

Abstract
The natural product α-hydroxytropolones manicol and β-thujaplicinol inhibit replication of herpes simplex viruses 1 and 2 (HSV-1 and HSV-2, respectively) at nontoxic concentrations. Because these were originally developed as divalent metal-sequestering inhibitors of the ribonuclease H activity of HIV-1 reverse transcriptase, α-hydroxytropolones likely target related HSV proteins of the nucleotidyltransferase (NTase) superfamily, which share an “RNase H-like” fold. One potential candidate is pUL15, a component of the viral terminase molecular motor complex, whose C-terminal nuclease domain, pUL15C, has recently been crystallized. Crystallography also provided a working model for DNA occupancy of the nuclease active site, suggesting potential protein–nucleic acid contacts over a region of ∼14 bp. In this work, we extend crystallographic analysis by examining pUL15C-mediated hydrolysis of short, closely related DNA duplexes. In addition to defining a minimal substrate length, this strategy facilitated construction of a dual-probe fluorescence assay for rapid kinetic analysis of wild-type and mutant nucleases. On the basis of its proposed role in binding the phosphate backbone, studies with pUL15C variant Lys700Ala showed that this mutation affected neither binding of duplex DNA nor binding of small molecule to the active site but caused a 17-fold reduction in the turnover rate (kcat), possibly by slowing conversion of the enzyme–substrate complex to the enzyme–product complex and/or inhibiting dissociation from the hydrolysis product. Finally, with a view of pUL15-associated nuclease activity as an antiviral target, the dual-probe fluorescence assay, in combination with differential scanning fluorimetry, was used to demonstrate inhibition by several classes of small molecules that target divalent metal at the active site.
Graphical abstract
Herpes simplex viruses 1 and 2 (HSV-1 and HSV-2, respectively) are closely related enveloped alphaherpes-viruses with large, double-stranded DNA genomes encoding ∼80 proteins. Etiologically, HSV-1 is associated with gingivostomatitis, herpetic stromal keratitis, and anogenital lesions, while HSV-2, the primary agent of ulcerative anogenital lesions, infects ∼20% of individuals in the United States and has been documented to increase the frequency of human immunodeficiency virus (HIV) acquisition.1 DNA polymerase has been the primary HSV antiviral target, exemplified by the deoxyguanosine analogue acyclovir (ACV), the drug of choice for almost 40 years.2 However, an increased incidence of ACV-refractory HSV keratitis, a leading cause of corneal morbidity in industrialized countries, has been reported.3 Recent reports from immunocompetent patients with recurrent herpetic keratitis receiving ACV treatment indicate that ACV resistance may be more common that previously recognized in this particular population.4 The emergence of viral resistance to this class of compounds thus suggests a need for alternative therapies with increased efficacy and improved pharmacokinetics.5 Current examples include the HIV-1 protease inhibitor nelfinavir, which Kalu et al. showed inhibits late-stage virion envelopment and release.6 Inhibition of the human cytomegalovirus kinase UL97 by maribavir7 suggests that its HSV ortholog, UL13, might also be targeted. The helicase-primase inhibitor pritelivir also provides a new class of antiviral compounds distinct from those in current use.8 Finally, Krawczyk et al. have suggested the use of humanized antibodies to counter drug-resistant HSV.9 On the basis of the effectiveness of combination antiretroviral therapy in treating HIV infection, a cocktail of agents for HSV showing acceptable pharmacodynamic profiles and distinct molecular targets would be expected to significantly reduce viral load and delay acquisition of drug resistance.
Tavis et al. recently reported inhibition of wild-type and ACV-resistant HSV-1 and HSV-2 replication by several natural product troponoids, including manicol and β-thujaplicinol.10 Although the viral target was not established, α-hydroxytropolones were previously shown to potently inhibit ribonuclease H (RNase H) activity of HIV-1 reverse transcriptase (RT) by sequestering the catalytically critical divalent metal at the active site.11–13 Thus, a structurally related HSV enzyme seems to be a plausible HSV target. As an example, infected cell protein 8 (ICP8), a DNA binding protein required for HSV replication, belongs to the superfamily of NTases sharing an “RNase H-like” fold,14 and Yan et al. have shown that raltegravir, an inhibitor of HIV-1 integrase (IN), another NTase, blocks replication of alpha-, beta-, and gammaherpesviruses.15 A second HSV candidate, pUL15, is a component of the terminase molecular motor complex that is responsible for mobilizing viral DNA into the capsid. The pUL15 C-terminal nuclease domain (pUL15C) uses a two-metal ion-mediated catalytic mechanism to cleave and package concatemeric DNA into the virus capsid,16 and its recently reported crystal structure17 also highlighted the RNase H-like fold common to NTases. Modeling studies with pUL15C17 suggested a binding cleft that could accommodate ∼14 bp of duplex DNA, within which residues Arg517, Arg695, Lys700, and Lys701 were proposed to interact with the phosphate backbone. Verifying features of the active site, as well as investigating whether pUL15 nuclease activity is a therapeutically accessible target, requires developing substrates that allow kinetic evaluation and at the same time can be adapted to a high-throughput screening (HTS) format. Use of the UL15 C-terminal domain for such studies is supported by data of Nadal et al., who used a similar domain of human cytomegalovirus (HCMV), UL89 (designated UL89-C), to demonstrate its sensitivity to raltegravir.18 However, the previous methodology for assaying nuclease activity required cleavage of supercoiled DNA and agarose gel electrophoresis, which is time-consuming, challenging to quantify, and impractical for HTS.
Here, we have used several short, closely related oligonucleotide duplexes (∼21 bp) to interrogate pUL15C nuclease activity, showing that relatively minor changes can affect both the kinetics and specificity of cleavage. Features of this assay were then incorporated into a simple and rapid dual-probe fluorescence assay that permitted a detailed kinetic evaluation. The dual-probe assay also demonstrated that a single Lys700Ala mutation severely reduced pUL15C nuclease activity, possibly by slowing its release from the product of duplex cleavage. Finally, by combining differential scanning fluorimetry (DSF) with the dual-probe fluorescence assay, we propose two facile screening tools for an HSV nuclease that can be extended to the orthologs of other herpesviruses.
Experimental Procedures
General Experimental
All reagents were purchased from commercial sources and used without further purification. Recombinant pUL15C was prepared as previously reported.17 α-Hydroxytropolones were dissolved in dimethyl sulfoxide (DMSO) and diluted in the same solvent. Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) with the exception of the dual-labeled HTS oligonucleotide, which was purchased from Biosearch Technologies (Petaluma, CA).
Assaying pUL15C Nuclease Activity on Defined DNA Duplexes
Cy5-labeled duplex DNAs were prepared by annealing a 5′ Cy5-labeled 21-nucleotide DNA oligonucleotide (Cy5-21D; 5′-ATGTATTTAGGATTGGGACTT-3′) to the following complementary strands: 33D, 5′-CACTGCTCAAGAAGTTCCAATCCTAAATACATA-3′; 28D1, 5′-CTCAAGAAGTTCCAATCCTAAATACATA-3′; 28D2, 5′-AAGTTCCAATCCTAAATACATACACTGG-3′; 22D, 5′-AAGTTCCAATCCTAAATACATA-3′; 16D1, 5′-CAATCCTAAATACATA-3′; 16D2, 5′-CCAATCCTAAATACAT-3′; 16D3, 5′-AAGTTCCAATCCTAAA-3′; 15D, 5′-CAATCCTAAATACAT-3′; and 14D, 5′-AATCCTAAATACAT-3′. This yielded duplexes 21D/ 33D, 21D/28D1, 21D/28D2, 21D/22D, 21D/16D1, 21D/ 16D2, 21D/16D3, 21D/15D, and 21D/14D. Annealing was performed by heating a mixture containing 10 μM Cy5-21D and complementary DNA (10 μM each) in 10 mM Tris-HCl (pH 7.6) and 25 mM NaCl to 80 °C and slowly cooling it to 4 °C. Nuclease digestion was initiated by incubating 0.1 μM duplex DNA in a solution of 20 mM Tris-HCl (pH 7.0), 1 mM MnCl2, 10 mM NaCl, and 0.3 μM pUL15C at 37 °C and terminated after 5 min by adding an equal volume of 8 M urea. Hydrolysis products were fractionated by 15% denaturing urea polyacrylamide gel electrophoresis [19:1 acrylamide:bis-(acrylamide)] and visualized by fluorescent imaging (Typhoon Trio+, GE Healthcare).
Dual-Probe Fluorescence Assay of pUL15C Nuclease Activity
The substrate for the dual-probe assay was a doubly labeled 42-nucleotide DNA oligonucleotide, Q670-42D-BHQ2plus, 5′-Q670-TATGTATTTAGGATTGGGATTATACCCAATCCTAAATACATA-BHQ2plus-3′, which forms a duplex structure with an intervening hairpin. Denaturation and reannealing were performed by heating a mixture containing 10 μM Q670-42D-BHQ2plus, 10 mM Tris-HCl (pH 7.6), and 25 mM NaCl to 80 °C and slowly cooling it to 4 °C. To a 96-well plate was added 1 μL of each inhibitor, followed by 89 μL of reaction buffer containing the substrate. Hydrolysis was initiated by adding 10 μL of 3 μM pUL15C. Final assay conditions were 20 mM Tris-HCl (pH 7.0), 10 mM NaCl, 1 mM MnCl2, 1% DMSO, 0.3 μM pUL15C, 250 nM DNA duplex, and increasing inhibitor concentrations. Wells containing only DMSO were used as a control. Plates were incubated at 37 °C in an Infinite M1000 PRO plate reader (Tecan) for 10 min, and fluorescence (λex = 646 nm; λem = 670 nm) was measured at 1 min intervals such that linear initial rates could be measured in the presence (νi) and absence (νo) of inhibitor. Percent inhibition was calculated as 100(νo – νi)/νo and plotted against log[I]. IC50 values were calculated using Prism6. All assays were performed in triplicate.
Differential Scanning Fluorimetry (DSF, Thermo-Fluor)
Thermal stability assays were performed according to the method of Nettleship et al.19 To each well of a LightCycler480 96-well plate (Roche) was added 1 μL of a 1 mM solution of each compound in DMSO, followed by 49 μL of DSF buffer containing 1.6 μM pUL15C, 20 mM HEPES (pH 7.5), 10 mM MgCl2, 100 mM NaCl, and a 1:2000 dilution of SYPRO Orange dye (Invitrogen). The mixture was heated from 30 to 80 °C in increments of 0.2 °C. The fluorescence intensity was measured using excitation and emission wavelengths of 483 and 568 nm, respectively. Compound-induced changes in thermal stability (ΔTm) were analyzed using LightCycler 480 Software. All assays were performed in duplicate.
Michaelis–Menten Kinetic Analysis
Inhibitory activities of compound 21 and raltegravir were measured with the dual-probe fluorescence assay in the presence of varying concentrations of substrate (50–2000 nM). Concentrations of compound 21 and raltegravir used were 0, 0.1, 0.2, and 0.4 μM and 0, 0.5, 1.0, and 2.0 μM, respectively. Wells containing only DMSO were used as a control. Initial velocities were obtained at an enzyme concentration (0.3 μM) that ensured a linear relationship between relative fluorescence unit (RFU) and incubation time. Kinetic parameters were determined using Prism6. Lineweaver–Burk plots were constructed by plotting reciprocals of initial velocity against the reciprocal of substrate concentration. All assays were performed in triplicate.
α-Hydroxytropolone IC50 Determination by Agarose Gel Electrophoresis
pUL15C nuclease activity was initiated by incubating 400 ng of supercoiled pCMV-Kan plasmid (a generous gift of B. Felber, National Cancer Institute, Bethesda, MD) in 10 μL of buffer containing 0.12 μM pUL15C, 20 mM Tris-HCl (pH 7.0), 1 mM MnCl2, 10 mM NaCl, 10% DMSO, and increasing concentrations of inhibitor at 37 °C and terminated after 10 min with 5 μL of 100 mM EDTA. Hydrolysis products were separated by 0.8% agarose gel electrophoresis and visualized by ethidium bromide staining. The proportion of supercoiled DNA breakage was quantified using ImageQuant (GE Healthcare), and IC50 values were calculated using Prism6 (GraphPad Software).
Yonetani–Theorell Analysis.20
To determine whether the binding sites for compound 21 and raltegravir overlap, nuclease activity was measured in the presence of varying concentrations of compound 21 (0–0.4 μM) and raltegravir (0–2.0 μM). Data analysis was performed using Prism6, and the following equation was used:
where νij is the enzyme velocity in the presence of both compounds at concentrations [I] and [J] and γ is the interaction term that defines the degree to which binding of one compound perturbs binding of the other. All assays were performed in triplicate.
Order-of-Addition Analysis
The inhibitory activity of compound 21 was evaluated under conditions where the overall composition of the reaction mixture was unchanged, but the order in which individual components (enzyme, divalent metal, nucleic acid, and inhibitor) were mixed was varied. Preincubation was conducted at room temperature for 10 min. Reaction mixtures were incubated at 37 °C in an Infinite M1000 PRO plate reader for 20 min, and fluorescence was measured at 1 min intervals.
α-Hydroxytropolone Synthesis
α-Hydroxytropolones 1–10 and 12–20 in Figure 2b were synthesized and characterized as previously described.21–23 Synthesis of compounds 11 and 21 is provided in the Supporting Information.
Figure 2.
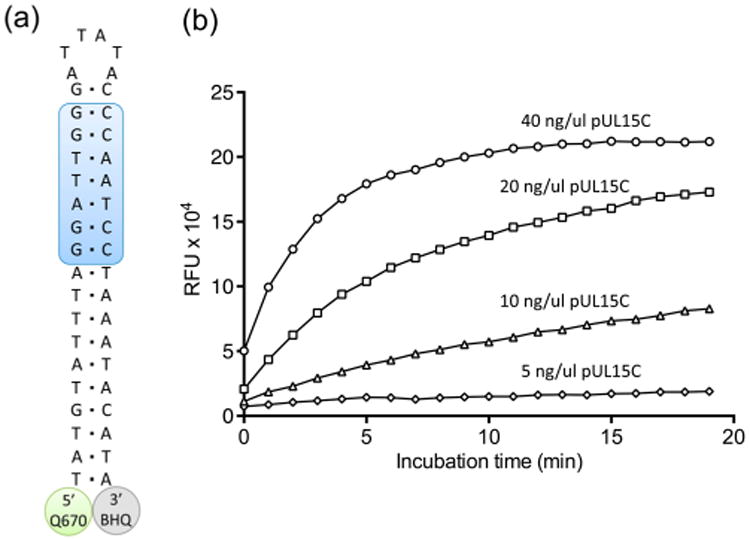
(a) Design of a dual-probe fluorescence substrate to evaluate pUL15C nuclease activity. The shaded region of the duplex represents cleavage sites defined by the substrate analysis of Figure 1. Abbreviations: Q670, Quasar 670 fluorescence donor; BHQ, Black Hole Quencher 2 Plus. (b) pUL15C-mediated cleavage of the dual-probe substrate as a function of increasing enzyme concentration.
Results
Defining a Minimal Substrate Length for pUL15C
Nonspecific cleavage of supercoiled DNA and fractionation of the hydrolysis products by agarose gel electrophoresis has been the standard method for studying the nuclease domains of herpesvirus terminases.17,18 However, because our goal was biochemical characterization of HSV pUL15C as a prerequisite to developing an HTS platform, defining a simple minimal-length substrate was important. In this respect, modeling studies with pUL15C17 propose the substrate binding cleft accommodates ∼14 bp of duplex DNA. With respect to cleavage specificity, pac motifs, which contribute to the genome cleavage/packaging process, have been characterized as containing adjacent G:C and A:T blocks.24 We therefore used this information to design the 21-nucleotide/33-nucleotide DNA duplex of Figure 1 whose lower, 33-nucleotide strand either varied in length or relocated the position of the single-stranded overhang.
Figure 1.
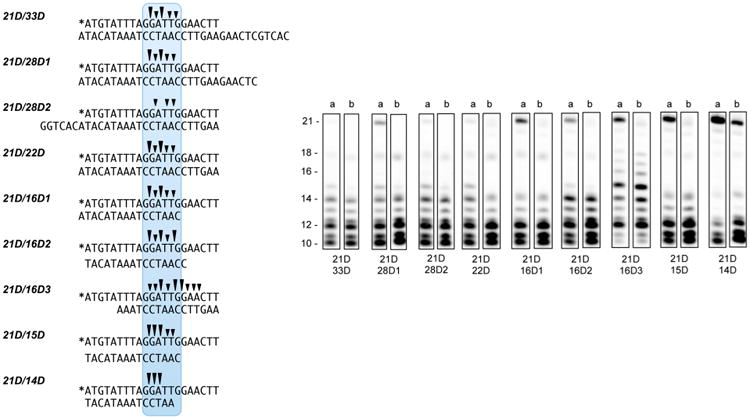
pUL15C cleavage of a 21-nucleotide/33-nucleotide DNA duplex and variants thereof. Sequence of duplex DNA substrates (left). Asterisks denote the positions of the Cy5 label. Cleavage patterns of specificity (right), following incubation with pUL15C for 5 and 30 min (lanes a and b, respectively). A summary of the primary cleavage sites (trianges) is illustrated above each duplex sequence.
pUL15C-mediated digestion of the test Cy5-21D/33D duplex yielded major products of 10 and 12 nucleotides and smaller amounts of 11-, 13-, and 14-nucleotide products, indicative of cleavage within the 5′-G-G-A-T-T-G-G-3′ sequence (Figure 1). Altering the length of the single-stranded overhang at the 5′ terminus (duplex 21D/28D1) or repositioning the overhang to the 3′ terminus (duplex 21D/ 28D2), while retaining a 21 bp duplex, did not affect the specificity of cleavage, although a slight decrease in cleavage kinetics was observed with 21D/28D1. A similar observation was made with duplex 21D/22D, suggesting terminal overhangs do not significantly contribute to substrate binding. In contrast, we observed significant alterations in cleavage kinetics as the length of the duplex region was progressively reduced. The lower strands of duplexes 21D/16D2, 21D/15D, and 21D/14D share the same 3′ termini, but their 5′ termini were deleted such that duplex length was reduced to 16, 15, and 14 bp, respectively. Consequently, the upper strand 5′-G-G-A-T-T-G-G-3′ motif of 21D/16D2 is fully base-paired but reduced to 5′-G-G-A-T-T-G-3′ and 5′-G-G-A-T-T-3′ on 21D/15D and 21D/14D, respectively. With duplexes 21D/15D and 21D/ 14D, we observed a gradual increase of the amount of uncleaved duplex, which was most pronounced with 21D/14D. These results indirectly suggest that a minimal duplex length of 14–15 bp is required for optimal positioning within the pUL15C active site. This notion of a minimal substrate size was strengthened by observations with duplex 21D/16D1. Although the lower-strand oligonucleotide is 16 nucleotides in length, it provided a 15 bp duplex together with a single-nucleotide 3′ overhang. As a result, hydrolysis kinetics of 21D/ 16D1 were also slightly reduced. In the model of pUL15C complexed with duplex DNA,17 two hairpins, comprising residues 620–633 and 684–704, are separated by ∼13 bp, which lends support to the data depicted in Figure 1. Finally, substrate 21D/16D3 maintains a duplex length of 16 bp but relocates this such that its 5′ terminus is blunt-ended. In this case, cleavage kinetics were reduced and accompanied by a relaxed hydrolysis profile, yielding products ranging from 10 to 18 nucleotides in length. Despite prolonged cleavage, levels of the 10- and 11-nucleotide product were significantly reduced on this substrate. Supplementary Figure 1 provides a similar analysis, but one performed on duplexes whose lower strand was 5′ 32P end-labeled. For duplex 21/33D*, cleavage was restricted primarily to the complementary 3′-C-C-T-A-A-C-C-5′ motif. Although cleavage kinetics of duplex 21/22D* were slightly reduced, a similar observation was made for duplexes 21/28D1*, 21/28D2*, and 21/22D*, suggestive of double-stranded scission throughout this 7 bp motif. Cleavage kinetics of duplexes 21/16D*, 21/15D*, and 21/14D* were again reduced, with specificity centering around the 3′-T-C-C-5′ sequence, supporting our contention that efficient pUL15C recognition requires duplex DNA longer than this minimal length. The rationale for enzyme “slippage” and broadened cleavage specificity is not immediately clear.
A Dual-Probe Fluorescence Assay of pUL15C Nuclease Activity
As discussed earlier, cleavage of supercoiled DNA followed by agarose gel electrophoresis has been used to evaluate nuclease activity of pUL15C and related terminases.17,18 As the significance of herpesvirus capsid assembly proteins as therapeutic targets grows,25 a gel-based approach to identifying small molecule antagonists would clearly be impractical. We therefore elected to take advantage of the duplex cleavage data depicted in Figure 1 to design a “minimal” substrate for pUL15C that would allow detailed kinetic evaluation and provide a simple HTS substrate.
Attempts to locate a fluorescence donor/quencher pair at either end of an 18 bp duplex were unsuccessful, possibly reflecting their separation distance or interaction with the viral nuclease (data not shown). Consequently, we designed the 42-nucleotide single-stranded DNA of Figure 2a containing adjacent G:C- and A:T-rich blocks. We elected to retain the neighboring A:T-rich portion of the duplex, reasoning that it separated the pUL15C recognition site from bulky modifications introduced at the DNA termini. These modifications included the indocarbocyanine Quasar 670 (Q670), which fluoresces in the red region of the visible spectrum, at the 5′ terminus and Black Hole Quencher 2 Plus (BHQ2plus) at the 3′ terminus. Following hydrolysis, we reasoned that residual duplex DNA containing the Q670/BHQ2plus pair would spontaneously dissociate at room temperature, allowing us to monitor hydrolysis via relief from quenching and increased fluorescence intensity. Our hypothesis was borne out by data depicted in Figure 2b, performed at different pUL15C concentrations. As a control, the signal from a complete DNase I digestion of the substrate was compatible with that achieved with pUL15C at 40 ng/μL (1.2 μM), showing that the reaction proceeded to completion (data not shown). To eliminate the possibility of low-level DNase I contamination, we exploited our observation that pUL15C was considerably more active in the presence of Mn2+ than in the presence of Mg2+. In Supplementary Figure 2, we compared a Mg2+-dependent pUL15C digest of substrate 21D/33D (lanes a–c) with that catalyzed by DNase I under the same condition (lanes d and e). The absence of hydrolysis products with the former despite prolonged incubation supports the notion that activity observed with the 21D/33D duplex indeed derived from pUL15C. In support of this, and in contrast to pUL15C, cleavage of substrate 21D/33D by DNase I in the presence of 1 mM Mn2+ was insensitive to inhibition by β-thujaplicinol at a concentration of 20 μM (Supplementary Figure 2b,c).
Characterization of Wild-Type and Mutant pUL15C
Modeling of duplex DNA into the active site of pUL15C suggests “loop 4” residues Arg695, Lys700, and Arg701 are sufficiently close to the phosphate backbone to engage in direct contact.17 To investigate this role for Lys700, the pUL15C variant containing a Lys700Ala mutation was purified and analyzed. We first assessed the integrity of wild-type and mutant protein by DSF (ThermoFluor), which monitors binding of the environmentally sensitive dye SYPRO orange as a function of temperature, providing a melting temperature (Tm).19 As HSV pUL15C belongs to the superfamily of NTases, which includes HIV RNase H, alterations to the thermal stability of both proteins were also determined in the presence of the RNase H active site inhibitor β-thujaplicinol.11 Because Lys700 does not constitute an active site residue, we expected both proteins to behave similarly in the absence and presence of the active site ligand, a notion that was confirmed by data depicted in panels a and b of Figure 3. In the absence of β-thujaplicinol, DSF yielded Tm values of 55.72 and 56.11 °C for wt and mutant protein, respectively, while incubation in the presence 20 μM β-thujaplicinol stabilized both proteins, increasing the Tm of the wt enzyme by 4.69 °C and that of the Lys700Ala mutant by 4.30 °C. Thus, to a first approximation, DSF confirmed that the active site architecture of Lys700Ala pUL15C was not compromised.
Figure 3.
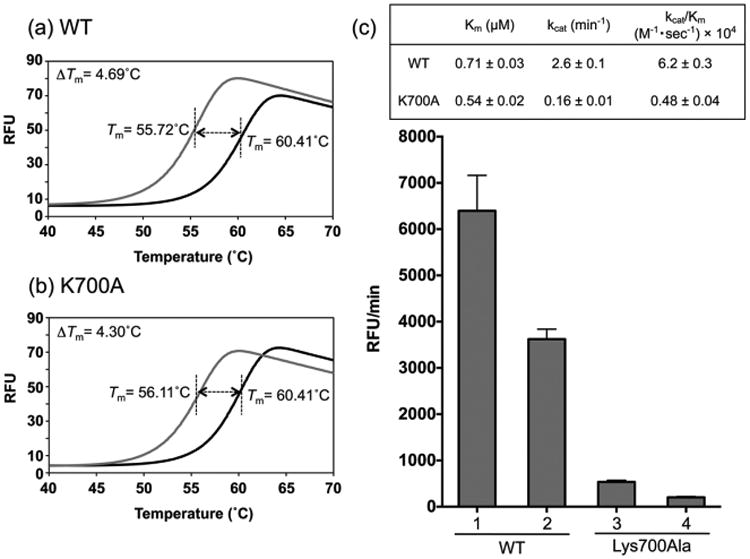
Charactarization of wild-type and Lys700Ala pUL15C. Differential scanning fluorimetry analysis of (a) wild-type and (b) mutant enzymes in the absence (gray trace) and presence of 20 μM β-thujaplicinol (black trace). (c) Duplex DNA cleavage activity and kinetic analysis of pUL15C using the dual-probe fluorescence assay. Enzyme activity was determined at protein concentrations of 0.6 μM (columns 1 and 3) and 0.3 μM (columns 2 and 4). Km, kcat, and kcat/Km values for both are provided in the inset.
We next used the dual-probe fluorescence assay to kinetically characterize the wild-type and mutant nucleases. As shown in Figure 3c, the activity of Lys700Ala pUL15C was reduced to ∼8% of that of the wild-type enzyme. Interestingly, Km values for both proteins were not significantly different (0.71 ± 0.03 μM for wt and 0.54 ± 0.02 μM for the Lys700Ala mutant), suggesting that loss of activity was not a result of a reduced affinity for substrate. In contrast, ∼17- and ∼13-fold differences were observed in the turnover number (kcat values of 2.6 ± 0.1 min−1 for wt and 0.16 ± 0.01 min−1 for Lys700Ala) and the catalytic efficiency [kcat/Km values of (6.2 ± 0.3) × 104 M−1 s−1 for wt and (0.48 ± 0.04) × 104 M−1 s−1 for Lys700Ala], respectively. Possible explanations for these differences are that altering Lys700 decreases the rate of conversion of the enzyme–substrate complex to the enzyme–product complex and/or affects the off rate of the mutant enzyme from the cleaved duplex.
α-Hydroxytropolone Inhibition of pUL15C
Tavis et al. have demonstrated that the natural product α-hydroxytropolones manicol and β-thujaplicinol suppress replication of wild-type and ACV-resistant HSV-1 and HSV-2 at noncytotoxic concentrations, illustrating their antiviral potential.10 On the basis of our interest in developing inhibitors directed toward a broader class of viral nucleotidyltransferases,26 the 21 novel β-thujaplicinol derivatives of Figure 4, substituted on their heptatriene ring while the metal binding pharmacophore is preserved, were synthesized. For comparative purposes, we initially monitored pUL15C inhibition by a subset of these compounds via agarose gel electrophoresis, the standard method of choice. This approach (Figure 5) yielded IC50 values ranging from 0.61 μM (compound 6) to 7.3 μM (compound 10), compared to 2.0 μM for the parent β-thujaplicinol. While agarose gel electrophoresis suggests that the bulkier substitution of compound 10 may sterically impede binding, quantification of such data is challenging, and its applicability to HTS is impractical. In contrast, the dual-probe fluorescence assay of Figure 6 circumvents these limitations. In this case, IC50 values ranged from 0.14 ± 0.01 μM (compound 5) to 49.1 ± 17.0 μM (compound 10). Although the submicromolar values obtained for compounds 1, 6, 14, and 21 were in keeping with data from agarose gel electrophoresis, there was significant variance in the absolute IC50 values, which we attributed to the difficulty in quantifying data from agarose gel electrophoresis. Likewise, the poor activity of compound 10 when analyzed by gel electrophoresis (IC50 = 7.3 μM) was confirmed by the dual-probe assay (IC50 = 49.1 ± 17.0 μM), although the absolute values differed almost 7-fold. Other possible reasons for these discrepancies might be the difference between using a large supercoiled DNA as opposed to a short duplex as a substrate, or the fact that supercoiled DNA provides significantly more pUL15C binding sites compared to the 15–20 bp substrates we have used.
Figure 4.
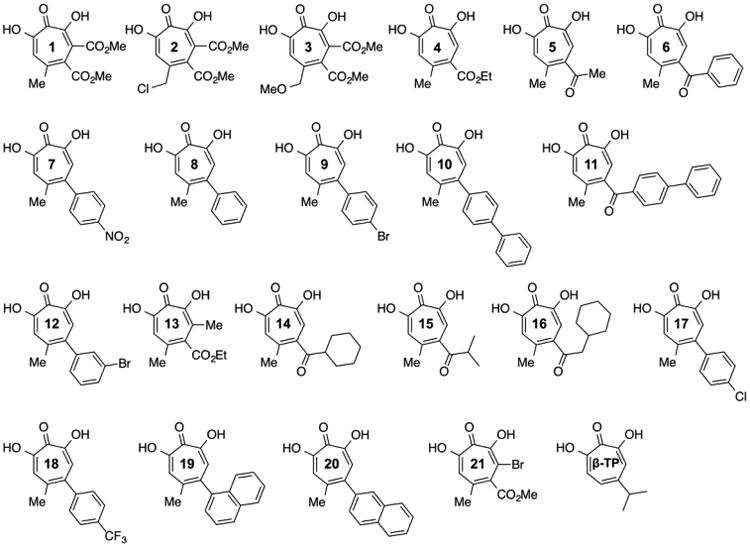
Structures of synthetic β-thujaplicinol derivatives. β-TP, β-thujaplicinol.
Figure 5.
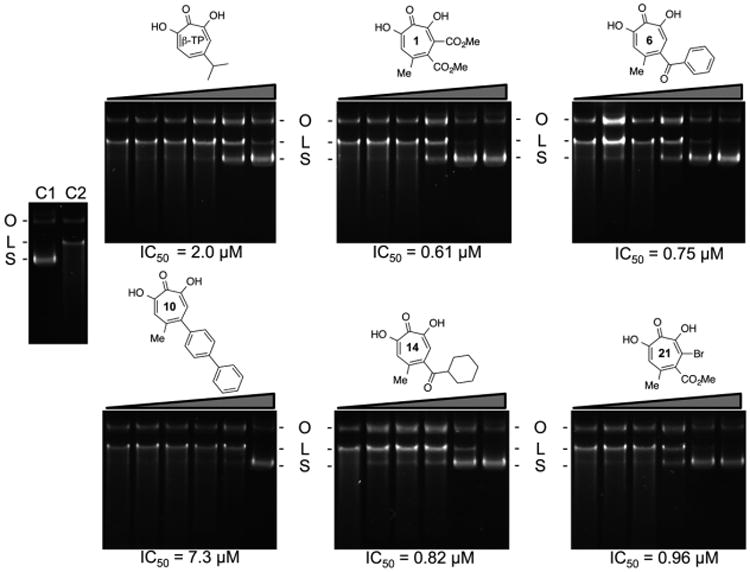
Inhibition of pUL15C nuclease activity by selected α-hydroxytropolones, analyzed by 1% agarose gel electrophoresis and ethidium bromide staining. Each compound was tested at a final concentration of 0.006, 0.032, 0.16, 0.80, 4.0, and 20.0 μM. The migration positions of supercoiled, open circular, and linear plasmid DNA are designated S, O, and L, respectively. Lane C1, no pUL15C; lane C2, pUL15C without inhibitor. IC50 values for all compounds are presented under their respective panel. β-TP, β-thujaplicinol.
Figure 6.

Analysis of α-hydroxytropolone inhibition of pUL15C nuclease activity using the dual-probe fluorescence assay. (a) Representative dose–response curves for β-thujaplicinol (Δ) and compound 21 (□). (b) Comparison of IC50 and ΔTm values of compounds 1–21 and β-thujaplicinol (β-TP).
Although the number of α-hydroxytropolones evaluated as pUL15C inhibitors may be too small to derive structure–activity relationships, a pattern emerges in which smaller ester or ketone substitutions at position 3 and/or 4 of the heptatriene ring (compounds 1–6, 13, 15, and 21) are significantly more potent than larger analogues (11 and 16) or those with direct aromatic substitutions (compounds 7–10 and 17–20). This trend suggests that steric problems associated with the latter impede the correct positioning of the metal binding pharmacophore at the nuclease active site. For those compounds with submicromolar activity, substituting position 5 of the heptatriene ring with -CH3 (1), -CH2Cl (2), and -CH2OCH3 (3) minimally affected the IC50, suggesting this position may be amenable to further modification to improve potency. Furthermore, the general trend of lower activity among those with direct aromatic substitution (7–10, 12, and 17–20) compared to the activity of those with ketones (5, 6, 11, and 14–16) or esters (1–4, 13, and 21) suggests that electronic effects of the substituents directly on the troponoid ring may be an important factor in determining potency. Indeed, comparing 4 with closely related compounds 1, 13, and 21 suggests that adding an electron-withdrawing group such as an ester (1) or bromide (21) directly to the tropolone enhances activity, whereas adding an electron-donating methyl group (13) decreases activity. Among analogues that have direct aromatic substitution (7–10, 12, and 17–20), compound 7, with a highly electron-withdrawing nitro group, is the most potent. Understanding the nuances of α-hydroxytropolone inhibition of pUL15C will require analysis of additional analogues, but the initial trends provide directions for further studies.
The ThermoFluor strategy that monitors protein stability in the presence of small molecule ligands is finding increased utility in drug discovery.27 As a complement to the IC50 values provided by the dual-probe fluorescence assay, we investigated how this correlated with ligand-induced alterations in pUL15C stability. As shown in Figure 6b, and consistent with reports on their interaction with retroviral RNase H,28 α-hydroxytropolones stabilized pUL15C against thermal denaturation. ΔTm values ranged from 2.35 °C (compound 10) to 8.70 °C (compound 21), which were among the least and most potent chemotypes, respectively. In general, we observed a good correlation between ΔTm and IC50 values, underscoring the value of differential scanning fluorimetry as a facile tool for ranking the efficacy of inhibitor binding. Mechanistically, an enhanced Tm may reflect decreased accessibility of the nucleic acid to the active site of the enzyme–inhibitor complex or stabilization of the enzyme–inhibitor–substrate complex in a configuration incompatible with hydrolysis.
Kinetic Analysis of pUL15C Inhibition
The robustness and reproducibility of our dual-probe fluorescence assay also allowed a kinetic evaluation of α-hydroxytropolone-mediated HSV pUL15C inhibition. The Lineweaver–Burke plot of Figure 7a suggests compound 21 is a noncompetitive inhibitor (Ki = 0.14 ± 0.01 μM), binding to the enzyme and the enzyme-substrate complex. This observation is significant because studies investigating α-hydroxytropolone-mediated inhibition of HIV-1 RNase H have indicated that binding to a preorganized enzyme-substrate complex is compromised, potentially limiting their therapeutic usefulness.29,30 Figure 7b presents a Yonetani–Theorell plot determined with compound 21 and the HIV-1 IN inhibitor raltegravir, a diketo acid that also sequesters divalent metal at the active site.31 The parallel nature of the plots indicates that the two molecules are mutually exclusive; i.e., they share a common binding site. Finally, Figure 8c examined whether the order in which enzyme (E), inhibitor (I), substrate (S), and divalent metal (M) were added affected pUL15C inhibition by compound 21. In the presence of an inhibitor, the rate of substrate degradation was unaffected regardless of the order of addition, confirming that the preformed enzyme–substrate complex could be accessed by the small molecule, in contrast to results with HIV-1 RNase H.
Figure 7.
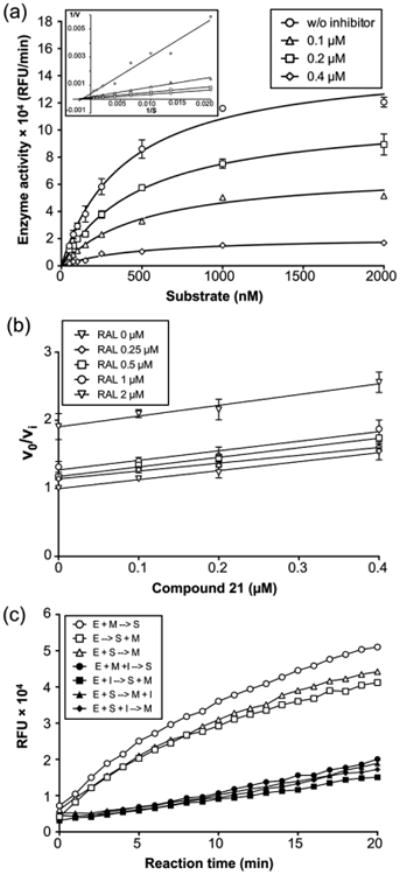
Kinetic analysis of pUL15C inhibition. (a) Michaelis–Menten kinetic plot of pUL15C nuclease activity in the presence of various compound 21 concentrations: 0.1 (△), 0.2 (□), and 0.4 μM (◇). Empty circles represent the no inhibitor control. The graph is representative of three independent experiments. A Lineweaver–Burke plot is presented in the inset, indicating compound 21 is a noncompetitive inhibitor. (b) Yonetani–Theorell plot for compound 21 and RAL. The inhibitory activity (defined as νo/νi) for each concentration of compound 21 (0, 0.1, 0.2, 0.4, and 0.8 μM) is plotted as a function of the concentration of RAL (0, 0.25, 0.5, 1, and 2 μM). The assay was performed at fixed concentrations of substrate (250 nM). Parallel lines for the two inhibitors indicate they are mutually exclusive. (c) Effect of the order of addition on the pUL15C activity of compound 21. The inhibitory activity of compound 21 was evaluated under seven different conditions, having the same composition but different order of addition of enzyme (E), substrate (S), Mn2+ (M), and inhibitor (I): preincubation of E and M, hydrolysis initiated with S (○); E alone, hydrolysis initiated with S and M (△); preincubation of E and S, hydrolysis initiated with M (□); preincubation of E, M, and I, hydrolysis initiated with S (●); preincubation of E and I, hydrolysis initiated with S and M (■); preincubation of E and S, hydrolysis initiated with M and I (▲); and preincubation of E, S, and I, hydrolysis initiated with M (◆). Preincubation was conducted at room temperature for 10 min. Reaction mixtures were incubated at 37 °C in an Infinite M1000 PRO plate reader for 20 min, and fluorescence was measured at 1 min intervals.
Figure 8.
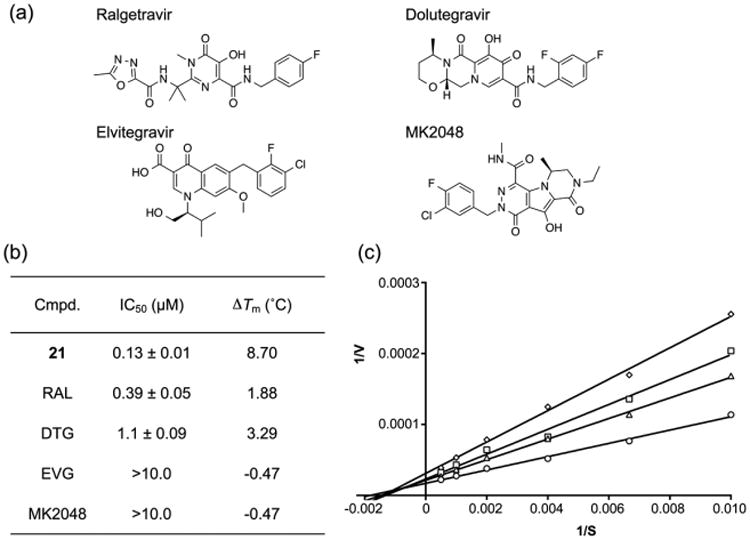
α-Hydroxytropolone vs diketo acid inhibition of pUL15C nuclease activity. (a) Chemical structures of HIV-1 IN inhibitors, raltegravir (RAL), dolutegravir (DTG), elvitegravir (EVG), and MK2048. (b) IC50 and ΔTm values of α-hydroxytropolone 21 and diketo acids. IC50 and ΔTm values are the average of triplicate and duplicate experiments, respectively. (c) Lineweaver–Burke plot of pUL15C nuclease activity in the presence of various RAL concentrations: 0.25 △, 0.5 (□), and 1.0 μM (◇). Empty circles represent the no inhibitor control. Nuclease activity of pUL15C was measured using the dual-probe fluorescence assay at varying substrate concentrations (100, 150, 250, 500, 1000, and 2000 nM). Similar intersection points on the x-axis in the absence and presence of RAL indicate noncompetitive inhibition. The graph is representative of three independent experiments.
Diketo Acid and Naphthyridinone Inhibition of pUL15C
In view of the common RNase H-like fold of herpesvirus NTases and HIV-1 IN, Nadal et al.18 and Yan et al.15 demonstrated inhibition of herpesvirus replication by different classes of diketo acid-based IN inhibitors. The dual-probe fluorescence assay therefore allowed us to make a direct comparison between these two inhibitor classes. For comparative analysis, we chose compound 21, which was one of the most potent α-hydroxytropolones. In the assay depicted in Figure 8a, its IC50 value of 0.13 ± 0.01 μM was slightly improved over that reported in Figure 6b. For diketo acids raltegravir (RAL) and dolutegravir (DTG), we calculated IC50 values of 0.39 ± 0.05 and 1.10 ± 0.09 μM, respectively. Figure 8b shows a Lineweaver–Burke plot determined in the presence of raltegravir, indicating that, similar to compound 21, it noncompetitively inhibits pUL15C (Ki = 1.0 ± 0.1 μM). Interestingly, data depicted in Figure 8b indicate that while these diketo acids also stabilized pUL15C against thermal denaturation, the magnitude of stabilization was significantly lower compared to that of compound 21. In contrast, elvitegravir (EVG) and MK2048, the latter of which is a second-generation diketo acid that is reportedly superior to raltegravir,32 were significantly less potent, having IC50 values in excess of 10 μM. Of note, these poorly active diketo acids had a destabilizing effect on pUL15C, in both instances decreasing the Tm by ∼0.5 °C.
Finally, the metal sequestering properties of naphthyridinone-based compounds have led to their investigation as inhibitors of HIV-1 RNase H and IN,22,33 as well as their potential repurposing as anticancer agents.34 We therefore tested this inhibitor class against pUL15C, the results of which are shown in Figure 9. IC50 values varied more that 40-fold, ranging from 0.12 ± 0.002 μM (compound 23) to 5.2 ± 0.4 μM (compound 25). While the requirement for the C4 OH remains to be validated, a clear pattern that emerges is reduced potency when it is replaced by bulkier substituents. Interestingly, and consistent with the data depicted in Figures 4 and 6, introducing an ester at position C3 is well tolerated, suggesting this position of the naphthyridine ring could be further derivatized without compromising its metal sequestering properties. In summary, data depicted in Figures 6–9 provide a strong case for targeting the catalytically critical divalent metal cofactor of pUL15C, for which the dual-probe fluorescence and ThermoFluor assays described here will find utility.
Figure 9.
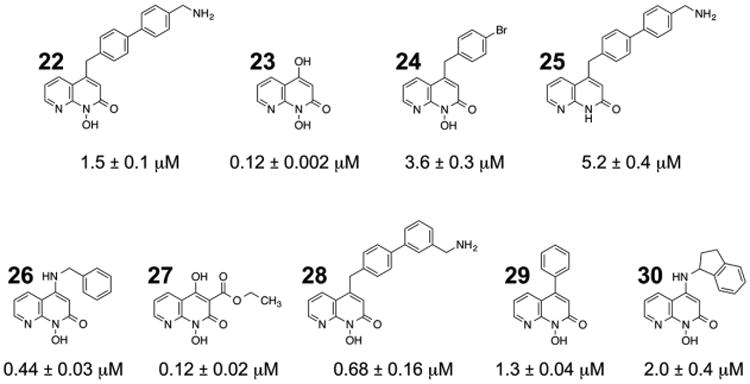
Napththyridinone inhibition of pUL15C nuclease activity. IC50 values are the average of triplicate experiments.
Discussion
In addition to the need for second-generation therapies with increased efficacy and improved pharmacokinetics for the treatment of drug resistance issues associated with HSV-1 and HSV-2 infections, a growing body of evidence suggests that HSV-2 infection can stimulate macrophages in vitro and induce HIV-1 replication in these cells. Developing a microbicide with efficient suppressive activity against HSV-2 for use in combination with an HIV-1 microbicide would therefore seem highly desirable. Progress in both areas would benefit from an improved understanding of herpesvirus proteins that mediate entry, genome replication, and capsid assembly. In this work, we have characterized pUL15C, the C-terminal nuclease domain of the viral terminase, with a view of targeting herpesvirus genome processing and packaging as an antiviral strategy. Because the parent protein, pUL15, and its homologues are highly conserved among all family members, small molecule antagonists evaluated here may have broader utility as antiviral agents for herpesvirus-associated disease.25
Central to our studies has been investigating substrate requirements for pUL15C; data depicted in Figure 1 illustrate the efficient cleavage of a “minimal” 14 bp duplex containing an A:T-rich segment flanked by G:C-rich segments. Although we must recognize that substrate length and/or sequence specificity may vary in the context of full-length pUL15, use of short duplexes such as those shown in Figure 1 allows alterations to sequence and/or structure to be analyzed by introducing targeted nucleoside analogue substitutions. Examples include (a) imposing increased rigidity or flexibility on the duplex (locked nucleic acids or pyrimidine isosteres, respectively), (b) charge neutralization via methylphosphonate linkages, or (c) removing nucleobases, leaving the sugar–phosphate backbone (abasic deoxyribosides). This approach has been successfully applied in analyzing substrate requirements of the reverse transcriptases of HIV-135,36 and the Saccharomyces cerevisiae LTR retrotransposon Ty3,37 as well as the cellular deaminase APOBEC3G.38 In the absence of a DNA-containing cocrystal, a nucleoside analogue strategy can provide important mechanistic details about the interaction of pUL15C with duplex DNA.
This possibility aside, an important outgrowth of our investigation has been development of a simple, inexpensive dual-probe fluorescence assay (Figure 2) for biochemical characterization of pUL15C as well as a robust HTS platform. Examples of the former are provided by kinetic analysis of the wild-type nuclease and a Lys700Ala mutant substituted at a residue implicated in contacting the DNA phosphate backbone, while use of the assay as an HTS tool is demonstrated by our investigation of α-hydroxytropolone, diketo acid, and naphthyridinone inhibition of pUL15C nuclease activity. The latter application of the dual-probe assay is particularly important, because cleavage of supercoiled DNA and fractionation of the products by agarose gel electrophoresis has been the general method of choice for studying the activity of herpesvirus nucleases. Adapting this or any related gel-based assay to an HTS format would present a significant practical obstacle, and comparison of the data depicted in Figures 5 and 6 shows that, for α-hydroxytropolones, the inhibitory trend observed by agarose gel electrophoresis is reproduced in the dual-probe fluorescence assay. Our fluorescence assay has been complemented by DSF, analyzing the effect of small molecule binding on pUL15C thermal stability. Data depicted in Figure 6 show that α-hydroxytropolone binding results in stabilization against thermal denaturation, with ΔTm values varying from 2.35 °C (compound 10) to 8.70 °C (compound 21). Equally important was the observation that ΔTm values correlate well with the inhibitory potency of these compounds (49.1 ± 17.0 μM for compound 10 vs 0.17 ± 0.002 μM for compound 21). Because DSF requires modest amounts of protein and utilizes common laboratory instrumentation, this provides a complementary, cost-effective alternative HTS strategy that should find use in evaluating related nucleases. Understanding the structural basis for ligand-induced stabilization, and its link to inhibitory potency, will require obtaining a cocrystal of pUL15C containing selected α-hydroxytropolones. Conceivably, this could occur through an increased number of contacts with divalent metal at the active site, providing a stabilizing effect on the protein while “freezing” its mobility, thereby interrupting catalysis.
Although we have evaluated a relatively small number of compounds, examining three structural classes of small molecules provides important insights into inhibition of pUL15 nuclease activity. For α-hydroxytropolones, relatively small substituents on the heptatriene ring appear to be most favorable, suggesting steric interference is caused by the bulkier substitutions. This notion can be extended to naphthyridinones, where bulkier aromatic substitutions again resulted in reduced potency. Although speculative, comparing IC50 values for compounds 23, 26, and 27 suggests that combining small substituents at positions C1 and C2 could provide increased potency. Finally, although our biochemical analysis suggests that a component of the viral terminase molecular motor is a target of α-hydroxytropolone inhibition in vitro, confirmation of this will require selection of a drug-resistant variant and mapping of the inactivating lesion. In this respect, Zhou et al. have documented mutations in 16 HSV-1 open reading frames following prolonged exposure to the HIV-1 IN inhibitor raltegravir.39 Of these, a Val296Ile mutation in UL42, encoding the DNA polymerase accessory factor, was sufficient to confer raltegravir resistance. Because crystallographic studies have not revealed the common RNase H-like fold of NTases for UL42, this could suggest an alternative mode for raltegravir-mediated HSV inhibition. At the same time, this suggests a cocktail of small molecule antagonists with divalent metal sequestering properties might provide an effective combination antiviral strategy.
Supplementary Material
Acknowledgments
We thank the Drug Synthesis & Chemistry Branch, DTP, NCI, for the supplies of β-thujaplicinol and manicol.
Funding: S.F.J.L.G., T.M., and J.A.B. are supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services. D.R.H., M.P.D., and C.M. are supported by National Institutes of Health Grant SC1GM111158 to R.P.M. H.Z. is supported by National Institutes of Health Grant R01GM090010 to L.T. J.B. is supported in part by National Institutes of Health Grant R01AI507421. A.G. was a recipient of a seed grant from the St. Louis University Department of Molecular Microbiology and Immunology.
Abbreviations
- DSF
differential scanning fluorimetry
- HSV
herpes simplex virus
- HIV
human immunodeficiency virus
- IN
integrase
- NTase
nucleotidyltransferase
- RAL
raltegravir
- RNase H
ribonuclease H
- HTS
high-throughput screening
- wt
wild type
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.5b01254.
pUL15C nuclease activity assay using 32P-labeled substrates (Supplementary Figure 1), nuclease activities in the presence of Mg2+ (Supplementary Figure 2), and experimental proceduers (Supplementay Methods) (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Schulte JM, Bellamy AR, Hook EW, 3rd, Bernstein DI, Levin MJ, Leone PA, SokolAnderson ML, Ewell MG, Wolff PA, Heineman TC, Belshe RB. HSV-1 and HSV-2 seroprevalence in the united states among asymptomatic women unaware of any herpes simplex virus infection (Herpevac Trial for Women) South Med J. 2014;107:79–84. doi: 10.1097/SMJ.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 2.Elion GB. Mechanism of action and selectivity of acyclovir. Am J Med. 1982;73:7–13. doi: 10.1016/0002-9343(82)90055-9. [DOI] [PubMed] [Google Scholar]
- 3.Remeijer L, Osterhaus A, Verjans G. Human herpes simplex virus keratitis: the pathogenesis revisited. Ocul Immunol Inflammation. 2004;12:255–285. doi: 10.1080/092739490500363. [DOI] [PubMed] [Google Scholar]
- 4.van Velzen M, van de Vijver DA, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J Infect Dis. 2013;208:1359–1365. doi: 10.1093/infdis/jit350. [DOI] [PubMed] [Google Scholar]
- 5.James SH, Prichard MN. Current and future therapies for herpes simplex virus infections: mechanism of action and drug resistance. Curr Opin Virol. 2014;8:54–61. doi: 10.1016/j.coviro.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Kalu NN, Desai PJ, Shirley CM, Gibson W, Dennis PA, Ambinder RF. Nelfinavir inhibits maturation and export of herpes simplex virus 1. J Virol. 2014;88:5455–5461. doi: 10.1128/JVI.03790-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prichard MN. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev Med Virol. 2009;19:215–229. doi: 10.1002/rmv.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biswas S, Sukla S, Goldner T, Field HJ, Kropeit D, Paulsen D, Welbers A, Ruebsamen-Schaeff H, Zimmermann H, Birkmann A. Pharmacokinetics-pharmacodynamics of the helicase-primase inhibitor pritelivir following treatment of wild-type or pritelivir-resistant virus infection in a murine herpes simplex virus 1 infection model. Antimicrob Agents Chemother. 2014;58:3843–3852. doi: 10.1128/AAC.02641-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krawczyk A, Arndt MA, Grosse-Hovest L, Weichert W, Giebel B, Dittmer U, Hengel H, Jager D, Schneweis KE, Eis-Hubinger AM, Roggendorf M, Krauss J. Overcoming drug-resistant herpes simplex virus (HSV) infection by a humanized antibody. Proc Natl Acad Sci U S A. 2013;110:6760–6765. doi: 10.1073/pnas.1220019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavis JE, Wang H, Tollefson AE, Ying B, Korom M, Cheng X, Cao F, Davis KL, Wold WS, Morrison LA. Inhibitors of nucleotidyltransferase superfamily enzymes suppress herpes simplex virus replication. Antimicrob Agents Chemother. 2014;58:7451–7461. doi: 10.1128/AAC.03875-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, Parniak MA, Crouch RJ, McMahon JB, Beutler JA, Le Grice SF. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, Dharia C, Clark AD, Jr, Ryan K, Hickey MJ, Love RA, Hughes SH, Bergqvist S, Arnold E. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 2009;17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung S, Himmel DM, Jiang JK, Wojtak K, Bauman JD, Rausch JW, Wilson JA, Beutler JA, Thomas CJ, Arnold E, Le Grice SF. Synthesis, activity, and structural analysis of novel alpha-hydroxytropolone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J Med Chem. 2011;54:4462–4473. doi: 10.1021/jm2000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majorek KA, Dunin-Horkawicz S, Steczkiewicz K, Muszewska A, Nowotny M, Ginalski K, Bujnicki JM. The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 2014;42:4160–4179. doi: 10.1093/nar/gkt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Z, Bryant KF, Gregory SM, Angelova M, Dreyfus DH, Zhao XZ, Coen DM, Burke TR, Jr, Knipe DM. HIV integrase inhibitors block replication of alpha-, beta-, and gammaherpesviruses. mBio. 2014;5:e01318–14. doi: 10.1128/mBio.01318-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgs MR, Preston VG, Stow ND. The UL15 protein of herpes simplex virus type 1 is necessary for the localization of the UL28 and UL33 proteins to viral DNA replication centres. J Gen Virol. 2008;89:1709–1715. doi: 10.1099/vir.0.2008/000448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, Tang L. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J Virol. 2013;87:7140–7148. doi: 10.1128/JVI.00311-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadal M, Mas PJ, Blanco AG, Arnan C, Sola M, Hart DJ, Coll M. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc Natl Acad Sci U S A. 2010;107:16078–16083. doi: 10.1073/pnas.1007144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nettleship JE, Brown J, Groves MR, Geerlof A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 2008;426:299–318. doi: 10.1007/978-1-60327-058-8_19. [DOI] [PubMed] [Google Scholar]
- 20.Yonetani T. The Yonetani-Theorell graphical method for examining overlapping subsites of enzyme active centers. Methods Enzymol. 1982;87:500–509. doi: 10.1016/s0076-6879(82)87028-6. [DOI] [PubMed] [Google Scholar]
- 21.Meck C, Mohd N, Murelli RP. An oxidopyrylium cyclization/ring-opening route to polysubstituted alpha-hydroxytropolones. Org Lett. 2012;14:5988–5991. doi: 10.1021/ol302892g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams YD, Meck C, Mohd N, Murelli RP. Triflic acid-mediated rearrangements of 3-methoxy-8-oxabicyclo[3.2.1]octa-3,6-dien-2-ones: synthesis of methoxytropolones and furans. J Org Chem. 2013;78:11707–11713. doi: 10.1021/jo401617r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirsch DR, Cox G, D'Erasmo MP, Shakya T, Meck C, Mohd N, Wright GD, Murelli RP. Inhibition of the ANT(2″)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic alpha-hydroxytropolones. Bioorg Med Chem Lett. 2014;24:4943–4947. doi: 10.1016/j.bmcl.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adelman K, Salmon B, Baines JD. Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc Natl Acad Sci U S A. 2001;98:3086–3091. doi: 10.1073/pnas.061555698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baines JD. Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol. 2011;19:606–613. doi: 10.1016/j.tim.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Lu G, Lomonosova E, Cheng X, Moran EA, Meyers MJ, Le Grice SF, Thomas CJ, Jiang JK, Meck C, Hirsch DR, D'Erasmo MP, Suyabatmaz DM, Murelli RP, Tavis JE. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity. Antimicrob Agents Chemother. 2015;59:1070–1079. doi: 10.1128/AAC.04617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huynh K, Partch CL. Analysis of protein stability and ligand interactions by thermal shift assay. Curr Protoc Protein Sci. 2015;79:28.9.1. doi: 10.1002/0471140864.ps2809s79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su HP, Yan Y, Prasad GS, Smith RF, Daniels CL, Abeywickrema PD, Reid JC, Loughran HM, Kornienko M, Sharma S, Grobler JA, Xu B, Sardana V, Allison TJ, Williams PD, Darke PL, Hazuda DJ, Munshi S. Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J Virol. 2010;84:7625–7633. doi: 10.1128/JVI.00353-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beilhartz GL, Ngure M, Johns BA, DeAnda F, Gerondelis P, Gotte M. Inhibition of the ribonuclease H activity of HIV-1 reverse transcriptase by GSK5750 correlates with slow enzyme-inhibitor dissociation. J Biol Chem. 2014;289:16270–16277. doi: 10.1074/jbc.M114.569707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beilhartz GL, Wendeler M, Baichoo N, Rausch J, Le Grice S, Gotte M. HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J Mol Biol. 2009;388:462–474. doi: 10.1016/j.jmb.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanco JL, Whitlock G, Milinkovic A, Moyle G. HIV integrase inhibitors: a new era in the treatment of HIV. Expert Opin Pharmacother. 2015;16:1313–1324. doi: 10.1517/14656566.2015.1044436. [DOI] [PubMed] [Google Scholar]
- 32.Leroy J. A Convenient Procedure for the Preparation of 3-Bromopropionic Esters. Synth Commun. 1992;22:567–572. [Google Scholar]
- 33.Johns BA, Kawasuji T, Weatherhead JG, Boros EE, Thompson JB, Koble CS, Garvey EP, Foster SA, Jeffrey JL, Fujiwara T. Naphthyridinone (NTD) integrase inhibitors: N1 protio and methyl combination substituent effects with C3 amide groups. Bioorg Med Chem Lett. 2013;23:422–425. doi: 10.1016/j.bmcl.2012.11.071. [DOI] [PubMed] [Google Scholar]
- 34.Zeng LF, Wang Y, Kazemi R, Xu S, Xu ZL, Sanchez TW, Yang LM, Debnath B, Odde S, Xie H, Zheng YT, Ding J, Neamati N, Long YQ. Repositioning HIV-1 integrase inhibitors for cancer therapeutics: 1,6-naphthyridine-7-carboxamide as a promising scaffold with drug-like properties. J Med Chem. 2012;55:9492–9509. doi: 10.1021/jm300667v. [DOI] [PubMed] [Google Scholar]
- 35.Dash C, Rausch JW, Le Grice SF. Using pyrrolo-deoxycytosine to probe RNA/DNA hybrids containing the human immunodeficiency virus type-1 3′ polypurine tract. Nucleic Acids Res. 2004;32:1539–1547. doi: 10.1093/nar/gkh307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rausch JW, Qu J, Yi-Brunozzi HY, Kool ET, Le Grice SF. Hydrolysis of RNA/DNA hybrids containing nonpolar pyrimidine isosteres defines regions essential for HIV type 1 polypurine tract selection. Proc Natl Acad Sci U S A. 2003;100:11279–11284. doi: 10.1073/pnas.1932546100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lener D, Kvaratskhelia M, Le Grice SF. Nonpolar thymine isosteres in the Ty3 polypurine tract DNA template modulate processing and provide a model for its recognition by Ty3 reverse transcriptase. J Biol Chem. 2003;278:26526–26532. doi: 10.1074/jbc.M302374200. [DOI] [PubMed] [Google Scholar]
- 38.Rausch JW, Chelico L, Goodman MF, Le Grice SF. Dissecting APOBEC3G substrate specificity by nucleoside analog interference. J Biol Chem. 2009;284:7047–7058. doi: 10.1074/jbc.M807258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou B, Yang K, Wills E, Tang L, Baines JD. A mutation in the DNA polymerase accessory factor of herpes simplex virus 1 restores viral DNA replication in the presence of raltegravir. J Virol. 2014;88:11121–11129. doi: 10.1128/JVI.01540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.