

Synthetic tissues with minimal cellular functionality can be externally controlled by light.
Abstract
We have previously used three-dimensional (3D) printing to prepare tissue-like materials in which picoliter aqueous compartments are separated by lipid bilayers. These printed droplets are elaborated into synthetic cells by using a tightly regulated in vitro transcription/translation system. A light-activated DNA promoter has been developed that can be used to turn on the expression of any gene within the synthetic cells. We used light activation to express protein pores in 3D-printed patterns within synthetic tissues. The pores are incorporated into specific bilayer interfaces and thereby mediate rapid, directional electrical communication between subsets of cells. Accordingly, we have developed a functional mimic of neuronal transmission that can be controlled in a precise way.
INTRODUCTION
Cell-free expression systems have been widely used in synthetic biology to create systems that can express functional proteins in a minimal cell-like environment (1–3). These systems have been used for in vitro selection and evolution of proteins (4–7) and for control of mammalian (8) and bacterial cells (9). This previous research was performed by encapsulating the cell-free expression system in a single lipid bilayer–bounded compartment, a synthetic cell. No systems have been created where multiple soft encapsulated synthetic cells can communicate with each other, although patterned two-dimensional (2D) solid-state microfluidic chambers containing cell-free expression medium can communicate through diffusion (10). Furthermore, no light-based method has been developed that can control protein expression inside synthetic cells. Here, we have created 3D synthetic tissues made up of hundreds of synthetic cells, using a water-in-oil droplet 3D printer (11). Additionally, we have developed a tightly regulated light-activated DNA (LA-DNA) promoter. By using these technologies in combination, light-activated electrical communication through the synthetic tissues has been achieved by expressing a transmembrane pore, α-hemolysin (αHL), in a subset of the synthetic cells, 3D-printed to form a conductive pathway that is a functional mimic of neuronal transmission.
RESULTS
Light-activated transcription and/or translation has been achieved previously; however, these systems either do not fully repress transcription in the off state (12) or cannot be encapsulated inside synthetic cells due to bursting of the lipid membranes (13, 14). We sought to develop an efficient light-activated T7 promoter (LA-T7 promoter) that could be placed upstream of any gene of interest so that no expression would occur until the DNA was illuminated (Fig. 1A). A complete off state is required so that no protein expression, and therefore no function, is observed without activation. To achieve this, C6-amino-dT–modified bases were incorporated across a single-stranded T7 promoter DNA sequence (Fig. 1B). A photocleavable (PC) biotin N-hydroxysuccinimide ester linker (15) was coupled to all the amines (fig. S1) so that, when the modified DNA was used as a polymerase chain reaction (PCR) primer with a gene of interest, PC biotin moieties would protrude from the major groove at the T7 polymerase binding site (Fig. 1A). The PC group was 2-nitrobenzyl, which allows rapid and efficient cleavage back to the original primary amine (15) to leave minimal scarring of the T7 promoter and allow similar expression to an unmodified T7 promoter. The LA-DNA was created by the addition of monovalent streptavidin (16) to the double-stranded DNA PCR product so that each biotin in the T7 promoter bound to a single monovalent streptavidin molecule (fig. S2). As a fully “photocleaved” control, we used amine-only DNA in which only the C6-amino groups are present in the T7 promoter and therefore does not bind streptavidin. We observed rapid and efficient photocleavage of the monovalent streptavidin with the biotin and linker group from the LA-DNA under a 365-nm ultraviolet (UV) light, as measured by gel electrophoresis (fig. S2). No binding of streptavidin was observed for the amine-only DNA (fig. S2).
Fig. 1. Construction and evaluation of a light-activated promoter.

(A) T7 RNA polymerase is blocked from binding to the LA-T7 promoter due to the presence of multiple monovalent streptavidins, bound to the DNA through biotinylated PC linkers. Following UV light cleavage of the linkers, T7 RNA polymerase can transcribe the downstream gene. (B) LA-T7 promoter sequence. Pink-colored thymines are replaced with amino-C6-dT modifications and the primary amines of the nucleobase coupled to the PC biotin group. (C) LA-DNA encoding for mVenus is only expressed upon UV irradiation. There is no significant difference between expression from the LA-DNA (+UV) and expression from the amine-only DNA construct. a.u., arbitary unit.
For cell-free expression of proteins, we used the “protein synthesis using recombinant elements” (PURE) system (17), which contains the minimal set of components required for protein expression (18). T7 RNA polymerase alone was used for transcription only. For our initial experiments, the LA-T7 promoter was placed upstream of a gene encoding the yellow fluorescent protein mVenus. RNA transcription (fig. S3) nd protein expression (Fig. 1C) experiments with the light-activatable mVenus DNA (LA-mVenus), demonstrated that there was minimal transcription/expression from the off state and that the on state was strongly activated (>130-fold increase in expression from off to on state). No inhibitory effects from the released monovalent streptavidin or the photocleaved biotin derivative were observed by comparison with the amine-only DNA (Fig. 1C). This demonstrates that the LA-DNA technology tightly regulates transcription of DNA by T7 polymerase.
To create synthetic cells, the PURE system components were mixed with LA-DNA, and the mix was encapsulated in lipid-coated water-in-oil droplets (Fig. 2A and fig. S4). Lipid-coated aqueous droplets in oil can be brought together to form a droplet interface bilayer (DIB) (19), which mimics the bilayer of a cell membrane. Networks of droplets joined by DIBs have been previously utilized as soft biodevices (20–22). However, DIB formation and stability between droplets that contain complex biological components have been a major hurdle to the creation of more complicated networks and biodevices (23). The high concentration of protein inside the droplets can promote droplet fusion. We have optimized a new lipid composition, a mixture of 10 or 15% 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DPPE-mPEG2000) in diphytanoyl phosphatidylcholine (DPhPC), which allows long-term bilayer stability in the presence of PURE components without affecting their function, as demonstrated by expression from LA-mVenus (Fig. 2, B and C) and a light-activated red fluorescent protein, mCherry (LA-mCherry; fig. S4). UV light had no effect on the expression of amine-only mVenus DNA in synthetic cells (fig. S5).
Fig. 2. Light-activated expression of LA-mVenus in synthetic cells and synthetic tissues.

(A) Schematic of a synthetic cell that will express mVenus protein upon light activation, where a single bilayer connects it to a neighboring synthetic cell. IVTT, in vitro transcription and translation. (B) Synthetic cells containing LA-mVenus DNA express mVenus protein (yellow) upon light activation. (C) Fluorescence intensity line profiles from (B). (D) Schematic of 3D-printed synthetic tissues containing hundreds of synthetic cells. A single lipid bilayer, as shown in (A), connects each cell with its neighbor. (E) Synthetic tissues containing LA-mVenus DNA express mVenus protein (yellow) upon light activation. (F) Fluorescence intensity line profiles from (E).
The gene encoding the heptameric membrane protein pore αHL was then placed downstream of the LA-T7 promoter and expressed in one droplet of a pair forming a DIB. As expected, αHL localized to the bilayer (Fig. 3). Because two synthetic cells could now be interfaced and a membrane protein expressed, communication between the two cells was examined to demonstrate that the αHL was functional. Natural cells communicate with each other through small-molecule effectors or electrical signals (24). We first tested for diffusion of a small-molecule fluorophore between two synthetic cells by light-activated insertion of αHL into the interface bilayer. Two synthetic cells were brought together; one contained a small-molecule dye, carboxytetramethylrhodamine, with no DNA and the other with LA-αHL DNA. Small-molecule transfer between the two synthetic cells was achieved when they were illuminated, but no transfer was observed without illumination (fig. S6). The small-molecule dye could only translocate across the bilayer into the second droplet by diffusion if the αHL formed a pore in the interface bilayer. We then tested for light-activated electrical signaling between two synthetic cells by electrical recording, following insertion of αHL into the bilayer. After forming a synthetic cell that contained LA-αHL DNA, it was activated by light, then an electrode was inserted into it and the cell was interfaced with another synthetic cell containing no DNA. Electrical communication, measured as an ionic current between the two synthetic cells, was achieved when the cell containing LA-αHL DNA had been illuminated, but no electrical signal was detected without illumination (Fig. 4). Electrical signals would only be transmitted between the synthetic cells if the αHL formed a functional pore in the membrane to allow ions to translocate in an applied potential. The communication between synthetic cells by a small molecule and by electrical signaling demonstrates that communication between soft biological compartments can be controlled by light.
Fig. 3. Light-activated expression from LA–αHL-GFP DNA in a pair of synthetic cells forming a DIB.
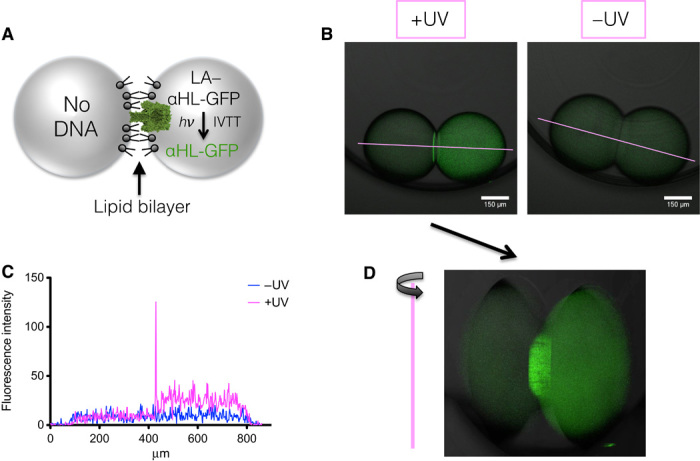
(A) Schematic of the synthetic cell pair. One cell contains LA–αHL–green fluorescent protein (GFP) DNA, the other contains no DNA. The αHL-GFP fusion pore protein will localize to the bilayer when expressed. (B) When LA–αHL-GFP DNA is activated in a synthetic cell neighboring another containing no DNA, the αHL-GFP fusion membrane pore protein locates to the bilayer. No expression is observed without light activation. (C) Fluorescence intensity line profiles from (B). (D) Rotated 3D projection of a z stack of αHL-GFP DNA, as expressed in (B), demonstrates that αHL-GFP becomes located throughout the flat interface bilayer.
Fig. 4. Light-activated electrical signal between synthetic cells.
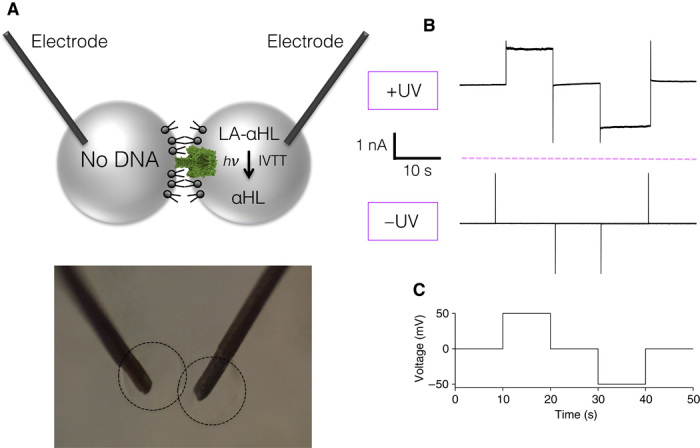
(A) Schematic of the synthetic cell pair. One cell contains LA-αHL DNA, the other contains no DNA. Both droplets have electrodes inserted within them to apply a potential and measure the ionic current. Below is an image of the experimental setup. (B) A current is detected only following the expression of αHL after light activation. (C) Voltage protocol used in (B).
We have previously made a 3D printer for water-in-oil droplets, which can create complex 3D-patterned functional soft structures composed of hundreds of aqueous compartments (11). Just as we found for individual DIBs, the droplets that formed the 3D networks fused in the presence of complex biological mixtures. Using our optimized lipid mixture, we were able to print 3D cuboids, containing hundreds of synthetic cells, which we refer to as synthetic tissues (Fig. 2D). Once printed, expression from LA-DNA was observed inside these synthetic tissues following illumination, but not without illumination (Fig. 2, E and F, and fig. S7). UV light had no effect on expression from amine-only DNA in synthetic tissues (fig. S8). It is important to be able to control the structure of the 3D-printed synthetic tissues. Packing of the droplets inside 3D networks is a function of the area of the bilayer between each droplet and can dictate network stability. We found that we could tune the bilayer size and packing of the synthetic cells inside the synthetic tissues by altering the composition of the lipids and the oil, without affecting protein expression (fig. S9).
To demonstrate electrical communication through synthetic tissues, synthetic cells that contained LA-αHL DNA were 3D-printed into tissues and studied by electrical recording (Fig. 5A). Once printed, the synthetic tissues were illuminated to trigger αHL protein expression. Electrodes were then placed on either side of the synthetic tissue cuboid to measure electrical communication. Electrical communication through the synthetic tissues could be observed following illumination, but no electrical signal was detected without illumination (Fig. 5, B and C).
Fig. 5. Recording of electrical communication in 3D-printed synthetic tissues mediated by LA-αHL DNA.
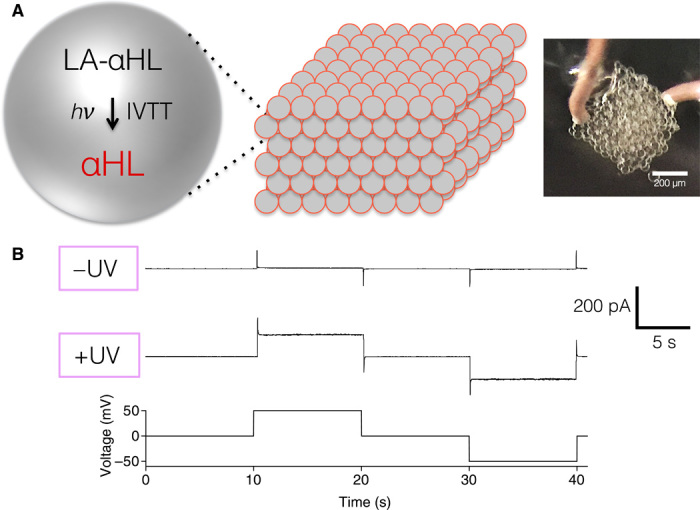
(A) Schematic of 3D-printed synthetic tissue containing LA-αHL DNA, which expresses the αHL membrane pore upon light activation. (B) Electrical recordings from a 3D-printed network demonstrate that a current through the synthetic tissues is only detected upon light activation. The voltage protocol used to detect αHL insertion into bilayer is also shown.
We also took advantage of the patterning capabilities of the 3D printer. Patterning was first evaluated by printing and then activating synthetic cells containing LA-mVenus DNA in programmed arrangements within synthetic tissues comprising cells lacking DNA (Fig. 6). Directional 3D electrical communication through the synthetic tissues was then tested by printing droplets containing LA-αHL DNA in defined 3D pathways within the tissues (Fig. 7A). Following printing, the synthetic tissues were illuminated, and αHL protein was expressed. Electrical communication was observed through the synthetic tissue by placing electrodes at the two ends of a pathway (Fig. 7B). When the electrodes were placed at other points on the synthetic tissue, no electrical signal was detected (Fig. 7, C to F). In this way, the accuracy and versatility of our 3D printer has been used to fabricate a synthetic tissue in which an electrical communication pathway, analogous to rapid directional neuronal transmission, can be switched on by an external stimulus.
Fig. 6. Light-activated patterned expression of mVenus from LA-mVenus DNA in a 3D-printed synthetic tissue.
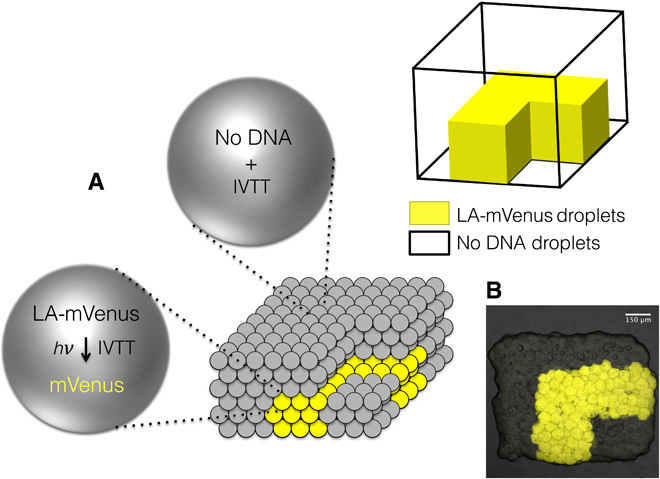
(A) Schematic showing a printed tissue with droplets containing LA-mVenus DNA (yellow) and droplets containing no DNA (gray). (B) After light activation, the mVenus-containing droplets become visible (yellow fluorescence).
Fig. 7. Electrical recordings from an L-shaped pathway formed by expression from LA-αHL DNA in a 3D-printed synthetic tissue.
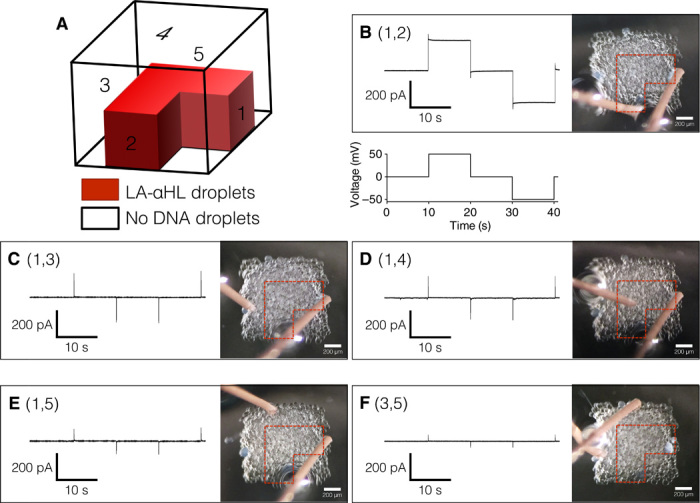
(A) Schematic of the printed tissue containing droplets with LA-αHL DNA (red) printed with droplets containing no DNA (clear region within black frame). Numbers represent sides of the cuboid where electrodes were placed to detect the conductive pathway. (B to F) Electrical recordings detect a current when the electrodes are at positions 1 and 2 (B), based on the voltage protocol in (B), but not when one or both of the electrodes are positioned off the pathway sides 1 and 3 (C), 1 and 4 (D), 1 and 5 (E), or 3 and 5 (F).
To fully exploit the use of light, we generated 2D pathways through 3D-printed synthetic tissues that contained LA-DNA in all the droplets. Using a field diaphragm on a fluorescence light microscope, we were able to illuminate a small circular area of the synthetic tissues (fig. S10). By illuminating multiple conjoined circular areas of a LA-mVenus DNA-containing synthetic tissue, protein expression was only activated in the illuminated droplets (fig. S11). Following this, LA-αHL DNA was activated in 2D pathways by light-patterning (fig. S12). Electrical communication, measured similarly to that in the 3D printer–patterned pathway, was observed both through the two ends of the light-patterned pathway (fig. S12B) and from the side to the top (fig. S12D). When the electrodes were placed at other points on the synthetic tissue, no electrical signal was detected (fig. S12, C to F). This illustrates that we can initiate rapid directional communication through light-patterned 2D pathways in synthetic tissues at a predetermined time.
DISCUSSION
Here, we have created 3D-printed and patterned synthetic tissues that contain hundreds of synthetic cells that express proteins under a tightly regulated LA-DNA promoter. Light-activated electrical communication was achieved through 3D-printed pathways in the synthetic tissues. This tightly regulated LA-DNA technology provides a precise means to control in vitro protein expression. Therefore, it is possible to create synthetic tissues, which comprise 3D-printed patterns of interconnected compartments, each with a minimal cellular functionality, which can be externally controlled by light. These synthetic tissues might be developed into controllable soft biodevices that could be interfaced with cellular tissue.
MATERIALS AND METHODS
Amino T7 oligo conjugation to PC biotin
Amino T7 oligo (5 μl, 100 μM; ATDBio) (Table 1) was reacted with PC biotin (25 μl, 10 mM in DMF; Ambergen) with 100 mM NaHCO3 in a total volume of 50 μl in a 0.5-ml Protein LoBind tube (Eppendorf). Reactions were held at room temperature in the dark for 1 hour with gentle vortexing and centrifugation every 15 min and then left at 4°C overnight. Reactions were stopped with the addition of 350 μl of 10 mM tris-HCl (pH 8.0; Sigma), and the derivatized oligo was purified with 3-kD Amicon Ultra columns according to the manufacturer’s protocol [Millipore; washed on the column four times with 350 μl of 10 mM tris-HCl (pH 8.0)].
Table 1. DNA sequences.
emGFP, emerald GFP; CT, control template.
| DNA name | DNA sequence | Modification |
| Amino T7 | GAATXAAXACGACXCACXAXAGGGXCXAG | X = C6-amino-dT |
| mV FRW | TTAACTTTAAGAAGGAGGTATACATATGGTGAGCAAGGGCGAGGAGCTGT | |
| mV REV | TACTCGAGAATTCCCGGGATCCTCATTACTTGTACAGCTCGTCCATGCCG | |
| CT-FP FRW | CGGCATGGACGAGCTGTACAAGTAATGAGGATCCCGGGAATTCTCGAGTA | |
| CT-FP REV | ACAGCTCCTCGCCCTTGCTCACCATATGTATACCTCCTTCTTAAAGTTAAAC | |
| mC FRW | TTAACTTTAAGAAGGAGGTATACATATGGTGAGCAAGGGCGAGGAGGATA | |
| mC REV | TACTCGAGAATTCCCGGGATCCTCATTACTTGTACAGCTCGTCCATGCCG | |
| HL FRW | TTAACTTTAAGAAGGAGGTATACATATGGCAGATTCTGATATTAATATTA | |
| HL-LINK REV | AGAGCCACCTCCGCCTGAACCGCCACCACCCGAATTTGTCATTTCTTCTTTTTCCCAA | |
| HL REV | TACTCGAGAATTCCCGGGATCCTCATTAATTTGTCATTTCTTCTTTTTCCCAA | |
| emGFP-LINK FRW | TCAGGCGGAGGTGGCTCTGGCGGTGGCGGATCGGTGAGCAAGGGCGAGGAGCTGT | |
| CT-HL FRW | TTGGGAAAAAGAAGAAATGACAAATTAATGAGGATCCCGGGAATTCTCGAGTA | |
| CT-HL REV | TAATATTAATATCAGAATCTGCCATATGTATACCTCCTTCTTAAAGTTAAAC | |
| CT-emGFP FRW | CGGCATGGACGAGCTGTACAAGTAATGAGGATCCCGGGAATTCTCGAGTA | |
| CT-seq FRW | GCTAGTGGTGCTAGCCCCGC | |
| CT-seq REV | GGGACCGCTAGCGCGGCCGC | |
| Fusion linker | TCGGGTGGTGGCGGTTCAGGCGGAGGTGGCTCTGGCGGTGGCGGATCG | |
| CT REV | GATATAGTTCCTCCTTTCAG |
Analytical high-performance liquid chromatography (HPLC) examination (Agilent 1260 Infinity) of the extent of the reaction was carried out with 1 μl of the amino T7–PC biotin reaction separated on a 5-μm Agilent Polaris 5 C18 (4.6 × 150–mm column) with a flow of 2 ml/min. Eluting buffers were buffer A [10 mM triethylammonium bicarbonate (pH 8.5); Sigma] and buffer B (acetonitrile). The gradient was 0 min, 2% B; 10 min, 35% B. Peaks were monitored by their absorbance at 260 nm.
The Amicon-purified amino T7–PC biotin was further purified by HPLC (Agilent 1260 Infinity) with a 10-μm Supelco Discovery BIO Wide Pore C18 (10 × 250–mm column) with a flow of 5 ml/min. Eluting buffers were buffer A [1 M ammonium acetate (pH 7)], buffer B (acetonitrile), and buffer C (H2O). Buffer A was held at 10% throughout the whole run, and the gradient for the remaining buffers was 0 min, 5% B; 40 min, 40% B. The method is adapted from a literature protocol (25).
HPLC-purified amino T7–PC biotin was lyophilized in Protein LoBind tubes, resuspended in 10 mM tris-HCl (pH 8.0), and concentrated with 3-kD Amicon Ultra columns before transfer to Protein LoBind tubes for storage.
Cloning of genes encoding mVenus, mCherry, and αHL-GFP into the PURExpress control template
Genes of interest were cloned into the PURExpress CT using homologous recombination (26, 27). mVenus (mVenus-N1 was a gift from M. Davidson; Addgene plasmid #54640), mCherry (provided by Wade-Martin’s laboratory, Department of Physiology and Anatomy, University of Oxford), αHL-NN mutant (Bayley’s laboratory, Chemistry Research Laboratory, University of Oxford), and emGFP (pRSET-EmGFP, Invitrogen) were amplified with PCR primers to form linear fragments that contained overlap regions to the CT at each end. The CT was also amplified into a linear PCR fragment that contained overlap regions to each gene of interest at each end. For the αHL-GFP construct, we added a linker region (fusion linker DNA sequence, Table 1) in between the two genes by adding overlaps of the fusion linker sequence to the end of the αHL gene and the beginning of the emGFP sequence (that is, a three-way recombination). All plasmids were digested to form linear fragments before PCR amplification, by using either Nde I (CT and mCherry) or Hind III (emGFP, αHL, and mVenus).
PCRs for homologous recombination were carried out with the Phusion High-Fidelity DNA Polymerase master mix (NEB). PCRs were made according to the manufacturer’s protocol in a total of 50 μl with 5 μl of each primer (10 μM; Sigma), and between 5 and 15 ng of digested plasmid template. The following thermal cycles were performed: 98°C for 30 s, 35× (98°C for 10 s, 55°C for 30 s, and 72°C for 30 or 60 s), and 72°C for 5 min. The extension times for each PCR fragment were as follows: CT (60 s), αHL (60 s), emGFP (60 s), mVenus (30 s), and mCherry (30 s).
In summary, plasmids were constructed so that they each contained a gene (mVenus, mCherry, αHL, and αHL-GFP) in place of the control dihydrofolate reductase gene in the PURE CT plasmid. Below are the PCR fragment combinations used to construct each plasmid, with the plasmid templates and primers (Table 1) used to produce each amplified product for homologous recombination:
Gene encoded in PURE CT = (template plasmid A: primer 1 + primer 2) + (template plasmid B: primer 3 + primer 4)
mVenus.CT = (mVenus N1: mV FRW + mV REV) + (CT: CT-FP FRW + CT-FP REV)
mCherry.CT = (mCherry: mC FRW + mC REV) + (CT: CT-FP FRW + CT-FP REV)
αHL.CT = (αHL: HL FRW + HL REV) + (CT: CT-HL FRW + CT-HL REV)
αHL-GFP.CT = (αHL: HL FRW + HL-LINK REV) + (CT: CT-emGFP FRW + CT-HL REV) + (emGFP: emGFP-LINK FRW + mV REV)
PCR products were not purified before homologous recombination. XL10-Gold competent Escherichia coli cells (NEB) were thawed on ice for 30 min. Between 1 and 5 μl of PCR product (100 ng of DNA based on agarose gel analysis) was added to 75 μl of cells, which was held on ice for an additional 30 min. The cells were then heat-shocked for 30 s at 42°C, and then held on ice for 2 min. LB (75 μl) was added to the cells, and they were plated on LB agar plates containing ampicillin (100 μg/ml). Colonies were grown in LB containing ampicillin (100 μg/ml), and the plasmids were purified with Thermo Scientific GeneJet Plasmid Miniprep kits. Sequences were verified with Sanger sequencing (Source BioScience) using the primers CT-seq FRW and CT-seq REV (Sigma).
PCR of genes of interest with amino T7–PC biotin primer
Linear DNA templates were constructed by PCR from the cloned plasmids (above). Each linear DNA template contained each of the genes of interest (mVenus, mCherry, αHL, and αHL-GFP) downstream of the amino T7–PC biotin oligo synthesized above, by using the HPLC-purified oligo as a PCR primer. PCRs were made with the DreamTaq DNA polymerase master mix (Life Technologies) according to the manufacturer’s protocol in a total of 50 μl with 1.25 μl of CT REV (10 μM; Sigma), 2.5 μl of amino T7–PC biotin (45 ng/μl), and 10 ng of Hind III–digested CT containing the desired gene of interest. The following thermal cycles were performed: 95°C for 3 min, 35× (95°C for 30 s, 52.5°C for 30 s, and 72°C for 1 min 15 s), and 72°C for 5 min. The extension time was increased to 2 min 15 s for the αHL-GFP template.
The PCR products were purified with the Thermo Scientific GeneJet PCR kits, and the DNA was precipitated in Protein LoBind tubes overnight at −80°C by first adding sodium acetate (1/10 volume, 3 M) and then ethanol (3 volumes). A DNA pellet was recovered by centrifugation at 4°C and 16,000 rcf for 30 min and washed twice with ice-cold 80% (v/v) ethanol. The DNA pellet was then dried in a SpeedVac Concentrator (Savant), resuspended in 10 mM tris-HCl (pH 8.0), and transferred to a new Protein LoBind tube.
These PCR-amplified genes now contain the amino T7–PC biotin promoter upstream of the coding sequences. The above protocol was repeated for mVenus using the amino T7 promoter (no PC biotin reaction performed). This template is the control amino-only DNA template.
LA-DNA formation with binding of monovalent streptavidin
To create the LA-DNA, monovalent streptavidin (provided by Howarth’s laboratory, Department of Biochemistry, University of Oxford) was bound to the PCR templates containing the amino T7–PC biotin promoter.
The PCR product (1 μg; final DNA concentration of 50 ng/μl) was incubated with a 50× molar excess of monovalent streptavidin in 10 mM tris-HCl (pH 8.0) in Protein LoBind tubes for 3 hours at room temperature and then overnight at 4°C. Amine-only DNA was also incubated with monovalent streptavidin.
Standard bulk solution UV photocleavage of LA-DNA
UV photocleavage of the LA-DNA was performed by using an LED Driver (set at 1.2 mA; LEDD1B, Thorlabs) connected to a 365-nm collimated LED (M365L2-C5, Thorlabs). A total of 10 μl of LA-DNA (50 ng/μl) was held either under ambient light or under the LED Driver (1/3 power setting) at a distance of 4.5 cm for 15 min, with UV directly illuminating the solution in the open tubes. This procedure was also performed with amine-only DNA. A total of 100 ng of each sample was run on a 1.5% (w/v) tris-acetate-EDTA (TAE) agarose gel with a 1-kb ladder (NEB).
T7 RNA transcription from LA-DNA
T7 RNA transcriptions (M0251, NEB) were performed according to the manufacturer’s protocol with the addition of a murine RNAse inhibitor (MB0314, NEB). The final concentration of LA-DNA or amine-only DNA (coding for mVenus) in the T7 RNA transcription was 8 ng/μl with a total reaction volume of 5 μl.
Following the assembly of the reaction mixes on ice, the samples were either held under ambient light or photocleaved under the same conditions as a standard bulk photocleavage. The reactions were then held at 37°C for 1 hour. DNAse I (M0303, NEB) was then added to the tubes, which were held at 37°C for an additional 20 min. EDTA was then added (5 mM final concentration), and the enzymes were denatured at 75°C for 10 min. The entire sample was then run on a 2% (w/v) TAE agarose gel with a single-stranded RNA ladder (N0364, NEB).
PURExpress protein expression in bulk from LA-DNA
Protein expression was performed with the PURExpress In Vitro Protein Synthesis kit (E6800, NEB) according to the manufacturer’s protocol with the addition of a murine RNAse inhibitor (MB0314, NEB). Final reaction volumes were between 2.5 and 4 μl. Poly(ethylene glycol) 4000 (Sigma) was added to the reactions at a final concentration of 1% (w/v). LA-DNA or amine-only DNA (coding for mVenus) was added to a final concentration of 5 to 10 ng/μl (depending on the activity of the batch of PURExpress).
Following the assembly of the reaction mixes on ice, the samples were either held under ambient light or photocleaved under the same conditions as a standard bulk photocleavage. The tubes were then held at 37°C for 3 hours. Aliquots (0.5 μl) were removed at 0, 1, 2, and 3 hours and worked up with 24.4 μl of 10 mM tris-HCl (pH 8.0). Triplicate reactions were performed for each DNA with and without UV irradiation.
The fluorescence intensity of these tris-HCl worked up aliquots was measured using a Cary Eclipse fluorescence spectrophotometer (Varian) with 30 μl of fluorescence cell (105.252-QS, Hellma Analytics). The settings on the fluorescence spectrophotometer were as follows: excitation at 490 nm, emission from 520 to 575 nm with an excitation slit of 20 nm, scan control at slow, and photomultiplier tube (PMT) detector voltage at high. Three scans were taken per sample. Mean fluorescence intensity at 530 nm (maximum for mVenus) of triplicate samples was plotted against time with the SD of each value.
Confocal microscopy
Fluorescence microscopy was performed with a confocal microscope (SP5, Leica) using a 10× objective lens (HC PL FLUOTAR, Leica) under the differential interference contrast polarizing setting. Image resolutions were set at 512 × 512 pixel2, and four frame averages were taken per image. The overall laser power was set to 20%, the smart offset was set to −1%, and the pinhole was held at 100 μm throughout.
The following settings were used for each fluorophore: mVenus: 514-nm laser at 30%, PMT detector between 525 and 575 nm, and smart gain at 800; mCherry: 543-nm laser at 40%, PMT detector between 600 and 700 nm, and smart gain at 1100; emGFP: 488-nm laser at 25%, PMT detector between 500 and 600 nm, and smart gain at 900; and TAMRA: 543-nm laser at 15%, PMT detector between 560 and 625 nm, and smart gain at 725.
All fluorescence images were analyzed using Fiji (ImageJ). Line profiles from images are shown in the same figures. Any brightness/contrast changes made to the images were the same for both +UV and −UV image sets.
Preparation of DPhPC and DPPE-mPEG2000 lipid-in-oil stocks
DPhPC (4ME 16:0 PC, Avanti) and DPPE-mPEG2000 (16:0 PEG2000 PE, Avanti) were weighed out, dissolved in chloroform, and then added to a new glass vial to give a total lipid amount of 2 × 10−6 mol. Two different molar fractions of the two lipids were used, 10 and 15% DPPE-mPEG2000. The chloroform was evaporated in a nitrogen stream, and the residue was desiccated for >3 hours before storage under argon gas at −20°C. Before use, the dried films were dissolved in hexadecane (Sigma), and silicone oil (AR-20, Sigma) was then added to a total volume of 2 ml, for a 1 mM total lipid concentration. Three different volume ratios of hexadecane and silicone oil were used: 50:50, 55:45, and 40:60 (hexadecane/silicone oil). Both oils were filtered with Millex-GP 0.22-μm filters (Millipore) before addition to the lipid.
General electrical recording protocol
Electrical recordings were conducted with a patch-clamp amplifier (Axopatch 200B, Axon Instruments) with its head stage, contained in a Faraday cage, connected to two Ag/AgCl wire electrodes (Sigma) 100 μm in diameter. The tips of both electrodes were coated with a hydrogel at 0.5% (w/v; low gelling agarose; Sigma). The electrodes were then inserted in droplets or placed on the sides of networks with the aid of micromanipulators (NMN-21, Narishige). A current signal was obtained by episodic acquisition and filtered at 2 kHz. Data were collected at 10 kHz at an interval of 100 μs and ×5 gain with a Digidata 1440A digitizer (Axon Instruments). Using an analog output, a waveform epoch with steps at 0, 50, 0, −50, and 0 mV, each with a duration of 10 s, was used to detect the current between droplet pairs or through networks. Data were analyzed by Clampfit (version 10.3; Axon Instruments), filtering with a low pass Bessel (8 pole), and −3-dB cutoff of 40 Hz. All electrical recordings were conducted at 22.0 ± 1.5°C.
PURExpress protein expression in droplets from LA-DNA
PURExpress reactions for droplet expression were prepared as described for bulk studies (above). Reactions were held on ice until droplet formation.
Poly(methyl methacrylate) chips were micromachined using a computer numerical control (CNC) milling machine (Modela MDX-40, Roland) to create arrays of circular wells 1.5 mm in diameter to which the lipid-in-oil stocks were added. Droplets (50 nl) were made into each of these wells using a 0.5-μl syringe (7000.5 KH, Hamilton) and were observed through a microscope (SZX10, Olympus).
To create a DIB of two synthetic cells, a single 50-nl droplet was placed in a well. Then, the second droplet was dropped on top or next to the first droplet, and the two droplets were left to incubate for at least 5 min. With the DPhPC and DPPE-mPEG2000 lipid mixes, the monolayers form almost instantaneously, so there was no need for incubation before bringing the two droplets together. Following droplet and DIB formation, all samples were held in a closed petri dish along with a smaller petri dish containing water to act as a hydration chamber.
Standard droplet/network UV photocleavage of LA-DNA
All droplets and networks under lipid-in-oil mixes were UV-treated with the same setup as the bulk solutions; however, the LED Driver (5/6 power setting) was set at a distance of 4.5 cm for 5 min. The UV setup illuminated the droplets/networks from above, so the UV only traveled through the lipid-in-oil before reaching the droplets/networks. Because all UV illumination was performed from above and confocal imaging was performed from below, the fluorescence signal from the confocal demonstrated that the UV penetrated all the way through the networks and activated the synthetic cells on the bottom of the networks.
LA-mVenus and LA-mCherry droplet experiments
Two droplets both containing either LA-mVenus or LA-mCherry DNA were incubated together to form a DIB in 10% DPPE-mPEG2000/DPhPC in 50:50 (v/v) hexadecane/silicone oil. These droplets were either held under ambient light or subjected to the standard droplet photocleavage. The droplets were then incubated at 25°C for 18 hours, and the fluorescence signal was imaged with the fluorescence confocal microscope under the settings for each fluorophore.
LA–αHL-GFP droplet experiment
Two droplets, one containing LA–αHL-GFP DNA and the second containing no DNA, were incubated together to form a DIB in 15% DPPE-mPEG2000/DPhPC in 50:50 (v/v) hexadecane/silicone oil. The droplets were either held under ambient light or subjected to the standard droplet photocleavage. The droplets were then incubated at 25°C for 18 hours, and the fluorescence signal was imaged with the fluorescence confocal microscope under the settings for each fluorophore.
LA-αHL and TAMRA droplet diffusion experiment
Two droplets, one containing LA-αHL DNA and the second containing TAMRA (20 μM), were incubated together to form a DIB in 15% DPPE-mPEG2000/DPhPC in 50:50 (v/v) hexadecane/silicone oil. The droplets were either held under ambient light or subjected to the standard droplet photocleavage. The droplets were then incubated at 25°C for 18 hours, and the fluorescence signal was imaged with the fluorescence confocal microscope under the settings for each fluorophore.
LA-αHL droplet electrical recording experiment
PURExpress reactions were made up as described previously with one exception, the final DNA concentration of LA-αHL was 0.5 ng/μl. Single droplets were made containing LA-αHL in 10% DPPE-mPEG2000/DPhPC in 50:50 (v/v) hexadecane/silicone oil. These droplets were either held under ambient light or subjected to the standard droplet photocleavage. These single droplets were incubated at 37°C for 10 min.
Electrical recording analysis
Using micromanipulators, hydrogel-coated electrodes were brought in contact with their respective droplets in the lipid-in-oil solution. Once a droplet spontaneously adheres to the hydrogel coating, the droplet is lifted off the surface of the chamber with the micromanipulators. As a result, the droplet hangs on the electrode’s tip. The two droplets, one attached to each electrode, were brought together to form a bilayer. To confirm that a bilayer was formed, we measured the capacitance during episodic stimulation with a triangular wave that oscillates between +15 and −15 mV every 30 ms. A capacitance of ≥20 pF demonstrates that a bilayer was formed. Once a bilayer is achieved, the droplets are evaluated as by the general electrical recording protocol.
3D printing networks of PURExpress droplets
PURExpress reactions for 3D network expression were prepared as described for bulk studies (above). The reaction mixes were held on ice until network printing. Apart from the optimization experiments, all 3D printing was performed in 10% DPPE-mPEG2000/DPhPC in 40:60 hexadecane/silicone oil.
3D droplet printing was performed as previously described (11), with important improvements. Before use, the handmade glass printing nozzles were oxygen plasma–treated (FEMTO version A, Diener Electronic) for 8 min (5 to 10 SCCM). Before loading each solution into a nozzle, it was washed with soap (Detsan neutral detergent, Bucks), water, and ethanol (with nitrogen gas used to remove each wash solution from the nozzle) and then left to air-dry for 10 min before use. The hexadecane plug and PURExpress solutions were loaded into the nozzle from a CNC-milled array with wells 1.5 mm in diameter. The networks were printed into truncated glass cuvettes (3.1125/SOG/10, Starna) containing the optimized lipid-in-oil solution. Maps representing each layer of each 3D-printed structure are described for each experiment. The piezo microcontroller was replicated but with a higher voltage output (±33 V) by the electronics workshop (Department of Chemistry, University of Oxford). The diameter of the droplets was tuned to ~75 μm, by altering the voltage and pulse of the piezo, before automated printing. Following network formation, all samples were held in a closed petri dish along with a smaller petri dish containing water to act as a hydration chamber.
LA-mVenus and LA-mCherry 3D network experiments
3D-printed droplet networks were created from solutions containing either LA-mVenus or LA-mCherry DNA. Maps for these structures were three (mCherry) or four (mVenus) layers of 7 × 8 droplets. Throughout printing, the PURExpress solution was held under ambient light in the nozzle. After printing, the droplet networks were either held under ambient light or subjected to the standard droplet photocleavage. The droplet networks were then incubated at 25°C for 18 hours, and the fluorescence signal was imaged with the fluorescence confocal microscope under the settings for the appropriate fluorophore.
LA-αHL 3D network electrical recording experiment
3D-printed droplet networks were created with PURExpress reactions that contained LA-αHL DNA at a final DNA concentration of 0.5 ng/μl. Maps for these structures were six layers of 7 × 8 droplets. Throughout printing, the PURExpress solution was held under ambient light in the nozzle. After printing, these droplet networks were either held under ambient light or subjected to the standard droplet photocleavage. These droplet networks were then incubated at 37°C for 10 min.
Electrical recording analysis
The tips of the electrodes were bent to aid their placement onto the sides of the networks before hydrogel coating. By using micromanipulators, the electrodes were brought in contact with a network, in the lipid-in-oil solution. The droplets on the sides of the networks spontaneously adhere to the electrodes. The capacitance is tested during episodic stimulation with a triangular wave that oscillates between +15 and −15 mV every 30 ms. A capacitance of ≥20 pF demonstrates that the droplets the electrodes are attached to, on each side of a single network, are connected by continuous droplet bilayers throughout the network. The networks were then evaluated as by the general electrical recording protocol.
LA-Venus 3D-printed pathway experiment
A 3D-printed network was created with an mVenus pathway through a network containing no DNA, using two PURE solutions containing LA-mVenus DNA and no DNA. Maps for each 2D layer of these structures are shown in fig. S13.
Initially, five layers of map A were printed with the LA-mVenus DNA solution. Then, four layers of map B were printed, with the no-DNA solution, around the L channel structure already printed. Immediately following this, four layers of map C were printed, with the same no-DNA solution, on top of the whole network structure.
Throughout printing, the PURExpress solution was held under ambient light in the nozzle. After printing, the droplet networks were subjected to the standard droplet photocleavage. The droplet networks were then incubated at 25°C for 18 hours, and the fluorescence signal was imaged with the fluorescence confocal microscope under the settings for the appropriate fluorophore.
LA-αHL patterned 3D channel experiment
A 3D-printed network was created with an αHL pathway through a network containing no DNA, using two PURE solutions containing LA-αHL DNA (at a final DNA concentration of 0.5 ng/μl) and no DNA. Maps for this structure are the same for the LA-mVenus 3D-printed pathway experiment.
Initially, five layers of map A were printed with the LA-αHL DNA solution. Then, four layers of map B were printed, with the no-DNA solution, around the L channel structure already printed. Immediately following this, four layers of map C were printed, with the same no-DNA solution, on top of the whole network structure.
Throughout printing, the PURExpress solution was held under ambient light in the nozzle. After printing, these droplet networks were subjected to the standard droplet photocleavage. The droplet networks were then incubated at 37°C for 10 min.
Electrical recording analysis
Electrical recording experiments were carried out on these networks in the same manner as the LA-αHL 3D network electrical recording experiment. Initially, electrical recording measurements were made across the two sides of the network connected with the LA-αHL pathway. Following this, one electrode was removed from the network and placed on a different side, using the micromanipulator. This was repeated until all the configurations shown in Fig. 7 were analyzed.
LA-mVenus and LA-αHL light-patterned 2D channel
3D-printed droplet networks were created from solutions containing either LA-mVenus (concentration as above) or LA-αHL DNA (at a final DNA concentration of 0.5 ng/μl). Maps for these structures were four layers of 9 × 11 droplets. Throughout printing, the PURExpress solution was held under ambient light in the nozzle.
After printing, shapes were illuminated onto the network using a Leica DMi8 wide-field light microscope, with an HCX PL FL L 40× lens and an illumination field diaphragm. Illumination was carried out for 1 min using a DAPI filter cube, 1/4 shutter intensity, 10% illumination intensity, and a circular illumination field setting of 2. The pathways were drawn into the networks by illuminating multiple conjoined restricted circles in the desired location.
The LA-mVenus droplet networks were then incubated at 25°C for 18 hours, and the fluorescence signal was imaged with the fluorescence confocal microscope under the settings for the appropriate fluorophore.
The LA-αHL droplet networks were incubated at 37°C for 10 min. Electrical recording experiments were then carried out on these networks in the same manner as the LA-αHL patterned 3D channel experiment.
Supplementary Material
Acknowledgments
We would like to thank M. Howarth for providing a sample of the monovalent streptavidin and R. Wade-Martins for providing the mCherry gene. Funding: This work was supported by a European Research Council Advanced Grant. M.J.B. is funded by Merton College, Oxford. A.D.G. was supported by a Biotechnology and Biological Sciences Research Council Doctoral Training Programme in Molecular Biochemistry and Chemical Biology studentship. Author contributions: M.J.B. and H.B. conceived the experiments. M.J.B. and V.R.S. performed the experiments. A.D.G. and S.N.O. assisted with instrument optimization. M.J.B and H.B. wrote the manuscript with contributions from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Supplementary information is available in the online version of the paper. Correspondence and requests for materials should be addressed to M.J.B. or H.B.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/4/e1600056/DC1
Fig. S1. Synthesis of LA-DNA from an amino T7 primer.
Fig. S2. Binding of monovalent streptavidin and photocleavage of LA-DNA.
Fig. S3. T7 RNA transcription from LA-DNA.
Fig. S4. Light-activated expression from LA-mCherry DNA in synthetic cells.
Fig. S5. Expression from amine-only mVenus DNA and LA-mVenus DNA in synthetic cells.
Fig. S6. Light-activated transfer of a small-molecule fluorophore between synthetic cells.
Fig. S7. Light-activated expression from LA-mCherry DNA in synthetic tissues.
Fig. S8. Expression from amine-only mVenus DNA in synthetic tissues.
Fig. S9. Tuning the packing of synthetic cells inside synthetic tissues by altering the bilayer size between synthetic cells.
Fig. S10. Restricted microscope illumination of a synthetic tissue.
Fig. S11. LA-mVenus protein expression in a light-patterned pathway.
Fig. S12. Electrical recordings from an L-shaped 2D pathway formed by light patterning of an LA-αHL DNA 3D-printed synthetic tissue.
Fig. S13. The three printing maps used to create the 3D printed pathway.
REFERENCES AND NOTES
- 1.Tawfik D. S., Griffiths A. D., Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 16, 652–656 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Noireaux V., Libchaber A., A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. U.S.A. 101, 17669–176674 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murtas G., Kuruma Y., Bianchini P., Diaspro A., Luisi P. L., Protein synthesis in liposomes with a minimal set of enzymes. Biochem. Biophys. Res. Commun. 363, 12–17 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Nishikawa T., Sunami T., Matsuura T., Ichihashi N., Yomo T., Construction of a gene screening system using giant unilamellar liposomes and a fluorescence-activated cell sorter. Anal. Chem. 84, 5017–5024 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Fujii S., Matsuura T., Sunami T., Kazuta Y., Yomo T., In vitro evolution of α-hemolysin using a liposome display. Proc. Natl. Acad. Sci. U.S.A. 110, 16796–16801 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichihashi N., Usui K., Kazuta Y., Sunami T., Matsuura T., Yomo T., Darwinian evolution in a translation-coupled RNA replication system within a cell-like compartment. Nat. Commun. 4, 2494 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Lu W.-C., Ellington A. D., In vitro selection of proteins via emulsion compartments. Methods 60, 75–80 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Kaneda M., Nomura S.-i. M., Ichinose S., Kondo S., Nakahama K.-i., Akiyoshi K., Morita I., Direct formation of proteo-liposomes by in vitro synthesis and cellular cytosolic delivery with connexin-expressing liposomes. Biomaterials 30, 3971–3977 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Lentini R., Santero S. P., Chizzolini F., Cecchi D., Fontana J., Marchioretto M., Del Bianco C., Terrell J. L., Spencer A. C., Martini L., Forlin M., Assfalg M., Dalla Serra M., Bentley W. E., Mansy S. S., Integrating artificial with natural cells to translate chemical messages that direct E. coli behaviour. Nat. Commun. 5, 4012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karzbrun E., Tayar A. M., Noireaux V., Bar-Ziv R. H., Programmable on-chip DNA compartments as artificial cells. Science 345, 829–832 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Villar G., Graham A. D., Bayley H., A tissue-like printed material. Science 340, 48–52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M., Asanuma H., Komiyama M., Azobenzene-tethered T7 promoter for efficient photoregulation of transcription. J. Am. Chem. Soc. 128, 1009–1015 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Estevez-Torres A., Crozatier C., Diguet A., Hara T., Saito H., Yoshikawa K., Baigl D., Sequence-independent and reversible photocontrol of transcription/expression systems using a photosensitive nucleic acid binder. Proc. Natl. Acad. Sci. U.S.A. 106, 12219–12223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diguet A., Yanagisawa M., Liu Y.-J., Brun E., Abadie S., Rudiuk S., Baigl D., UV-induced bursting of cell-sized multicomponent lipid vesicles in a photosensitive surfactant solution. J. Am. Chem. Soc. 134, 4898–4904 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olejnik J., Sonar S., Krzymañska-Olejnik E., Rothschild K. J., Photocleavable biotin derivatives: A versatile approach for the isolation of biomolecules. Proc. Natl. Acad. Sci. U.S.A. 92, 7590–7594 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howarth M., Chinnapen D. J.-F., Gerrow K., Dorrestein P. C, Grandy M. R., Kelleher N. L., El-Husseini A., Ting A. Y., A monovalent streptavidin with a single femtomolar biotin binding site. Nat. Methods 3, 267–273 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu Y., Inoue A., Tomari Y., Suzuki T., Yokogawa T., Nishikawa K., Ueda T., Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751–755 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Shimizu Y., Kanamori T., Ueda T., Protein synthesis by pure translation systems. Methods 36, 299–304 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Bayley H., Cronin B., Heron A., Holden M. A., Hwang W. L., Syeda R., Thompson J., Wallace M., Droplet interface bilayers. Mol. Biosyst. 4, 1191–1208 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holden M. A., Needham D., Bayley H., Functional bionetworks from nanoliter water droplets. J. Am. Chem. Soc. 129, 8650–8655 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Hwang W. L., Holden M. A., White S., Bayley H., Electrical behavior of droplet interface bilayer networks: Experimental analysis and modeling. J. Am. Chem. Soc. 129, 11854–11864 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Villar G., Heron A. J., Bayley H., Formation of droplet networks that function in aqueous environments. Nat. Nanotechnol. 6, 803–808 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Syeda R., Holden M. A., Hwang W. L., Bayley H., Screening blockers against a potassium channel with a droplet interface bilayer array. J. Am. Chem. Soc. 130, 15543–15548 (2008). [DOI] [PubMed] [Google Scholar]
- 24.B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts, P. Walter, Molecular Biology of the Cell (Garland Science, New York, ed. 3, 2015), pp. 1464. [Google Scholar]
- 25.Sanzone A. P., El-Sagheer A. H., Brown T., Tavassoli A., Assessing the biocompatibility of click-linked DNA in Escherichia coli. Nucleic Acids Res. 40, 10567–10575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bubeck P., Winkler M., Bautsch W., Rapid cloning by homologous recombination in vivo. Nucleic Acids Res. 21, 3601–3602 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y., Muyrers J. P., Testa G., Stewart A. F., DNA cloning by homologous recombination in Escherichia coli. Nat. Biotechnol. 18, 1314–1317 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/4/e1600056/DC1
Fig. S1. Synthesis of LA-DNA from an amino T7 primer.
Fig. S2. Binding of monovalent streptavidin and photocleavage of LA-DNA.
Fig. S3. T7 RNA transcription from LA-DNA.
Fig. S4. Light-activated expression from LA-mCherry DNA in synthetic cells.
Fig. S5. Expression from amine-only mVenus DNA and LA-mVenus DNA in synthetic cells.
Fig. S6. Light-activated transfer of a small-molecule fluorophore between synthetic cells.
Fig. S7. Light-activated expression from LA-mCherry DNA in synthetic tissues.
Fig. S8. Expression from amine-only mVenus DNA in synthetic tissues.
Fig. S9. Tuning the packing of synthetic cells inside synthetic tissues by altering the bilayer size between synthetic cells.
Fig. S10. Restricted microscope illumination of a synthetic tissue.
Fig. S11. LA-mVenus protein expression in a light-patterned pathway.
Fig. S12. Electrical recordings from an L-shaped 2D pathway formed by light patterning of an LA-αHL DNA 3D-printed synthetic tissue.
Fig. S13. The three printing maps used to create the 3D printed pathway.