

Abstract
The primary objectives of this study were to evaluate the treatment effect of D-tagatose on glycemic control, determined by a statistically significant decrease in hemoglobin A1c (HbA1c), and safety profile of D-tagatose compared to placebo. The secondary objectives were to evaluate the treatment effects on fasting blood glucose, insulin, lipid profiles, changes in BMI, and the proportion of subjects achieving HbA1c targets of <7%. Type 2 diabetic patients not taking any blood glucose lowering medications were administered either 15 g of D-tagatose dissolved in 125–250 ml of water three times a day or placebo with meals. Reduction in HbA1c was statistically significant compared to placebo at all post-baseline time points in the ITT population. Additionally, secondary endpoints were achieved in the ITT population with regard to LDL, total cholesterol, fasting blood glucose, and proportion of subjects achieving HbA1c targets of <7%. D-tagatose was unable to lower triglycerides or raise HDL compared to placebo. A subgroup LOCF analysis on the ITT US population showed a greater and statistically significant LS mean reduction in HbA1c in the D-tagatose group at all post-baseline visits. Based on these results it is concluded that in the ITT population D-tagatose is an effective single agent at treating many of the therapy targets of type 2 diabetes including lowering fasting blood glucose and HbA1c, and lowering of LDL and total cholesterol.
Keywords: HbA1c, Triglycerides, Insulin, LDL, HDL, Cholesterol, Blood glucose
INTRODUCTION
According to the American Diabetes Association, diagnosed diabetes cost Americans $245 billion in 2012. This includes $176 billion for direct medical costs as well as $69 billion in reduction of productivity [1]. The National Diabetes Statistics Report, 2014, estimates that 29.1 million people, or 9.3% of the United States population, have diabetes (an estimated 8.1 million of these were undiagnosed) [2]. From 2009–2012, The Centers for Disease Control and Prevention determined that 37% of U.S. adults aged 20 years or older had prediabetes, as did 51% of adults aged 65 years or older (based on fasting glucose or hemoglobin A1c levels). Extrapolating this percentage to the entire U.S. population during that time, the prevalence of prediabetes can be estimated to be around 86 million American adults [2].
Two abnormalities that typically characterize the pathogenesis of type 2 diabetes are peripheral insulin resistance and progressive failure of pancreatic β-cell function leading to loss of insulin secretion. These complications eventually lead to chronic hyperglycemia and associated long-term disease complications [3]. Insulin resistance does not progress in parallel with loss of β-cell function, but occurs first. Most type 2 diabetes patients who are initially treated by diet management or oral blood glucose lowering agents eventually require insulin as loss of β-cell function progresses. None of the therapies currently available appear to be able to slow or stop the progression of β-cell loss once it begins. Therefore, there is clearly a need for therapies for the long-term treatment of type 2 diabetes that can prevent patients from reaching this current “point of no return” of loss of β-cell function.
Currently the approach to the treatment of type 2 diabetes is stepwise and systematic with early treatment consisting of having patients alter and manage their diet, adhere to a regular exercise program and, if overweight, control their weight. If the disease progresses and blood glucose remains uncontrolled, then pharmacologic therapy is initiated, usually with one or two oral anti-hyperglycemic drugs. Additional medications or insulin may be added if the disease continues to progress. Many, if not most, patients with type 2 diabetes will eventually require insulin as a primary therapy with or without adjuvant drug therapy in order to control their blood glucose [4].
Despite the introduction in recent years of new drugs for the treatment of diabetes, glucose control in many patients remains unsatisfactory. Metformin remains the first drug of choice for the treatment of type 2 diabetes. A recent review that included 140 controlled trials and 26 observational studies comparing diabetes medications, both as monotherapy and in two-drug combinations, concluded that the long-term benefits and harms of current drug treatments remain unclear [5]. The study concluded that there was not enough evidence to clearly support the use of one drug or drug combination over another for stemming the complications of diabetes, including macrovascular and microvascular complications, and for mortality. Metformin remains the initial drug of choice, however, because compared to the newer drugs it has the highest benefit-to-risk ratio for intermediate outcomes, such as moderate HbA1c reduction, less weight gain and less risk of hypoglycemia. Many of the other therapies have limiting side effects such as weight gain, hypoglycemia, and edema, and have restrictions for use [6].
This information illustrates the limitations of drug therapies currently available for the progression of diabetes. Further, even with aggressive intervention, it is estimated that 60% of diabetics do not achieve target blood sugar levels with their current treatment regimen [4]. Moreover, current drugs available for treatment of diabetes may result in unwanted weight gain (long and rapid acting Insulin, sulfonylureas, thiazolidinediones, repaglinide, nateglinide), hypoglycemia (insulin, sulfonylureas), gastrointestinal distress (metformin, α-glucosidase inhibitor, amylin mimetics, bile acid sequestrant, bromocriptine), or more serious adverse events such as pancreatitis (short and long-acting glucagon-like peptide-1 (GLP-1) agonists and dipeptidyl peptidase-4 (DDP-4) inhibitors) [7,8]. From the statistics showing a high percentage of diabetics are not able to consistently control their blood sugar levels within recommended limits, it is evident that there is a medical need for a drug that can slow and/or halt the progression of diabetes. Preferably, such a drug should exhibit a unique mode of action to enable additive or synergistic use with current therapies; produce no weight gain, hypoglycemia, or other limiting or unmanageable side effects; preserve or enhance β-cell function; and reduce cardiovascular risk factors that potentially lead to morbidity and mortality.
D-tagatose is an isomer of fructose that is ~90% as sweet as sucrose. In 2001 D-tagatose was designated as a Generally Recognized as Safe (GRAS) product by the United States Food and Drug Administration, and subsequently has been used as a nutritive or low-calorie sweetener [9]. Currently, D-tagatose may be used as a sweetener in diet beverages, light ice creams or yogurts, and regular or dietetic hard candies [10]. Subsequently it was hypothesized from observations in food use, and then demonstrated experimentally that, when consumed, D-tagatose functions as a “sugar blocker” and inhibits lipid formation from carbohydrates without stimulation of pancreatic beta cells for insulin production or secretion [11]. Preliminary animal and pre-clinical studies of D-tagatose demonstrated its ability to lower blood glucose and lipoprotein levels. D-tagatose has been shown to reduce total cholesterol and VLDL and LDL-cholesterol when compared to sucrose [12], and increase HDL-cholesterol levels [13]. A number of clinical trials demonstrating the ability of D-tagatose to blunt postprandial rises in blood glucose and reduce HbA1c have been conducted in both healthy subjects and diabetic patients [13–18]. A phase 2 study designed to estimate the lowest dose of D-tagatose capable of lowering HbA1c found that the dose was 5 g TID [18]. Single-dose and repeated-dose studies in healthy and diabetic human subjects have shown that the predominant adverse effects associated with excessive consumption of D-tagatose are gastrointestinal disturbances attributed to osmotic effects from incomplete absorption [13–17]. Such effects are also commonly associated with excessive consumption of other poorly digestible carbohydrates including polyols. In short, D-tagatose provides glycemic and lipoprotein control through a mechanism of action unlike any agent that is currently available on the market in the United States.
Here we report the results of a phase 3 clinical trial with D-tagatose and demonstrate statistically significant reductions in hemoglobin A1c levels (HbA1c) in patients with mild type 2 diabetes. HbA1c levels are an indicator of glycolated hemoglobin in the blood and reflect a patient’s glycemic control over a six to twelve week period [19]. If blood glucose levels have been elevated over recent weeks, there will likely be a concomitant rise in HbA1c levels.
The primary objective of this Phase 3 clinical trial was to evaluate the placebo-controlled effect of D-tagatose on glycemic control and safety in subjects with type 2 diabetes over the course of a 10-month treatment. The secondary objectives of this clinical trial were to evaluate the placebo-controlled effects of D-tagatose on fasting blood glucose, insulin, lipid profiles, and changes in BMI. D-tagatose effectively lowered HbA1c levels in type 2 diabetic patients compared to placebo; thus, results of this phase 3 clinical trial illustrate the potential for D-tagatose to fulfill a need in current diabetes treatment.
PATIENTS AND METHODS
Ethical Conduct of the Study
The protocol and protocol amendment(s) were reviewed and approved by an Institutional Review Board (IRB) before the study was initiated. This trial was conducted in accordance with regulations governing clinical trials including the US Code of Federal Regulations (CFR), Title 21, Part 50; regulations governing IRBs, Title 21, Part 56; and the Declaration of Helsinki concerning medical research in humans (Recommendations Guiding Physicians in Biomedical Research Involving Human Patients: adopted by the 18th World Medical Assembly (WMA), Helsinki, Finland, June 1964 and amended by the 29th WMA, Tokyo, Japan, October 1975, the 35th WMA, Venice, Italy, October 1983, the 41st WMA, Hong Kong, September 1989, the 48th WMA, Somerset West, Republic of South Africa, October 1996, and the 52nd WMA, Edinburgh, Scotland, October 2000). Additional governing regulations included US CFR Title 21, Part 54 and US CFR Title 21, Part 312. This study was also conducted according to International Conference on Harmonization (ICH) Good Clinical Practices (GCP).
Eligibility
Subjects were required to meet the following criteria for inclusion in this study: male or female between the ages of 18 and 75 diagnosed with type 2 diabetes (according to WHO criteria) who were being treated with diet and exercise alone, and not on any medication for diabetes; a HbA1c level at screening and baseline greater than 6.6% and less than 9.0%; a fasting glucose concentration less than 240 mg/dL (13.3 mmol/l); a BMI of less than or equal to 45 kg/m2; and a stable weight (±10%) for 3 months prior to entry into the study. Exclusion criteria included: treatment with any sulfonylureas, or other antidiabetic medications (e.g., thiazolidinediones, metformin, acarbose, exenatide, or insulin) within the prior 3 months; chronic (lasting longer than 14 consecutive days) systemic glucocorticoid treatment within 4 weeks of the baseline visit; use of any weight loss drugs within the prior 3 months; proliferative retinopathy; known or suspected abuse of alcohol or narcotics; any experience with hypoglycemic unconsciousness; impaired hepatic, renal or cardiac function; uncontrolled hypertension; pregnancy, breastfeeding, or intention of becoming pregnant or judged to be using inadequate contraception; documented gastrointestinal disease, or taking of medications to alter gut motility or absorption; and treatment with any investigational drug within 30 days of the screening visit.
Three populations were evaluated:
The Intent-to-Treat (ITT) population consisted of all randomized subjects who received at least one dose of their randomized treatment and had at least one post-treatment visit evaluating efficacy.
The Per Protocol (PP) population consisted of all ITT subjects who had at least 80% compliance with medication for 75% of the dosing time points and had no major protocol violations. Subjects who had a screening and visit 2 HbA1c value ≤ 6.6% and ≥ 9.0% and who were put on other diabetes medications were excluded from PP population.
The Safety population consisted of all randomized subjects who received at least one dose of their randomized treatment and had at least one post-treatment visit evaluating safety.
Treatment protocol
This was a Phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy, safety, and tolerability of D-tagatose.
Subjects were screened for eligibility using physical examination and clinical laboratory tests. The basic physical examination included physical measurements, general examination by observation (inspection), palpation, percussion, auscultation, blood pressure measure, and heart rate check. The clinical laboratory tests included: (1) hematology (hematocrit, hemoglobin, MCH, MCHC, MCV, total white blood cells, platelets, and differential); (2) clinical chemistry (sodium, chloride, potassium, carbon dioxide, BUN, uric acid, albumin, creatinine clearance, SGOT, SGPT, bilirubin (total and direct), phosphorus, calcium, alkaline phosphatase, total protein, and glucose (fasting); (3) HbA1c; (4) serum lipid profile including total cholesterol, HDL, LDL, and triglycerides; (5) urinalysis (appearance, volume, specific gravity, pH, glucose, protein, and microscopic evaluation of urinary sediment). Those determined to be provisionally eligible participated in an 8-week lead-in period prior to the start of the study during which diabetes education was provided and diet and exercise treatment stabilized.
Patients recorded their food intake and exercise in nutrition diaries. After the 8 week lead-in period, fasting (minimum of 8 hours) subjects returned to the study sites and underwent medical history review followed by baseline tests including ECG (or EKG), pregnancy test for females, and hematology, clinical chemistry, and urinalysis tests. The clinical laboratory tests were the same as those performed during the screening visit. Subjects continued on a weight-maintaining diet plus exercise under physician’s recommendation. In addition, subjects received their randomized study treatment and detailed instructions about its use. A total of 494 subjects were randomized into the study. Randomization was stratified according to screening HbA1c values (<7.5% and ≥7.5%) to achieve a balanced distribution of subjects across two arms (treatment and placebo).
The treatment period consisted of 12 monthly visits, the first (Visit 2, designated month 0 in figures) of which was used to gather the baseline data for the efficacy and safety parameters and also included the first distribution of test and placebo treatments. Baseline was defined as the last available value before the first randomized treatment. Subsequent visits occurred monthly and were of two types: (1) Supply Visits and (2) Supply and Procedures Visits. HbA1c was monitored at baseline (Visit 2) and every 2 months thereafter as were the secondary end-point parameters (described below). Efficacy analyses were conducted on data from 2, 6, and 10 months.
There were two treatment groups in this trial: drug (D-tagatose), and placebo (Splenda). The dose of D-tagatose was 15 g dissolved in 125 to 250 mL of water TID; the dose of placebo was 1.5 g dissolved in 125 to 250 mL of water TID. The placebo amounts were chosen to match sweetness for blinding. The powder packets were the same size and bore the same labeling with the exception of the designation “Substance A” or Substance B”. The entire drug/placebo solution was consumed prior to each main meal (breakfast, lunch, dinner). If severe gastrointestinal (GI) effects were seen, the tagatose dosage was reduced to 10 g TID temporarily and further reduced to 5 g TID if severe GI side effects were not resolved within 24 hours. The dosage of D-tagatose would then be increased to 10 g TID and further increased to 15 g TID when patients had adapted to the treatment (i.e., GI effects reduced to mild). The placebo dosage would be reduced in a similar fashion if severe GI effects were seen. That is, the dose could be reduced to 1.0 g TID temporarily and then further reduced to 0.5 g TID if severe GI side effects were not resolved within 24 hours. The dosage of placebo was then to be increased to 1.0 g TID and further increased to 1.5 g TID when patients had adapted to the treatment (i.e., GI effects reduced to mild).
Removal of Subjects from Therapy or Assessment
Premature end-point
A premature end-point of the efficacy analysis for the trial occurred when additional antidiabetic medications were prescribed to a patient at the sole discretion of the their primary care physician. However, patients were advised to continue in the trial if an antidiabetic medication was added after the start of the trial, even though the HbA1c data were not used in efficacy analyses at the time points subsequent to the initiation of the additional medication(s). The continuation of the trial was solely for the safety analysis of D-tagatose and patient’s data were withdrawn from the efficacy analysis.
Treatment failures
Subjects were categorized as treatment failures if both of the following criteria were met:
HbA1c change of +1.0% from baseline at any clinic visit or an HbA1c ≥10% or required additional antidiabetic medication for glycemic control;
Received at least 80% of all study drug doses within the initial 3 month dosing period.
The need for additional antidiabetic medication was determined by an elevated fasting blood glucose value (>240 mg/dL) not secondary to a readily identified illness or pharmacological treatment. Investigators were to withdraw subjects from study treatment (and therefore the evaluable population for assessment of efficacy as measured by HbA1c) after additional antidiabetic medication had been prescribed. However, subjects were advised to continue the rest of the trial procedures for the assessment of safety parameters.
Criteria for Evaluation
Efficacy
The primary efficacy variable was the change from baseline in HbA1c level when measured at pre-specified time points. Efficacy analyses were conducted on data from 2, 6 and 10 months (visit 4, 8 and 12). Statistical analyses were conducted on the 2-month, 6-month and 10-month data separately. The primary efficacy endpoint was at 2 months in the Intent-To-Treat population. Secondary efficacy endpoints for change in HbA1c were at 6 and 10 months. Additional secondary efficacy end-points were measured at the same time points and included changes from baseline in (1) fasting blood glucose, (2) serum insulin levels, (3) lipid profiles (LDL, triglycerides, total cholesterol, HDL), and (4) BMI, as well as the proportion of subjects achieving HbA1c targets of <7%.
Safety
The safety parameters assessed for each subject included adverse events, serious adverse events, vital signs, results of physical exams, 12-lead ECG, and clinical laboratory tests (hematology, chemistry, and urinalysis).
Statistical Methods
Three analysis populations were evaluated: (1) The Intent-to-Treat (ITT) population, (2) the Per Protocol (PP) population, and (3) the Safety population. The ITT population was the focus of the efficacy analyses for regulatory purposes, with the PP population being used in supportive analyses. For the ITT population, missing data (including missing values at intermediate visits) were to be imputed from scheduled visits using the last-observation-carried-forward (LOCF) method.
The Safety population was used for analyses of all safety parameters. Secondary efficacy end-points were evaluated using the closed test procedure (in order to preserve the type I error rate, α, of 0.05). Accordingly, the analysis of end-points was conducted in the following order: (1) body mass index, (2) triglycerides, (3) LDL, (4) total cholesterol, (5) fasting blood glucose, (6) proportion of subjects with HbA1c concentration of <7%, (7) insulin, and (8) HDL. Analysis of continuous efficacy variables used mixed-model repeated-measures analysis of covariance controlling for covariates and stratification factors identified as significant in preliminary explorations. Categorical efficacy variables were analyzed using general estimating equation models.
RESULTS
Study population
There were 494 subjects randomized, 185 subjects in the US and 309 subjects in India. Of these, 480 were treated, 248 with placebo and 232 with D-tagatose (Table 1). The mean age of subjects in the ITT population was 52 with an age range between 22 and 74. The ITT population was approximately evenly divided between males and females and the racial distribution was 72% Asian, 12% Caucasian, 11% Latino and 5% Black, with approximately equivalent distributions in the D-tagatose and placebo groups. At baseline, approximately 90% of subjects in both treatment groups were controlling their diet and exercising in order to control their diabetes.
Table 1.
Analysis Population.
| Population | Placebo N =253 n (%) |
D-tagatose N = 241 n (%) |
Total N = 494 n (%) |
|---|---|---|---|
| ITT | 184 (72.7%) | 172 (71.4%) | 356 (72.1%) |
| Per Protocol | 119 (47.0%) | 85 (35.3%) | 204 (41.3%) |
| Safety | 207 (81.8%) | 185 (76.8%) | 392 (79.4%) |
Efficacy Results
Primary Endpoint Results: Decrease in Hemoglobin A1c Level
The primary efficacy variable was the change from baseline in HbA1c level when measured at pre-specified time points. An efficacy analysis was conducted on data from 2, 6 and 10 months (Visits 4, 8 and 12). The primary efficacy end-point, set at the 2-month time point in the ITT population, showed a statistically significant difference (p = 0.0198) between D-tagatose and the placebo (Figure 1A). In the ITT population, the active treatment group, i.e., subjects who received D-tagatose, showed greater and statistically significant reductions in HbA1c levels at all post-baseline visits when compared to the placebo group (Figure 1B). The same mixed model analysis conducted with the PP population data showed similar results insofar as the population receiving D-tagatose always showed greater decreases in HbA1c compared to placebo (Figure 1C). The differences in the PP population did not achieve statistical significance until month 6, however, but then remained significant throughout the remainder of the study.
Figure 1.
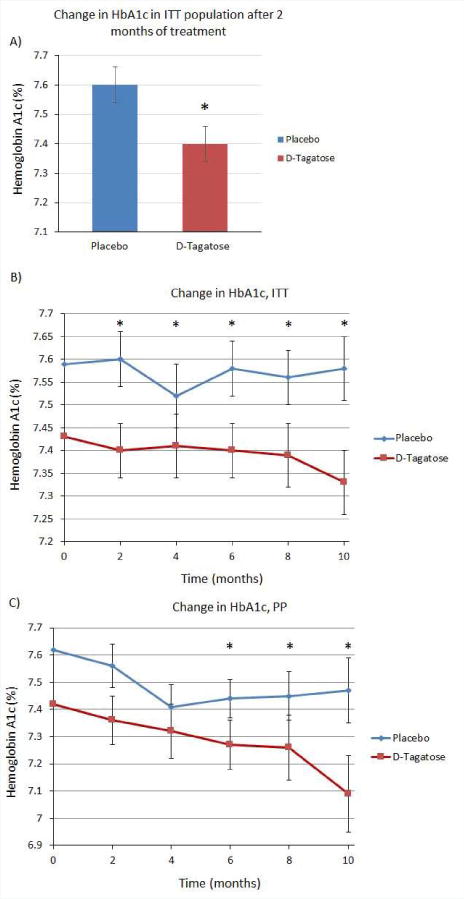
(A) Change in HbA1c in ITT population after 2 months of treatment. The primary efficacy endpoint set at 2 months in the ITT population showed a significant difference between placebo and D-tagatose groups (p=0.0198) (placebo n=182, D-tagatose n=172). (B) Change in HbA1c in ITT population. Significant differences were seen at 2 (p=0.0198), 4 (p=0.0160), 6 (p=0.0015), 8 (p=0.0002), and 10 months (p=<0.0001). (C) Change in HbA1c in PP population. Significant differences were seen at 6 (p=0.0343), 8 (p=0.0148), and 10 months (p=0.0021). Zero time points plotted as means, remaining time points plotted as least squares means ±SEM.
Analysis of subgroups
In light of the above results, subgroup analyses were conducted to examine whether starting HbA1c levels (< 7.5% or ≥ 7.5%) influenced the effectiveness of D-tagatose. The results of these analyses for the ITT population (LOCF) are shown in Figure 2A and 2B. A significant difference between D-tagatose- and placebo-treated subjects was seen earlier in the subgroup entering with a baseline HbA1c < 7.5%., although both subgroups achieved greater changes from baseline than those observed in the placebo groups.
Figure 2.
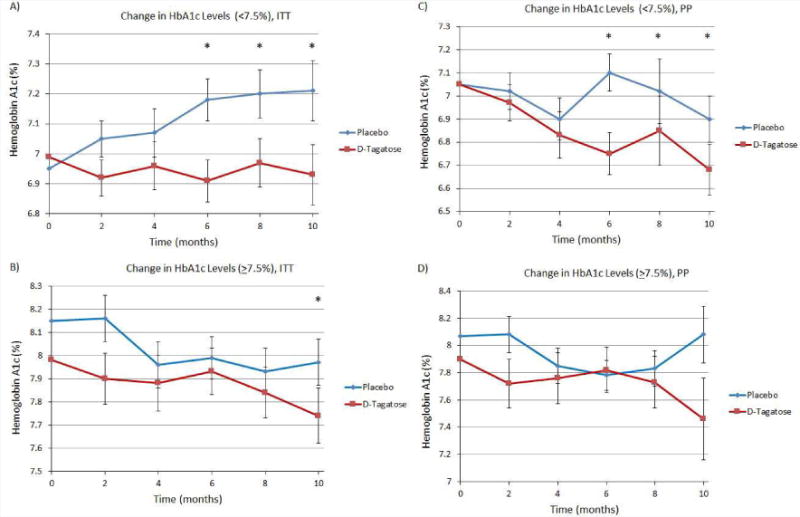
(A) Results of analyses for the ITT subgroup with starting HbA1c baseline < 7.5%. Significant differences between D-tagatose and placebo were seen at 6 (p=0.0030), 8 (p=0.0004), and 10 months (p=<0.0001). (B) Change in HbA1c in ITT subgroup with baseline >7.5%. A significant difference between D-tagatose and placebo was seen at 10 months (p=0.0277). (C) Change in HbA1c in PP subgroup with baseline HbA1c <7.5%, (D) Change in HbA1c in PP subgroup with baseline >7.5%. Zero time points plotted as means, remaining time points plotted as least squares means ±SEM.
In the PP population the D-tagatose treatment for both subgroups demonstrated greater reductions in HbA1c compared to placebo (with the exception at 6 months in the ≥ 7.5% subgroup) (Figure 2C and 2D). However, a statistically significant difference between the D-tagatose and placebo-treatment groups was only achieved in the <7.5% subgroup (Figure 2C). Statistical significance between D-tagatose and placebo groups was achieved after 6 months of treatment in subjects with a baseline HbA1c of <7.5% (Figure 2C, p-values of 0.0497, 0.029, and 0.0121 after 6, 8, and 10 months of treatment, respectively), whereas statistical significance between D-tagatose and placebo groups was not achieved in patients with a higher baseline HbA1c at any time point (Figure 2D).
Further subgroup analyses were conducted to compare the ability of D-tagatose to effectively decrease HbA1c in the United States (US) population of subjects versus subjects of Indian origin. Interestingly, the effect on lowering HbA1c was more pronounced in US compared to Indian subjects (Figure 3). A subgroup analysis on the ITT US population (Figure 3A) showed a greater and statistically significant LS mean reduction in HbA1c in the D-tagatose groups than in the placebo group at all post-baseline visits: month 2 (reduction of 0.23 vs. an increase of 0.07, Δ = 0.3 and p = 0.0273), month 4 (reduction of 0.15 vs. an increase of 0.28, Δ = 0.4 and p = 0.0011), month 6 (reduction of 0.17 vs. an increase of 0.17, Δ = 0.3 and p = <0.0001), month 8 (reduction of 0.10 vs. an increase of 0.29, Δ = 0.4 and p = <0.0001), and month 10 (reduction of 0.07 vs. an increase of 0.37, Δ = 0.4 and p = <0.0001). Analysis of the LOCF ITT data showed statistically significant reductions in the India population only at month 10 (Δ = 0.2, p = 0.0187) (Figure 3B). PP analyses for the US population produced similar results with statistically significant differences being noted at all post-baseline time points (Figure 3C). The effect of D-tagatose in reducing HbA1c was evident by 2 months of treatment in the US subjects. However, the effect of D-tagatose in reducing HbA1c in the Indian subjects was not significantly different than placebo at any time point in the PP population (Figure 3D).
Figure 3.
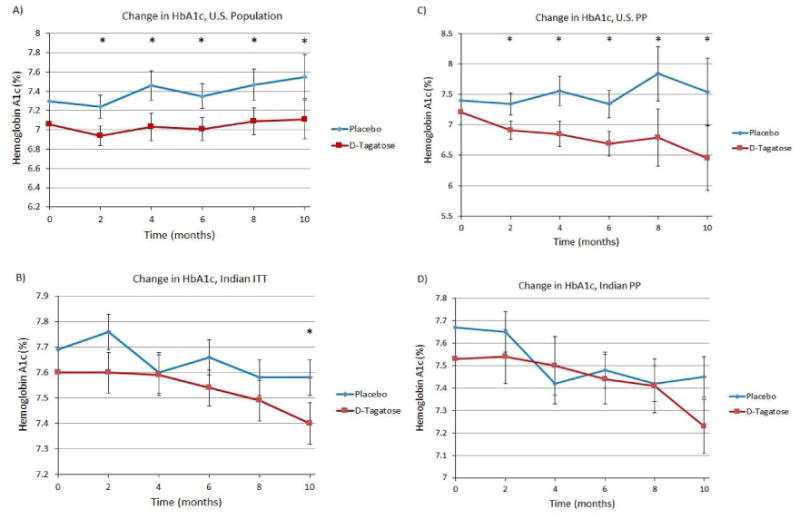
(A) Change in HbA1c U.S. subjects, ITT population. (B) Change in HbA1c, Indian subjects, ITT population. (C) Change in HbA1c U.S. subjects, PP population. p-values for 2, 4, 6, 8, and 10 months of treatment are 0.0435, 0.0016, <0.0001, <0.0001, and <0.0001, respectively. (D) Change in HbA1c, Indian subjects, PP population. Zero time points plotted as means, remaining time points plotted as least squares means ±SEM.
Proportion of Subjects Achieving HbA1c < 7%
The data from the ITT population with LOCF indicated that the D-tagatose group had a greater proportion of subjects achieving an HbA1c level of less than 7% at all post baseline time points compared to placebo, with the results being statistically significant at post-baseline months 6, 8 and 10 (Table 2). Thus, the percent of responders in the D-tagatose group was higher than in the placebo group at 6 months (38.37% vs 22.4%), 8 months (40.7% vs 25.68%) and 10 months (43.02% vs 26.23%, respectively). Similar results were observed in the analyses using the PP population except that in the latter case statistical significance was observed at months 6 and 10 (Table 3).
Table 2.
Proportion of Subjects Achieving HbA1c <7%, ITT Population.
| Month | Statistic | Placebo | D-tagatose | Delta | P-values |
|---|---|---|---|---|---|
| 2 | Responder Non-responder |
49 (26.92%) 133 (73.08%) |
65(38.01%) 106 (61.99%) |
11.09 | 0.0913 |
| 4 | Responder Non-responder |
53 (28.96%) 104(60.47%) |
68 (39.53%) 84 (59.57%) |
10.57 | 0.1580 |
| 6 | Responder Non-responder |
41 (22.40%) 106(61.63%) |
66 (38.37%) 76 (60.32%) |
15.97 | 0.0048 |
| 8 | Responder Non-responder |
47 (25.68%) 136 (74.32%) |
70 (40.70%) 102 (59.30%) |
15.01 | 0.0133 |
| 10 | Responder Non-responder |
48 (26.23%) 135 (73.77%) |
74 (43.02%) 98 (56.98%) |
16.79 | 0.0052 |
Table 3.
Proportion of Subjects Achieving HbA1c <7%, PP Population.
| Month | Statistic | Placebo | D-tagatose | Delta | P-values |
|---|---|---|---|---|---|
| 2 | Responder Non-responder |
31(26.50%) 86(73.50%) |
32(38.10%) 52(61.90%) |
11.06 | 0.3787 |
| 4 | Responder Non-responder |
31(30.39%) 71(69.61%) |
29(43.94%) 37(56.06%) |
13.55 | 0.5260 |
| 6 | Responder Non-responder |
19(21.11%) 71(78.89%) |
27(48.21%) 29(51.79%) |
27.10 | 0.0073 |
| 8 | Responder Non-responder |
21(28.00%) 54(72.00%) |
21(46.67%) 24(53.33%) |
18.67 | 0.1052 |
| 10 | Non-responder Responder |
14(25.93%) 40(74.07%) |
22(57.89%) 16(42.11%) |
31.97 | 0.0044 |
Secondary Endpoint Results: Changes in BMI, fasting blood glucose, insulin, blood lipids
D-tagatose was also evaluated for its placebo-controlled treatment effects on BMI, fasting blood glucose, insulin, blood cholesterol, and triglyceride levels. There was no observed effect of D-tagatose treatment on changes in body weight or BMI (body mass index) compared to placebo in either the ITT or PP populations.
For the ITT population better reductions in fasting blood glucose levels from baseline were observed in the D-tagatose group compared to the placebo group at all post baseline time points (Figure 4A), with the differences in LS mean being statistically significant for post-baseline month 6 and beyond, i.e., month 6 (reduction of 2.0 vs. an increase of 4.1, Δ = 6.0 and p = 0.0440), month 8 (reduction of 0.44 vs. an increase of 2.5, Δ 2.9 and p = 0.0340), and month 10 (reduction of 0.25 vs. an increase of 6.6, Δ = 6.9 and p = 0.0079). Reductions were also seen in PP population analyses; however, none were statistically significant (Figure 4B). Regardless of the statistically significant difference in fasting blood glucose between placebo and D-tagatose groups, there was no detectable consistent change in serum insulin concentrations in this clinical trial (data not shown).
Figure 4.
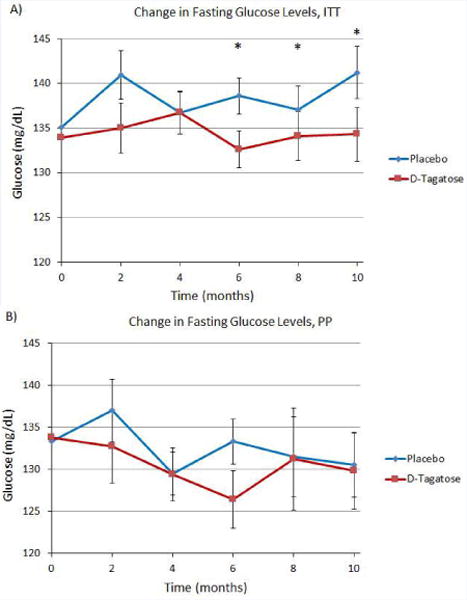
(A) Change in fasting blood glucose, ITT population. D-tagatose significantly decreased fasting blood glucose compared to placebo after 6 months of treatment. (B) Change in fasting blood glucose levels, PP population. No significant difference between placebo and D-tagatose groups was observed at any time point. Zero time points plotted as means, remaining time points plotted as least squares means ±SEM.
Particularly interesting findings of this phase 3 clinical trial include the effect of D-tagatose to significantly reduce total cholesterol and LDL-cholesterol compared to placebo. The D-tagatose group (ITT population) showed better reductions in total cholesterol from baseline compared to placebo at all post baseline time points (Figure 5A), with the differences becoming statistically significant after 4 months of treatment. This effect was maintained for the duration of the trial (differences in LS means being statistically significant starting from month 4 5.4 to 5.3, month 6 (reductions of 6.9 3.8 to 3.7 and p = 0.0217), month 8 (reductions of 8.1 vs. 5.3, Δ = 2.8 and p = 0.0157), and month 10 1.6 to 1.5. In the PP population analyses, reductions were also seen; however, none were statistically significant (Figure 5B).
Figure 5.
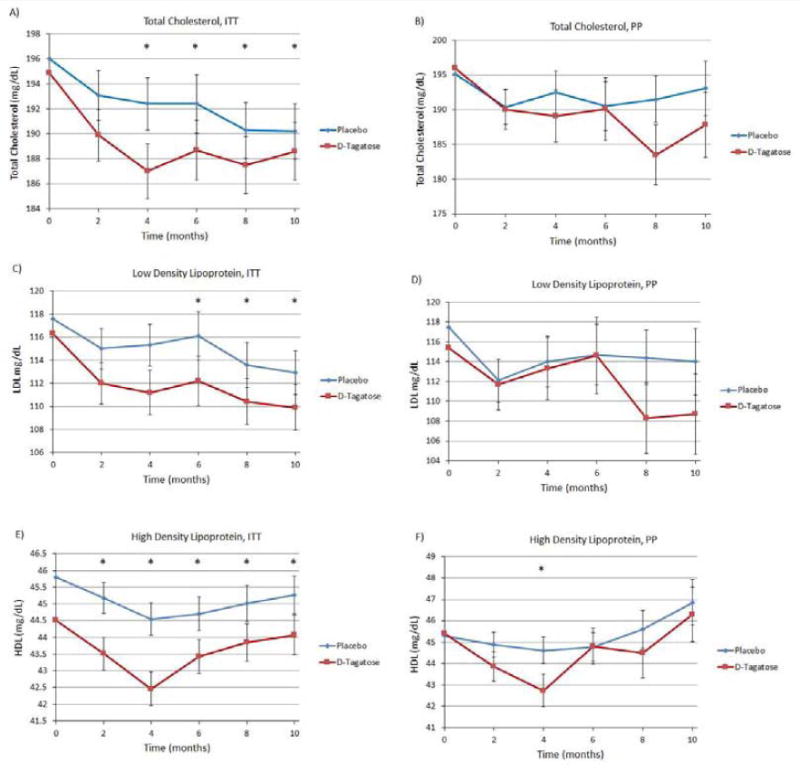
(A) Total cholesterol, ITT population. (B) Total cholesterol, PP population. (C) LDL, ITT population. (D) LDL, PP population. (E) HDL, ITT population. Significant differences between D-tagatose and placebo were seen at 2 (p=0.0126), 4 (p=0.00040, 6 (p=0.0002), 8 (p=0.0003), and 10 months (p=0.0008). (F) HDL, PP population. The only significant difference was at 4 months (p=0.0397). Zero time points plotted as means, remaining time points plotted as least squares means ±SEM.
Better reductions from baseline in LDL levels were observed in the D-tagatose-treated group (ITT population) than the placebo at post baseline time points (Figure 5C), with the differences in LS mean being at month 6 a reduction of 4.1 to 3.8; at month 8 a reduction of 3.1 to 3.0.; and at month 10 a reduction of 7.1 vs. 4.2, Δ = 2.9 and p = 0.0057. Smaller reductions were seen in the PP population analyses and none were statistically significant (Figure 5D).
The results of analyses of serum HDL concentrations in the ITT population showed greater decreases in the D-tagatose group than in the placebo group with the differences being statistically significant at all post-treatment time points in the ITT population and at the month 4 time point in the PP population (Figure 5E and 5F). However, the mean HDL values always remained above the target value of 40 mg/dL, so it is unclear whether the reduction in HDL concentrations observed in the D-tagatose group has any clinical significance.
Triglyceride levels showed no benefit from D-tagatose treatment, i.e., the placebo group showed greater reductions in triglyceride levels than the D-tagatose group with the differences becoming statistically significant from month 8 onward (i.e., at month 8 a reduction of 12.7 vs. an increase of 1.46, Δ = 14.2 and p = 0.0301; and at month 10 a reduction of 10.8 vs. an increase of 11.2, Δ = 22 and p = 0.0048 (Figure 6A)). No statistically significant differences were seen in the PP population analyses (Figure 6B). Additionally, subgroup analyses for subjects who had baseline triglyceride levels 1) less than 200 mg/dL, 2) between 200 and 500 mg/dL, and 3) above 500 mg/dL, revealed no statistically significant differences between the D-tagatose and placebo treatment groups (data not shown).
Figure 6.
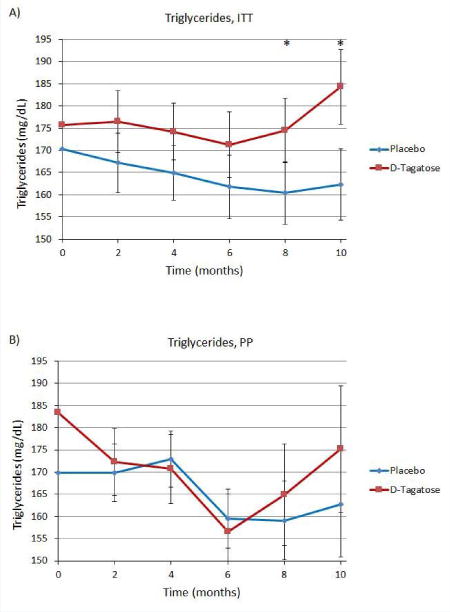
Serum triglycerides. (A) ITT population. (B) PP population. Zero time points plotted as means, remaining time points plotted as least squares means ±SEM.
Safety Results
D-tagatose was reasonably well tolerated by the subjects in this trial with most of the adverse events (AEs) experienced being mild to moderate in intensity. Adverse events in the D-tagatose treatment group were similar to that of the placebo group, with most instances noted as GI disturbances. Importantly, there were no reported episodes of hypoglycemia or pancreatitis.
The incidence of SAEs in the D-tagatose group was about one-half that seen in the placebo group. The incidences of AEs by system organ class were generally similar between the placebo and D-tagatose groups, the one exception being AEs in the gastrointestinal disorders class. Most of the subjects in both treatments experienced GI symptoms; however, the incidence was higher in the D-tagatose group, and the GI AEs were more frequently severe in the D-tagatose group. There were no remarkable effects in other safety parameters with very few clinically significant changes in safety laboratory values, ECG parameters, vital signs or physical examinations.
DISCUSSION
This trial met its primary objective of demonstrating that D-tagatose was effective at reducing the HbA1c level when administered for two months at doses of 15 g TID. The secondary end-point of significant reductions in the the HbA1c level at six and ten months were also met. Mixed model analyses using the ITT population and LOCF imputation of missing values indicated significantly greater reductions in the mean HbA1c levels in the D-tagatose group than in the placebo group at all post-baseline visits. The same mixed model analysis conducted with the PP population data showed similar results insofar as the population receiving D-tagatose always showed greater decreases in HbA1c compared to placebo. Additionally, subgroup analyses were done for the primary end-point on different subgroups of subjects to see if the reduction in HbA1c is significantly different between the treatment groups in the different subgroups i.e.
1) subjects whose baseline HbA1c level was less than 7.5%, 2) subjects whose baseline HbA1c level was greater than or equal to 7.5%, 3) subjects who were randomized in India, and 4) subjects who were randomized in US. In these analyses the results were generally agreement with the primary results although in the India subgroup the results were statistically significant at far fewer time points. Additionally it was observed that a greater proportion of subjects in the D-tagatose group had achieved HbA1c targets of <7%; this difference was also found to be statistically significant for the majority of the post-baseline time points. Regardless of the subgroup analyzed, the effect of D-tagatose to decrease HbA1c levels was robust and could be seen with differing analytical approaches. These results demonstrate the ability of D-tagatose to effectively aid in the control of blood sugar levels for type 2 diabetics and suggest D-tagatose as a potential drug therapy in this area.
Statistically significant differences were observed between the D-tagatose and placebo treatment groups for the secondary end-points LDL, total cholesterol, and fasting blood glucose when examining the ITT population. Reductions were also seen in PP population analyses however, none were statistically significant. Numerous studies have demonstrated that increased total cholesterol and particularly elevated LDL-cholesterol are associated with amplified risk for the development of cardiovascular disease. Thus, this finding is especially important for type 2 diabetic patients that may also battle dyslipidemia and/or the metabolic syndrome [20].
There were also two end-points, triglycerides and HDL levels, where the D-tagatose treatment produced greater and statistically significant increases in the case of triglycerides and decreases in the case of HDL levels compared to the placebo group in the ITT population. The PP population however, showed no significant difference in triglycerides or HDL between placebo and D-tagatose. Of note, a recent study by Donner et al. [13] demonstrated a striking and significant effect of D-tagatose to increase HDL from a baseline level of 30.5 ± 15.8 to 41.7 ± 12.1 mg/dL (p<0.001) after 10 months of treatment in 6 subjects who did not take lipid medications during the study. Beginning HDL levels in the current study were higher (mean placebo 45.8, D-tagatose 44.5 in ITT and mean placebo 45.5, D-tagatose 45.5 in PP) than in the Donner study. The differences in results between these studies could be from this initial HDL level discrepancy, and any elevations in HDL that D-tagatose is able to provide may only manifest if HDL levels are below recommended values. Consequently, further studies are needed to elucidate the direct effects of D-tagatose on HDL cholesterol levels. HDL-cholesterol levels are inversely related to risk of developing cardiovascular disease.
There was no observed effect of D-tagatose treatment on changes in body weight or BMI (body mass index) compared to placebo in either the ITT or PP populations. In contrast to these findings, previous studies in humans [13] and rodents [12] have demonstrated significant weight loss and a reduction in body weight gain with D-tagatose, respectively. The mean body weight of subjects (both D-tagatose and placebo groups) included in this phase 3 clinical trial at baseline was 73.8 kg, or 162 pounds; with an average BMI of 28.3 mg/m2 (overweight category). However, in previous studies which have demonstrated an effect of D-tagatose to promote weight loss, the mean body weight of all subjects at baseline was much higher (mean body weight at baseline ~109 kg, or 240 pounds [13]. Thus, it is plausible that the subject population within this phase 3 clinical trial was not adequately overweight to demonstrate an effect of D-tagatose to promote weight loss.
D-tagatose appeared to be reasonably well tolerated by the subjects in this trial with most of the AEs experienced being mild to moderate in intensity. The incidence of SAEs in the D-tagatose group was about one-half that seen in the placebo group. The incidences of AEs by system organ class were generally similar between the placebo and D-tagatose group, the one exception being AEs in the gastrointestinal disorders class. Most of the subjects in both treatments experienced GI symptoms; however, the incidence was generally higher in the D-tagatose group, and the GI AEs were more frequently severe in the D-tagatose group. There were no remarkable effects in other safety parameters with very few clinically significant changes in safety laboratory values, ECG parameters, vital signs or physical examinations. Previous single-dose and repeated-dose studies in healthy and diabetic human subjects showed that the predominant adverse effects associated with consumption of high levels D-tagatose were gastrointestinal disturbances attributed to osmotic effects from incompletely absorbed tagatose [13–18, 21–23]. At single doses of up to 25 g D-tagatose per meal, flatulence was generally the only side effect, with nausea, borborygmi (i.e., rumbling or gurgling noises, colic, and laxation noted at higher doses). Such effects are also commonly associated with excessive consumption of other poorly digestible carbohydrates.
In summary, results of this phase 3 clinical trial demonstrate an effect of D-tagatose to significantly decrease HbA1c levels over time compared to placebo. The longer subjects remained on the treatment, the further HbA1c was reduced. D-tagatose treatment also reduced fasting blood glucose levels and total and LDL-cholesterol concentrations.
CONCLUSIONS
D-tagatose was effective at lowering HbA1c levels when administered at a daily dose of 15 g in 125–250 mL of water three times per day just prior to meals.
Unlike many other diabetes drugs, the longer a patient is on D-tagatose therapy in compliance with instructions, the better the efficacy.
The effect on HbA1c levels was robust and could be demonstrated in both subgroups, various subpopulations, and with differing analytical approaches.
D-tagatose appeared to be reasonably well tolerated with most of the AEs experienced in both treatment groups being mild or moderate in severity.
Bloomgarden et al.’s [24] analysis of 61 studies shows that the degree of decrease in HbA1c produced by drugs for treating type 2 diabetes is dependent on the baseline HbA1c; the higher the baseline the greater the decrease. When the baseline HbA1c was <8.0%, the reduction from active therapy is only 0.1–0.2% greater than in the control group. When the HbA1c was >8.0 to 11.8 the reduction in HbA1c was between −0.6 and −1.2%, and the greater the HbA1c, the greater the reduction overall.
In this study the mean HbA1c baselines were 7.6% ± 0.75 (± Stdev) and 7.4% ± 0.59 for placebo and D-tagatose groups respectively in the ITT population, and 7.7% ± 0.64 and 7.5% ± 0.53 for placebo and D-tagatose groups respectively in the PP population. Based on Bloomgarden’s study (see Table 1 in Bloomgarden), other drugs show an average of only half the efficacy of D-tagatose in similar patients. Overall results of this phase 3 clinical trial suggest a strong potential for D-tagatose as an adjunct in the management of type 2 diabetes.
Acknowledgments
CREDITS
This research was supported in part by the Biospherics subsidiary of Spherix Incorporated. The project described was also supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR000117. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
ABBREVIATIONS
- AE
adverse event
- BMI
body mass index
- BUN
blood urea nitrogen
- ECG (or EKG)
electrocardiogram
- GI
gastrointestinal
- GRAS
generally recognized as safe
- HbA1c
glycosylated hemoglobin A1c
- HDL
high-density lipoprotein
- ITT
intent-to-treat
- LDL
low density lipoprotein
- LSMean
least squares mean
- LOCF
Last Observation Carried Forward
- MCH
mean corpuscular hemoglobin
- MCHC
mean corpuscular hemoglobin concentration
- MCV
mean corpuscular volume
- PP
per protocol
- SAE
serious adverse event
- SEM
standard error of the mean
- SGOT
serum glutamic oxaloacetic transaminase
- SGPT
serum glutamic pyruvic transaminase
- TID
three times daily
- VLDL
very low density lipoprotein.
Footnotes
Conflict of Interest
Robert Lodder was president of Spherix at the time the clinical data were collected.
References
- 1.Diabetes statistics. http://www.diabetes.org/diabetes-basics/diabetes-statistics.
- 2.Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA: U.S. Department of Health and Human Services; 2014. [Google Scholar]
- 3.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA. 2004;291:335–342. doi: 10.1001/jama.291.3.335. [DOI] [PubMed] [Google Scholar]
- 5.Bennett WL, Maruthur N, Bolen S, Singh S, Chatterjee R, Marinopoulos SS, Puhan MA, et al. Oral Diabetes Medications for Adults With Type 2 Diabetes: An Update. Rockville, MD: Agency for Healthcare Research and Quality; (Comparative Effectiveness Review No. 27. (Prepared by Johns Hopkins University Evidence-based Practice Center under Contract No. 290-02-0018.). AHRQ Publication No. 11-EHC038-EF). [PubMed] [Google Scholar]
- 6.Inzucchi SE. Oral antihyperglycemic therapy for type 2 diabetes: scientific review. JAMA. 2002;287:360–372. doi: 10.1001/jama.287.3.360. [DOI] [PubMed] [Google Scholar]
- 7.Nathan DM. Thiazolidinediones for initial treatment of type 2 diabetes? N Engl J Med. 2006;355:2477–2480. doi: 10.1056/NEJMe068264. [DOI] [PubMed] [Google Scholar]
- 8.Nathan DM. Rosiglitazone and cardiotoxicitys-weighing the evidence. N Engl J Med. 2007;357:64–66. doi: 10.1056/NEJMe078117. [DOI] [PubMed] [Google Scholar]
- 9.Levin GV. Tagatose, the new GRAS sweetener and health product. J Med Food. 2002;5:23–36. doi: 10.1089/109662002753723197. [DOI] [PubMed] [Google Scholar]
- 10.Rulis Agency response letter GRAS notice, http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/NoticeInventory/ucm245241.htm, retrieved October 10 2014.
- 11.Lu Y, Levin GV, Donner TW. Tagatose, a new antidiabetic and obesity control drug. Diabetes Obes Metab. 2008;10:109–134. doi: 10.1111/j.1463-1326.2007.00799.x. [DOI] [PubMed] [Google Scholar]
- 12.Police SB, Harris JC, Lodder RA, Cassis LA. Effect of diets containing sucrose vs. D-tagatose in hypercholesterolemic mice. Obesity (Silver Spring) 2009;17:269–275. doi: 10.1038/oby.2008.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donner TW, Magder LS, Zarbalian K. Dietary supplementation with d-tagatose in subjects with type 2 diabetes leads to weight loss and raises high-density lipoprotein cholesterol. Nutr Res. 2010;30:801–806. doi: 10.1016/j.nutres.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 14.Buemann B, Toubro S, Astrup A. D-Tagatose, a stereoisomer of D-fructose, increases hydrogen production in humans without affecting 24-hour energy expenditure or respiratory exchange ratio. J Nutr. 1998;128:1481–1486. doi: 10.1093/jn/128.9.1481. [DOI] [PubMed] [Google Scholar]
- 15.Buemann B, Toubro S, Astrup A. Human gastrointestinal tolerance to D-tagatose. Regul Toxicol Pharmacol. 1999;29:S71–77. doi: 10.1006/rtph.1998.1265. [DOI] [PubMed] [Google Scholar]
- 16.Buemann B, Toubro S, Raben A, Astrup A. Human tolerance to a single, high dose of D-tagatose. Regul Toxicol Pharmacol. 1999;29:S66–70. doi: 10.1006/rtph.1998.1252. [DOI] [PubMed] [Google Scholar]
- 17.Saunders JP, Donner TW, Sadler JH, Levin GV, Makris NG. Effects of acute and repeated oral doses of D-tagatose on plasma uric acid in normal and diabetic humans. Regul Toxicol Pharmacol. 1999;29:S57–65. doi: 10.1006/rtph.1998.1264. [DOI] [PubMed] [Google Scholar]
- 18.Ensor M, Williams J, Smith R, Banfield A, Lodder RA. Effects of three low-doses of D-tagatose on glycemic control over six months in subjects with mild type 2 diabetes mellitus under control with diet and exercise. J Endocrinol Diabetes Obes. 2014;2:1057. , in press. [PMC free article] [PubMed] [Google Scholar]
- 19. (NIH Publication No. 14-7816).The A1C Test and Diabetes. 2014 Mar; [Google Scholar]
- 20.Malik S, Wong ND, Franklin SS, Kamath TV, L’Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004 Sep 7;110(10):1245–50. doi: 10.1161/01.CIR.0000140677.20606.0E. 2004. [DOI] [PubMed] [Google Scholar]
- 21.Donner T, Wilber J, Ostrowski D. D-Tagatose: a novel therapeutic adjunct for non-insulin-dependent diabetes. Diabetes. 1996;45(Suppl. 2):125A. [Google Scholar]
- 22.Donner TW, Wilber JF, Ostrowski D. D-tagatose, a novel hexose: acute effects on carbohydrate tolerance in subjects with and without type 2 diabetes. Diabetes Obes Metab. 1999;1:285–291. doi: 10.1046/j.1463-1326.1999.00039.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee A, Storey DM. Comparative gastrointestinal tolerance of sucrose, lactitol, or D-tagatose in chocolate. Regul Toxicol Pharmacol. 1999;29:S78–82. doi: 10.1006/rtph.1998.1255. [DOI] [PubMed] [Google Scholar]
- 24.Bloomgarden ZT, Dodis R, Viscoli CM, Holmboe ES, Inzucchi SE. Lower baseline glycemia reduces apparent oral agent glucose-lowering efficacy: a meta-regression analysis. Diabetes Care. 2006 Sep;29(9):2137–9. doi: 10.2337/dc06-1120. [DOI] [PubMed] [Google Scholar]