

Abstract
Objective:
To investigate the influence of common and low-frequency genetic variants on the risk of ischemic stroke (all IS) and etiologic stroke subtypes.
Methods:
We meta-analyzed 12 individual genome-wide association studies comprising 10,307 cases and 19,326 controls imputed to the 1000 Genomes (1 KG) phase I reference panel. We selected variants showing the highest degree of association (p < 1E-5) in the discovery phase for replication in Caucasian (13,435 cases and 29,269 controls) and South Asian (2,385 cases and 5,193 controls) samples followed by a transethnic meta-analysis. We further investigated the p value distribution for different bins of allele frequencies for all IS and stroke subtypes.
Results:
We showed genome-wide significance for 4 loci: ABO for all IS, HDAC9 for large vessel disease (LVD), and both PITX2 and ZFHX3 for cardioembolic stroke (CE). We further refined the association peaks for ABO and PITX2. Analyzing different allele frequency bins, we showed significant enrichment in low-frequency variants (allele frequency <5%) for both LVD and small vessel disease, and an enrichment of higher frequency variants (allele frequency 10% and 30%) for CE (all p < 1E-5).
Conclusions:
Our findings suggest that the missing heritability in IS subtypes can in part be attributed to low-frequency and rare variants. Larger sample sizes are needed to identify the variants associated with all IS and stroke subtypes.
Stroke is a leading cause of disability in Western countries and among the most common causes of premature death worldwide.1,2 Ischemic stroke (IS) accounts for up to 85% of all stroke cases. Evidence for a substantial genetic contribution to IS risk comes from twin and family history studies and the discovery of risk loci for IS through genome-wide association studies (GWAS).3–7 Most previously identified associations have been confined to etiologic stroke subtypes, which include large vessel disease (LVD), cardioembolic stroke (CE), and small vessel disease (SVD). Despite these discoveries, a significant proportion of heritability remains unexplained.3,6–8
Prior GWAS in IS have been based on genetic data imputed to versions of the HapMap panel9 with training sets of up to 2.5 million single nucleotide polymorphisms (SNPs) of which 85% are common variants (minor allele frequency [MAF] > 5%). Since then, the 1000 Genomes (1 KG) Project10 has considerably expanded the coverage of human genetic variation especially for low-frequency (MAF 1%–5%) variants. We thus performed an extended meta-analysis informed by 1 KG including low-frequency variants in the human genome not assessed in the previous METASTROKE collaboration to determine whether these variants mediate risk for ischemic stroke.
We assembled 10,307 Caucasian cases and 19,326 Caucasian controls from 12 studies for a GWAS meta-analysis of IS based on the 1 KG phase I imputation training set. After quality control, 8.3 million SNPs and 1 million indels were available for analysis. Promising signals were replicated both in Caucasian and non-Caucasian populations.
METHODS
Overall study design.
The discovery stage consisted of a meta-analysis of 12 case-control studies of IS with previously genotyped data (table e-1 on the Neurology® Web site at Neurology.org). For each set of cases, population-matched controls were recruited from studies with existing genotyping data (details of study cohorts and controls are given in the supplementary material). For both sample sets, raw autosomal data were imputed to approximately 9 million SNPs using 1 KG phase I data as a reference panel. Genome-wide logistic regression analysis was performed independently in all samples, summary statistics were shared, and meta-analysis was performed centrally for all datasets. Covariates were not considered as they were not equally available over all study sets. Subsequently, the top SNPs (p < 1E-5) from the discovery meta-analysis were tested for replication in 3 independent samples (figure 1 and table e-2): (1) 5,137 de novo genotyped stroke samples from Europe and the United States and 2,040 controls; (2) genome-wide data from 8,298 Caucasian stroke patients and 27,229 controls recruited through the Cervical Artery Dissection and Ischemic Stroke Patients (CADISP) and National Institute of Neurological Disorders and Stroke–Stroke Genetics Network (NINDS-SiGN) networks11,12; and (3) genome-wide data from South Asian patients recruited through the Risk Assessment of Cardiovascular Events (RACE) study phase 1 and 2.13
Figure 1. Study profile.

Study profile summarizing the study samples and analytical strategy.
Standard protocol approvals, registrations, and patient consents.
Written or oral informed consent was obtained from all participants, and the study was approved by the respective research ethics committees.
Discovery-stage genotyping.
Genotyping was performed individually for all sites and quality control was performed as described previously.14
Replication-stage genotyping.
The first part of de novo genotyping was done at the Helmholtz Center Munich using iPlex Gold (Sequenom, San Diego, CA) methodology. Amplification reactions and parameters were based on the manufacturer's instructions. Spectrocaller software supplied by the manufacturer was used for automatic genotype calling. Clusters were checked manually, and all doubtful calls were evaluated. Sex was checked to remove any sample misidentifications.
The second part of de novo genotyping was performed at the Psychiatric & Neurodevelopmental Genetics Unit, Boston, Massachusetts, using the Sequenom iPLEX Gold chemistry and the MassARRAY system. Genotypes were called using SpectroCHIP array and matrix-assisted laser desorption/ionization–time of flight mass spectrometry. Genotype clusters were checked manually, and all doubtful calls were evaluated.
Imputation.
We performed imputation separately for each study (table e-1) using the algorithms IMPUTEv215 and MACH16 with standard parameters. We removed SNPs with an imputation quality (info) score <0.3, leaving approximately 8 million variants per individual study.
GWAS and meta-analysis.
We performed GWAS on the combined phenotype (all ischemic stroke), as well as for etiologic stroke subtypes classified according to Trial of Org 10172 in Acute Stroke Treatment (TOAST) criteria.17 TOAST subtyping was available for all but 2 studies (Heart Protection Studyand Vitamin Intervention for Stroke Prevention; see table e-1). To ensure high quality of the resulting GWAS data, we calculated λ for the overall discovery sample (figure e-1) and for each study individually (figure e-2). We further calculated lambda on sets of SNPs stratified by frequency and imputation quality to determine whether particular bins of SNPs were susceptible to genomic inflation (figure 2). SNPs with frequency <1%, introduced during imputation, showed high levels of genomic inflation and were thus excluded from the transethnic meta-analysis. 1 KG phase I samples were used to calculate reference frequencies for European samples. SNPs with a frequency difference >30% from 1 KG in controls or a difference >30% with any other study in the meta-analysis were removed from the data, as were SNPs with missing p values and SNPs that only produced p values in <50% of the studies.
Figure 2. Quantile-quantile plots for different allele frequency bins.
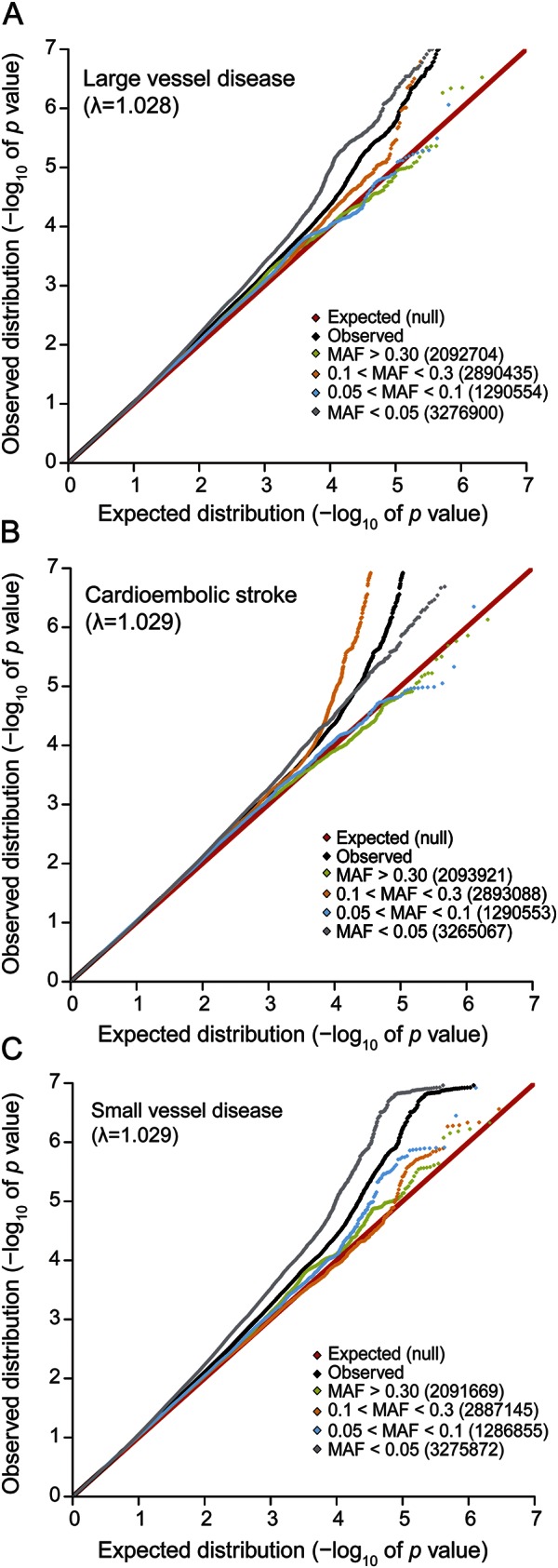
Shown is the distribution of p values for different allele frequency bins: (A) large vessel disease, (B) cardioembolic stroke, (C) small vessel disease. The red line displays the expected (null) distribution of the statistic. The black line shows the observed distribution of all variants studied. Frequency bins are depicted in different colors: green (>30% minor allele frequency [MAF]), orange (10%–30% MAF), blue (5%–10% MAF), and gray (1%–5% MAF). The number in parentheses shows the number of SNPs that were included in the respective bins. Statistical significance was tested using a 2-sample Kolmogorov-Smirnov test.28
Finally, λ was calculated across the cleaned set of association results, and the results were combined to perform fixed-effects inverse variance weighted meta-analysis using METAL.18 During meta-analysis, SNPs were analyzed across cohorts to ensure that effect alleles were consistent and that alleles matched those reported in the 1 KG phase I. Outliers in these analyses were excluded from further processing. We used genomic control to correct for incidental inflation of test statistics. Upon completion of the meta-analyses, we again confirmed that the genomic inflation was well behaved (all λ <1.03).
All SNPs with p < 1E-5 in any of the performed GWAS (all IS and subtypes) and a median imputation quality >0.7 were selected for downstream replication. This resulted in sets of SNPs analyzed for the following traits: all IS, LVD, SVD, and CE. The replication strategy consisted of 3 parts (figure 1). We first performed replication in the de novo genotyped wet-laboratory studies using Sequenom technology.
Summary statistics for each replication sample were produced using logistic regression with the phenotype of interest as outcome. Model 1 was calculated without covariates; model 2 included sex as a covariate. Since the results did not differ between the 2 models and to ensure consistency between the discovery and replication phase, results are reported only for model 1. Cases from Leuven were folded into the German sample using the German controls. Results were summarized using fixed-effects inverse-variance meta-analysis. We next performed in silico replication in the CADISP11 and NINDS-SiGN12 sample. Overlapping cases and controls between the NINDS-SiGN sample and METASTROKE were identified and removed and summary statistics from NINDS-SiGN were recalculated for the replication SNPs.
For both meta-analyses, fixed-effects inverse variance models were used. The first and second replication steps were combined to form the Caucasian replication set. Third, multiethnic meta-analysis was performed by integrating in silico lookup data from RACE1 and RACE2 (forming the South Asian replication set) using METASOFT. We used Han and Eskin's19 random effects model to maximize power under heterogeneity. Combination of the discovery, the Caucasian replication set, and the South Asian replication set using multiethnic analysis formed the final results (figure 1). Any SNPs with p < 5E-8 were considered to be genome-wide significant. Any SNPs with 5E-8 < p < 1E-5 were considered to have suggestive evidence for association.
Pathway analysis.
Pathway analysis was performed using Meta-Analysis Gene-set Enrichment of Variant Associations (MAGENTA).20 We used all available databases and 10,000 permutations to select statistically significant pathways and processes. We deemed a false discovery rate (FDR) q value of q < 0.05 or a Bonferroni corrected p value of p < 0.05 as significant. Bonferroni correction was performed on the number of gene sets in a pathway.
RESULTS
A total of 10,307 cases and 19,326 controls from 12 studies were investigated in the discovery analysis. Data on etiologic stroke subtypes were available for 10 of the 12 studies (table e-1). A set of 15,820 IS cases (3,808 LVD, 3,697 CE, and 2,206 SVD) and 34,462 controls was available for replication (see Methods and table e-2). The overall genomic inflation factors (λ) for the meta-analyses of IS, LVD, CE, and SVD were 1.015, 1.028, 1.029, and 1.029, respectively, indicating minimal inflation due to population stratification or due to cases and controls who had undergone separate genotyping (figure 2). Manhattan plots for the discovery sample are shown in figures e3-e6. QQ plots for all IS and subtypes are depicted in figure e-1 and for individual studies in figure e-2.
Established risk loci for ischemic stroke.
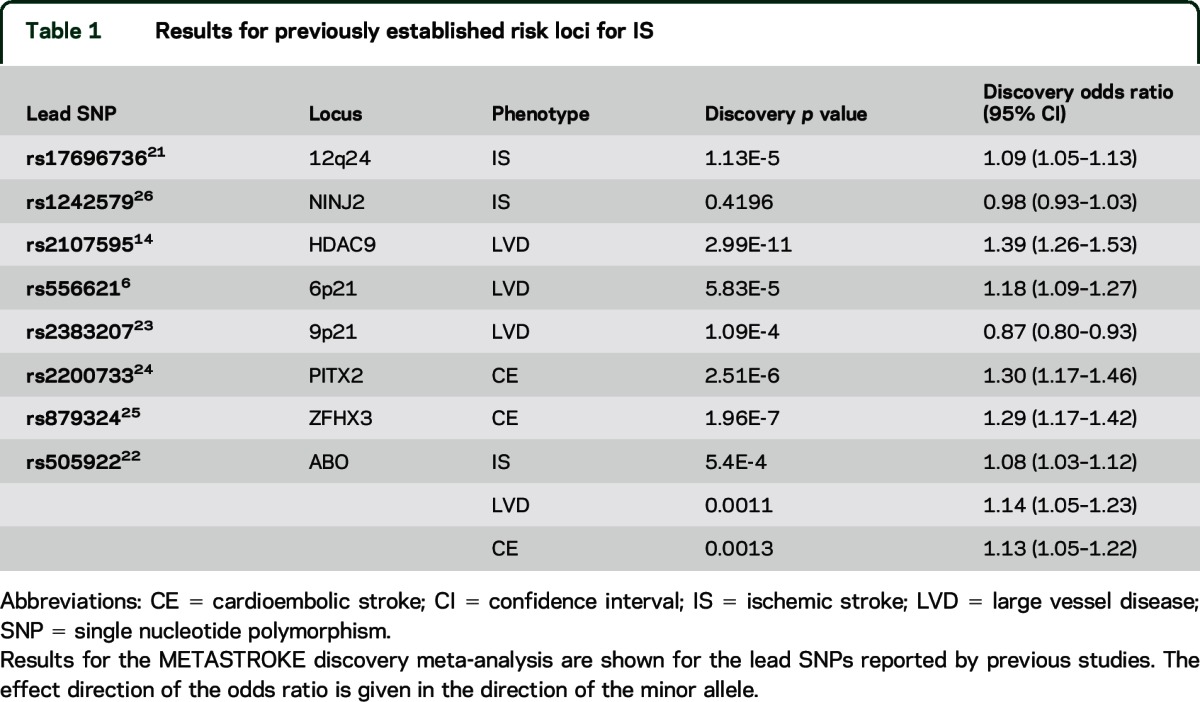
We first examined the lead SNPs of established risk loci for IS and subtypes derived from GWAS in our discovery meta-analysis (table 1). The previously identified lead signal for HDAC9 (LVD)5 was associated on a genome-wide level in the discovery analysis. We also observed p values < 1E-3 for association at chr12q2421 (all IS, p = 1.13E-5), ABO22 (all IS, p = 5.40E-4), chr6p216 (LVD, p = 5.83E-5), chr9p2123 (LVD, p = 1.09E-4), PITX224 (CE, p = 2.51E-6), and ZFHX325 (CE, p = 6.73E-5). In contrast, we found no association of NINJ226 (all IS, p = 0.4196). For full results, see table 1 and figure e-7.
Table 1.
Results for previously established risk loci for IS
Novel risk loci for ischemic stroke.
Our discovery analysis yielded 4 new and independent loci that exceeded the threshold for genome-wide significance of 5E-8 (table 2) as well as 25 additional loci with association p values < 1E-5 (table e-3). This list included 7 loci (13 variants) for all IS, 8 loci (13 variants) for LVD, 7 loci (11 variants) for CE, and 7 loci (13 variants) for SVD, all of which were selected for replication (figure 1). In the Caucasian replication, 3 SNPs for 3 loci (ABO, PITX2, and ZFHX3) were nominally associated after Bonferroni correction (p < 6.3E-3, table 2). The results of a transethnic meta-analysis yielded 4 loci genome-wide significant, all of which have been reported previously. The lead SNPs for these loci were rs532436 for ABO (all IS, overall p = 4.28E-8), rs2107595 for HDAC9 (LVD, p = 2.99E-11), rs2723334 for PITX2 (CE, p = 8.37E-24), and rs12932445 for ZFHX3 (CE, p = 1.20E-8). Association signals for the indels did not achieve genome-wide or near genome-wide significance.
Table 2.
Meta-analysis results for all IS and subtypes: Listed are all SNPs reaching genome-wide significance in the transethnic meta-analysis

Fine mapping of risk loci for ischemic stroke.
In order to refine the association signals at confirmed, previously published loci, we took advantage of the denser imputation panel provided by the 1 KG consortium to produce association signals for an enlarged set of low-frequency variants (figure e-7). We discovered a new peak association for PITX2 (rs2723334), which is only in moderate linkage disequilibrium (LD) with the previously published lead signal for CE (rs2200733, r2 = 0.45, figure e-7C) and with the previously published lead signal for atrial fibrillation (AF)27 (rs6817105, r2 = 0.46). Furthermore, our novel lead SNP (rs532436) for ABO is only in moderate LD with the previously published variant (rs505922, r2 = 0.53, figure e-7A). In contrast, we found the lead SNPs for HDAC9 and ZHFX to be identical or in high LD (r2 > 0.9) with the lead SNPs reported by prior studies (rs2107595 for HDAC9, and rs879324 for ZFHX3, figure e-7, B and D; table 1). Functional annotation of all lead SNPs is presented in table e-4.
Role of allele frequency bins.
To study the contribution of low-frequency alleles and common alleles to individual stroke subtypes, we further investigated the p value distribution across bins of variants categorized according to their minor allele frequencies (>30%; 10–30%; 5–10%; <5%) for all IS and stroke subtypes (figure 2). There was no enrichment of specific bins of allele frequencies for all IS. However, we found an enrichment in low-frequency variants (<5%) for both LVD and SVD. In contrast, CE showed an enrichment of variants between 10% and 30%. The enrichment of these specific variant bins was significant when compared to all SNPs or any other frequency bin using a 2-sample Kolmogorov-Smirnov test (all p < 1E-5).28 Of note, the distribution of observed vs expected p values was well-behaved in all analyses (figure 2) and we did not observe a systematic bias towards low-frequency variants that could have been introduced through imputation artifacts.
Pathway analysis.
Applying a Bonferroni corrected threshold of p < 0.05 we found several pathways for all IS and IS subtypes (table e-5). In total, there were 136 nominally associated pathways for CE, 84 for all IS, 86 for LVD, and 55 for SVD. The following terms showed the highest degree of association: germ cell development (CE), microtubule (IS), mitochondrial envelope (LVD), and SH3 domain binding (SVD). When using a predefined FDR cutoff of q < 0.05, we observed a single association of natural killer (NK) cell signaling with all IS.
DISCUSSION
Adopting a classical GWAS approach based on 1 KG imputed data with replication in both de novo and in silico genotype data, we found no novel locus reaching genome-wide significance for ischemic stroke or its subtypes. However, for the first time, we report genome-wide significance for association of the ABO locus with all IS and were able to fine-map 2 known stroke loci (PITX2 and ABO) by making use of the expanded 1 KG panel. We further found enrichment of association of low-frequency alleles with both LVD and SVD, and of higher frequency variants (10%–30%) with CE. This finding has important implications for future studies, as design and analysis strategies may differ for low-frequency and common variants.
ABO has previously been shown to be genome-wide associated with circulating levels of von Willebrand factor and factor VIII.22 An assessment of these signals in IS cohorts showed a nominal replication for the lead SNP rs505922 in LVD and CE, but not for SVD.22 Our findings extend this observation by demonstrating that a different variant in the ABO gene (rs532436, p = 4.30E-8) is genome-wide associated with all IS and that the association with ABO is strictly confined to LVD (p = 0.0029) and CE (p = 0.0011) with no signal with SVD (p = 0.53). Together, these findings emphasize a role of ABO in thrombosis and associated stroke phenotypes.
Although not reaching genome-wide significance, there are 3 novel loci that deserve attention. GUCY1A3, which showed suggestive association with LVD (p = 8.25E-6) in the current study, has recently been reported as a risk gene for early-onset myocardial infarction in a family-based study.29 The allele frequency of the lead SNP in our study was 1.5%. Thus, the low-frequency nature of this variant together with the lower power for association detection in LVD might have hindered our ability to detect a genetic association.
The second locus is TNFSF11 (RANKL), which showed suggestive evidence for association with CE (p = 1.03E-7). The allele frequency of the lead SNP in our study was 24%. TNFSF11, a major player in bone remodeling and part of the RANK/RANKL/OPG pathway, has repeatedly been reported in the pathogenesis of AF and as a predictor of IS in patients with nonvalvular AF.30–32 Of note, however, variants in or near this gene have not emerged from prior GWAS of AF, thus highlighting the need for sample expansion in future GWAS.
The third locus is GCH1, which showed a p value of 3.31E-5 for association with SVD. The allele frequency of the lead SNP in our study was 1.5%. GCH1 encodes for GTP cyclohydrolase 1, a rate-limiting factor in the tetrahydrobiopterin (BH4) biosynthesis.33 BH4 is an essential cofactor for nitric oxide (NO) synthases in endothelial cells and has been shown to enhance NO bioavailability.34 Supplementation with a synthetic BH4 analog has previously been tested in a trial in monogenic SVD.35
Aside from GUCY1A3 and GCH1, we found other low-frequency variants with p values < 1E-4 for association and consistent effect directions across all samples. These include NACC2 as well as an intergenic locus near KCNN2 (both LVD), TMEM108 (SVD), and CBFA2T3 (CE). More work is needed to determine the potential role of these low-frequency variants in stroke subtypes. Our findings on low-frequency variants together with the observed enrichment of association of low-frequency alleles with both LVD and SVD supports the notion that parts of the missing heritability in IS are explained by rare and low-frequency variation. Next-generation sequencing studies and targeted resequencing efforts of known risk loci together with larger sample sizes for stroke subtypes are needed to capture this missing heritability and to depict the heritability of ischemic stroke more precisely.
Previous GWAS have revealed that associations with ischemic stroke are largely confined to etiologic stroke subtypes. We found the ABO locus to be associated with all IS on a genome-wide level, which is primarily due to its association with LVD and CE. Another major locus that has been reported to be associated with multiple stroke subtypes is the chr12q24 region, which has recently been shown to be implicated in LVD, CE, and SVD.21 New results, however, show association restricted to SVD without evidence for association in any other subtype.12 Conceivably, shared associations in conjunction with subtype-specific signals may provide insights into stroke mechanisms.
Our pathway analysis revealed several novel pathways for all IS and etiologic stroke subtypes. The strongest association was seen for all IS and NK cell signaling. It was recently shown that NK cells promote neuronal death in experimental stroke.36 However, additional work is needed to fully explore the role of this and other candidate pathways in IS. Combining pathway analysis with more detailed phenotyping may provide further insight into specific stroke subtypes. It is well-known that genes do not act in isolation, but rather in complex molecular networks that are often involved in disease susceptibility and progression. Pathway analysis has promise in other diseases like coronary artery disease37 where canonical pathways like inflammation and lipid metabolism had been identified as key players in disease development. This information is highly valuable in a context of mechanistic and functional studies to elucidate the biological processes in disease development. Further, it also provides potential mechanisms in gene–environment interactions, which are mostly unexplored in the cardiovascular disease context. An additional point to consider is the potential use of such pathways in the discovery of biomarkers. Finally, it may provide researchers with therapeutic targets that could ultimately prove to be of high relevance.
A methodologic strength of our approach is the replication of signals in a wet laboratory environment with de novo genotyping in addition to in silico replication. Prior results have shown that signals confirmed in an in silico setting may not necessarily replicate in a de novo genotyping environment.38,39 Hence, we have minimized the risk of false-positive reporting by our study design. By integrating genome-wide data from non-Caucasian populations and performing a transethnic meta-analysis, we maximized the chance of detecting association signals across different ethnicities while preserving the power in our dataset. Thus for example, we saw nominally significant replication p values for GUCY1A3 in LVD in the South Asian samples (p = 0.012), pointing towards shared risk in Caucasian and South Asian populations. Discoveries of both shared and ethnicity-specific genetic risk factors will be further facilitated by recently completed GWAS studies in non-Caucasian populations.40 In the pathway analysis, we made use of the MAGENTA software, which is tailored towards elucidating pathways in a GWAS setting. MAGENTA has been shown to be superior to other pathway analysis tools; for one, MAGENTA accounts for inherent difficulties in the assignment of SNP data to gene/gene products. It ensures that the results are comparable and that there is no inherent bias in the final outcome. Second, it accounts for important confounders on the association scores of genes and gene sets, which cannot be performed by other pathway analysis tools. Our study was limited by the relatively low power for detecting associations in stroke subtypes, especially for low-frequency variants and the heterogeneity of the imputation accuracy, particularly for lower frequency variants, which may have been introduced by decentralized imputation.
Aside from providing new insights into the genetic architecture of IS, this large meta-analysis of 1 KG imputed data provides a valuable resource for even larger meta-analyses with recently published GWAS12 and provides additional insight into the genetic architecture of ischemic stroke. The complete summary statistics of this analysis are available upon request through the METASTROKE Web site (www.strokegenetics.org).
Supplementary Material
ACKNOWLEDGMENT
Australian Stroke Genetics Collaboration (ASGC) Australian population control data were derived from the Hunter Community Study. The authors thank the University of Newcastle for funding and the men and women of the Hunter region who participated in this study.
GLOSSARY
- AF
atrial fibrillation
- CADISP
Cervical Artery Dissection and Ischemic Stroke Patients
- CE
cardioembolic stroke
- FDR
false discovery rate
- GWAS
genome-wide association studies
- IS
ischemic stroke
- LD
linkage disequilibrium
- LVD
large vessel disease
- MAF
minor allele frequency
- MAGENTA
Meta-Analysis Gene-set Enrichment of Variant Associations
- NINDS-SiGN
National Institute of Neurological Disorders and Stroke–Stroke Genetics Network
- NK
natural killer
- NO
nitric oxide
- RACE
Risk Assessment of Cardiovascular Events
- SNP
single nucleotide polymorphism
- SVD
small vessel disease
- TOAST
Trial of Org 10172 in Acute Stroke Treatment
Footnotes
Supplemental data at Neurology.org
AUTHOR AFFILIATIONS
From the Institute for Stroke and Dementia Research, Klinikum der Universität München (R.M., M.D.), Ludwig-Maximilians University, Munich, Germany; Department of Clinical Neurosciences (M.T., S.B., H.S.M.), University of Cambridge, UK; Department of Medical Genetics (S.L.P.), Center for Molecular Medicine, University Medical Center Utrecht, the Netherlands; CTSU (J.C.H., R.C.), Nuffield Department of Population Health, University of Oxford, UK; Hunter Medical Research Institute (E.G.H., J.R.A.), Public Health Research Program, Newcastle, Australia; Department of Genetics (W.Z., D.S.), Perelman School of Medicine, University of Pennsylvania, PA; Instituto de Medicina Molecular (P. Abrantes, S.A.O.), Faculdade de Medicina, Universidade de Lisboa (J.M.F.); Instituto Gulbenkian de Ciência (IGC) (P. Abrantes, S.A.O., A.M.V.), Oeiras, Portugal; Inserm U1167 (P. Amouyel), Pasteur Institute Lille, France; Department of Public Health (P. Amouyel), Lille University Hospital, University of Lille Nord de France; Division of Neurocritical Care and Emergency Neurology (T.W.K.B., C.E.K., G.J.F., J.R.), Department of Neurology (T.W.K.B., G.J.F., C.E.K., J.R.), Center for Human Genetic Research (T.W.K.B., G.J.F., C.E.K., J.R.), and J. Philip Kistler Stroke Research Center (G.J.F.), Massachusetts General Hospital, Boston; Institute of Epidemiology and Social Medicine (K.B.), University of Münster, Germany; Department of Cerebrovascular Diseases (G.B.B.), Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy; Team Neuroepidemiology (G.C., S.D.), Inserm Research Center for Epidemiology and Biostatistics (U897), Bordeaux, France; University of Bordeaux (G.C., S.D.), France; Department of Medicine (Y.-C.C., B.D.M.), University of Maryland School of Medicine (S.J.K.), Baltimore; Center for Public Health Genomics and Cardiovascular Research Center (W.-M.C., B.B.W.), University of Virginia, Charlottesville; Institute of Cardiovascular Research Royal Holloway (I.C., P.S.), University of London (ICR2UL), UK; Program in Medical and Population Genetics (G.J.F., C.E.K., J.R.), Broad Institute, Cambridge, MA; Department of Neurosciences (J.M.F.), Neurology, Hospital Santa Maria, Lisbon, Portugal; Department of Neurology (D.M.G., J.F.M.), Mayo Clinic, Jacksonville, FL; Department of Clinical Sciences Lund (A.I., A.L.), Neurology, Lund University, Sweden; Department of Neurology (S.J.K.), Veterans Administration Medical Center, Baltimore, MD; Department of Neurosciences, Experimental Neurology, and Leuven Research Institute for Neuroscience and Disease (LIND) (R.L., V.T.), KU Leuven, University of Leuven; Laboratory of Neurobiology (R.L., V.T.), VIB, Vesalius Research Center, Leuven, Belgium; Brain and Mental Health Program (C.R.L.), Hunter Medical Research Institute, Newcastle, Australia; Helmholtz Zentrum München and Technische Universität München (P.L.), Institut für Humangenetik, Munich, Germany; Fred Hutchinson Cancer Research Center (J.L., A.P.R.), Seattle, WA; Department of Neurology (J.P., A.S.), Jagiellonian University Medical College, Krakow, Poland; Stroke Prevention Research Unit (P.M.R.), Nuffield Department of Clinical Neurosciences, University of Oxford, UK; Ashford & St Peters NHS Foundation Trust (P.S.), Surrey; Division of Clinical Neurosciences (C.L.M.S.), University of Edinburgh, UK; Department of Neurology (V.T.), Austin Health, Heidelberg, Australia; Florey Institute for Neuroscience and Mental Health (V.T.), University of Melbourne, Heidelberg, Australia; Institute of Neuroscience and Physiology (T.T.), Sahlgrenska Academy at University of Gothenburg; Department of Neurology (T.T.), Sahlgrenska University Hospital, Gothenburg, Sweden; Departamento Promoção da Saúde e Doenças Crónicas (DPSDC) (A.M.V.), Instituto Nacional de Saúde Dr. Ricardo Jorge (INSA), Lisbon; Center for Biodiversity, Functional & Integrative Genomics (BIOFIG) (A.M.V.), Lisbon, Portugal; Department of Neurology and Rehabilitation (D.W.), University of Cincinnati College of Medicine, OH; Department of Neurology (S.S.), Boston University School of Medicine, MA; Departments of Neurology and Public Health Sciences (B.B.W.), University of Virginia Health System, Charlottesville; and Munich Cluster for Systems Neurology (SyNergy) (M.D.), Munich, Germany.
AUTHOR CONTRIBUTIONS
Rainer Malik: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Matthew Traylor: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Sara L. Pulit: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Steve Bevan: drafting/revising the manuscript for content, study concept or design, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Jemma C. Hopewell: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Elizabeth G. Holliday: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Wei Zhao: analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Philippe Amouyel: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Patricia Abrantes: acquisition of data, accepts responsibility for conduct of research and final approval. John R. Attia: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Thomas W.K. Battey: acquisition of data, accepts responsibility for conduct of research and final approval. Klaus Berger: acquisition of data, accepts responsibility for conduct of research and final approval. Giorgio B. Boncoraglio: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Ganesh Chauhan: drafting/revising the manuscript for content, analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Yu-Ching Cheng: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Wei-Min Chen: drafting/revising the manuscript for content, analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Robert Clarke: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Ioana Cotlarciuc: drafting/revising the manuscript for content, analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Stephanie Debette: drafting/revising the manuscript for content, acquisition of data, accepts responsibility for conduct of research and final approval. Guido J. Falcone: drafting/revising the manuscript for content, analysis or interpretation of data, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Jose M. Ferro: acquisition of data, accepts responsibility for conduct of research and final approval. Dale M. Gamble: analysis or interpretation of data, statistical analysis, accepts responsibility for conduct of research and final approval. Andreea Ilinca: acquisition of data, accepts responsibility for conduct of research and final approval. Steven J. Kittner: drafting/revising the manuscript for content, acquisition of data, accepts responsibility for conduct of research and final approval. Christina E. Kourkoulis: acquisition of data, accepts responsibility for conduct of research and final approval. Robin Lemmens: acquisition of data, accepts responsibility for conduct of research and final approval. Christopher R. Levi: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Peter Lichtner: acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Arne Lindgren: drafting/revising the manuscript for content, acquisition of data, accepts responsibility for conduct of research and final approval. Jingmin Liu: analysis or interpretation of data, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. James F. Meschia: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Braxton D. Mitchell: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Sofia A. Oliveira: acquisition of data, accepts responsibility for conduct of research and final approval. Joana Pera: drafting/revising the manuscript for content, acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Alex P. Reiner: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Peter M. Rothwell: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Pankaj Sharma: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Agnieszka Slowik: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Cathie L.M. Sudlow: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Turgut Tatlisumak: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Vincent Thijs: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Astrid M. Vicente: acquisition of data, statistical analysis, accepts responsibility for conduct of research and final approval. Daniel Woo: acquisition of data, accepts responsibility for conduct of research and final approval. Sudha Seshadri: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Danish Saleheen: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Jonathan Rosand: drafting/revising the manuscript for content, study concept or design, acquisition of data, study supervision or coordination, accepts responsibility for conduct of research and final approval. Hugh S. Markus: drafting/revising the manuscript for content, study concept or design, acquisition of data, study supervision or coordination, accepts responsibility for conduct of research and final approval. Bradford B. Worrall: drafting/revising the manuscript for content, study concept or design, acquisition of data, accepts responsibility for conduct of research and final approval. Martin Dichgans: drafting/revising the manuscript for content, study concept or design, acquisition of data, study supervision or coordination, accepts responsibility for conduct of research and final approval.
STUDY FUNDING
This research was funded by grants from the Australian National and Medical Health Research Council (NHMRC project grant ID: 569257), the Australian National Heart Foundation (NHF project grant ID: G 04S 1623), the University of Newcastle, the Gladys M. Brawn Fellowship scheme, and the Vincent Fairfax Family Foundation in Australia. Elizabeth G. Holliday was supported by a Fellowship from the National Heart Foundation and National Stroke Foundation of Australia (ID: 100071). Bio-Repository of DNA in Stroke (BRAINS) is partly funded by a Senior Fellowship from the Department of Health (UK) to P. Sharma, the Henry Smith Charity, and the UK-India Education Research Institutive (UKIERI) from the British Council. Genetics of Early Onset Stroke (GEOS) Study, Baltimore, Maryland, USA, was supported by the NIH Genes, Environment and Health Initiative (GEI) grant U01 HG004436, as part of the GENEVA consortium under GEI, with additional support provided by the Mid-Atlantic Nutrition and Obesity Research Center (P30 DK072488), the Office of Research and Development, Medical Research Service, and the Baltimore Geriatrics Research, Education, and Clinical Center of the Department of Veterans Affairs. Genotyping services were provided by the Johns Hopkins University Center for Inherited Disease Research (CIDR), which is fully funded through a federal contract from the NIH to the Johns Hopkins University (contract number HHSN268200782096C). Assistance with data cleaning was provided by the GENEVA Coordinating Center (U01 HG 004446; PI Bruce S. Weir). Study recruitment and assembly of datasets were supported by a cooperative agreement with the Division of Adult and Community Health, Centers for Disease Control and Prevention, and by grants from NINDS and the NIH Office of Research on Women's Health (R01 NS45012, U01 NS069208-01). Heart Protection Study (HPS) (ISRCTN48489393) was supported by the UK Medical Research Council (MRC), British Heart Foundation, Merck and Co. (manufacturers of simvastatin), and Roche Vitamins Ltd. (manufacturers of vitamins). Genotyping was supported by a grant to Oxford University and CNG from Merck and Co. Jemma C. Hopewell acknowledges support from the British Heart Foundation (FS/14/55/30806). Ischemic Stroke Genetics Study (ISGS)/Siblings With Ischemic Stroke Study (SWISS) was supported in part by the Intramural Research Program of the NIA, NIH project Z01 AG-000954-06. ISGS/SWISS used samples and clinical data from the NIH-NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds), human subjects protocol numbers 2003-081 and 2004-147. ISGS/SWISS used stroke-free participants from the Baltimore Longitudinal Study of Aging (BLSA) as controls. The inclusion of BLSA samples was supported in part by the Intramural Research Program of the NIA, NIH project Z01 AG-000015-50, human subjects protocol number 2003-078. The ISGS study was funded by NIH-NINDS grant R01 NS-42733 (J.F. Meschia). The SWISS study was funded by NIH-NINDS grant R01 NS-39987 (J.F. Meschia). This study used the high-performance computational capabilities of the Biowulf Linux cluster at the NIH (http://hpc.nih.gov). MGH Genes Affecting Stroke Risk and Outcome Study (MGH-GASROS) was supported by NINDS (U01 NS069208), the American Heart Association/Bugher Foundation Centers for Stroke Prevention Research 0775010N, the NIH and NHLBI's STAMPEED genomics research program (R01 HL087676), and a grant from the National Center for Research Resources. The Broad Institute Center for Genotyping and Analysis is supported by grant U54 RR020278 from the National Center for Research resources. Milano–Besta Stroke Register Collection and genotyping of the Milan cases within CEDIR were supported by annual research funding of the Italian Ministry of Health (grant numbers: RC 2007/LR6, RC 2008/LR6, RC 2009/LR8, RC 2010/LR8), and FP6 LSHM-CT-2007-037273 for the PROCARDIS control samples. Wellcome Trust Case-Control Consortium 2 (WTCCC2) was principally funded by the Wellcome Trust, as part of the Wellcome Trust Case Control Consortium 2 project (085475/B/08/Z and 085475/Z/08/Z and WT084724MA). The Stroke Association provided additional support for collection of some of the St George's, London cases. The Oxford cases were collected as part of the Oxford Vascular Study, which is funded by the MRC, Stroke Association, Dunhill Medical Trust, National Institute of Health Research (NIHR), and the NIHR Biomedical Research Centre, Oxford. The Edinburgh Stroke Study was supported by the Wellcome Trust (clinician scientist award to C. Sudlow) and the Binks Trust. Sample processing occurred in the Genetics Core Laboratory of the Wellcome Trust Clinical Research Facility, Western General Hospital, Edinburgh. Much of the neuroimaging occurred in the Scottish Funding Council Brain Imaging Research Centre (www.sbirc.ed.ac.uk), Division of Clinical Neurosciences, University of Edinburgh, a core area of the Wellcome Trust Clinical Research Facility and part of the SINAPSE (Scottish Imaging Network–A Platform for Scientific Excellence) collaboration (www.sinapse.ac.uk), funded by the Scottish Funding Council and the Chief Scientist Office. Collection of the Munich cases and data analysis was supported by the Vascular Dementia Research Foundation. M. Farrall and A. Helgadottir acknowledge support from the BHF Centre of Research Excellence in Oxford and the Wellcome Trust core award (090532/Z/09/Z). Leuven Stroke Study is funded by personal research funds from the Department of Neurology, University Hospital Leuven, Belgium. V. Thijs and R. Lemmens are supported by Fundamental Clinical Investigatorships from FWO. Flanders Lund Stroke Register (LSR) was supported by the Swedish Research Council (K2007-61X-20378-01-3, K2010-61X-20378-04-3), the Swedish Heart and Lung Foundation, Skåne University Hospital, Region Skåne, the Freemasons Lodge of Instruction EOS in Lund, King Gustaf V and Queen Victoria's Foundation, Lund University, and the Swedish Stroke Association. DNA extraction and preparation for LSR was performed by the SWEGENE Resource Center for Profiling Polygenic Disease, Skåne University Hospital, Malmö, Sweden. Biobank services were provided by Region Skåne Competence Centre (RSKC Malmö), Skåne University Hospital, Malmö, Sweden, and Biobank, Labmedicin, Skåne University and Regional Laboratories Region Skåne, Sweden. Munster (Westphalian Stroke Cases and Controls from the Dortmund Health Study, Germany): Case ascertainment in the Westphalian Stroke Register was part of the German Competence Net Stroke, supported by the German Federal Ministry of Education and Research (01GI9909/3). Blood collection in the Dortmund Health Study was done through funds from the Institute of Epidemiology and Social Medicine University of Muenster. The collection of sociodemographic and clinical data in the Dortmund Health Study was supported by the German Migraine and Headache Society (DMKG) and by unrestricted grants of equal share from Almirall, Astra Zeneca, Berlin Chemie, Boehringer, Boots Health Care, Glaxo-Smith-Kline, Janssen Cilag, McNeil Pharma, MSD Sharp & Dohme, and Pfizer to the University of Muenster. Portugal: S.A.O., J.M.F., and A.M.V. thank the Portuguese study participants, the genotyping unit at the Instituto Gulbenkian de Ciência, and the Portuguese study neurologists and nurses for their contributions. This work was supported by the PTDC/SAU-GMG/64426/2006 grant, a Cilência 2008 contract (S.A.O.), doctoral fellowships, and an Investigator-FCT contract (S.A.O.) from the Portuguese Fundaçaão para a Ciência e a Tecnologia. Poland: The study was supported by a grant from the Jagiellonian University, Krakow, Poland: K/ZDS/002848. VISP: The GWAS component of the VISP study was supported by the United States National Human Genome Research Institute (NHGRI), grant U01 HG005160 (PI Michèle Sale and Bradford Worrall), as part of the Genomics and Randomized Trials Network (GARNET). Genotyping services were provided by the Johns Hopkins University Center for Inherited Disease Research (CIDR), which is fully funded through a federal contract from the NIH to the Johns Hopkins University. Assistance with data cleaning was provided by the GARNET Coordinating Center (U01 HG005157; PI Bruce S. Weir). Study recruitment and collection of datasets for the VISP clinical trial were supported by an investigator-initiated research grant (R01 NS34447; PI James Toole) from the United States Public Health Service, NINDS, Bethesda, Maryland. Control data were obtained through the database of genotypes and phenotypes (dbGAP) maintained and supported by the United States National Center for Biotechnology Information, US National Library of Medicine. WHI Funding support for WHI-GARNET was provided through the NHGRI GARNET (grant number U01 HG005152). Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the GARNET Coordinating Center (U01 HG005157). Funding support for genotyping, which was performed at the Broad Institute of MIT and Harvard, was provided by the NIH Genes, Environment, and Health Initiative (GEI; U01 HG004424). The Stroke Genetics Network (SiGN) study was funded by a cooperative agreement grant from the National Institute of Neurological Disorders and Stroke (NINDS) U01 NS069208. Genotyping services were provided by the Johns Hopkins University Center for Inherited Disease Research (CIDR), which is fully funded through a federal contract from the NIH to the Johns Hopkins University (contract no. HHSN268200782096C). The Biostatistics Department Genetics Coordinating Center at the University of Washington, Seattle, provided more extensive quality control of the genotype data through a subcontract with CIDR. Additional support to the Administrative Core of SiGN was provided by the Dean's Office, University of Maryland School of Medicine. This work was supported by grants received from the German Federal Ministry of Education and Research (BMBF) in the context of the e:Med program (e:AtheroSysMed), the FP7 European Union project CVgenes@target (261123), the DFG as part of the CRC 1123 (B3), the Corona Foundation, and the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Bonita R. Epidemiology of stroke. Lancet 1992;339:342–344. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380:2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falcone GJ, Malik R, Dichgans M, Rosand J. Current concepts and clinical applications of stroke genetics. Lancet Neurol 2014;13:405–418. [DOI] [PubMed] [Google Scholar]
- 4.Dichgans M. Genetics of ischaemic stroke. Lancet Neurol 2007;6:149–161. [DOI] [PubMed] [Google Scholar]
- 5.International Stroke Genetics Consortium, Wellcome Trust Case Control Consortium, , Bellenguez C, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet 2012;44:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holliday EG, Maguire JM, Evans TJ, et al. Common variants at 6p21.1 are associated with large artery atherosclerotic stroke. Nat Genet 2012;44:1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holliday EG, Traylor M, Malik R, et al. Genetic overlap between diagnostic subtypes of ischemic stroke. Stroke 2015;46:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bevan S, Traylor M, Adib-Samii P, et al. Genetic heritability of ischemic stroke and the contribution of previously reported candidate gene and genomewide associations. Stroke 2012;43:3161–3167. [DOI] [PubMed] [Google Scholar]
- 9.International HapMap Consortium, Frazer KA, Ballinger DG, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007;449:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1000 Genomes Project Consortium, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debette S, Kamatani Y, Metso TM, et al. Common variation in PHACTR1 is associated with susceptibility to cervical artery dissection. Nat Genet 2015;47:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.NINDS Stroke Genetics Network (SiGN); International Stroke Genetics Consortium (ISGC). Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study. Lancet Neurol Epub 2015 Dec 8. [DOI] [PMC free article] [PubMed]
- 13.Saleheen D, Bukhari S, Haider SR, et al. Association of phosphodiesterase 4D gene with ischemic stroke in a Pakistani population. Stroke 2005;36:2275–2277. [DOI] [PubMed] [Google Scholar]
- 14.Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol 2012;11:951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 2012;44:955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2010;34:816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams HP, Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke: definitions for use in a multicenter clinical trial: TOAST: Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35–41. [DOI] [PubMed] [Google Scholar]
- 18.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet 2011;88:586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segrè AV, Groop L, Mootha VK, et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 2010;6:e1001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilarski LL, Achterberg S, Devan WJ, et al. Meta-analysis in more than 17,900 cases of ischemic stroke reveals a novel association at 12q24.12. Neurology 2014;83:678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams FM, Carter AM, Hysi PG, et al. Ischemic stroke is associated with the ABO locus: the EuroCLOT study. Ann Neurol 2013;73:16–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gschwendtner A, Bevan S, Cole JW, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol 2009;65:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gretarsdottir S, Thorleifsson G, Manolescu A, et al. Risk variants for atrial fibrillation on chromosome 4q25 associate with ischemic stroke. Ann Neurol 2008;64:402–409. [DOI] [PubMed] [Google Scholar]
- 25.Gudbjartsson DF, Holm H, Gretarsdottir S, et al. A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat Genet 2009;41:876–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikram MA, Seshadri S, Bis JC, et al. Genomewide association studies of stroke. N Engl J Med 2009;360:1718–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012;44:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schork AJ, Thompson WK, Pham P, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet 2013;9:e1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erdmann J, Stark K, Esslinger UB, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 2013;504:432–436. [DOI] [PubMed] [Google Scholar]
- 30.Ferro D, Loffredo L, Polimeni L, et al. Soluble CD40 ligand predicts ischemic stroke and myocardial infarction in patients with nonvalvular atrial fibrillation. Arterioscler Thromb Vasc Biol 2007;27:2763–2768. [DOI] [PubMed] [Google Scholar]
- 31.Lip GY, Patel JV, Hughes E, Hart RG. High-sensitivity C-reactive protein and soluble CD40 ligand as indices of inflammation and platelet activation in 880 patients with nonvalvular atrial fibrillation: relationship to stroke risk factors, stroke risk stratification schema, and prognosis. Stroke 2007;38:1229–1237. [DOI] [PubMed] [Google Scholar]
- 32.Xi L, Cao H, Chen Y. OPG/RANK/RANKL axis in atrial fibrillation. Cardiology 2013;125:174–175. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Rao F, Zhang K, et al. Discovery of common human genetic variants of GTP cyclohydrolase 1 (GCH1) governing nitric oxide, autonomic activity, and cardiovascular risk. J Clin Invest 2007;117:2658–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 2004;24:413–420. [DOI] [PubMed] [Google Scholar]
- 35.De Maria R, Campolo J, Frontali M, et al. Effects of sapropterin on endothelium-dependent vasodilation in patients with CADASIL: a randomized controlled trial. Stroke 2014;45:2959–2966. [DOI] [PubMed] [Google Scholar]
- 36.Gan Y, Liu Q, Wu W, et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. Proc Natl Acad Sci USA 2014;111:2704–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet 2001;29:306–309. [DOI] [PubMed] [Google Scholar]
- 39.Liu YJ, Papasian CJ, Liu JF, Hamilton J, Deng HW. Is replication the gold standard for validating genome-wide association findings? PLoS One 2008;3:e4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carty CL, Keene KL, Cheng YC, et al. Meta-analysis of genome-wide association studies identifies genetic risk factors for stroke in African Americans. Stroke 2015;46:2063–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.