol. 2010;25(Suppl 1):S90–4. doi: 10.1111/j.1440-1746.2009.06213.x. [DOI] [PubMed] [Google Scholar]
- 7.Jefferson KK. What drives bacteria to produce a biofilm? FEMS Microbiol Lett. 2004;236:163–73. doi: 10.1016/j.femsle.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Fux C, Costerton J, Stewart P, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13:34–40. doi: 10.1016/j.tim.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Gião MS, Azevedo N, Wilks SA, Vieira M, Keevil CW. Persistence of Helicobacter pylori in heterotrophic drinking-water biofilms. Appl Environ Microbiol. 2008;74:5898–904. doi: 10.1128/AEM.00827-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carron MA, Tran VR, Sugawa C, Coticchia JM. Identification of Helicobacter pylori biofilms in human gastric mucosa. J Gastrointest Surg. 2006;10:712–7. doi: 10.1016/j.gassur.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 11.Coticchia JM, Sugawa C, Tran VR, Gurrola J, Kowalski E, Carron MA. Presence and density of helicobacter pylori biofilms in human gastric mucosa in patients with peptic ulcer disease. J Gastrointest Surg. 2006;10:883–9. doi: 10.1016/j.gassur.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Yonezawa H, Osaki T, Kurata S, Fukuda M, Kawakami H, Ochiai K, et al. Outer membrane vesicles of Helicobacter pylori TK1402 are involved in biofilm formation. BMC Microbiol. 2009;9:197. doi: 10.1186/1471-2180-9-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falsafi T, Favaedi R, Mahjoub F, Najafi M. Application of stool-PCR test for diagnosis of Helicobacter pylori infection in children. World J Gastroenterol. 2009;15:484–8. doi: 10.3748/wjg.15.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grande R, Di Campli E, Di Bartolomeo S, Verginelli F, Di Giulio M, Baffoni M, et al. Helicobacter pylori biofilm: A protective environment for bacterial recombination. J Appl Microbiol. 2012;113:669–76. doi: 10.1111/j.1365-2672.2012.05351.x. [DOI] [PubMed] [Google Scholar]
- 15.O'Riordan AA, Morales VA, Mulligan L, Faheem N, Windle HJ, Kelleher DP. Alkyl hydroperoxide reductase: A candidate Helicobacter pylori vaccine. Vaccine. 2012;30:3876–84. doi: 10.1016/j.vaccine.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and Grading of Gastritis. The Updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161–81. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Chen XY, van der Hulst RW, Bruno MJ, van der Ende A, Xiao SD, Tytgat GN, et al. Interobserver variation in the histopathological scoring of Helicobacter pylori related gastritis. J Clin Pathol. 1999;52:612–5. doi: 10.1136/jcp.52.8.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akhiani AA, Stensson A, Schön K, Lycke NY. IgA antibodies impair resistance against Helicobacter pylori infection: Studies on immune evasion in IL-10-deficient mice. J Immunol. 2005;174:8144–53. doi: 10.4049/jimmunol.174.12.8144. [DOI] [PubMed] [Google Scholar]
- 19.Falsafi T, Lavasani P, Basardeh I, Massarrat S, Landarani Z. Evaluation of an Iranian Home Helicobacter pylori Stool Antigen ELISA Kit. Jundishapur J Microbiol. 2014;7:e10629. doi: 10.5812/jjm.10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogers SL, Rogers GC. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat Protoc. 2008;3:606–11. doi: 10.1038/nprot.2008.18. [DOI] [PubMed] [Google Scholar]
- 21.Yang FL, Hassanbhai AM, Chen HY, Huang ZY, Lin TL, Wu SH, et al. Proteomannans in Biofilm of Helicobacter pylori ATCC 43504. Helicobacter. 2011;16:89–98. doi: 10.1111/j.1523-5378.2010.00815.x. [DOI] [PubMed] [Google Scholar]
- 22.Costerton JW, Lappin-Scott HM. Introduction to microbial biofilms. In: Lappin- Scott HM, Costerton JW, editors. Microbial Biofilms. Cambridge: Cambridge University Press; 1995. pp. 1–11. [Google Scholar]
- 23.O'Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Annu Rev Microbiol. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- 24.Milyani RM. Cytopathic effect of coccoid forms of Helicobacter pylori in Albino rats and Swiss mice. J Am Sci. 2011;7:1087–92. [Google Scholar]
- 25.Narikawa S, Kawai S, Aoshima H, Kawamata O, Kawaguchi R, Hikiji K, et al. Comparison of the nucleic acids of helical and coccoid forms of Helicobacter pylori. Clin Diagn Lab Immunol. 1997;4:285–90. doi: 10.1128/cdli.4.3.285-290.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayakawa Y, Fox J, Gonda T, Worthley D, Muthupalani S, Wang T. Mouse models of gastric cancer. Cancers (Basel) 2013;5:92–130. doi: 10.3390/cancers5010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos. 1995;16:351–80. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 28.Jabbari Moghaddam Y, Rafeey M, Radfar R. Comparative assessment of Helicobacter pylori colonization in children tonsillar tissues. Int J Pediatr Otorhinolaryngol. 2009;73:1199–201. doi: 10.1016/j.ijporl.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Aslan S, Yilmaz I, Bal N, Sener M, Butros R, Demirhan B, et al. Investigation of Helicobacter pylori in tonsillary tissue with Pronto Dry® test and pathologic examination. Auris Nasus Larynx. 2007;34:339–42. doi: 10.1016/j.anl.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez O, Akamatsu T, Cardona H, Graham DY, El-Zimaity HM. Helicobacter pylori and hetertopic gastric mucosa in the upper esophagus (the inlet patch) Am J Gastroenterol. 2003;98:1266–70. doi: 10.1111/j.1572-0241.2003.07488.x. [DOI] [PubMed] [Google Scholar]
- 31.Kadurugamuwa JL, Sin L, Albert E, Yu J, Francis K, DeBoer M, et al. Direct continuous method for monitoring biofilm infection in a mouse model. Infect Immun. 2003;71:882–90. doi: 10.1128/IAI.71.2.882-890.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cammarota G, Sanguinetti M, Gallo A, Posteraro B. Review article: Biofilm formation by Helicobacter pylori as a target for eradication of resistant infection. Aliment Pharmacol Ther. 2012;36:222–30. doi: 10.1111/j.1365-2036.2012.05165.x. [DOI] [PubMed] [Google Scholar]
- 33.Yonezawa H, Osaki T, Hanawa T, Kurata S, Ochiai K, Kamiya S. Impact of Helicobacter pylori biofilm formation on clarithromycin susceptibility and generation of resistance mutations. PLoS One. 2013;8:e73301. doi: 10.1371/journal.pone.0073301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cammarota G, Branca G, Ardito F, Sanguinetti M, Ianiro G, Cianci R, et al. Biofilm demolition and antibiotic treatment to eradicate resistant Helicobacter pylori: A clinical trial. Clin Gastroenterol Hepatol. 2010;8:817–20.e3. doi: 10.1016/j.cgh.2010.05.006. [DOI] [PubMed] [Google Scholar]