

Abstract
Fifteen patients with treatment-refractory colorectal cancer were enrolled on a phase 1b study of Pexa-Vec (pexastimogene devacirepvec; JX-594), an oncolytic and immunotherapeutic vaccinia designed to selectively replicate in cancer cells. Pexa-Vec was administered intravenously every 14 days, at dose levels of 1 × 106, 1 × 107, or 3 × 107 plaque-forming units (pfu)/kg. The primary endpoint was to determine the maximum tolerated dose. Secondary endpoints were pharmacokinetics and pharmacodynamics as well as antitumor activity. Patients were heavily pretreated (mean 4.5 lines of therapy). All patients received at least two Pexa-Vec doses (median = 4; range = 2–4). No dose-limiting toxicities were reported, and the maximum tolerated dose was not reached. The most common adverse events were grade 1/2 flu-like symptoms, generally lasting <24 hours. During the first and last cycles, genome pharmacokinetics were unchanged. Infectious pfu could be detected in plasma up to 2 hours after cycle 1 and up to 30 minutes after cycle 4 (when antivaccinia antibody titers are known to have peaked). Ten patients (67%) had radiographically stable disease. Given the acceptable safety profile of multiple intravenous Pexa-Vec infusions in patients with treatment-refractory colorectal cancer, further trials evaluating efficacy of intravenous Pexa-Vec, as monotherapy or in combination with chemotherapeutic agents, is warranted in this patient population.
Introduction
Patients with metastatic colorectal cancer can be eligible for multiple lines of chemotherapy. While most patients will receive all available chemotherapeutic agents (fluoropyrimidines, irinotecan, and oxaliplatin) as well as bevacizumab and/or anti-epidermal growth factor receptor (EGFR) antibodies (cetuximab and panitumumab) during their first two lines of treatment, there are little remaining options for third-line treatment and beyond. Anti-EGFR antibodies demonstrated clinical activity in third-line setting,1,2 but the benefits are limited to the patients with KRAS wild-type tumors3 and the oral multikinase inhibitor regorafenib was demonstrated to provide a 1.4-month overall survival improvement in the third-line setting.4
Oncolytic immunotherapy represents a novel therapeutic platform for the treatment of cancer with unique attributes compared with conventional chemotherapy.5 Oncolytic viruses have tumor selectivity, while sparing normal cells thus leading to a broad therapeutic range between cancer and normal tissues. Pexa-Vec (pexastimogene devacirepvec; JX-594) is a thymidine kinase gene-inactivated oncolytic vaccinia virus engineered for the expression of transgenes encoding human granulocyte-macrophage colony-stimulating factor (GM-CSF) and β-galactosidase.6 In phase 1/2 trials of intratumoral injection into liver cancers,7,8 Pexa-Vec was well-tolerated, and associated with tumor responses and a dose-related survival benefit. Of note, the safety and tolerability of intratumoral Pexa-Vec were maintained following systemic dissemination that occurred hours to weeks after an injection.
However, for patients with refractory, widespread metastatic tumors, drug delivery through blood with a high dose of Pexa-Vec may be required to achieve systemic oncolytic tumor responses. In a phase 1 trial of a single intravenous infusion of Pexa-Vec in 23 patients with advanced solid tumors,9 dose-related antitumor activity was observed and normal tissues were not affected clinically. Although repeated intravenous efficacy of Pexa-Vec was superior to single dose efficacy in animal models (unpublished data), and increasing the total dose of intravenous Pexa-Vec in patients with large tumor burdens may lead to an overall increase in tumor dose, it could theoretically also result in increased toxicity.
On the basis of promising antitumor activity seen with earlier trials, and encouraging safety data, this study was conducted to determine the maximum-tolerated dose (MTD) and/or maximum feasible dose (MFD) of biweekly (every 2 weeks) intravenous infusion of Pexa-Vec in patients with refractory, cetuximab-resistant (or KRAS mutant), metastatic colorectal cancer. Secondary endpoints included pharmacokinetic and pharmacodynamic effects, and antitumor activity. The starting dose in this trial was 1 × 106 plaque-forming units (pfu)/kg based on toxicology data analyses, and because the cumulative total dose after four cycles (4 × 106 pfu/kg) was roughly equivalent to the total dose safely administered at once in the single dose intravenous infusion study.9 Three cohorts were planned; the highest dose level (cohort 3, 3 × 107 pfu/kg) was the same as the highest dose in a good laboratory practice animal toxicology study.
Results
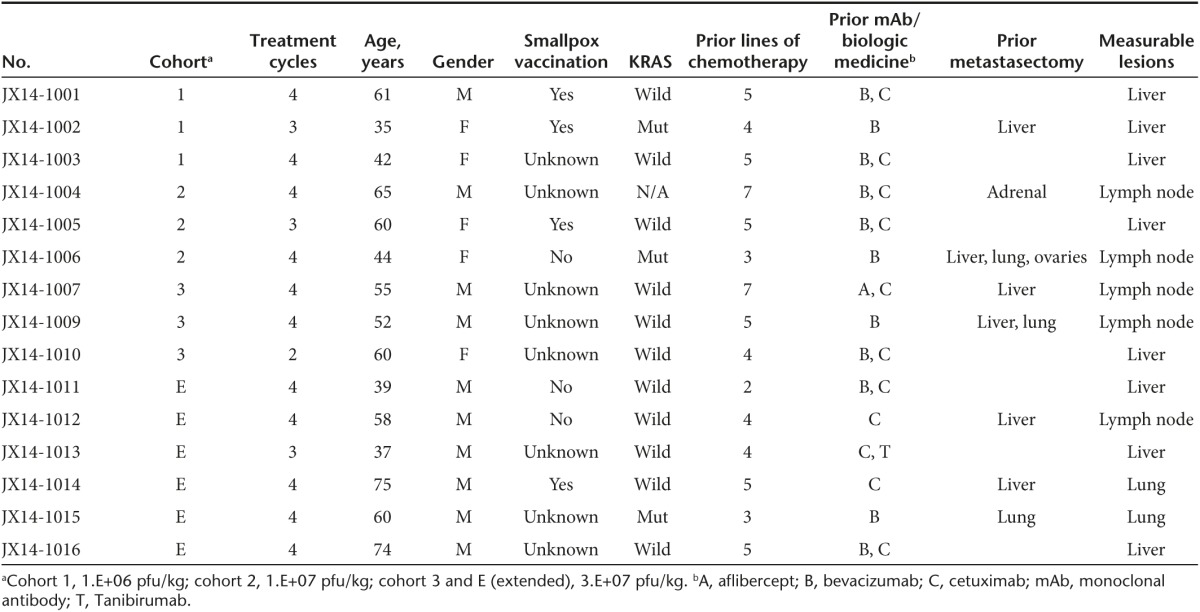
Eighteen patients were screened for study eligibility. Fifteen patients (median age, 58 years) were entered onto the trial between September 2010 and July 2012 (Supplementary Figure S1). All patients had refractory, metastatic colorectal cancer and documented disease progression after two or more lines of chemotherapy. All 15 patients received at least one dose of intravenous Pexa-Vec and all were evaluable for safety and efficacy. Important patient and treatment characteristics of these patients are listed in Table 1. Except for three patients with KRAS mutant tumors, all patients were previously treated with anti-EGFR antibodies. Karnofsky performance status was 80 or 90 in all patients.
Table 1. Patient characteristics.
Safety
Three patients each were treated at three escalating intravenous Pexa-Vec dose levels: 1 × 106, 1 × 107, and 3 × 107 pfu/kg. Since no MTD was reached for any dose levels, the MFD was defined at 3 × 107 pfu/kg for biweekly intravenous infusion of Pexa-Vec; this cohort was expanded to recruit six additional patients. The mean and range for each treatment and total exposure by cohort are presented in Table 2. Eleven patients (73%) completed all four planned infusions of Pexa-Vec over 8 weeks, while four patients (27%) did not achieve the full duration of treatment. These patients who came off study early discontinued study treatment due to disease progression. More specifically, patients JX-14–1002 and JX14-1005 developed disease progression by Response Evaluation Criteria In Solid Tumors after the completion of day 29 treatment. One patient (JX14-1010) needed palliative radiotherapy after day 15 and discontinued treatment at the investigator's discretion. Another patient (JX14-1013) complained of rapid symptomatic deterioration prior to day 43 infusion and was found to have disease progression. Overall, the median number of Pexa-Vec intravenous infusions was 4 (range, 2–4). No patient discontinued treatment due to an adverse event.
Table 2. Drug exposure.

Intravenous infusion of Pexa-Vec was well-tolerated in all patients, with no clinically significant adverse events during entire study period (Table 3 and Supplementary Table S1). There were no treatment-related serious adverse events, no dose-limiting toxicities (DLTs), no infusion-related reactions, and no fatal events related to the study treatment. All patients experienced mild and generally brief episodes (<24 hours) of pyrexia and/or chills after Pexa-Vec infusion. Since no patients developed Pexa-Vec infusion-related reactions, corticosteroids or antihistamine were not given to any patient. Other common events were headache (60%), nausea (53%), anorexia (33%), rash (27%), and vomiting (27%). Pexa-Vec related skin pustules (Grade 1 papulopustular rash) developed on days 3–7 of cycle 1 only in 7 of 9 patients at high dose, and 0 of 6 at lower doses (47% incidence overall). Pustules resolved without complications within 5–26 days (Figure 1a,b). No pustules developed after subsequent cycles. Locations included palms, soles, oral mucosa, and lips. Pustules on the lips or oral mucosa were associated with other symptoms including pain or discomfort without note of an impact on oral intake.
Table 3. Adverse events observed during study period (all-causality).
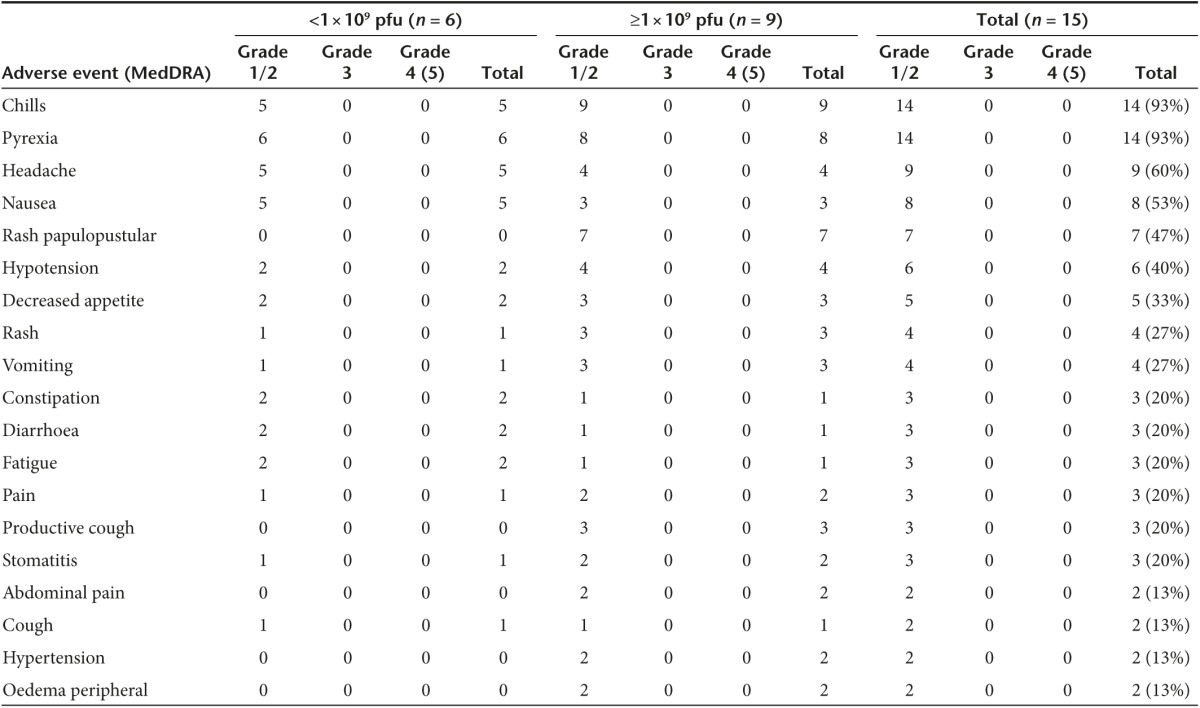
Figure 1.

Pexa-Vec-related skin pustules. Pexa-Vec related pustule onset and resolution of two pustules in patient JX14-1009 (a) and one pustule in patient JX14-1007 (b).
Hypotension had previously been noted after intravenous Pexa-Vec infusion,9 therefore patients were instructed to hydrate by taking ≥ 2 l of fluids orally over the course of 24 hours prior to each infusion as well as ≥ 2 l of fluids orally within 24 hours post-treatment. Six patients (40%) had mild (Grade 1/2), transient (generally <30 minutes) episodes of hypotension. The decrease in blood pressure (systolic blood pressure <90 mmHg) was recorded approximately 4–8 hours postinfusion, and recovered spontaneously without corticosteroids or other medical support.
Productive cough was observed in three patients (20%) but none developed clinically relevant or symptomatic decreases in oxygen saturation. The only significant laboratory abnormalities were Grade 3 aspartate transaminase and alanine transaminase increases in patient JX14-1002 (Cohort 1) patient. These Grade 3 events were deemed unrelated to Pexa-Vec treatment.
Pharmacokinetics and shedding
Pharmacokinetics were dose-related, and were unchanged over four cycles (Figure 2a). Plasma from high-dose patients was tested at the same time points in cycles 1 and 4 for biologically-active pfu. All samples taken during infusion were positive in cycles 1 (Figure 2b) and 4 (Figure 2c). Six of nine patients were still positive 30 minutes after infusion completion on cycles 1, and four were positive at the 1–2-hour time points. Pexa-Vec concentration in plasma was generally lower during cycle 4 versus cycle 1.
Figure 2.

Pexa-Vec pharmacokinetics following multiple IV infusions. (a) Quantitative polymerase chain reaction detection of Pexa-Vec genomes in blood after cycles 1–4. Mean genomes/ml were calculated after removing individual patient values that were below the limit of detection (LOD). The solid marker indicates that the mean value at a time point is based on three samples for Cohorts 1 and 2 and nine samples (D1 and D15), eight samples (D29), or seven samples (D43) for Cohort 3. The “x” marker indicates that there were only two samples above the LOD used to calculate the mean value. The unfilled marker indicates that the value of the time point is based on only one sample above the LOD. (b) Pexa-Vec pfu detection in plasma after cycle 1. (c) Pexa-Vec pfu detection in plasma after cycle 4.
Pexa-Vec shedding was assessed in patients over time via throat swab and urine culture. No urine shedding was demonstrated. Throat swabs were positive for pfu between days 5–8 in one of six patients at low doses, and five of nine at the highest dose level. All high-dose patients with positive throat swabs had active oral or lip pustules at the time.
Cytokine and white blood cell induction
Inflammatory cytokines were induced within 8 hours in a dose-related fashion. Several cytokines were elevated at 3 and/or 8 hours after dosing; elevations were transient. Overall, the peak concentration for IL-6, MIP-1α, MCP-1, IL-8, IL-18, MIP-1β, and TNF-α was generally highest after the first dose and was lower after the second dose (on day 15) (Figure 3a–g). In contrast, concentrations for IL-2, IL-10, and IFN-γ were generally higher after cycle 2–4 than cycle 1 (Figure 3h–j). GM-CSF was also induced acutely after each infusion, though it can not be discerned whether this is due to induction of endogenous GM-CSF or due to early gene expression from Pexa-Vec infected cells (Figure 3k).
Figure 3.

Cytokine detection in plasma. Cytokine concentrations were measured in plasma before and after each cycle of Pexa-Vec treatment. (a) IL-6, (b) MIP-1α, (c) MCP-1, (d) IL-8, (e) IL-18, (f) MIP-1β, (g) TNF-α, (h) IL-2, (i) IL-10, (j) IFN-γ, (k) GM-CSF.
In a previous clinical trial, we demonstrated that replication could result in detectable GM-CSF in blood when other inflammatory cytokines had returned to baseline (days 4–15),7 despite the relatively short half-life of GM-CSF (< 2 hours).10 Therefore, detection of GM-CSF in blood is indicative of high-level GM-CSF expression, and may be a specific but insensitive marker of transgene expression. Four patients (27%) had significant increases from baseline in plasma GM-CSF concentrations at day 5 (all other inflammatory cytokines returned to baseline in < 24 hours). Neutrophil induction (defined as at least 40% induction within the first 15 days) was noted in six patients (40%).
Tumor response, progression-free survival, and overall survival
A waterfall plot showing best objective response at Day 29 (change in the sum of diameters of measurable lesions) assessed by independent radiographic review is depicted in Figure 4. There were no objective radiologic responses, but 10 patients (67%) had radiologically stable disease. Interestingly, among patients with a total Pexa-Vec dose <3 × 109 pfu/kg (n = 6), two (33%) had stable disease; whereas eight out of nine patients (89%) with a total dose ≥3 × 109 pfu/kg achieved stable disease (P = 0.09; Fisher's exact test). KRAS mutational status was evaluated in 15 patients (94%). Eleven (69%) patients were KRAS wild-type and 3 (19%) patients were KRAS mutant. Among 11 KRAS wild-type patients, 8 (73%) exhibited stable disease at day 29. Among three KRAS mutant patients, one (33%) exhibited stable disease at day 29. Finally, one patient whose KRAS mutational status was unknown exhibited stable disease at day 29. Progression-free survival and overall survival of all eligible patients were 61 days and 10.3 months, respectively.
Figure 4.

Waterfall plot showing maximum percent changes of the sum of longest diameters of target lesions compared to baseline. X-axis: patient numbers.
Discussion
This was the first clinical trial to use repeated doses of Pexa-Vec (pexastimogene devacirepvec; JX-594), or any other vaccinia or poxvirus, by intravenous infusion to treat patients with refractory, metastatic colorectal cancer. A biweekly schedule was chosen for the following reasons. First, preclinical pharmacology and toxicology studies supported the safety and efficacy of this regimen.6 Second, clinical data support safety of vaccinia viruses in patients upon repeat intratumoral dosing every 2 weeks. A subset of patients treated by intratumoral injection were shown to have subsequent intravenous exposure of Pexa-Vec due to replication in the tumor approximately a week after dosing.7,8 In addition, it is desirable to administer multiple doses prior to development of neutralizing antibodies, which have been shown to occur within the first month of treatment.9 The safety and activity of more frequent dosing are unknown at this time in humans.
Vaccinia virus shows a natural selectivity toward tumor relative to normal tissues after intravenous administration.6,11 This inherent selectivity appears to be due to three underlying biological characteristics of tumors: their “leaky” blood vessels allow for deposition of this relatively large virus (for example, due to vascular endothelial growth factor (VEGF) effects), their proliferative and metabolic state, and the expression and/or activity of the EGFR and VEGFR pathways. As tumors grow, new blood vessel formation is required to sustain the cancerous growth. The process of angiogenesis creates “leaky” blood vessels that are susceptible to breach by vaccinia, allowing the virus to gain entry into surrounding cancerous cells. This leaky vasculature is limited to areas of cancer cell growth and certain other tissues in the body, such as the corpus luteum, primarily due to VEGF expression. In addition, highly proliferative epithelial tissues such as cancers are preferred targets for vaccinia because their cell cycle status naturally produces large nucleotide pools that are necessary for viral replication. Finally, vaccinia virus replication and spread is facilitated by the secretion of vaccinia growth factor. This protein interacts with the EGFR commonly expressed in tumor cells, thereby stimulating their proliferation and priming them for subsequent vaccinia infection and replication. Therefore, vaccinia virus preferentially targets and replicates in tumors.
Our study demonstrated that intravenous infusion of Pexa-Vec is a safe and well-tolerated agent that was associated with only a few grade 1 or 2 drug-related adverse events, but no DLTs or infusion-related reactions. As none of nine patients at 3 × 107 pfu/kg biweekly intravenous infusion experienced DLT, we were not able to formally define the MTD of Pexa-Vec based on clinical toxicity. Further dose escalation at this time was not feasible due to clinical trial material constraints; manufacturing process improvements may allow further dose escalation in the future. The MFD was 3 × 107 pfu/kg, repeated biweekly for four cycles, based on the established safety of this regimen. With regard to safety, the most commonly observed treatment-related adverse events were flu-like symptoms (in all 15 patients). The symptoms were easily relieved with acetaminophen or over-the-counter nonsteroidal anti-inflammatory drugs. Considering that increased cytokine induction temporally correlates with observed symptoms and may represent an underlying mechanism, it is conceivable that the flu-like symptoms could be considered an indication of biologic activity of oncolytic viruses; future studies will be necessary to determine this association. Grade 1/2 hypotension (systolic blood pressure < 90 mmHg) was noted in 6 of 15 patients on this study. These patients received hydration pre- and post-treatment for blood pressure support and no other medical intervention was necessary. Blood pressure will be closely monitored on future intravenous studies of Pexa-Vec. Pexa-Vec-related skin pustules were noted in patients treated at high-dose. Though swab samples of pustules were not collected on this study, data from other trials have shown that Pexa-Vec pfu can be recovered from Pexa-Vec related pustules (unpublished data).
Although the sample size was limited, determining antitumor activity of intravenous Pexa-Vec was a secondary endpoint of the present phase 1 study. The lack of complete or partial responses in a refractory and heavily pretreated cancer population may not be surprising, but the higher rate of disease stabilization noted in patients treated with high-dose Pexa-Vec merit confirmation in further phase 2 studies.
Pharmacokinetic and GM-CSF blood concentration profiles demonstrate continued activity and biodistribution over the course of four cycles. Anti-Pexa-Vec antibodies are induced over several weeks following multiple doses.8,9 Therefore, these results represent a major milestone in the field of IV cancer therapy with oncolytic viruses, and specifically with vaccinia. Pexa-Vec was engineered from vaccinia virus as described by Kim et al.6,8 because the extracellular enveloped virus form is able to traffic in the blood while avoiding inactivation by complement and antibodies. Repeat IV dosing with Pexa-Vec (JX-594) was feasible in animals and can be enhanced through extracellular enveloped virus.12 Reproducible IV delivery and safe repeat IV dosing are important features for further clinical development.
In conclusion, Pexa-Vec administered as biweekly intravenous infusion was safe and well-tolerated. Therefore, one way to reasonably move forward with this agent would be to test intravenous Pexa-Vec as monotherapy at 3 × 107 pfu/kg repeated doses in a conventional phase 2 study. Alternatively, a new phase 1/2 study to establish the optimal safe dosing of intravenous Pexa-Vec in combination with chemotherapy in earlier stages of disease would also be a rational approach, borrowing from preclinical data showing synergism with this combination in in vivo models.13 As proof of this concept, a phase 1/2 study of Pexa-Vec in combination with irinotecan is currently underway in patients with metastatic colorectal cancer. Given the safety of repeat IV dosing described here, along with manufacturing improvements, further dose escalation may also be explored in the future.
Materials and Methods
This was a single-center, open-label phase 1b study, registered in advance to ClinicalTrials.gov (NCT01469611). The study protocol and consent forms were approved by the Korean Food and Drug Administration and the Samsung Medical Center Institutional Review Board (Seoul, Korea).
Patients. All patients had histologically confirmed measurable colorectal adenocarcinoma. They had failed all approved chemotherapeutic agents, including irinotecan and oxaliplatin. If a patient had KRAS wild-type tumors, documentation of prior failure to treatment with anti-EGFR antibodies was required. Other criteria for eligibility included age of 18 years or older, Karnofsky performance status of at least 70, no antitumor therapy within 4 weeks, normal hepatic, renal and marrow functions, and at least one measurable tumor mass by Response Evaluation Criteria In Solid Tumors. Patients with the following were excluded: significant immunodeficiency state, tumors invading a major vascular structure or in location that would potentially result in significant clinical adverse effects if post-treatment tumor swelling were to occur, history of exfoliative skin conditions requiring systemic therapy, serious or unstable cardiac disease, history of a serious systemic reaction to previous smallpox vaccination, HIV positivity, systemic corticosteroids or other immunosuppressive medication in use, or with clinically significant active infection or medical conditions. All patients gave written informed consent according the Declaration of Helsinki and the principles of Good Clinical Practice.
Treatment, evaluation, and toxicity criteria. Three patients each were planned for entry at three escalating intravenous Pexa-Vec (JX-594) dose levels: 1 × 106, 1 × 107, and 3 × 107 pfu/kg. Pexa-Vec production, transport, storage, and administration were as previously described.6,9 In brief, Pexa-Vec was administered via intravenous infusion over 60 minutes, with adequate pre- and post-treatment hydration, during which vital signs and oxygen saturation were monitored. Premedication with acetaminophen was allowed, and corticosteroids and antihistamine were provided if required. Treatment was given on days 1, 15, 29, and 43. Vital signs were taken at the following time-points: prior to infusion, 15, 30, and 45 minutes after the start of infusion, end of infusion (time 0); and 15 minutes, 30 minutes, 60 minutes, 2 hours, 3 hours, 4 hours post-treatment. Thereafter, vitals were taken every 2 hours between 4–12 hours post-treatment and every 4 hours between 12–24 hours post-treatment. Adverse events were recorded and scored according to the National Cancer Institute criteria (CTCAE v3). DLT was defined as any grade 4 adverse events or any grade 3 nonhematologic toxicity persisting for >7 days, except for flu-like symptoms that respond to standard therapies. At a given dose level, rules for dose advancement were as follows: If there were no DLTs at the dose level, advancement to the next level would be permitted. If DLT was noted in one of three patients, up to three additional patients were added at that level. If none of these three new patients experienced a DLT, then the next dose level would be opened. MTD was defined as the dose immediately preceding the dose resulting in two patients with DLT. MFD was defined as the top dose level if no MTD was defined. Once the MTD/MFD was defined, the dose cohort was expanded to recruit six additional patients. Intrapatient dose escalation was not allowed.
At baseline, patients underwent a history and physical examination, vital signs, Karnofsky performance status, an electrocardiogram, blood counts, serum chemistries, coagulation profiles, and computed tomography scans of chest, abdomen and all affected sites. Safety evaluations were performed on days 1, 5, 8, 15, 22, 29, 36, 43, 50, and 57. These safety assessments consisted of history and physical examination, vital signs, hematology, and chemistries. Disease evaluations were done by computed tomography on day 29, every 6 weeks thereafter. Appropriate tumor markers (CEA, MIA, CA19-9) were collected at baseline and weekly thereafter. Clinical activity of Pexa-Vec based on computed tomography scans were summarized according to Response Evaluation Criteria In Solid Tumors.
Patients who completed the study, with stable disease or a response to treatment and had not gone on to other cancer therapy, returned to the clinic for follow-up visits every 6 weeks for the first 6 months. Thereafter, patients were followed a minimum of every 3 months or according to current standard of care guidelines until patients had documented tumor progression, went on to other cancer therapy, or died. If a measurable radiological response was observed at any time, the same response assessment was repeated after 4 weeks to confirm the response. After disease progression or initiation of new cancer therapy, patients were monitored for survival and for information relating to subsequent therapies, and their general safety and efficacy. Radiology assessments of subsequent therapies, if done, were reviewed.
Genome qunatitative polymerase chain reaction in blood over time. Quantitative polymerase chain reaction was used for measuring Pexa-Vec genomes in blood. Pexa-Vec DNA purification and quantitative polymerase chain reaction was run as described previously.7,14
Detection of pfu in plasma and throat swabs over time. Plasma was processed from whole blood collected in Vacuette K3E EDTA K3 from patients of highest dose level (cohort 3) on day 1 and day 43 (prior to treatment, after start of infusion at the following time-points: 15 and 30 minutes, and at the end of infusion at the following time points: (time 0); 30, 60, 120 (2 hours), and 240 (4 hours) minutes. Plasma samples were prepared by three rounds of centrifugation and resuspended in equal volume of Tris buffer containing 10% fetal bovine serum.
Throat swabs were collected at multiple times after single or repeat dosing by swabbing the back of patients' throats with polyester-tipped swabs, placing swab tips in 3 ml of BDUM, vortexing rigorously, and saving the BDUM for testing. PFU of Pexa-Vec in plasma and in throat swabs was analyzed by a qualified plaque assay method, which follows standard plaque assay procedures but includes staining for β-gal. Monolayers of U-2 OS cells in six-well tissue culture plates were incubated with 10-fold dilutions of human sample containing Pexa-Vec. After 2 hours absorption, the inoculum was removed, and medium containing 1.5% carboxymethyl cellulose was added. The incubation was continued for 72 hours at 37 °C. Plaques were stained in two steps using neutral red and x-gal (5-bromo-4-chloro-indolyl-β-D-galactopyranoside) in an agarose solution. Neutral red stains live cells red and the β-gal expressed in Pexa-Vec cleaves x-gal to form 5-bromo-4-chloro-3-hydroxyindole which stains Pexa-Vec-infected plaques blue. Other viral plaques (not containing β-gal) remain unstained and form a clear plaque (white). Blue and white plaques were counted.
GM-CSF detection. Samples were collected for detection of hGM-CSF protein concentrations in plasma on day 1 (predose, 3 and 8 hours postdose), day 5, day 8, day 15 (predose, 3 and 8 hours postdose), day 22, day 29 (predose, 3 and 8 hours postdose), day 36, day 43 (predose, 3 and 8 hours postdose), day 50 and day 57. hGM-CSF concentrations in plasma were determined by solid-phase sandwich enzyme-linked immunosorbent assay using the Quantikine hGM-CSF Kit (R&D Systems, Minneapolis, MN) as directed by the manufacturer.
Plasma cytokine detection. Blood was collected and processed to plasma on day 1 (predose, 3 and 8 hours postdose), day 5; and day 15 (predose, 3 and 8 hours postdose), day 29 (predose, 3 and 8 hours postdose), and at day 43(predose, 3 and 8 hours postdose). Plasma was assayed at Myriad Rules Based Medicine (Austin, TX) using a validated Luminex Human Cytokine assay MAP A v1.0 panel consisting of 16 cytokines that included IFN-γ, IL-2, IL-3, IL-4, IL-5, IL-6, Il-7, IL-8, IL-10, IL-18, MIP-1α, MIP-1β, MCP-1, TNF-α, and TNF-β.
Statistics. The primary endpoints of this study were to determine MTD/MFD and safety and tolerability of repeated doses of intravenous Pexa-Vec in patients with colorectal cancer. Secondary endpoints were to characterize pharmacokinetics and pharmacodynamics as well as antitumor activity. Patient demographics and other baseline characteristics were summarized using conventional descriptive statistics. Descriptive statistics were also employed to list adverse events. Continuous variables are presented as mean or median values, and categorical variables as frequencies (percentage), unless otherwise stated. Statistical analyses were performed using SPSS 13 for Windows (SPSS, Chicago, IL) and GraphPad Prism 5 (GraphPad, La Jolla, CA).
SUPPLEMENTARY MATERIAL Figure S1. Patient flow. Table S1. Adverse events observed during study period (treatment-related).
Acknowledgments
This study was supported by Jennerex Inc (now SillaJen Biotherapeutics). Additional support was provided by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C1951) and a grant from the 20 by 20 project of Samsung Medical Center (Seoul, Korea; GF01140111). C.J.B., A.M., E.M.S., L.M.A., A.P., J.B., and T.-H.H. are employees of SillaJen. D.K. owns SillaJen stock options.
Supplementary Material
References
- Douillard, JY, Siena, S, Cassidy, J, Tabernero, J, Burkes, R, Barugel, M et al. (2010). Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28: 4697–4705. [DOI] [PubMed] [Google Scholar]
- Peeters, M, Price, TJ, Cervantes, A, Sobrero, AF, Ducreux, M, Hotko, Y et al. (2010). Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28: 4706–4713. [DOI] [PubMed] [Google Scholar]
- Lièvre, A, Bachet, JB, Le Corre, D, Boige, V, Landi, B, Emile, JF et al. (2006). KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66: 3992–3995. [DOI] [PubMed] [Google Scholar]
- Grothey, A, Van Cutsem, E, Sobrero, A, Siena, S, Falcone, A, Ychou, M et al.; CORRECT Study Group. (2013). Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381: 303–312. [DOI] [PubMed] [Google Scholar]
- Parato, KA, Breitbach, CJ, Le Boeuf, F, Wang, J, Storbeck, C, Ilkow, C et al. (2012). The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther 20: 749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, JH, Oh, JY, Park, BH, Lee, DE, Kim, JS, Park, HE et al. (2006). Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther 14: 361–370. [DOI] [PubMed] [Google Scholar]
- Park, BH, Hwang, T, Liu, TC, Sze, DY, Kim, JS, Kwon, HC et al. (2008). Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 9: 533–542. [DOI] [PubMed] [Google Scholar]
- Heo, J, Reid, T, Ruo, L, Breitbach, CJ, Rose, S, Bloomston, M et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach, CJ, Burke, J, Jonker, D, Stephenson, J, Haas, AR, Chow, LQ et al. (2011). Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477: 99–102. [DOI] [PubMed] [Google Scholar]
- Hovgaard, D, Mortensen, BT, Schifter, S and Nissen, NI (1992). Clinical pharmacokinetic studies of a human haemopoietic growth factor, GM-CSF. Eur J Clin Invest 22: 45–49. [DOI] [PubMed] [Google Scholar]
- McCart, JA, Ward, JM, Lee, J, Hu, Y, Alexander, HR, Libutti, SK et al. (2001). Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res 61: 8751–8757. [PubMed] [Google Scholar]
- Kirn, DH, Wang, Y, Liang, W, Contag, CH and Thorne, SH (2008). Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res 68: 2071–2075. [DOI] [PubMed] [Google Scholar]
- Heinemann, L, Simpson, GR, Boxall, A, Kottke, T, Relph, KL, Vile, R et al. (2011). Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer 11: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, TC, Hwang, T, Park, BH, Bell, J and Kirn, DH (2008). The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther 16: 1637–1642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.