

Abstract
During the first 100 years of Alzheimer's disease research, this devastating and intractable disorder has been characterized at the clinical, histological, and molecular levels. Nevertheless, many key mechanistic questions remain unanswered. Here we will emphasize the importance of the cell biology of Alzheimer's disease, reviewing the relevant literature that has expanded our mechanistic understanding, with a particular focus on pathways regulating protein sorting. Accumulated evidence indicates that sorting pathways may be uniquely vulnerable to disease pathogenesis, and recent studies have begun to reveal disease-related defects in the regulation of protein sorting.
One hundred years ago, Alois Alzheimer examined the brain of Auguste D., a demented woman who began developing cognitive deficits in her late 40s, and thereby established that dementia can be associated with neurofibrillary tangles and amyloid plaques (Alzheimer et al., 1995 [an English translation of 1907 article by Alzheimer et al.]). In 1910, Emile Kraepelin defined “Alzheimer's disease” as a distinct dementing illness, which, he maintained, provided evidence that dementia can have an “organic” etiology (Schorer, 1985). Yet, for decades thereafter, the major dementia of late life was either dismissed as the inevitable endstage of normal aging or else attributed to “cerebral atherosclerosis” (Katzman, 1986). Over fifty years would pass before investigators began appreciating that late-onset (“senile”) dementia and early-onset (“pre-senile”) Alzheimer's disease are phenotypically indistinguishable under the microscope, prompting the unification of the disorders under the same name (Amaducci et al., 1986).
Nevertheless, despite a shared name and overlapping features, the early-onset and late-onset forms of Alzheimer's disease are epidemiologically and etiologically distinct (Mayeux, 2003; Bertram and Tanzi, 2005). The early-onset form is extremely rare and is caused by monogenic defects that follow an autosomal-dominant pattern (Mayeux, 2003; Bertram and Tanzi, 2005). Like other autosomal-dominant diseases, early-onset Alzheimer's disease has proven amenable to linkage analysis, and many pathogenic mutations have been isolated. In contrast, late-onset Alzheimer's disease, which typically manifests after the sixth decade, accounts for over 95% of all cases and has a complex etiology (Mayeux, 2003; Bertram and Tanzi, 2005). Like other complex disorders, the cause of late-onset Alzheimer's disease has been difficult to pinpoint. Indeed, the late-onset form has historically been called “sporadic” Alzheimer's disease to reflect the elusiveness of its etiology. Not only is late-onset disease the more common form of Alzheimer's disease, but with increased longevity, late-onset Alzheimer's disease has emerged as one of the most common disorders of the brain (Small and Mayeux, 2000).
Whereas the 20th century began with the histological description of Alzheimer's disease, the century ended with its genetic and molecular characterization. This era began in 1984 when amyloid-β (Aβ) was isolated as the core peptide of amyloid plaques (Glenner and Wong, 1984; Masters et al., 1985) and entered full swing when, in 1986, the gene encoding the peptide's parent protein, the amyloid precursor protein (APP), was identified (Goldgaber et al.,1987; Kang et al., 1987; Robakis et al., 1987; Tanzi et al., 1987). APP turned out to be a type-I transmembrane protein (Kang et al., 1987), and fine chromosomal mapping of APP revealed its localization to the Down's syndrome region of chromosome 21 (Tanzi et al., 1987), a particularly intriguing discovery since trisomy 21 (Down's syndrome) invariably causes early-onset Alzheimer's disease (Wisniewski et al., 1978). Then, in 1990 and 1991, linkage analyses revealed that mutations in APP can cause either early-onset Alzheimer's disease (Goate et al., 1991) or hereditary cerebrovascular amyloidosis (Dutch type) (Levy et al., 1990), and missense mutations were localized within or adjacent to the Aβ domain of APP (Levy et al., 1990). Although disease-causing mutations in APP occur in only a handful of families, these findings provided the strongest evidence that the disease can be initiated by abnormalities in APP processing and the accumulation of Aβ peptide (reviewed in Hardy and Allsop, 1991; Hardy and Higgins, 1992; Haass and Selkoe, 1993; and Tanzi and Bertram, 2005).
Further reaffirming the relevance of APP processing to the disease, in 1995, additional mutations causing early-onset Alzheimer's disease were isolated in two unknown genes encoding complex, polytopic proteins (Sherrington et al., 1995; Rogaev et al., 1995; Levy-Lahad et al., 1995a, 1995b). Since these genes cause early-onset, “pre-senile,” Alzheimer's disease, they were named presenilin 1 and presenilin 2, and they were localized to chromosome 14 and chromosome 1, respectively. Mutations in presenilins 1 and 2 modify APP processing by shifting the relative generation of Aβ species so as to increase the Aβ42/Aβ40 ratio (where the number indicates the amino-acid length of each peptide species) (Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996; Citron et al., 1997). Aβ42 oligomerizes more rapidly than other forms of Aβ (Hilbich et al., 1991; Burdick et al., 1992; Jarrett et al., 1993), and Aβ oligomers appear particularly toxic to neurons (reviewed in Klein et al., 2001; see also Lesne et al., 2006). Finally, in 1999–2000 this productive era was capped off with the isolation of β-site APP cleaving enzyme (BACE), a transmembrane protease that governs the first enzymatic step in APP processing, known as β-secretase cleavage or simply β-cleavage (Vassar et al., 1999; Sinha et al., 1999; Lin et al., 2000).
When the knowledge of the pathogenic mutations for early-onset disease were taken together with other discoveries related to APP metabolism, the basic biochemical formula for Aβ production was established (Figure 1A) (for review, see Gandy, 2005). The Aβ domain is located within APP at the junction between the intralumenal and transmembrane domains. Two enzymatic steps liberate Aβ from APP. In the first, “β-cleavage” step, BACE cleaves APP at or near the N terminus of the Aβ peptide; then, in the second, or “γ-cleavage” step, the membrane-bound C-terminal APP fragment (CTF) generated by BACE goes on to be cleaved by the γ-secretase, a multimeric complex thought to be made up of an essential quartet of transmembrane proteins—presenilin 1 (or 2), nicastrin, APH1, and PEN2 (Edbauer et al., 2003; Kimberly et al., 2003; Takasugi et al., 2003). Recent evidence indicates that a fifth molecule, TMP21, is also associated with the γ-secretase complex and plays a regulatory role in γ-cleavage (Chen et al., 2006).
Figure 1.
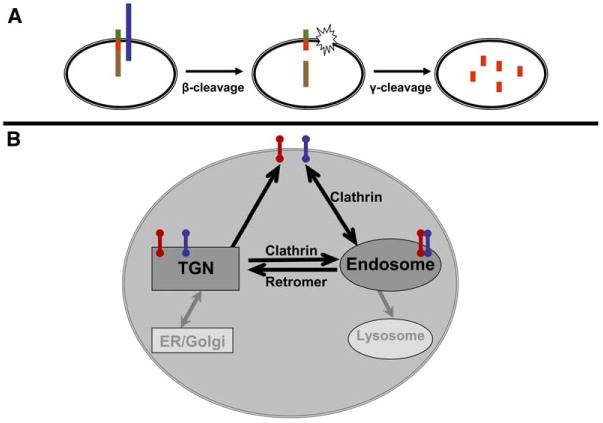
Protein Sorting as a Key Mechanism in Alzheimer's Cell Biology, (A) Aβ (red bar) is liberated from its parent protein, APP (multicolor bar), in two enzymatic steps. In the first β-cleavage step, BACE (blue bar) splits full-length APP into an sAPPβ fragment (brown bar) and a C-terminal fragment (CTFβ, red/green bar). Then, in the γ-cleavage step, the γ-secretase (star) splits CTFβ into Aβ (red bar) and amyloid intracellular domain (AICD, green bar). β-cleavage is the committed step in APP processing and may be upregulated in late-onset Alzheimer's disease.
(B) Both APP (red bar) and BACE (blue bar) are type-I transmembrane proteins that are sorted through multiple membranous compartments of the cell. The sorting triangle that interconnects the trans-Golgi network (TGN), cell surface, and the endosome is critically important for APP and BACE sorting. As indicated, clathrin is the coat complex that regulates transport from the cell surface and the TGN to the endosome, while the retromer is the coat complex that regulates transport from the endosome back to the TGN. A convergence of findings suggests that APP and BACE are most likely to interact within the membranes of the endosomal system, initiating the potentially amyloidogenic pathway.
Knowing the biochemical formula for Aβ production is sufficient to explain the Aβ accumulation observed in early-onset Alzheimer's disease. Nevertheless, these molecular defects do not exist in late-onset Alzheimer's disease and therefore cannot explain the accumulation of both Aβ40 and Aβ42 that characterize the common form of the disease. In reality, of course, Aβ production occurs in living neurons, not in a cell-free biochemical reaction. Pursuing the cell biology of how and where in the cell APP is processed has expanded the list of possible mechanisms that regulate Aβ production and has suggested pathogenic possibilities that extend beyond simple molecular defects in APP or the secretases. In this regard, it is notable that APP and the secretases are all integral transmembrane proteins; that they are dynamically sorted through the plasma membrane and the membranes of intracellular organelles; and that the liberation of Aβ involves a transmembrane secretase enzyme acting on a transmembrane APP CTF substrate. Thus, from a cell biology perspective, sorting mechanisms that cause APP and the secretases to colocalize in the same membranous compartment would be expected to play important roles in the regulation of Aβ production.
Over 30% of all proteins are transmembrane proteins (Cobbold et al., 2003), and most are typically sorted via the secretory and endocytic pathways (Figure 1B) (Le Borgne and Hoflack, 1998; Harter and Reinhard, 2000). During the last two decades, great strides have been made in understanding the molecular mechanisms that govern transmembrane protein sorting. The trans-Golgi network (TGN) and the endosome have emerged as especially key “waystation” organelles, where the complex movement of transmembrane proteins is orchestrated, particularly in polarized cells such as neurons (Ponnambalam and Baldwin, 2003; Gleeson et al., 2004; Rodriguez-Boulan and Musch, 2005). Contributing to their importance, a bidirectional pathway exists between these two organelles, providing the only direct intracellular link between the secretory and endocytic pathways (Figure 1) (Le Borgne and Hoflack, 1998). (Note: the “endosome” is, of course, not a singular organelle, but rather is made up of multiple subtypes—including the early endosome, the sorting endosome, the recycling endosome, the late endosome, and the multivesicular body. Nevertheless, for purposes of clarity, unless otherwise specified, we will use the term “endosome” to encompass all subtypes.)
Because of the importance of the trafficking pathways interconnecting the TGN, cell surface, and the endosome, investigators have sought to elucidate many of the basic mechanisms governing protein transport through this itinerary (for review, see Bonifacino and Rojas, 2006). In particular, the sorting signals contained within transmembrane proteins and the specific coat complexes that initiate transport through each leg of this sorting itinerary have been characterized (Kreis and Pepperkok, 1994). The discovery of key findings about the cell biology of protein sorting has chronologically tracked with molecular discoveries related to Alzheimer's disease. For example, in 1990, the asparagiNe-Proline-any-tYrosine (N-P-X-Y) motif in the cytoplasmic tail of type-I transmembrane proteins was identified as an evolutionarily conserved sorting signal that targets proteins for clathrin pit localization and transport via clathrin-coated vesicles from the cell surface to the endosome (Chen et al., 1990) (Figure 2). APP, cloned 4 years earlier, was found to contain this motif, immediately generating specific predictions about its sorting itinerary. In a similar fashion, the sorting of BACE was informed by later reports showing that the D-X-X-L-L dileucine motif, present in the cytoplasmic tail of BACE, is a separate type of sorting signal involved with clathrin-coat transport between the TGN and endosome (He et al., 2005; Wahle et al., 2005). Most recently, at the turn of the century and about the same time that BACE was cloned, cell biological studies characterized a novel coat complex, the retromer, involved primarily in retrograde transport from the endosome (Seaman, 2004; Bonifacino and Rojas, 2006), and the retromer has recently been implicated in APP and BACE sorting (Small and Kim, 2003; Small et al., 2003, 2005; He et al., 2005).
Figure 2.
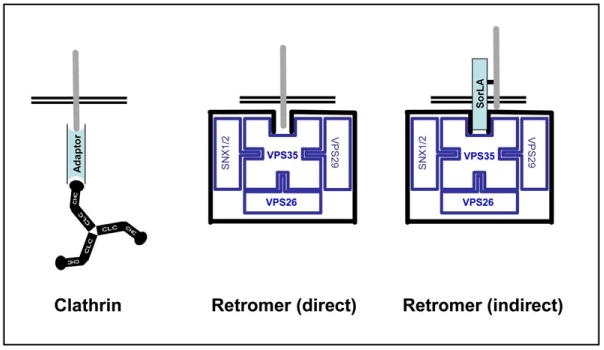
Coat Complexes in Alzheimer's Disease, The clathrin coat (left). The clathrin coat complex transports type-I transmembrane proteins to the endosome from the TGN or from the cell surface. Each clathrin molecule is made up of three clathrin light chains (CLC) and three clathrin heavy chains (CHC). The terminal domains of CHC binds adaptor proteins (blue), which in turn bind the to-be-sorted transmembrane cargo proteins (gray bar). The adaptor proteins impose cargo and itinerary specificity. The dileucine motif found in BACE typically binds GGAs, a family of adaptor proteins engaged in clathrin-mediated transport from the TGN to the endosome. The NPXY motif found in APP binds a wide-range of adaptor proteins (as discussed in the text) engaged in clathrin-mediated transport from the cell surface to the endosome.
The retromer (middle and right panels). The retromer coat complex transports type-I transmembrane proteins from the endosome to the TGN. VPS35 is the core of the retromer and binds directly to the to-be-transported type-I transmembrane protein cargo (gray bar). VPS26, VPS29, and sortin nexins (SNX) 1 or 2 assemble onto VPS35 to generate the complete functional retromer complex. Recent findings suggest that the retromer sorts APP or BACE, either by direct binding to the retromer or by indirect binding to the transmembrane adaptor protein sorLA (blue).
The dovetailing of studies elucidating the general mechanisms that regulate transmembrane protein sorting with studies isolating the specific transmembrane proteins related to Alzheimer's disease has suggested potential pathogenic pathways and forms the basis of this review. We will first attempt to reconcile sometimes conflicting findings and pinpoint the membranous compartments in which neuronal APP is most commonly cleaved to generate discrete fragments. Next, we will review the known or postulated sorting mechanisms that regulate the transport of APP and the transmembrane secretase enzymes through relevant pathways of the neuron. In so doing, we will generate a list of sorting molecules that play roles in Aβ biogenesis and are therefore potential points for disease origins. Finally, we will end by reviewing recent findings showing that defects in some of these sorting pathways exist in late-onset Alzheimer's disease and may contribute to its pathogenesis.
The Membranous Compartments in which APP Is Cleaved
The First Enzymatic Step in the Amyloidogenic Pathway: β-Cleavage
The earliest studies of β-cleavage were conducted in the early 1990s, when β-cleavage and Aβ generation were discovered to accompany normal cellular metabolism (Shoji et al., 1992; Seubert et al., 1992, 1993; Haass et al., 1992). Both APP and BACE are sorted through the secretory and endocytic pathways and reside, at least transiently, in almost all membranous compartments of these pathways. A number of early cell biology studies suggested that the endocytic process and the endosomal/lysosomal pathways contributed significantly to Aβ production (Caporaso et al., 1992a; Gandy et al., 1992; Nordstedt et al., 1993; Caporaso et al., 1994; Perez et al., 1996; Ono et al., 1997; Marquez-Sterling et al., 1997; Soriano et al., 1999).
Madin-Darby canine kidney (MDCK)-based systems were applied to the study of polarization of APP trafficking and sorting, as models for similar events that go on in neuronal cells (Haass et al., 1994, 1995a; Capell et al., 2002). In MDCK cells, apical sorting is often associated with proteins destined for axonal transport, while basolateral sorting is often associated with somatodendritic destinations (Ahn et al., 1996). In the MDCK system, there is selective basolateral secretion of sAPP and Aβ, both of which are generated intracellularly (Ahn et al., 1996). BACE, however, is apically sorted, where Aβ generation is limited (Capell et al., 2002). When a relative excess of APP is provided to the apical surface, a substantial fraction of that APP is converted to Aβ (Capell et al., 2002). This situation was considered potentially relevant to neurons since APP was known to be highly targeted for axonal transport (Koo et al., 1990). These data from MDCK cells tended to predict that, since APP and BACE may be targeted for axonal transport to nerve terminals, the presynaptic nerve terminal might be an especially efficient site for Aβ generation. The identity of the particular β-cleavage-competent organelle most relevant to the pathogenesis of Alzheimer's disease has been a matter of debate. Kindling this debate were apparently discrepant findings in studies reporting that Aβ production could be demonstrated in the cultured cell to occur, alternately, in the ER, in the TGN, or in the endosome (Haass et al., 1995b; Chyung et al., 1997; Cook et al., 1997; Greenfield et al., 1999; Skovronsky et al., 2000a; Petanceska et al., 2000).
Many factors contributed to the difficulty in resolving which of these compartments was most likely responsible for generation of the Aβ that causes pathology. Since BACE was only isolated and cloned in 1999, the task of localizing where BACE activity resides in the cell in studies from the 1990s could only be inferred based on indirect evidence. An example is the chemical poisoning of specific organelles with compounds such as brefeldin A and monensin and then determining how this manipulation affected the profile of intracellular and secreted APP fragments (Caporaso et al., 1992a, 1992b, 1994).
More generally, however, since they are both transmembrane proteins, APP and BACE do in fact reside, at least temporarily, in multiple organelles. It is important to emphasize that the mechanisms that sort transmembrane proteins are very much dependent on specific experimental conditions, and thus methodological differences across studies might shift the sites where APP and BACE interact. Methodological differences, therefore, probably account for some apparently discrepant findings in early cell culture studies investigating the cell biology of APP processing. One important source of variability is that studies often differed in whether wild-type or Swedish (K670N/M671L) mutant APP was transfected into cells under investigation. Since the Swedish mutation in particular enhances the efficiency of BACE cleavage of APP (Citron et al., 1992), metabolism of this mutant yields particularly robust generation of Aβ. For this reason, many investigators, past and present, have studied the cell biology of Swedish mutant APP. However, as compared to wild-type APP, Swedish APP is cleaved by BACE in more proximal compartments that are probably not identical, at least on a quantitative stoichiometric basis, to those where wild-type APP is cleaved by BACE (Haass et al., 1995b; Perez et al., 1996, 1999; Sodhi et al., 2004). Thus, while there was early agreement that most Swedish APP is β-cleaved in the TGN, there remained uncertainty about where most wild-type APP was β-cleaved. Furthermore, there was always the concern that artificially overexpressing APP or BACE might artifactually shift the locus of APP processing.
The isolation and cloning of BACE provided a much-needed experimental handle with which the cell biology of its interaction with APP could be investigated. A detailed analysis of the sorting itinerary of BACE provided clear evidence that mature BACE is found predominantly in the endosome, with relatively lower levels distributed among the cell surface and the TGN (Huse et al., 2000). Virtually no mature BACE is found in the lysosome or in the endoplasmic reticulum (Huse et al., 2000). The relative absence of BACE in the lysosome was important, since biochemical studies suggested that BACE activity is maximized in an acidic environment. Thus, the possibility that the lysosome was an important site for β-cleavage was excluded.
Based on the cellular localization of BACE alone, the site of β-cleavage was narrowed down significantly, leaving open the possibility that important β-cleavage occurs in the TGN, the cell surface, the endosome, or in the sorting pathways that connect these sites. Among the remaining possibilities, the endosome and the endocytic pathway are the most acidic, making these sites best suited for BACE activity. Nevertheless, the relatively subtle pH difference between endosomes and the TGN does not by itself provide sufficient evidence to resolve this important issue. In support of earlier findings, recent studies have shown that blocking endocytosis reduces Aβ production (Carey et al., 2005), whereas enhancing endocytosis favors its production (Grbovic et al., 2003), thus confirming that the endocytic pathway can play an important role in APP processing. However, these studies do not rule out the importance of the TGN and the secretory pathway for β-cleavage.
Only by explicitly comparing each of the three membrane compartments can a more definitive answer be provided. Indeed, the results of a number of recent studies seem to converge on the endocytic pathway as the one in which most β-cleavage of wild-type APP is most likely to occur. A recent study that directly tested for sites of BACE-APP interactions employed fluorescence resonance energy transfer (FRET) analysis, arguably the best technique for studying protein-protein interactions in living cells (Kinoshita et al., 2003). This study showed that wild-type APP binds BACE with greatest efficiency in the endosome, less so on the cell surface, and to a negligible degree in the TGN and secretory pathway (Kinoshita et al., 2003). Interestingly, an increase of APP:BACE binding was observed in the TGN when the Swedish mutant form of APP was investigated, agreeing with the earlier evidence that β-cleavage of Swedish mutant APP occurs preferentially within the TGN (Haass et al., 1995b).
In summary, although the interaction of APP with BACE can, in principle, occur in the ER, in the TGN, on the cell surface, in the endosome, or along the sorting pathways interconnecting them, the preponderance of circumstantial and direct evidence suggests that, for wild-type APP, β-cleavage probably occurs most commonly in the endosome and the endocytic pathway.
The Second Enzymatic Step in the Amyloidogenic Pathway: γ-Cleavage
Identifying the membranous site in which γ-secretase cleaves the carboxyl-terminal fragment of APP, the final step in liberating the Aβ peptide, has been even more controversial than localizing the cleavage site for β-cleavage. As discussed above, the fact that β-cleavage turned out to be governed by a monomeric protein greatly simplified the search for its site of action. In contrast, not only is the γ-secretase a multimeric complex, made up of multiple proteins or protein fragments, but there is still debate about whether its full complement of component elements has been established. Although most studies agree that the presenilins are components of the γ-secretase, there are a number of experimental and biological reasons to explain why mapping the cellular location of the presenilins might not necessarily indicate the site of γ-cleavage. First, compared to endogenous patterns, overexpressing exogenous presenilins appears to shift their subcellular location (Thinakaran et al., 1996; Lee et al., 1997; Lah et al., 1997). Second, there is coordinated regulation of trafficking of various γ-secretase components (Zhang et al., 2005; Niimura et al., 2005). As a result, expressing presenilins with and without the other putative elements of the γ-secretase complex (nicastrin, APH1, PEN2) also shifts the membranous compartment in which presenilins or the other γ-secretase components are concentrated (Zhang et al., 2005; Niimura et al., 2005). Third, γ-secretase activity is dependent on membrane-bound cleaved fragments of presenilin, and presenilin fragments may exist in different compartments compared to the holoprotein (Petanceska et al., 2000), which is itself a zymogen and therefore is inactive until it is cleaved.
Thus, since simple protein localization studies are difficult to interpret, localizing γ-secretase activity, while indirect, may be more informative in identifying sites in which γ-cleavage occurs. In this regard, it is notable that a number of studies have established that γ-cleavage can occur in the presynaptic terminal of the neuron (Kamenetz et al., 2003). Components of the TGN are specifically excluded from the axon and axon hillock (Caporaso et al., 1994). Therefore, among the organelles implicated in γ-cleavage, the endosome is the only key Aβ-generating intracellular organelle that exists in the presynaptic terminal, therefore suggesting that endosomes, or the transport pathways into endosomes, contain the molecular machinery necessary for both β- and γ-cleavage (Koo et al., 1990; Ikin et al., 1996; Marquez-Sterling et al., 1997; Lah et al., 1997; Buxbaum et al., 1998a). Pathogenically, a convergence of findings suggests that Aβ biogenesis in the nerve terminal is the processing site of APP that is most relevant to the pathophysiology of Alzheimer's disease in patients (Arnold et al., 1991; Beach and McGeer, 1992; Rogers and Morrison, 1985) and in mouse models of disease (Buxbaum et al., 1998a; Lazarov et al., 2002; Sheng et al., 2002; Lee et al., 2005). Thus, based on the site within the neuron in which both β- and γ-cleavage occurs, it can be inferred that the γ-secretase is active, in a pathogenically relevant manner, in the endosome or endocytic pathway (Lah and Levey, 2000; Fukumori et al., 2006). Indeed, recent studies using a reporter system that couples γ-secretase cleavage to generation of a fluorescent signal shows unequivocally that, in cultured cells, most γ-cleavage, and hence, most Aβ generation, occurs in endosomes (Kaether et al., 2006).
Lipids appear to play important roles in APP processing. Lipid rafts, perhaps within endosomal membranes, may act by coordinating the proximity of APP, BACE, and γ-secretase (Riddell et al., 2001; Ehehalt et al., 2003; Cordy et al., 2003; Vetrivel et al., 2004, 2005). The role of cholesterol in APP metabolism has received much attention, and a proper review of that literature would occupy far more space than is allotted here. Suffice it to say that there are many important observations relating cholesterol to APP metabolism, but elucidation of the basis for that link has been challenging and frustrating. Polymorphisms in the major cholesterol transport protein, apolipoprotein E, specify risk for Alzheimer's in as many as 25%–30% of cases of the common form of Alzheimer's (Strittmatter et al., 1993; Corder et al., 1993; Schmechel et al., 1993). Cholesterol-lowering drugs decrease Aβ levels released from cultured neurons (Simons et al., 1998) and attenuate Aβ accumulation in the brains of plaque-forming transgenic mice (Refolo et al., 2000, 2001). Surprisingly, however, the use of genetic manipulation to lower circulating cholesterol levels has no effect on brain Aβ (Fagan et al., 2004). Clearly, additional investigation will be required before one can develop a satisfactory and parsimonious model for the relationship between cholesterol and Aβ. From a clinical perspective, the link between cholesterol and Alzheimer's is being tested in a large, placebo-controlled, double-blind clinical trial in which the potential for the cholesterol-lowering drug simvastatin to slow the progression of Alzheimer's is being evaluated. Certainly, if the simvastatin trial is positive, this will provide enormous impetus for a redoubled effort toward explaining the “Alzheimer-cholesterol connection.”
Sorting Mechanisms in Alzheimer's Disease
Delivering transmembrane proteins to their precise intracellular destinations is of critical importance to the biology of the cell, and it is therefore not surprising that a growing number of sorting mechanisms are being described, dedicated to the faithful execution of this complex process (van Vliet et al., 2003). Defects in the sorting protein sorCS1 have recently been associated with increasing risk for another complex disorder, type-2 diabetes mellitus (Clee et al., 2006). For the next section of this review, we will be guided by the sorting itinerary proven important for APP processing. As a result, we will restrict our focus to the sorting mechanisms governing transport among the TGN, cell surface, and endosome. Specifically, since they are monomeric proteins, the sorting of APP and BACE is best understood. Therefore, we will review known or postulated sorting mechanisms involved in the transport of AP and BACE.
Vesicular Transport of APP and BACE
Coat complexes play critical roles in the budding of transport vesicles off donor membrane compartments that initiates the process of transmembrane protein sorting. Based on the sorting motifs expressed in APP and BACE, these proteins were predicted to be transported by the clathrin coat complex (Bonifacino and Traub, 2003). Clathrin proteins are constructed from three heavy chains and three light chains (Hirst and Robinson, 1998). Clathrin does not bind directly to its cargo, but rather, the globular tail of each clathrin heavy chain binds “adaptor” proteins, which, in turn, bind to the cytoplasmic tails of the transmembrane protein cargo (Hirst and Robinson, 1998). Clathrin coats are involved in two main sorting routes—the endocytic pathway connecting the cell surface to the endosome, and the pathway connecting the TGN to the endosome (Traub, 2005) (Figure 1). While identical clathrin proteins are involved in different sorting pathways and in sorting different transmembrane proteins, it is the variety of adaptor proteins that is believed to be responsible for imposing sorting specificity at the stage of cargo selection (Hirst and Robinson, 1998).
As mentioned above, the cytoplasmic tail of APP contains the NPXY amino acid motif, a motif that is evolutionarily conserved and which governs the targeting of cargo to clathrin-coated vesicles (Chen et al., 1990) (Figure 2). Motifs such as this are important for generating predictions about sorting events. However, direct experimental proof of these predictions is required before these sorting itineraries are established. Indeed, mutagenesis studies have been used to assess and confirm that each of the amino acids within the NPXY motif plays some role in the process of transporting APP from the cell surface to the endosome (Chen et al., 1990). A number of families of adaptor proteins have been found to mediate the binding of the NPXY motif to clathrin coats. Accordingly, a growing number of clathrin adaptor proteins have been found to bind APP's NPXY motif and therefore serve as regulators of APP endocytosis. These adaptor proteins include the autosomal-recessive hypercholesterolemia (ARH) protein (Noviello et al., 2003), disabled family member (Dab) 1 (Homayouni et al., 1999), c-Jun N-terminal kinase interaction (Jip1b) protein (Inomata et al., 2003; Matsuda et al., 2001; Scheinfeld et al., 2002), members of the Fe65 family (including Fe65, Fe65L1, and Fe65L2) (Fiore et al., 1995; Borg et al., 1996; Bressler et al., 1996; Guenette et al., 1996; Zambrano et al., 1997; Duilio et al., 1998; Tanahashi and Tabira, 1999), and members of the X11 family of proteins, in particular X11a (Borg et al., 1996; McLoughlin et al., 1999; Tomita et al., 1999). In many instances, these adaptors reduce generation of Aβ by causing APP to spend more residence time at the plasma membrane or within the ER (Watanabe et al., 1999; Sabo et al., 1999).
In contrast to the NPXY signal within the APP cytoplasmic tail, BACE expresses a dileucine sorting motif in the corresponding domain (Rapoport et al., 1998). This sorting motif also binds the clathrin coat complex; however, transmembrane proteins that express the dileucine motif are typically transported through the pathway that directly connects the TGN and the endocytic system (Figure 2) (Boman, 2001; Bonifacino, 2004). The reason for this different sorting itinerary is attributed to the Golgi-localized γ-ear-containing ARF-binding (GGA) family of adaptor proteins, including GGA1, GGA2, and GGA3 (Boman, 2001; Bonifacino, 2004). GGA1 appears to be the GGA subtype that controls BACE transport from the endosome to the TGN. GGA proteins express a VPS-27, Hrs, and STAM (VHS) domain that binds the dileucine motif of BACE (Boman, 2001; Bonifacino, 2004). Localized mainly in the TGN and less so in endosomes, GGAs are involved in sorting transmembrane proteins away from the TGN and toward the endosome. Somewhat surprisingly, some, though not all, studies suggest that BACE is transported from the endosome to the TGN through GGA-mediated sorting (He et al., 2005).
The retromer is the second coat complex implicated in the transport of BACE (Figure 2) and perhaps APP as well (Small and Kim, 2003; Small et al., 2003, 2005; He et al., 2005) (Figure 2). First described in yeast, the retromer is a newly characterized coat complex, made up of VPS35, VPS26, VPS29, VPS5, and VPS17 (VPS = Vacuolar Protein Sorting), which, in yeast, serves to sort the type-I transmembrane protein VPS10 from the endosome back to the TGN (Seaman et al., 1998; Edgar and Polak, 2000; Haft et al., 2000; Nothwehr et al., 2000; Pfeffer, 2001; Reddy and Seaman, 2001; Seaman and Williams, 2002). The name “retromer” is derived from the role of this coat in retrograde sorting itineraries. VPS35 serves as the core of the retromer complex, binding not only the other retromer elements, but also binding the cytoplasmic tail of the transmembrane protein cargo that is being sorted (Figure 2) (Reddy and Seaman, 2001).
Except for VPS17, mammalian (including human) homologs of retromer molecules have been identified (Haft et al., 2000) and are highly expressed in the brain. VPS35, VPS26, and VPS29 have single mammalian homologs, which are known by the same names (Haft et al., 2000). Recent studies suggest that VPS26 has two isoforms, termed VPS26a and VPS26b (Kerr et al., 2005). VPS5 has two mammalian homologs, known as sorting nexin 1 and sorting nexin 2 (Haft et al., 2000). Because of these homologies, the retromer was predicted to mediate the sorting of type-I transmembrane proteins in mammals. In 2004, this prediction was confirmed when a number of groups established that the retromer is involved in transporting type-I transmembrane proteins such as the cationic-independent mannose-6-phosphate receptor (Arighi et al., 2004; Seaman, 2004) and the polymeric immunoglobulin G receptor (Verges et al., 2004). Furthermore, these studies suggested that another molecule, known as sortilin, is also transported by the mammalian retromer (Seaman, 2004). This observation is particularly interesting since sortilin is one of the five known type-I transmembrane proteins that express VPS10, the primary cargo of the yeast retromer (Hampe et al., 2001), suggesting functional homologies between the yeast and mammalian retromer. The other members of this family are known as sorLA (also known as LR11), sorCS1, sorCS2, and sorCS3 (Hampe et al., 2001).
Although the retromer is highly expressed in the brain, until recently, the type-I transmembrane proteins transported by the neuronal retromer were unknown. The first clues about which type-I transmembrane proteins might be sorted by the neuronal retromer came from a study exploring microarray data generated from human brain tissue (Small and Kim, 2003; Small et al., 2003). Among a list of possible retromer cargo molecules, this study demonstrated that sorLA and BACE were among the type-I transmembrane molecules whose expression levels cross-correlated most strongly with levels of neuronal VPS35 (Small and Kim, 2003; Small et al., 2003). Although this cross-correlation approach has been successfully used in microarray studies to identify molecules that are candidates for interacting with one another (Bhardwaj and Lu, 2005), these predictions can, of course, only be used as a screen whose results require confirmation by direct experimental investigation. Direct evidence that the retromer plays a role in BACE sorting has recently emerged from a study that used siRNA to knock down VPS26 in cell culture (He et al., 2005). This manipulation, which was originally used in the studies implicating the retromer in mannose-6-phosphate receptor sorting, reduces both VPS26 and VPS35 levels, causing retromer dysfunction (Arighi et al., 2004; Seaman, 2004; Verges et al., 2004). By knocking down VPS26, retromer dysfunction was shown to missort BACE as manifested by an increase in endosomal BACE (He et al., 2005). Experimental confirmation regarding interaction of VPS35 with sorLA has not yet been provided. Nevertheless, since sorLA has close homologies to sortilin, its VPS10-containing family member, one would plausibly speculate that sorLA is also sorted by the neuronal retromer. Interestingly, a number of studies have shown that sorLA binds APP and BACE (Andersen et al., 2005; Offe et al., 2006). Therefore, sorLA and perhaps other VPS10-containing molecules might function as the equivalent of an adaptor protein, binding the retromer complex to cargo proteins such as APP (Figure 2). Of note, although some studies suggest that GGAs transport BACE along the same sorting pathway—from the endosome to the TGN—there is no evidence that GGA and retromer sorting interact or compete with each other.
Whereas coat complexes begin the process of transmembrane sorting by forming transport vesicles at the donor membrane compartment, the process ends, of course, when the cargo travels over its route and successfully arrives at its destination membrane (Bonifacino and Glick, 2004). It is worth noting that transport vesicles typically travel over microtubule-supported routes, and, therefore, any process that interrupts the integrity of microtubules might affect transmembrane protein sorting. One major neuropathological feature of Alzheimer's disease that is not discussed in detail here involves accumulation of intracellular precipitates of the cytoskeleton and formation of structures known as neurofibrillary tangles (Alzheimer et al., 1995). While a discussion of neurofibrillary tangle pathogenesis is beyond the scope of this review, the fact that the major tangle protein, tau, is a microtubule-binding protein merits mention in this review of protein sorting (see Geschwind, 2003, for review).
The fusion of a transport vesicle to the membrane of the targeted organelle completes the process of transmembrane protein sorting, and this fusion step is governed by three main families of proteins—soluble N-ethylmaleimide sensitive factor-attachment protein receptor (SNARE) proteins (Rothman, 1994), Rab (a subfamily of the Ras superfamily of low-molecular-mass GTPases) proteins (Novick and Zerial, 1997), and Sec1/Munc18 (SM) proteins (Toonen and Verhage, 2003). Membrane fusion and cargo delivery occur when SNAREs expressed in the membranes of the transport vesicle (e.g., syntaxins and vesicle-associated membrane proteins [VAMPs]) interact with a single SNARE protein expressed in the membrane of the recipient or-ganelles, while the Rabs and SM proteins play important roles in mediating this interaction. The fidelity of vesicular transport, in which vesicles are differentially delivered to their proper destinations, is governed by the specific combination of SNARES, Rabs, and SM proteins found in the receptor membranes. Although among these families of proteins, the specific combinations have not yet been identified for the transport of APP and BACE through the cell surface, endosome, and TGN, a select group of these fusing proteins have been implicating in this sorting itinerary. This list includes syntaxins 5, 6, 7, 8, 13, and 16 (Prekeris et al., 1999); VAMPs 5, 7, and 8 (Prekeris et al., 1999); Rabs 5 and 11 (Pfeffer, 1999); the SM protein VPS45, Munc 13, and Munc 18 (Toonen and Verhage, 2003; Rossner et al., 2004).
Phosphorylation-State-Specific Sorting of APP and BACE
The phosphorylation states of substrate proteins and lipids also play roles in APP and BACE transport. The phosphorylation states of membrane proteins such as APP or BACE, and/or the phosphorylation states of their specific interacting proteins, provide for dynamic regulation of signal transduction and protein sorting on a moment-to-moment basis, thereby integrating protein sorting and neurotransmission (Mostov and Cardone, 1995; Clague and Urbe, 2001; Bonifacino and Traub, 2003). A striking example is that of regulated ectodomain shedding of APP (Buxbaum et al., 1990, 1992; Caporaso et al., 1992b; Nitsch et al., 1992; Gillespie et al., 1992; Pedrini et al., 2005). During regulated shedding, first messengers such as neurotransmitters and hormones (Buxbaum et al., 1992; Nitsch et al., 1992; Jaffe et al., 1994; Xu et al., 1998; Qin et al., 2006) impinge upon neurons and direct APP toward the cell surface and away from the TGN and endocytic pathways (Xu et al., 1995), and hence away from BACE. At the cell surface, APP can be processed by a nonamyloidogenic pathway, known as the α-secretase pathway and defined by the metallo-proteinases ADAM-9, ADAM-10, and ADAM-17 (Buxbaum et al., 1998b; Esler and Wolfe, 2001; Allinson et al., 2003; Postina et al., 2004; Kojro and Fahrenholz, 2005). ADAM is an acronym derived from “a disintegrin and metalloproteinase.” The molecular mechanism of regulated shedding remains to be fully elucidated but appears to involve phosphorylation of components of the TGN vesicle biogenesis machinery (thereby increasing APP delivery to the cell surface; Xu et al., 1995) as well as phosphorylation of protein components of the endocytic system (thereby blocking APP internalization; Chyung and Selkoe, 2003; Carey et al., 2005). The phosphorylation states of APP and BACE do not appear to be involved in this process (Gandy et al., 1988; Oishi et al., 1997; da Cruz e Silva et al., 1993; Jacobsen et al., 1994; Pastorino et al., 2002). With regard to Aβ generation, this phenomenon is noteworthy because hyperactivation of the α pathway (e.g., with a combination of simultaneous protein kinase activation and protein phosphatase inhibition) can lead to relatively greater cleavage of APP by α-secretase(s) (Caporaso et al., 1992b; Gillespie et al., 1992), thereby reducing or completely abolishing Aβ generation (Buxbaum et al., 1993; Gabuzda et al., 1993; Hung et al., 1993). Interest in this phenomenon has recently been revived with the demonstration that microdialysis techniques can be used to demonstrate and quantify regulated shedding and regulated Aβ generation in the brains of living experimental animals (Cirrito et al., 2005; Selkoe, 2006).
Recent evidence suggests that axonal transport of APP (Lee et al., 2003) and perhaps also prolyl isomerization might be modulated by the state of phosphorylation of the APP cytoplasmic tail at threonine-668 (Pastorino et al., 2006). APP is axonally transported in holoprotein form (Koo et al., 1990; Buxbaum et al., 1998a); hence, the phosphorylation of threonine-668 was proposed to serve as a “tag,” targeting phospho-forms of APP for delivery to the nerve terminal (Lee et al., 2003). However, recent evidence calls into question the proposal that the phosphorylation state of threonine-668 plays a major physiological role in APP localization or Aβ generation, since threonine-to-alanine-668 knock-in mice show normal levels and subcellular distributions of APP and its metabolites, including Aβ (Sano et al., 2006). There is compelling evidence, however, that, once at the nerve terminal, APP is processed, generating Aβ locally at the terminal and releasing Aβ at, near, or into the synapse (Kamenetz et al., 2003).
The cytoplasmic tail of BACE also undergoes reversible phosphorylation, and that event appears to specify its recycling (von Arnim et al., 2004; He et al., 2005). In cell lines, the dephospho- and phospho-forms of BACE appear to perform with similar efficiencies in generating Aβ40 and Aβ42 (Pastorino et al., 2002), but this has not been evaluated in primary neuronal cultures. This failure of Aβ generation to be regulated by BACE recycling is somewhat unexpected since, as reviewed above, most Aβ is believed to arise from the endocytic pathway. Hence, one would expect that increasing BACE concentration in the endocytic pathway would increase generation of Aβ. One explanation for this unexpected result is that substrate may be limiting in post-TGN compartments, and therefore increased levels of BACE are unable to raise Aβ generation. This notion agrees with the proposal mentioned above that regulated shedding acts at the TGN to divert APP molecules toward the plasma membrane as a means of lower generation of Aβ at least in part because a limited pool of APP is transported out of the TGN (Buxbaum et al., 1993; Skovronsky et al., 2000b). Indeed, in some neuronal-like cell types, over 80% of the newly synthesized moles of APP are degraded without generating obvious discrete metabolic fragments (Caporaso et al., 1992b).
Clathrin-independent endocytosis of transmembrane proteins is regulated by protein phosphorylation (Robertson et al., 2006). Further, two components of the endocytosis machinery, dynamin and amphiphysin, control clathrin-mediated endocytosis in a fashion that is sensitive to their direct phosphorylation by the protein kinase cdk5 (Tomizawa et al., 2003; Nguyen and Bibb, 2003). Retromer function is regulated by a separate complex of molecules known as “complex II” (Burda et al., 2002). Complex II includes several catalytic functions that direct retromer action. The phosphoinositide kinase VPS34 binds the protein kinase VPS15, and then, secondarily, VPS30 and VPS38 are recruited and the four molecules comprise the complete complex II (Burda et al., 2002). Thus, complex II action is modulated not only by protein phosphorylation but also by lipid phosphorylation (Stack et al., 1995). Some investigators have proposed that the PI 3-kinase component of complex II directs synthesis of a specific pool of endosomal PI3, which, in turn, activates or stimulates assembly of the retromer complex, thereby ensuring efficient endosome-to-Golgi retrograde transport (Stack et al., 1995). These regulatory mechanisms may have implications for Aβ generation, but such a connection, if one exists, remains to be elucidated. Clearly, much remains to be studied about the intricacies of retromer function and dysfunction.
Presenilins and APP Sorting
Soon after the discovery of presenilins, gene targeting experiments were performed in mice in order to investigate the essential bioactivities of these complex, polytopic, molecules, especially presenilin 1 (PS1) (Wong et al., 1997; Naruse et al., 1998). In cells from PS1-deficient mice, delivery of multiple type-I proteins to the cell surface was observed to be disturbed; APP and the p75 neurotrophin receptor were among those missorted proteins (Naruse et al., 1998). This work was somewhat overshadowed, however, when cells from PS1-deficient mice were demonstrated to be incapable of generating Aβ (De Strooper et al., 1998). This observation placed APP and PS1 on a common metabolic pathway for the first time and was rapidly followed by demonstration that PS1 did, indeed, contain the catalytic site of γ-secretase, as established by cross-linking of γ-secretase inhibitors to PS1 (Li et al., 2000a, 2000b).
The unusual intramembranous localization of two aspartate residues led to the postulation that these amino acids were forming the active site of an aspartyl proteinase (Wolfe et al., 1999). This dovetailed with the apparent fact that APP CTFs were cleaved by regulated intramembranous proteolysis (RIP), and when the aspartates were mutated to alanines, γ-secretase activity was abolished (Wolfe et al., 1999). RIP was, at the time, a relatively recently recognized phenomenon, and conventional wisdom up to that point had held that the hydrophobicity of membranes would preclude the entry of water into the lipid bilayer to enable hydrolysis of peptide bonds. Even to this day, the mechanism that provides the capability for surmounting that energy barrier is poorly understood. The popular formulation at that point was that PS1 was a proteinase, and the notion that PS1 was a trafficking factor was underemphasized. The possibility was also raised that perhaps aberrant trafficking in PS1-deficient cells was due to the inability of some unidentified PS1 substrate trafficking factor to function properly in its uncleaved state since its cognate protease (PS1) was absent.
Beginning in the last few years, experiments in cultured cells and cell-free assays have begun to yield compelling evidence that PS1 serves a trafficking function in addition to its catalytic function. However, these recent experiments have a common potential alternative interpretation: i.e., the exact same processing effects would be predicted in the event that key trafficking proteins were important substrates for cleavage by PS1. Therefore, when PS1 is deficient, post-TGN trafficking of membrane protein cargos would become abnormal due to deficient processing of the trafficking factor/PS substrate (Kaether et al., 2002; Wang et al., 2004; Wood et al., 2005; Rechards et al., 2006).
Most PS1-deficient mice and cells are highly compromised and resemble Notch-deficient mice and cells (Wong et al., 1997). This is not entirely unexpected since Notch is a substrate for cleavage by γ-secretase as are another several dozen type-I transmembrane proteins, including cadherin, erb-b4, and the p75 NGF receptor (De Strooper et al., 1999; Struhl and Greenwald, 1999; for review, see Fortini, 2002). Therefore, PS1 deficiency can lead to dysfunction of a host of proteins whose physiological function requires cleavage by RIP in order to release their cytoplasmic domains. In many examples, the cytoplasmic domain released by γ-secretase appears to diffuse rapidly to the nucleus, where these intracellular domains (ICDs) such as Notch intracellular domain (NICD) modulate gene transcription (Cupers et al., 2001; Fortini, 2002; Cao and Sudhof, 2001).
PS1-mediated trafficking appears to localize to post-TGN steps of trafficking of type-I transmembrane proteins (Annaert et al., 1999; Kaether et al., 2002; Cai et al., 2003; Wang et al., 2004, 2006; Wood et al., 2005; Zhang et al., 2006; Cai et al., 2006a, 2006b; S.G. et al., unpublished data). This role for PS1 in regulation of APP trafficking has been implicated in both cell culture and cell-free in vitro reconstitution studies (Annaert et al., 1999; Kaether et al., 2002; Cai et al., 2003; Wang et al., 2004, 2006; Wood et al., 2005; Zhang et al., 2006; Cai et al., 2006a, 2006b; S.G. et al., unpublished data). Pathogenic PS1 mutations retard egress of APP from the TGN by a mechanism that appears to involve phospholipase D (Cai et al., 2006a, 2006b), a known TGN budding modulator (Kahn et al., 1993). It is clear that the mutations that have been tested so far increase the residence time at the TGN while also increasing the Aβ42/40 ratio (Kahn et al., 1993). Recent data suggest that TGN retention per se can increase generation of Aβ42/40 in cerebral neurons in vivo, indicating that abnormal post-TGN trafficking of APP might be sufficient to initiate Aβ accumulation (S.G. et al., unpublished data).
The pathogenic PS1 defect can be corrected in cell culture and in cell-free systems following supplementation of the budding factor phospholipase D (PLD) (Cai et al., 2003, 2006a, 2006b). The molecular details of how PS1 and PLD are connected remain obscure; however, as cargos other than APP are found to be missorted, including, e.g., tyrosinase (Wang et al., 2006), the notion that PS1 has a protein trafficking function has become more widely appreciated and accepted. Now, the challenge is to identify at the molecular level those factors that selectively favor cleavage at the Aβ42-43 scissile bond.
PS1 has also been implicated in trafficking of APP and perhaps its carboxyl-terminal fragments out of the endosome (Zhang et al., 2006). Thus, PS1 dysfunction could also result in retention of APP and CTFs within the endocytic compartment, which, in turn, would favor Aβ generation. Thus, accumulating evidence implicates PS1 in the regulation of APP trafficking. The possibility exists that the local environment within the TGN or the endocytic system contributes to misalignment of mutant PS1 and APP carboxyl-terminal fragments, thereby favoring generation of Aβ42. Such a mechanism has been implicated in other diseases (e.g., cystic fibrosis) that are also caused by missense mutations in polytopic proteins (Gentzsch et al., 2004).
Sorting Defects in Late-Onset Alzheimer's Disease
As reviewed, the sorting of APP and BACE among the membranous compartments of the TGN, cell surface, and the endosome is a dynamic and highly regulated process. Thus, even subtle sorting defects that would slow the trafficking in and out of any individual membranous compartment could, in principle, increase Aβ generation. Guided by the sorting mechanisms that transport APP and BACE from the surface to the endosome, and from the endosome back to the TGN, a list of candidate molecules can be generated in which an abnormality could, in theory, contribute to the accumulation of Aβ found in late-onset Alzheimer's disease (Table 1).
Table 1. Molecules that Participate in Sorting APP and/or BACE.
| Coat complexes | clathrin heavy chains, clathrin light chains, VPS35, VPS26A, VPS26B, VPS29, sorting nexin 1, sorting nexin 2, ARF |
|---|---|
| Coat adaptors | sorLA, sortilin, ARH, Numb, Dab1, Dab2, Jip1b, Fe65, Fe65L1, Fe65L2, X11α, GGA1, GGA2, GGA3 |
| Factors for docking and/or fusion | syntaxin 5, 6, 7, 8, 13, 16; VAMP 5, 7, 8; Rab 5, 11; SM protein VPS45, Munc 13, 18 |
| Protein or lipid kinases, phosphatases | PKA, PKC, ERK, cdk5, GSK3, ROCK1, PP1, PP2A, PI3K, Akt, VPS30 (beclin)-VPS34-PI3K |
| Other factors | presenilin 1, presenilin 2, nicastrin, APH1, PEN2, lipid rafts, caveolin, flotillin, cholesterol |
In principle, manipulating any molecule on this list might modulate Aβ generation. Indeed, this predicted effect has already been shown for many of the listed molecules (e.g., Ho et al., 2002; Hill et al., 2003). To date, however, only a few of these molecules have been found to be defective in brain tissue harvested from patients with late-onset Alzheimer's disease. VPS35, VPS26, and Beclin (the mammalian homolog of VPS30) were recently implicated in a study that relied on brain imaging to construct an a priori model predicting how pathogenic molecules should behave—spatially and over time (Small et al., 2005). This model was used as a guide, first in generating gene-expression profiles with microarray technology from areas of the brain most vulnerable (the entorhinal cortex) and relatively resistant (the dentate gyrus) to late-onset Alzheimer's disease, and second in analyzing the complex microarray dataset. As discussed in a recent review, imaging-guided microarray is an effective approach for addressing many of the analytic limitations inherent to profiling gene expression patterns generated from postmortem brains (Lewandowski and Small, 2005). sorLA (also known as LR11), a fourth molecule on the list, was implicated in a study that first used microarray technology to profile the gene expression patterns of lymphocytes of patients with late-onset Alzheimer's disease and then found that sorLA levels are reduced in affected brains (Scherzer et al., 2004). Subsequently, experimental evidence directly established a role for sorLA in APP trafficking and Aβ generation in endosomal compartments (Andersen et al., 2005; Offe et al., 2006).
Although some of these identified molecules serve multiple functions, they are all unified in playing a role in the retromer trafficking pathway. As reviewed, VPS35 and VPS26 are core molecules of the retromer complex itself (Seaman, 2005); VPS30 interacts with retromer-related phosphoinositide kinases, whose activity mediates the binding of the retromer complex to the endosomal membrane (Seaman, 2005); and, sorLA is a putative cargo of the neuronal retromer (Small and Kim, 2003; Small et al., 2003).
Taken together, these studies suggest that the retromer trafficking pathway might play a primary role in Aβ generation and the pathogenesis of late-onset Alzheimer's disease. Furthermore, since APP and/or BACE (directly or indirectly via sorLA) (Figure 2) might be trafficked by the retromer, this generates a specific hypothesis for how Aβ might be overproduced in the absence of defects in APP, BACE, or components of the γ-secretase. Namely, defects in the retromer trafficking pathway are predicted to increase the residence time of APP and/or BACE in the endosome, thereby increasing the β-cleavage step of APP processing (Figure 3). Indeed, a number of studies have suggested that β-cleavage is enhanced in late-onset disease (Fukumoto et al., 2002; Holsinger et al., 2002; Johnston et al., 2005; Li et al., 2004; Tyler et al., 2002; Yang et al., 2003).
Figure 3.
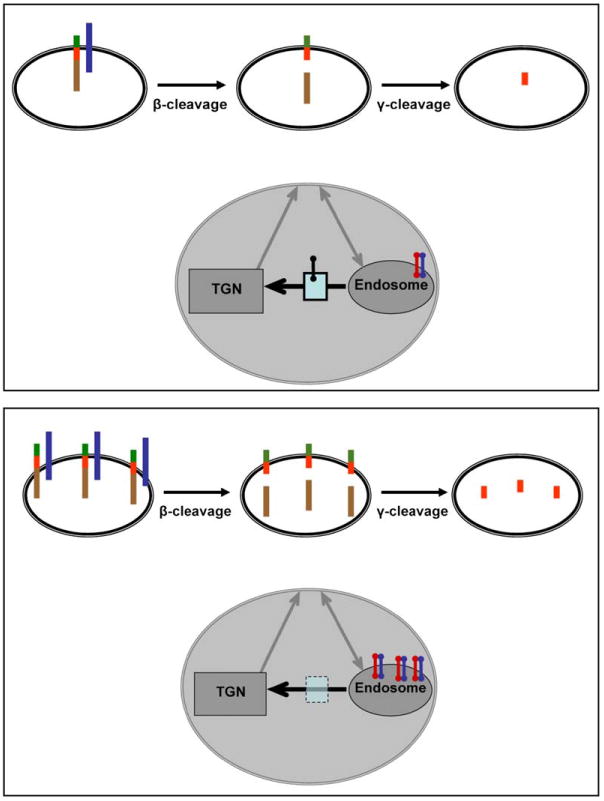
Sorting Defects in Late-Onset Alzheimer's Disease, To date, a handful of sorting molecules involved in APP and BACE transport (see Table 1) have been found to be downregulated in the brains of patients with late-onset Alzheimer's disease. Although serving multiple functions, the identified molecules all play potential roles in retromer-mediated transport (blue box, retromer; black bar, type-I transmembrane cargo protein transported from the endosome to the TGN), suggesting a model whereby endosome-to-TGN transport is a sorting itinerary particularly vulnerable to disease. When compared to normal neurons (upper panel), neurons of Alzheimer's patients (lower panel) are predicted to have diminished transport of APP and/or BACE (black bar) out of the endosome, thereby increasing the colocalization of APP and BACE in endosomes. This, in turn, could lead to excess β-cleavage and excess generation of Aβ.
Nevertheless, many questions remain about the retromer trafficking pathway and Alzheimer's disease. For example, further confirmation is required to establish whether the retromer affects the sorting of APP and/or BACE and whether it does so by directly binding either of these type-I transmembrane proteins. Also, although studies agree that sorLA modulates the production of Aβ and binds APP, there are discrepancies in the currently existing data as to whether sorLA downregulation missorts APP to the TGN or to the endosome (Offe et al., 2006; Spoelgen et al., 2006). Finally, the upstream mechanisms that cause the reductions in VPS35, VPS26, VPS30, and sorLA have not yet been established. In this regard, the reported mismatch between the levels of VPS35 mRNA and protein (Small et al., 2005) suggests that retromer elements might undergo accelerated degradation in the disease state. Although a recent report did not find disease-related polymorphisms for VPS26 (Riemenschneider et al., 2006), a pattern of accelerated degradation is consistent with a genetic defect, suggesting that polymorphisms in other retromer elements might exist (Lewandowski and Small, 2005).
More generally, beyond implicating a specific pathway, the recent convergence of evidence provides proof-of-principle that protein sorting is a cell biological mechanism that can contribute to late-onset Alzheimer's disease. As a complex disorder, many molecular defects are expected to contribute to late-onset Alzheimer's disease, and other sorting molecules on the list are predicted to contribute to disease pathogenesis.
Conclusions
Transmembrane protein sorting is a fundamental property of normal cellular function, and it is therefore not surprising that a growing number of diseases are found to be associated with, or directly caused by, defects in sorting molecules (Olkkonen and Ikonen, 2000; Cobbold et al., 2003; Clee et al., 2006). On theoretical grounds, because APP and its cleaving enzymes are transmembrane proteins, and because Aβ liberation is a membrane-dependent event, protein sorting is a cell biological process uniquely vulnerable to Alzheimer's disease pathogenesis. Specifically, the trafficking of APP and BACE among the TGN, cell surface, and endosome are predicted to be the sorting pathways most relevant to Alzheimer's disease. Indeed, recent studies have begun confirming this prediction, isolating disease-related defects in protein sorting, and future studies are expected to identify additional defects that regulate the transport of APP and its cleaving enzymes through its sorting itinerary (Table 1).
Elucidating the mechanisms that sort APP and the secretases through the TGN, cell surface, and endosome has significantly expanded the understanding of Alzheimer's disease cell biology. More importantly, isolating specific defects in protein sorting opens up unexplored therapeutic avenues, which optimistically, may accelerate the development of effective treatments for this devastating and intractable disease.
Acknowledgments
The authors would like to acknowledge the support of the McKnight Foundation (S.A.S.), the McDonnell Foundation (S.A.S.), the Farber Family Foundation (S.G.), and the National Institutes of Health, including P50 AG08702 and RO1 AG025161 (S.A.S.), R01 AG023611 (S.G.), R01 NS41017 (S.G.), and P01 AG10491 (S.G.). We also thank Huaxi Xu, Rick Kahn, and Allan Levey for helpful discussions, and we thank Jennifer Gropper for administrative support.
References
- Ahn J, Mundigl O, Muth TR, Rudnick G, Caplan MJ. Polarized expression of GABA transporters in Madin-Darby canine kidney cells and cultured hippocampal neurons. J Biol Chem. 1996;271:6917–6924. doi: 10.1074/jbc.271.12.6917. [DOI] [PubMed] [Google Scholar]
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res. 2003;74:342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, “Uber eine eigenartigeErkankung der Hirnrinde. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Amaducci LA, Rocca WA, Schoenberg BS. Origin of the distinction between Alzheimer's disease and senile dementia: how history can clarify nosology. Neurology. 1986;36:1497–1499. doi: 10.1212/wnl.36.11.1497. [DOI] [PubMed] [Google Scholar]
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2005;38:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B. Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol. 1999;147:277–294. doi: 10.1083/jcb.147.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol. 2004;165:123–133. doi: 10.1083/jcb.200312055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- Beach TG, McGeer EG. Senile plaques, amyloid beta-protein, and acetylcholinesterase fibres: laminar distributions in Alzheimer's disease striate cortex. Acta Neuropathol (Berl) 1992;83:292–299. doi: 10.1007/BF00296792. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–1457. doi: 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N, Lu H. Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics. 2005;21:2730–2738. doi: 10.1093/bioinformatics/bti398. [DOI] [PubMed] [Google Scholar]
- Boman AL. GGA proteins: new players in the sorting game. J Cell Sci. 2001;114:3413–3418. doi: 10.1242/jcs.114.19.3413. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS. The GGA proteins: adaptors on the move. Nat Rev Mol Cell Biol. 2004;5:23–32. doi: 10.1038/nrm1279. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans Golgi network. Nat Rev Mol Cell Biol. 2006;7:568–579. doi: 10.1038/nrm1985. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressler SL, Gray MD, Sopher BL, Hu Q, Hearn MG, Pham DG, Dinulos MB, Fukuchi K, Sisodia SS, Miller MA, et al. cDNA cloning and chromosome mapping of the human Fe65 gene: interaction of the conserved cytoplasmic domains of the human beta-amyloid precursor protein and its homologues with the mouse Fe65 protein. Hum Mol Genet. 1996;5:1589–1598. doi: 10.1093/hmg/5.10.1589. [DOI] [PubMed] [Google Scholar]
- Burda P, Padilla SM, Sarkar S, Emr SD. Retromer function in endosome-to-Golgi retrograde transport is regulated by the yeast Vps34 PtdIns 3-kinase. J Cell Sci. 2002;115:3889–3900. doi: 10.1242/jcs.00090. [DOI] [PubMed] [Google Scholar]
- Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer beta/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc Natl Acad Sci USA. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Koo EH, Greengard P. Protein phosphorylation inhibits production of Alzheimer amyloid beta/A4 peptide. Proc Natl Acad Sci USA. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998a;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998b;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- Cai D, Zhong M, Wang R, Netzer WJ, Shields D, Zheng H, Sisodia SS, Foster DA, Gorelick FS, Xu H, Greengard P. Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer's disease-linked presenilin-1 mutant neurons. Proc Natl Acad Sci USA. 2006a;103:1936–1940. doi: 10.1073/pnas.0510710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Netzer WJ, Zhong M, Lin Y, Du G, Frohman M, Foster DA, Sisodia SS, Xu H, Gorelick FS, Greengard P. Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci USA. 2006b;103:1941–1946. doi: 10.1073/pnas.0510708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Capell A, Meyn L, Fluhrer R, Teplow DB, Walter J, Haass C. Apical sorting of beta-secretase limits amyloid beta-peptide production. J Biol Chem. 2002;277:5637–5643. doi: 10.1074/jbc.M109119200. [DOI] [PubMed] [Google Scholar]
- Caporaso GL, Gandy SE, Buxbaum JD, Greengard P. Chloroquine inhibits intracellular degradation but not secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci USA. 1992a;89:2252–2256. doi: 10.1073/pnas.89.6.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Gandy SE, Buxbaum JD, Ramabhadran TV, Greengard P. Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc Natl Acad Sci USA. 1992b;89:3055–3059. doi: 10.1073/pnas.89.7.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P. Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci. 1994;14:3122–3138. doi: 10.1523/JNEUROSCI.14-05-03122.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RM, Balcz BA, Lopez-Coviella I, Slack BE. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC Cell Biol. 2005;11:30. doi: 10.1186/1471-2121-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, Gu Y, Sanjo N, Glista M, Rogaeva E, et al. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature. 2006;440:1208–1212. doi: 10.1038/nature04667. [DOI] [PubMed] [Google Scholar]
- Chyung JH, Selkoe DJ. Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid beta-protein at the cell surface. J Biol Chem. 2003;278:51035–51043. doi: 10.1074/jbc.M304989200. [DOI] [PubMed] [Google Scholar]
- Chyung AS, Greenberg BD, Cook DG, Doms RW, Lee VM. Novel beta-secretase cleavage of beta-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J Cell Biol. 1997;138:671–680. doi: 10.1083/jcb.138.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;370:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Clague MJ, Urbe S. The interface of receptor trafficking and signalling. J Cell Sci. 2001;114:3075–3081. doi: 10.1242/jcs.114.17.3075. [DOI] [PubMed] [Google Scholar]
- Clee SM, Yandell BS, Schueler KM, Rabaglia ME, Richards OC, Raines SM, Kabara EA, Klass DM, Mui ET, Stapleton DS. Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat Genet. 2006;38:688–693. doi: 10.1038/ng1796. [DOI] [PubMed] [Google Scholar]
- Cobbold C, Monaco AP, Sivaprasadarao A, Ponnambalam S. Aberrant trafficking of transmembrane proteins in human disease. Trends Cell Biol. 2003;13:639–647. doi: 10.1016/j.tcb.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci USA. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupers P, Orlans I, Craessaerts K, Annaert W, De Strooper B. The amyloid precursor protein (APP)-cytoplasmic fragment generated by gamma-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J Neurochem. 2001;78:1168–1178. doi: 10.1046/j.1471-4159.2001.00516.x. [DOI] [PubMed] [Google Scholar]
- da Cruz e Silva OA, Iverfeldt K, Oltersdorf T, Sinha S, Lieberburg I, Ramabhadran TV, Suzuki T, Sisodia SS, Gandy S, Greengard P. Regulated cleavage of Alzheimer beta-amyloid precursor protein in the absence of the cytoplasmic tail. Neuroscience. 1993;57:873–877. doi: 10.1016/0306-4522(93)90031-a. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Pereztur J, Hut-ton M, Buee L, Harigaya Y, Yager D, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Duilio A, Faraonio R, Minopoli G, Zambrano N, Russo T. Fe65L2: a new member of the Fe65 protein family interacting with the intracellular domain of the Alzheimer's beta-amyloid precursor protein. Biochem J. 1998;330:513–519. doi: 10.1042/bj3300513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Edgar AJ, Polak JM. Human homologues of yeast vacuolar protein sorting 29 and 35. Biochem Biophys Res Com-mun. 2000;277:622–630. doi: 10.1006/bbrc.2000.3727. [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler WP, Wolfe MS. A portrait of Alzheimer secretases–new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Christopher E, Taylor JW, Parsadanian M, Spinner M, Watson M, Fryer JD, Wahrle S, Bales KR, Paul SM, Holtzman DM. ApoAI deficiency results in marked reductions in plasma cholesterol but no alterations in amyloid-beta pathology in a mouse model of Alzheimer's disease-like cerebral amyloidosis. Am J Pathol. 2004;165:1413–1422. doi: 10.1016/s0002-9440(10)63399-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- Fortini ME. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol. 2002;3:673–684. doi: 10.1038/nrm910. [DOI] [PubMed] [Google Scholar]
- Fukumori A, Okochi M, Tagami S, Jiang J, Itoh N, Nakayama T, Yanagida K, Ishizuka-Katsura Y, Morihara T, Kamino K, et al. Presenilin-dependent gamma-secretase on plasma membrane and endosomes is functionally distinct. Biochemistry. 2006;45:4907–4914. doi: 10.1021/bi052412w. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Gabuzda D, Busciglio J, Yankner BA. Inhibition of beta-amyloid production by activation of protein kinase C. J Neurochem. 1993;61:2326–2329. doi: 10.1111/j.1471-4159.1993.tb07479.x. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy S, Czernik AJ, Greengard P. Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci USA. 1988;85:6218–6221. doi: 10.1073/pnas.85.16.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy SE, Buxbaum JD, Suzuki T, Ramabhadran TV, Caporaso GL, Nairn AC, Greengard P. The nature and metabolism of potentially amyloidogenic carboxyl-terminal fragments of the Alzheimer beta/A4-amyloid precursor protein: some technical notes. Neurobiol Aging. 1992;13:601–603. doi: 10.1016/0197-4580(92)90063-4. [DOI] [PubMed] [Google Scholar]
- Gentzsch M, Chang XB, Cui L, Wu Y, Ozols VV, Choudhury A, Pagano RE, Riordan JR. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. 2004;15:2684–2696. doi: 10.1091/mbc.E04-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH. Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- Gillespie SL, Golde TE, Younkin SG. Secretory processing of the Alzheimer amyloid beta/A4 protein precursor is increased by protein phosphorylation. Biochem Biophys Res Commun. 1992;187:1285–1290. doi: 10.1016/0006-291x(92)90442-n. [DOI] [PubMed] [Google Scholar]
- Gleeson PA, Lock JG, Luke MR, Stow JL. Domains of the TGN: coats, tethers and G proteins. Traffic. 2004;5:315–326. doi: 10.1111/j.1398-9219.2004.00182.x. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci USA. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenette SY, Chen J, Jondro PD, Tanzi RE. Association of a novel human FE65-like protein with the cytoplasmic domain of the beta-amyloid precursor protein. Proc Natl Acad Sci USA. 1996;93:10832–10837. doi: 10.1073/pnas.93.20.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- Haass C, Koo EH, Teplow DB, Selkoe DJ. Polarized secretion of beta-amyloid precursor protein and amyloid beta-peptide in MDCK cells. Proc Natl Acad Sci USA. 1994;91:1564156–1564158. doi: 10.1073/pnas.91.4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Koo EH, Capell A, Teplow DB, Selkoe DJ. Polarized sorting of beta-amyloid precursor protein and its proteolytic products in MDCK cells is regulated by two independent signals. J Cell Biol. 1995a;128:537–547. doi: 10.1083/jcb.128.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995b;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- Haft CR, de la Luz Sierra M, Bafford R, Lesniak MA, Barr VA, Taylor SI. Human orthologs of yeast vacuolar protein sorting proteins Vps26, 29, and 35: assembly into multimeric complexes. Mol Biol Cell. 2000;11:4105–4116. doi: 10.1091/mbc.11.12.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe W, Rezgaoui M, Hermans-Borgmeyer I, Schaller HC. The genes for the human VPS10 domain-containing receptors are large and contain many small exons. Hum Genet. 2001;108:529–536. doi: 10.1007/s004390100504. [DOI] [PubMed] [Google Scholar]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Harter C, Reinhard C. The secretory pathway from history to the state of the art. Subcell Biochem. 2000;34:1–38. doi: 10.1007/0-306-46824-7_1. [DOI] [PubMed] [Google Scholar]
- He X, Li F, Chang WP, Tang J. GGA proteins mediate the recycling pathway of memapsin 2 (BACE) J Biol Chem. 2005;280:11696–11703. doi: 10.1074/jbc.M411296200. [DOI] [PubMed] [Google Scholar]
- Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Aggregation and secondary structure of synthetic amyloid beta A4 peptides of Alzheimer's disease. J Mol Biol. 1991;218:149–163. doi: 10.1016/0022-2836(91)90881-6. [DOI] [PubMed] [Google Scholar]
- Hill K, Li Y, Bennett M, McKay M, Zhu X, Shern J, Torre E, Lah JJ, Levey AI, Kahn RA. Munc18 interacting proteins: ADP-ribosylation factor-dependent coat proteins that regulate the traffic of beta-Alzheimer's precursor protein. J Biol Chem. 2003;278:36032–36040. doi: 10.1074/jbc.M301632200. [DOI] [PubMed] [Google Scholar]
- Hirst J, Robinson MS. Clathrin and adaptors. Biochim Biophys Acta. 1998;1404:173–193. doi: 10.1016/s0167-4889(98)00056-1. [DOI] [PubMed] [Google Scholar]
- Ho CS, Marinescu V, Steinhilb ML, Gaut JR, Turner RS, Stuenkel EL. Synergistic effects of Munc18a and X11 proteins on amyloid precursor protein metabolism. J Biol Chem. 2002;277:27021–27028. doi: 10.1074/jbc.M201823200. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J Biol Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000;275:33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- Ikin AF, Annaert WG, Takei K, De Camilli P, Jahn R, Greengard P, Buxbaum JD. Alzheimer amyloid protein precursor is localized in nerve terminal preparations to Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem. 1996;271:31783–31786. doi: 10.1074/jbc.271.50.31783. [DOI] [PubMed] [Google Scholar]
- Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N. A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and its association with kinesin light chain 1. J Biol Chem. 2003;278:22946–22955. doi: 10.1074/jbc.M212160200. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Spruyt MA, Brown AM, Sahasrabudhe SR, Blume AJ, Vitek MP, Muenkel HA, Sonnenberg-Reines J. The release of Alzheimer's disease beta amyloid peptide is reduced by phorbol treatment. J Biol Chem. 1994;269:8376–8382. [PubMed] [Google Scholar]
- Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. Estrogen regulates metabolism of Alzheimer amyloid beta precursor protein. J Biol Chem. 1994;269:13065–13068. [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Liu WW, Todd SA, Coulson DT, Murphy S, Irvine GB, Passmore AP. Expression and activity of beta-site amyloid precursor protein cleaving enzyme in Alzheimer's disease. Biochem Soc Trans. 2005;33:1096–1100. doi: 10.1042/BST20051096. [DOI] [PubMed] [Google Scholar]
- Kaether C, Lammich S, Edbauer D, Ertl M, Rietdorf J, Capell A, Steiner H, Haass C. Presenilin-1 affects trafficking and processing of betaAPP and is targeted in a complex with nicastrin to the plasma membrane. J Cell Biol. 2002;158:551–561. doi: 10.1083/jcb.200201123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaether C, Schmitt S, Willem M, Haass C. Amyloid precursor protein and notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic. 2006;7:408–415. doi: 10.1111/j.1600-0854.2006.00396.x. [DOI] [PubMed] [Google Scholar]
- Kahn RA, Yucel JK, Malhotra V. ARF signaling: a potential role for phospholipase D in membrane traffic. Cell. 1993;75:1045–1048. doi: 10.1016/0092-8674(93)90314-g. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;6106:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Katzman R. Alzheimer's disease. N Engl J Med. 1986;314:964–973. doi: 10.1056/NEJM198604103141506. [DOI] [PubMed] [Google Scholar]
- Kerr MC, Bennetts JS, Simpson F, Thomas EC, Flegg C, Gleeson PA, Wicking C, Teasdale RD. A novel mammalian retromer component, Vps26B. Traffic. 2005;6:991–1001. doi: 10.1111/j.1600-0854.2005.00328.x. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA. 2003;100:6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci. 2003;116:3339–3346. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Pepperkok R. Coat proteins in intracellular membrane transport. Curr Opin Cell Biol. 1994;6:533–537. doi: 10.1016/0955-0674(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Levey AI. Endogenous presenilin-1 targets to endocytic rather than biosynthetic compartments. Mol Cell Neurosci. 2000;16:111–126. doi: 10.1006/mcne.2000.0861. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R, Hoflack B. Protein transport from the secretory to the endocytic pathway in mammalian cells. Biochim Biophys Acta. 1998;1404:195–209. doi: 10.1016/s0167-4889(98)00057-3. [DOI] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, et al. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3:756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Troja-nowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD. A familial Alzheimer's disease locus on chromosome 1. Science. 1995a;269:970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995b;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Lewandowski NM, Small SA. Brain microarray: finding needles in molecular haystacks. J Neurosci. 2005;25:10341–10346. doi: 10.1523/JNEUROSCI.4006-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA. 2000a;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000b;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci USA. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci USA. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez-Sterling NR, Lo AC, Sisodia SS, Koo EH. Trafficking of cell-surface beta-amyloid precursor protein: evidence that a sorting intermediate participates in synaptic vesicle recycling. J Neurosci. 1997;17:140–151. doi: 10.1523/JNEUROSCI.17-01-00140.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, et al. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J Neurosci. 2001;21:6597–6607. doi: 10.1523/JNEUROSCI.21-17-06597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- McLoughlin DM, Irving NG, Brownlees J, Brion JP, Leroy K, Miller CC. Mint2/X11-like colocalizes with the Alzheimer's disease amyloid precursor protein and is associated with neuritic plaques in Alzheimer's disease. Eur J Neurosci. 1999;11:1988–1994. doi: 10.1046/j.1460-9568.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- Mostov KE, Cardone MH. Regulation of protein traffic in polarized epithelial cells. Bioessays. 1995;17:129–138. doi: 10.1002/bies.950170208. [DOI] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, et al. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Nguyen C, Bibb JA. Cdk5 and the mystery of synaptic vesicle endocytosis. J Cell Biol. 2003;163:697–699. doi: 10.1083/jcb.200310038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura M, Isoo N, Takasugi N, Tsuruoka M, Ui-Tei K, Saigo K, Morohashi Y, Tomita T, Iwatsubo T. Aph-1 contributes to the stabilization and trafficking of the gamma-secretase complex through mechanisms involving intermolecular and intramolecular interactions. J Biol Chem. 2005;280:12967–12975. doi: 10.1074/jbc.M409829200. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- Nothwehr SF, Ha SA, Bruinsma P. Sorting of yeast membrane proteins into an endosome-to-Golgi pathway involves direct interaction of their cytosolic domains with Vps35p. J Cell Biol. 2000;151:297–310. doi: 10.1083/jcb.151.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Zerial M. The diversity of Rab proteins in vesicle transport. Curr Opin Cell Biol. 1997;9:496–504. doi: 10.1016/s0955-0674(97)80025-7. [DOI] [PubMed] [Google Scholar]
- Noviello C, Vito P, Lopez P, Abdallah M, D'Adamio L. Autosomal recessive hypercholesterolemia protein interacts with and regulates the cell surface level of Alzheimer's amyloid beta precursor protein. J Biol Chem. 2003;278:31843–31847. doi: 10.1074/jbc.M304133200. [DOI] [PubMed] [Google Scholar]
- Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, Lah JJ. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi M, Nairn AC, Czernik AJ, Lim GS, Isohara T, Gandy SE, Greengard P, Suzuki T. The cytoplasmic domain of Alzheimer's amyloid precursor protein is phosphorylated at Thr654, Ser655, and Thr668 in adult rat brain and cultured cells. Mol Med. 1997;3:111–123. [PMC free article] [PubMed] [Google Scholar]
- Olkkonen VM, Ikonen E. Genetic defects of intracellular-membrane transport. N Engl J Med. 2000;343:1095–1104. doi: 10.1056/NEJM200010123431507. [DOI] [PubMed] [Google Scholar]
- Ono Y, Kinouchi T, Sorimachi H, Ishiura S, Suzuki K. Deletion of an endosomal/lysosomal targeting signal promotes the secretion of Alzheimer's disease amyloid precursor protein (APP) J Biochem (Tokyo) 1997;121:585–590. doi: 10.1093/oxfordjournals.jbchem.a021625. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Ikin AF, Nairn AC, Pursnani A, Buxbaum JD. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total A(beta) Mol Cell Neurosci. 2002;19:175–185. doi: 10.1006/mcne.2001.1065. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Pedrini S, Carter TL, Prendergast G, Petanceska S, Ehrlich ME, Gandy S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005;2:e18. doi: 10.1371/journal.pmed.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Squazzo SL, Koo EH. Enhanced release of amyloid beta-protein from codon 670/671 “Swedish” mutant beta-amyloid precursor protein occurs in both secretory and endocytic pathways. J Biol Chem. 1996;271:9100–9107. doi: 10.1074/jbc.271.15.9100. [DOI] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Petanceska SS, Seeger M, Checler F, Gandy S. Mutant presenilin 1 increases the levels of Alzheimer amyloid beta-peptide Abeta42 in late compartments of the constitutive secretory pathway. J Neurochem. 2000;74:1878–1884. doi: 10.1046/j.1471-4159.2000.0741878.x. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. Transport-vesicle targeting: tethers before SNAREs. Nat Cell Biol. 1999;1:E17–E22. doi: 10.1038/8967. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. Membrane transport: retromer to the rescue. Curr Biol. 2001;11:R109–R111. doi: 10.1016/s0960-9822(01)00042-2. [DOI] [PubMed] [Google Scholar]
- Ponnambalam S, Baldwin SA. Constitutive protein secretion from the trans-Golgi network to the plasma membrane. Mol Membr Biol. 2003;20:129–139. doi: 10.1080/0968768031000084172. [DOI] [PubMed] [Google Scholar]
- Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1456–1464. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris R, Foletti DL, Scheller RH. Dynamics of tubulovesicular recycling endosomes in hippocampal neurons. J Neurosci. 1999;19:10324–10337. doi: 10.1523/JNEUROSCI.19-23-10324.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Thiyagarajan M, Macgrogan D, Rodgers JT, Puigserver P, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer's disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Rapoport I, Chen YC, Cupers P, Shoelson SE, Kirchhausen T. Dileucine-based sorting signals bind to the beta chain of AP-1 at a site distinct and regulated differently from the tyrosine-based motif-binding site. EMBO J. 1998;17:2148–2155. doi: 10.1093/emboj/17.8.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechards M, Xia W, Oorschot V, van Dijk S, Annaert W, Selkoe DJ, Klumperman J. Presenilin-1-mediated retention of APP derivatives in early biosynthetic compartments. Traffic. 2006;7:354–364. doi: 10.1111/j.1600-0854.2006.00388.x. [DOI] [PubMed] [Google Scholar]
- Reddy JV, Seaman MN. Vps26p, a component of retromer, directs the interactions of Vps35p in endosome-to-Golgi retrieval. Mol Biol Cell. 2001;12:3242–3256. doi: 10.1091/mbc.12.10.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2001;8:890–899. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- Riemenschneider M, Schoepfer-Wendels A, Friedrich P, Konta L, Laws SM, Mueller JC, Kurz A, Forstl H. No association of vacuolar protein sorting 26 polymorphisms with Alzheimer's disease. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.07.016. in press Published online September 18, 2006. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol. 2001;11:1288–1293. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM. Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci USA. 1987;84:4190–4194. doi: 10.1073/pnas.84.12.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SE, Setty SR, Sitaram A, Marks MS, Lewis RE, Chou MM. Extracellular signal-regulated kinase regulates clathrin-independent endosomal trafficking. Mol Biol Cell. 2006;17:645–657. doi: 10.1091/mbc.E05-07-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Musch A. Protein sorting in the Golgi complex: shifting paradigms. Biochim Biophys Acta. 2005;1744:455–464. doi: 10.1016/j.bbamcr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Rogers J, Morrison JH. Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer's disease. J Neurosci. 1985;5:2801–2808. doi: 10.1523/JNEUROSCI.05-10-02801.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossner S, Fuchsbrunner K, Lange-Dohna C, Hartlage-Rubsamen M, Bigl V, Betz A, Reim K, Brose N. Munc13-1-mediated vesicle priming contributes to secretory amyloid precursor protein processing. J Biol Chem. 2004;279:27841–27844. doi: 10.1074/jbc.C400122200. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Lanier LM, Ikin AF, Khorkova O, Sahasrabudhe S, Greengard P, Buxbaum JD. Regulation of beta-amyloid secretion by FE65, an amyloid protein precursor-binding protein. J Biol Chem. 1999;274:7952–7957. doi: 10.1074/jbc.274.12.7952. [DOI] [PubMed] [Google Scholar]
- Sano Y, Nakaya T, Pedrini S, Takeda S, Iijima-Ando K, Iijima K, Mathews PM, Itohara S, Gandy S, Suzuki T. Physiological mouse brain A-beta levels are not related to the phosphorylation state of threonine-668 of Alzheimer's APP. PloS ONE. 2006;1 doi: 10.1371/journal.pone.0000051. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D'Adamio L. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer's beta-amyloid precursor protein (APP) J Biol Chem. 2002;277:3767–3775. doi: 10.1074/jbc.M108357200. [DOI] [PubMed] [Google Scholar]
- Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, Schaller C, Bujo H, Levey AI, Lah JJ. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol. 2004;61:1200–1205. doi: 10.1001/archneur.61.8.1200. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorer CE. Historical essay: Kraepelin's description of Alzheimer's disease. Int J Aging Hum Dev. 1985;21:235–238. doi: 10.2190/gnq1-gdux-eptl-0f2l. [DOI] [PubMed] [Google Scholar]
- Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN. Recycle your receptors with retromer. Trends Cell Biol. 2005;15:68–75. doi: 10.1016/j.tcb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Seaman MN, Williams HP. Identification of the functional domains of yeast sorting nexins Vps5p and Vps17p. Mol Biol Cell. 2002;13:2826–2840. doi: 10.1091/mbc.02-05-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, McCaffery JM, Emr SD. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J Cell Biol. 1998;142:665–681. doi: 10.1083/jcb.142.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. The ups and downs of Abeta. Nat Med. 2006;12:758–759. doi: 10.1038/nm0706-758. [DOI] [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, et al. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- Seubert P, Oltersdorf T, Lee MG, Barbour R, Blomquist C, Davis DL, Bryant K, Fritz LC, Galasko D, Thal LJ, et al. Secretion of beta-amyloid precursor protein cleaved at the amino terminus of the beta-amyloid peptide. Nature. 1993;361:260–263. doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Abeta amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Pijak DS, Doms RW, Lee VM. A distinct ER/IC gamma-secretase competes with the proteasome for cleavage of APP. Biochemistry. 2000a;39:810–817. doi: 10.1021/bi991728z. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000b;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- Small SA, Kim TW. Vps35-based assays and methods for treating alzheimer's disease. U.S. Patent Serial No 60/518, 250 (filed Nov 7, 2003; USPTO 514044000; A61K048/00 Intl Class) 2003
- Small SA, Mayeux R. Delirium and Dementia. In: Rowland LP, editor. Merritt's Neurology. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 3–6. [Google Scholar]
- Small SA, Peirce AL, Kent K, Leung C, Honig L, Vonsattel JP. Paper presented at: Annual Meeting of the Society for Neuroscience (New Orleans) 2003. Combining functional imaging with microarray; identifying an unexplored cellular pathway implicated in sporadic Alzheimer's disease. [Google Scholar]
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- Sodhi CP, Rampalli S, Perez RG, Koo EH, Quinn B, Gottardi-Littell NR. The endocytotic pathway is required for increased A beta 42 secretion during apoptosis. Brain Res Mol Brain Res. 2004;128:201–211. doi: 10.1016/j.molbrainres.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Soriano S, Chyung AS, Chen X, Stokin GB, Lee VM, Koo EH. Expression of beta-amyloid precursor protein-CD3gamma chimeras to demonstrate the selective generation of amyloid beta(1-40) and amyloid beta(1-42) peptides within secretory and endocytic compartments. J Biol Chem. 1999;274:32295–32300. doi: 10.1074/jbc.274.45.32295. [DOI] [PubMed] [Google Scholar]
- Spoelgen R, von Arnim CA, Thomas AV, Peltan ID, Koker M, Deng A, Irizarry MC, Andersen OM, Willnow TE, Hyman BT. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J Neurosci. 2006;26:418–428. doi: 10.1523/JNEUROSCI.3882-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack JH, Horazdovsky B, Emr SD. Receptor-mediated protein sorting to the vacuole in yeast: roles for a protein kinase, a lipid kinase and GTP-binding proteins. Annu Rev Cell Dev Biol. 1995;11:1–33. doi: 10.1146/annurev.cb.11.110195.000245. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Tanahashi H, Tabira T. Molecular cloning of human Fe65L2 and its interaction with the Alzheimer's beta-amyloid precursor protein. Neurosci Lett. 1999;261:143–146. doi: 10.1016/s0304-3940(98)00995-1. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Tomita S, Ozaki T, Taru H, Oguchi S, Takeda S, Yagi Y, Sakiyama S, Kirino Y, Suzuki T. Interaction of a neuron-specific protein containing PDZ domains with Alzheimer's amyloid precursor protein. J Biol Chem. 1999;274:2243–2254. doi: 10.1074/jbc.274.4.2243. [DOI] [PubMed] [Google Scholar]
- Tomizawa K, Sunada S, Lu YF, Oda Y, Kinuta M, Ohshima T, Saito T, Wei FY, Matsushita M, Li ST, et al. Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biol. 2003;163:813–824. doi: 10.1083/jcb.200308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol. 2003;13:177–186. doi: 10.1016/s0962-8924(03)00031-x. [DOI] [PubMed] [Google Scholar]
- Traub LM. Common principles in clathrin-mediated sorting at the Golgi and the plasma membrane. Biochim Biophys Acta. 2005;1744:415–437. doi: 10.1016/j.bbamcr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Tyler SJ, Dawbarn D, Wilcock GK, Allen SJ. alpha- and beta-secretase: profound changes in Alzheimer's disease. Biochem Biophys Res Commun. 2002;299:373–376. doi: 10.1016/s0006-291x(02)02635-9. [DOI] [PubMed] [Google Scholar]
- van Vliet C, Thomas EC, Merino-Trigo A, Teasdale RD, Gleeson PA. Intracellular sorting and transport of proteins. Prog Biophys Mol Biol. 2003;83:1–45. doi: 10.1016/s0079-6107(03)00019-1. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Verges M, Luton F, Gruber C, Tiemann F, Reinders LG, Huang L, Burlingame AL, Haft CR, Mostov KE. The mammalian retromer regulates transcytosis of the polymeric immunoglobulin receptor. Nat Cell Biol. 2004;6:763–769. doi: 10.1038/ncb1153. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–44954. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–25900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Arnim CA, Tangredi MM, Peltan ID, Lee BM, Irizarry MC, Kinoshita A, Hyman BT. Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J Cell Sci. 2004;117:5437–5445. doi: 10.1242/jcs.01422. [DOI] [PubMed] [Google Scholar]
- Wahle T, Prager K, Raffler N, Haass C, Famulok M, Walter J. GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol Cell Neurosci. 2005;29:453–461. doi: 10.1016/j.mcn.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Wang H, Luo WJ, Zhang YW, Li YM, Thinakaran G, Greengard P, Xu H. Presenilins and gamma-secretase inhibitors affect intracellular trafficking and cell surface localization of the gamma-secretase complex components. J Biol Chem. 2004;279:40560–40566. doi: 10.1074/jbc.M404345200. [DOI] [PubMed] [Google Scholar]
- Wang R, Tang P, Wang P, Boissy RE, Zheng H. Regulation of tyrosinase trafficking and processing by presenilins: partial loss of function by familial Alzheimer's disease mutation. Proc Natl Acad Sci USA. 2006;103:353–358. doi: 10.1073/pnas.0509822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Sukegawa J, Sukegawa I, Tomita S, Iijima K, Oguchi S, Suzuki T, Nairn AC, Greengard P. A 127-kDa protein (UV-DDB) binds to the cytoplasmic domain of the Alzheimer's amyloid precursor protein. J Neurochem. 1999;72:549–556. doi: 10.1046/j.1471-4159.1999.0720549.x. [DOI] [PubMed] [Google Scholar]
- Wisniewski K, Howe J, Williams DG, Wisniewski HM. Precocious aging and dementia in patients with Down's syndrome. Biol Psychiatry. 1978;13:619–627. [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wood DR, Nye JS, Lamb NJ, Fernandez A, Kitzmann M. Intracellular retention of caveolin 1 in presenilin-deficient cells. J Biol Chem. 2005;280:6663–6668. doi: 10.1074/jbc.M410332200. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Xu H, Greengard P, Gandy S. Regulated formation of Golgi secretory vesicles containing Alzheimer beta-amyloid precursor protein. J Biol Chem. 1995;270:23243–23245. doi: 10.1074/jbc.270.40.23243. [DOI] [PubMed] [Google Scholar]
- Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, et al. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat Med. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Zambrano N, Buxbaum JD, Minopoli G, Fiore F, De Candia P, De Renzis S, Faraonio R, Sabo S, Cheetham J, Sudol M, Russo T. Interaction of the phosphotyrosine interaction/phosphotyrosine binding-related domains of Fe65 with wild-type and mutant Alzheimer's beta-amyloid precursor proteins. J Biol Chem. 1997;272:6399–6405. doi: 10.1074/jbc.272.10.6399. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Luo WJ, Wang H, Lin P, Vetrivel KS, Liao F, Li F, Wong PC, Farquhar MG, Thinakaran G, Xu H. Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma-secretase components. J Biol Chem. 2005;280:17020–17026. doi: 10.1074/jbc.M409467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Haapasalo A, Kim DY, Ingano LA, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006;20:1176–1178. doi: 10.1096/fj.05-5531fje. [DOI] [PubMed] [Google Scholar]