

Abstract
S100A1, a 21-kDa dimeric Ca2+-binding protein, is an enhancer of cardiac Ca2+ release and contractility and a potential therapeutic agent for the treatment of cardiomyopathy. The role of S100A1 in skeletal muscle has been less well defined. Additionally, the precise molecular mechanism underlying S100A1 modulation of sarcoplasmic reticulum Ca2+ release in striated muscle has not been fully elucidated. Here, utilizing a genetic approach to knock out S100A1, we demonstrate a direct physiological role of S100A1 in excitation-contraction coupling in skeletal muscle. We show that the absence of S100A1 leads to decreased global myoplasmic Ca2+ transients following electrical excitation. Using high speed confocal microscopy, we demonstrate with high temporal resolution depressed activation of sarcoplasmic reticulum Ca2+ release in S100A1−/− muscle fibers. Through competition assays with sarcoplasmic reticulum vesicles and through tryptophan fluorescence experiments, we also identify a novel S100A1-binding site on the cytoplasmic face of the intact ryanodine receptor that is conserved throughout striated muscle and corresponds to a previously identified calmodulin-binding site. Using a 12-mer peptide of this putative binding domain, we demonstrate low micromolar binding affinity to S100A1. NMR spectroscopy reveals this peptide binds within the Ca2+-dependent hydrophobic pocket of S100A1. Taken together, these data suggest that S100A1 plays a significant role in skeletal muscle excitation-contraction coupling, primarily through specific interactions with a conserved binding domain of the ryanodine receptor. This warrants further investigation into the use of S100A1 as a therapeutic target for the treatment of both cardiac and skeletal myopathies.
The S100 family of proteins, so named because of their solubility in 100% ammonium sulfate, are small (16–26 kDa), acidic, Ca2+-binding proteins that possess no intrinsic enzymatic activity (1). Instead, S100 proteins function through interaction with target proteins, usually via a Ca2+-dependent mechanism, to elicit biological responses (2). At least 21 different S100 proteins are known in humans, and they are expressed in a highly tissue-specific manner (3, 4). S100A1 is most highly expressed in cardiac and skeletal muscle and is present in lower levels in the brain (5). The structures of both apoS100A1 and Ca2+-S100A1 have been solved via NMR spectroscopy techniques (6, 7). These reveal that S100A1 exists as a homodimer, with each subunit consisting of two EF-hand motifs connected by a loop, termed the hinge region. When S100A1 binds to Ca2+, one of the EF hands, consisting of helices 3 and 4, undergoes a large conformational change, exposing a previously buried hydrophobic pocket made up of residues from the hinge region, helix 3, and helix 4 (6, 7). This hydrophobic pocket has been shown to be the binding interface between S100A1 and a small peptide from the CapZ protein, and it is generally thought to be the target protein interacting region (8, 9).
S100A1 regulates Ca2+ signaling and contractility in cardiac muscle. S100A1 interacts with the ryanodine receptor (RyR)6 and the phospholamban/sarcoplasmic reticulum (SR) Ca2+-ATPase complex, and presumably modulates Ca2+ cycling through interactions with these proteins, leading to faster and more robust myocyte contraction (10–12). More recently, S100A1 has been linked to a protective role against cardiomyopathy; S100A1−/− mice demonstrate an increased incidence of heart failure in stressed hearts, and S100A1 expression levels are decreased in cardiomyopathic conditions (13, 14). As proof of concept, adenoviral gene delivery of S100A1 in a heart failure model restores Ca2+ cycling and markers of cardiac contractility (15). These and similar studies suggest a potential therapeutic role for S100A1 in the treatment of cardiomyopathy. However, the specifics of how S100A1 achieves this beneficial effect are uncertain; the precise region on RyR that S100A1 binds and whether this event is the basis for increased cardiomyocyte contraction have not been fully explored (16–18).
Although a relatively large body of knowledge has accumulated about how S100A1 behaves in cardiac myocytes, skeletal muscle research has lagged behind. Addition of S100A1 has been shown to increase caffeine-evoked force transients in skinned skeletal muscle fibers (19). S100A1 has also been shown to bind the skeletal muscle RyR isoform (RyR1), to potentiate the open probability of RyR1 in lipid bilayer experiments, and to modulate ryanodine binding activity at high and low Ca2+ in SR terminal cisternae (20). Thus, S100A1 may regulate Ca2+ signaling and contractility through similar mechanisms of RyR modulation in cardiac and skeletal muscle. However, the particulars of how S100A1 alters the kinetics of Ca2+ release and the physiological relevance of this mechanism to excitation-contraction (EC) coupling in skeletal muscle have not been well studied.
In an attempt to delve further into how S100A1 modulates EC coupling in skeletal muscle, we examined the effect of S100A1 on the electrically evoked Ca2+ transient in an endogenous experimental setting using enzymatically dissociated, intact, flexor digitorum brevis (FDB) muscle fibers. We demonstrate here the novel finding that the Ca2+ transient following action potential (AP) stimulation is depressed in S100A1−/− skeletal muscle fibers. Using both whole cell epifluorescent and high speed confocal microscopy, we show that the peak amplitude of the Ca2+ transient and the rising rate of the Ca2+ transient are significantly decreased in the absence of S100A1. We also demonstrate rescue of this depressed Ca2+ release phenotype via adenoviral expression of S100A1 in S100A1−/−muscle fibers. To elucidate the biophysical interface between S100A1 and RyR1, we have performed competition experiments between calmodulin (CaM) and S100A1 on intact RyR1 in SR vesicles and located the CaM-binding site on RyR1 as an S100A1 interaction domain. Furthermore, we show that Ca2+-S100A1 binds to a peptide from this domain with low micro-molar affinity. Utilizing NMR spectroscopy we have probed the molecular interaction between these two proteins on a residue by residue basis. Based on these data, we suggest that S100A1 regulates Ca2+ release through coupling to this discrete area on RyR. This domain of interest is conserved between RyR1 and RyR2, and thus we postulate that the molecular mechanism by which S100A1 modulates the Ca2+ release apparatus may be conserved between cardiac and skeletal muscle.
EXPERIMENTAL PROCEDURES
Materials
All chemical reagents were ACS grade or higher and purchased from Sigma unless otherwise indicated. All buffers were passed through Chelex-100 resin (Bio-Rad) to remove trace metals. Perdeuterated Tris, d11-Tris (1 M solution in D2O >98.7 atom % deuterium) was purchased from C/D/N Isotopes Inc. (Pointe-Claire, Quebec, Canada), and D2O (100.0 at % deuterium) was purchased from Aldrich. 15NH4Cl (>99%) and 13C-labeled glucose were purchased from Cambridge Isotope Laboratories (Woburn, MA).
Generation of S100A1−/− Mice
Hybridization screening was used to isolate an EcoRI fragment containing the S100A1 gene from a P1 mouse 129 genomic DNA library (Genome Systems, St. Louis). The targeting vector was constructed by replacing a portion of the S100A1 locus with a DNA cassette containing a bacterial β-galactosidase reporter gene with a nuclear localization signal and a neomycin resistance gene under the control of a phosphoglycerokinase promoter (Fig. 1A). The β-galactosidase reporter gene coding sequence was inserted in-frame at the A8 codon in exon 2 of the S100A1 gene. The targeting vector was electroporated into W4/129S6 embryonic stem cells (Taconic Farms Inc., Germantown, NY), and drug-resistant colonies were selected by growth in G418-containing media (21). Colonies containing homologous recombination events were identified by Southern blot analysis using a BamHI/SmaI fragment located outside the targeting construct (Fig. 1B). Gene-targeted clones were injected into C57BL/6 blastocysts (The Jackson Laboratories, Bar Harbor, ME), and the resulting chimeric male offspring were mated with C57BL/6 females to obtain S100A1+/− progeny. The line was maintained on a mixed C57BL/6–129S6 line. All procedures and breeding were performed in accordance with the National Institutes of Health Guide for the Use and Care of Laboratory Animals and institutional regulations at Texas A&M University.
FIGURE 1. Generation of S100A1LacZ/LacZ knock-out mice.
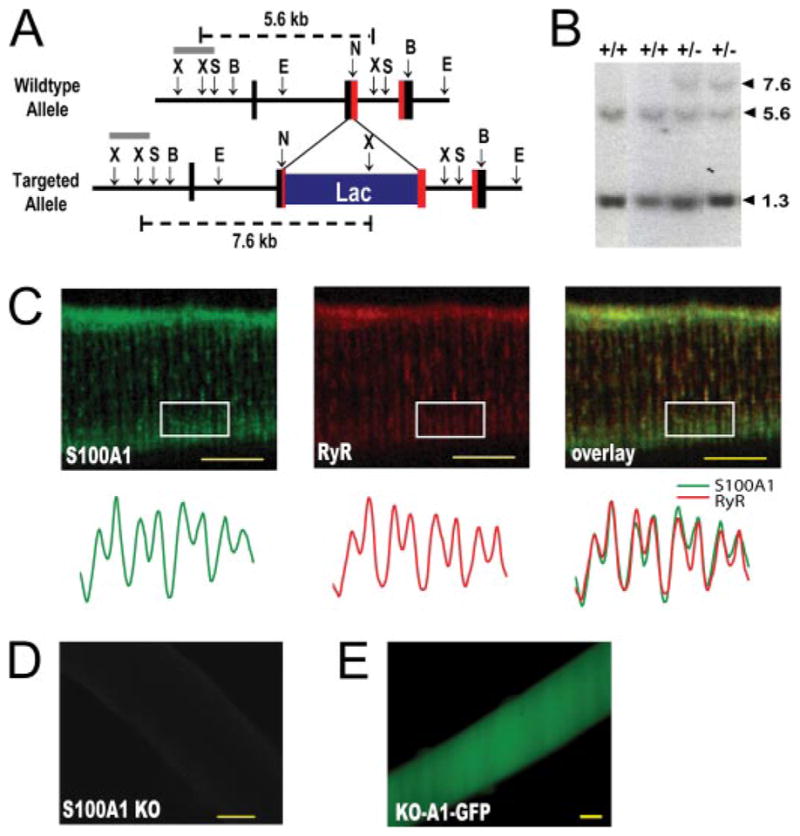
A, structure of the murine S100A1 gene and targeted alleles. The boxed regions represent exons I–III of the S100A1 gene; protein coding sequences are shown in red and untranslated regions in black. The gray line denotes the hybridization probe. Restriction enzyme sites are as follows: N, NcoI; X, XbaI; S, SmaI; B, BglIB; E, EcoRI. B, representative Southern blot for wild type (+/−) and heterozygous (+/+) offspring. The probe and predicted sizes of the hybridizing fragments are shown in A. C, S100A1 localizes near the skeletal muscle Ca2+ release channel (RyR1) in murine FDB fibers, visualized through overlay (right) of anti-S100A1 (left) and anti-RyR (middle) immunofluorescence images. Below, plot profiles of fluorescence intensity from representative boxed regions demonstrate S100A1 fluorescence in two discrete peaks per sar-comere. These peaks align with RyR fluorescent peaks in doublet staining pattern characteristic of RyR1 localization to T-tubule structures in skeletal muscle (bottom right). D, no S100A1 staining is visible in S100A1 KO fibers. E, S100A1 KO fiber 48 h following infection with S100A1-GFP adenovirus. Scale bar denotes 10 μm.
Genotyping
Tail biopsies were digested in proteinase K mixture (10 mM Tris, pH 7.8, 75 mM NaCl, 25 mM EDTA, 1% SDS, and 0.5 mg/ml proteinase K). After phenol/chloroform (1:1 v/v) extraction, genomic DNAs were ethanol-precipitated and resuspended in water. The same Southern strategy that was used to identify homologous recombination events in embryonic stem cells was used to identify F1 founders. Subsequent generations were genotyped using a three-allele PCR strategy. Amplification of a 500-bp product using PCR primers for the mouse β-casein gene (forward primer 5′ GAT GTG CTC CAG GCT AAA GTT 3′and reverse primer 5′AGA AAC GGA ATG TTG TGG AGT 3′) was used to assess genomic DNA quality. Amplification of an 800-bp band using oligonucleotide primers for the β-galactosidase reporter gene (forward primer 5′ GAC ACC AGA CCA ACT GGT AGC GAC 3′ and reverse primer 5′ GCA TCG AGC TGG GTA ATA AGG GTT GGC AAT 3′) indicated the presence of a mutated allele. Amplification of a 1300-bp band using oligonucleotide primers for the wild type S100A1 gene (forward primer 5′ CAG TTT GTA GCA GGT CTT TCA GCT C 3′ and reverse primer 5′ AAA CAC AAA CCA GCC CTA GCC TGC TCC 3′) indicated the presence of a wild type allele. All PCR programs consisted of a 5-min denaturation at 95 °C, followed by 30 cycles consisting of 94 °C for 1 min, 60 °C for 2 min, and 72 °C for 3 min and a final extension step consisting of 72 °C for 7 min.
FDB Fiber Preparation
Fibers were prepared using enzymatic dissociation of FDB muscles of 4–6-week-old C57 ×129 WT and S100A1 KO mice and were cultured as described previously (22). Six mouse pairs from four different litters were used to ensure robustness and meaningful statistical significance. Isolated fibers were cultured on laminin-coated glass bottom Petri dishes. Fibers were cultured in minimal essential medium (MEM) containing 10% fetal bovine serum and 50 μg/ml gentamicin sulfate at 37 °C in a 5% CO2 incubator. After ~24 h in MEM, fiber culture medium was changed to 2 ml of Ringer’s solution (135 mM NaCl, 4 mM KCl, 1 mM MgCl2, 10 mM HEPES, pH 7.4, 10 mM glucose, 1.8 mM CaCl2).
Immunocytochemistry and Analysis
Following enzymatic dissociation and ~24 h in culture, fibers were rinsed three times in Ringer’s solution at room temperature. Fibers were then fixed with 4% paraformaldehyde at room temperature for 10 min and then permeabilized with 0.2% Triton X-100 in phosphate-buffered saline for 20 min. Nonspecific binding sites were blocked by incubation with 4% normal horse serum. Subsequently fibers were incubated for 48 h at 4 °C with either mouse monoclonal antibody against S100A1 (1:200; Sigma) or goat monoclonal antibody against RyR (1:200; Santa Cruz Bio-technology, Santa Cruz, CA). This was followed by overnight incubation at 4 °C with Cy5-conjugated donkey anti-goat or fluorescein isothiocyanate-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch), respectively. These chambers were then mounted to an Olympus IX70 inverted microscope equipped with an Olympus FluoView 500 laser scanning confocal imaging system, using an excitation wavelength of 488 or 543 nm. Fibers were viewed with an Olym-pus 60×/1.2 NA water immersion objective and scanned at ×4 zoom using constant laser power. The gain between the knockout and wild type S100A1 staining was kept constant. Background fluorescence and noise filtering of images were performed using ImageJ software (National Institutes of Health).
Indo-1AM Ratiometric Recordings—
Cultured fibers were transferred to Ringer’s solution and loaded for 30 min with 1 μM Indo-1 AM in 0.1% Me2SO, rinsed three times with Ringer’s solution, and equilibrated for 30 min before recording. The culture chamber was mounted on an Olympus IX71 inverted microscope and viewed with an Olympus 60×/1.20 NA water immersion objective. Fibers were illuminated at 360 nm, and the fluorescence emitted at 405 and 485 nm was detected simultaneously. The emission signal was digitized and sampled repetitively at 0.5-ms intervals with the data collection program LabView 7.1 (National Instruments, Austin, TX), and the fluorescence ratio F405/F485 was calculated. Two parallel platinum electrodes were used to stimulate fibers with a single pulse of 1-ms duration. The stimulation voltage was adjusted to give observed fiber contraction in all cases (10–12 V). Selection criteria for fibers analyzed included morphological integrity, consistent response to twitch stimulation, and reproducibility of transient responses. Fibers were stimulated three times, and responses were averaged. Analysis of emission ratio recordings was performed using OriginPro 7.5.
Adenoviral Expression of S100A1-GFP
Both adenoviral constructs (Ad-GFP, Ad-S100A1) were graciously supplied by Walter J. Koch, Thomas Jefferson University, Philadelphia. Generation of these constructs has been described previously (15), as has infection protocol for FDB fibers (23). Briefly, ~24 h following enzymatic dissociation of FDB fibers, WT and KO dishes received 20 μl of Ad-S100A1 or Ad-GFP in MEM without serum. Fibers were incubated for 1 h with adenovirus, and then the medium was changed to MEM containing both fetal bovine serum and gentamicin to stop infection and for continuous culture. 48 h following infection fibers were imaged on an Olympus IX71 inverted microscope with 470 ± 10 nm excitation filter and 505LP emission filter to detect GFP fluorescence.
Fluo-4 AM Fluorescence Recordings
Cultures were loaded with 2 μM Fluo-4 AM in 0.1% Me2SO for 30 min, rinsed three times with Ringer’s solution, and equilibrated for 30 min before recording. The culture chamber was mounted on a Zeiss Axio-vert 200M inverted microscope on a Zeiss LSM 5 Live confocal system and viewed with a ×63, NA water immersion objective. Excitation was provided by a 488 nm 100-milliwatt diode laser with emission collected using an LP 505 filter. Fibers were stimulated as detailed above at a set time after the start of a confocal scan. Fibers were selected based on the above inclusion criteria as well as an additional criterion of minimum fiber movement in the line scan plane to prevent movement artifacts in recordings. The confocal system was operated in line scan x-t mode, with images collected at either 50 or 100 μs/line for 500-ms acquisition time using Zeiss LSM 5 live software. Three trials were taken for each fiber at each of two regions of interest. In separate experiments, fibers were imaged on an Olympus IX71 inverted microscope using a 60×/1.20 NA water immersion objective coupled to a Bio-Rad Cell Map laser scanning confocal system. The system was again operated in line scan x-t mode at a scanning speed of 2 ms/line for 512 ms. Line scan images were processed and fluorescent recordings were converted to ΔF/Fo. Images were then analyzed using OriginPro 7.5 to characterize the rising and decaying phase of the fluorescence recording.
Statistical Analysis
All statistical analysis was performed using OriginPro7.5. All data are presented as mean ± S.E. unless otherwise noted. Normality of data sets was ensured for each statistical calculation. All significance tests were done using Student’s t test, and significance was set at p < 0.05.
Isolation of SR Vesicles
SR was isolated from a New Zealand White rabbit. The method of MacLennan (24) was used with some modifications. Following euthanasia, white muscle from hind legs and back of the rabbit was isolated, cut into small pieces, and transferred into a working buffer containing NaCl (120 mM), imidazole (10 mM), DTT (0.1 mM), and EGTA (0.1 mM) at pH 7.4 in the presence of the protease inhibitor leupep-tin. The muscle was blended into a fine homogenate and centrifuged in a large Fiberlite F10-6x500y GSA rotor for 10 min at 3300 rpm. The fat layer at the top of the centrifuge tubes was removed, and the supernatant was strained through four layers of cheesecloth. The pH of the supernatant was adjusted to 7.4 with dry imidazole. Mitochondria were removed by centrifuging the supernatant at 7700 rpm for 15 min in the Fiberlite GSA rotor. This supernatant was centrifuged in a Beckman Ti-19 rotor at 19,000 rpm for 70 min. The resulting pellet was transferred into a homogenizer. Myosin was removed by spinning the homogenate in a Ti-50.2 rotor at 10,500 rpm for 10 min. The final spin yielded SR pellets by spinning the supernatant in a Ti-50.2 rotor at 32,000 rpm for 30 min. SR pellets were transferred into a homogenizer and resuspended using a resuspension buffer (100 mM KCl and 20 mM HEPES at pH 7.0) to a final working concentration of 13.0 mg/ml protein.
Western Blots
5 μl of CaM-linked agarose beads (Sigma) were washed three times with binding buffer that included bovine serum albumin (15 μM), NaCl (50 mM), CaCl2 (100 nM to 2 mM), TES (5 mM), pH 7.5. Higher bovine serum albumin and NaCl concentrations, along with 1 mM MgCl2, were also tested and did not affect the outcome of the experiments. Various concentrations of S100A1 or CaM were added to the beads and mixed with 2 μl of the SR vesicle preparation for a final volume of 50 μl. After a 2-h incubation period at 4 °C, the solutions were spun down; the supernatant was removed, and the beads were washed three times in the binding buffer including the appropriate amounts of S100A1 or CaM. The beads were then boiled, run on a 4–12% acrylamide gel for 3 h, and transferred to a polyvinylidene difluoride membrane. A mouse anti-RyR monoclonal antibody (34C) (1:5000; Alexis, San Diego) was used to visualize the RyR band, which was observed to run on the gel with a molecular weight of ~550 kDa. ImageJ software was used to quantify the intensity of each band.
Tryptophan Fluorescence
Ca2+-S100A1 was added slowly to a 20 μM RyRP12 peptide solution containing 15 mM NaCl, 10 mM CaCl2, 15 mM DTT, 15 mM Tris, pH 7.2, at 37 °C. Fluorescence intensity was monitored during the titrations upon complex formation using excitation at 280 nm and measuring the tryptophan emission at 350 nm.
S100A1 NMR Sample Preparation
The RyRP12 peptide (KKAVWHKLLSKQ) was purified via high performance liquid chromatography as described previously (25). Unlabeled, 15N-, and 13C,15N-labeled S100A1 protein was purified using standard purification procedures (6). The identity and purity of S100A1 were confirmed by amino acid analysis and electro-spray mass spectrometry. The samples used for NMR spectroscopy were passed through an additional G-25 gel filtration column, and the final NMR sample contained 13C,15N-labeled or 15 N-labeled S100A1 (0.5–1.0 mM), unlabeled RyRP12 peptide (1.0–3.0 mM), d11-Tris pH 7.2 (20 mM), CaCl2 (10 mM), NaN3 (0.35 mM), NaCl (20 mM), DTT (20 mM), and D2O (10%). Samples prepared in this manner were stable for at least 2 weeks at 37 °C as judged by no changes in their NMR spectra.
NMR Spectroscopy
NMR spectra were collected at 37 °C with a Bruker DMX600 NMR spectrometer (600.13 MHz for protons) and a Bruker AVANCE 800 NMR spectrometer (800.27 MHz for protons) equipped with four frequency channels and a triple-resonance z axis gradient 5 mm cryogenic probe head. Unless otherwise stated, multidimensional NMR data were collected in the indirect dimensions using States-TPPI phase cycling using a 1-s relaxation delay (26). For most experiments, initial delays in the indirect dimension were set to give zero- and first-order phase corrections of 90 and 180°, respectively (27). Sequential backbone and side chain assignments of S100A1 were obtained using standard heteronuclear multidimensional NMR spectroscopy as described previously (28). In general, pulse field gradients were included to purge undesired transverse magnetization, and the WATERGATE technique was used in most cases to suppress the solvent signal in samples dissolved in H2O. In samples dissolved in D2O, low power pre-saturation of the HDO resonance was used. The backbone coupling constants (3JNH-Hα) of S100A1 were measured using the HNHA experiment and evaluated as described (29). The data were processed on Linux Fedora 3.0 with the software package NMRPIPE (30), and all proton chemical shifts are reported with respect to the H2O or HDO signal, which is taken as 4.658 ppm downfield from external TSP (0.00 ppm) at 37 °C. The 13C and 15N chemical shifts were indirectly referenced using the following ratios of the zero-point frequencies at 37 °C: 0.10132905 for 15N-1H and 0.25144953 for 13C-1H (31–33).
Uniformly 15N-labeled S100A1 was used to collect the two-dimensional 1H, 15N fast HSQC (34), three-dimensional 15N, 15N-edited HMQC-NOESY-HSQC (35), three-dimensional HNHA (36), and three-dimensional 15N-edited NOESY-HSQC spectra. The 13C, 15N-labeled S100A1 was used to collect three-dimensional CBCA(CO)NH (37), three-dimensional H(CCO)NH (38), three-dimensional HNCACB (39), three-dimensional HNCO (40), three-dimensional HNCA (34), three-dimensional C(CO)NH (37), four-dimensional 13C,15N-edited NOESY-HSQC (41), and four-dimensional 13C,13C-edited NOESY-HSQC spectra.
RESULTS
Generation/Characterization of S100A1−/− Mice
The mutated S100A1 locus exhibited a Mendelian inheritance pattern as follows: 19% wild type (+/+), 56% heterozygous (+/−), and 25% homozygous (−/−). Homozygous mice exhibited no detectable S100A1 protein in tissues processed for immunohis-tochemistry (Fig. 1D) or immunodot blots (data not shown). In addition, the S100A1 locus was identified as a significantly down-regulated gene product in gene expression profiling (microarray) experiments comparing wild type and knock-out mouse tissues (data not shown). Heterozygous and homozygous mice exhibited normal fertility, general appearance, general behavior, and life span. These findings are consistent with previous reports of no obvious overt phenotype in other S100A1 knock-out mouse models (42); this knock-out mouse model is further described in the on-line data base managed by the Trans-NIH Mouse Initiative, Deltagen, and Lexicon Knockout Mice, and Phenotypic Data.
Localization of S100A1 and RyR1 in Isolated FDB Muscle Fibers
Although previous reports have demonstrated colocal-ization of S100A1 and RyR2 at distinct sarcomeric structures in cardiac muscle (43, 44), and S100A1 has been shown to associate with the SR and membrane fractions in slow twitch muscle (5), it was important to verify their subcellular localization in fast-twitch skeletal muscle fibers. To this end isolated FDB fibers were double immunostained for S100A1 and RyR. As visualized in Fig. 1C, S100A1 and RyR expressed similar staining patterns with a notable absence of staining at the Z-line and increased staining at the junction of A bands and I bands. S100A1 fluorescence intensity accurately reflects the doublet staining pattern in each sarcomere characteristic of RyR1 localization to T-tubule/SR triad junctions in mammalian skeletal muscle. The large troughs in fluorescence intensity of both RyR and S100A1 correspond to decreased expression at M-band structures, with the two discrete peaks representing increased expression at A/I junctions on adjacent sides of the intervening Z-disk (45). T-tubule structures are known to invaginate from the sarcolemma at A/I junctions in mammalian skeletal muscle. Thus, S100A1 localizes near the skeletal muscle RyR at triad structures in FDB muscle fibers.
S100A1−/− Muscle Fibers Exhibit Normal Resting [Ca2+]i, but Decreased Peak Amplitude of the Ca2+Transient Following Single Action Potential Stimulation
S100A1 has been shown to increase caffeine-evoked force transients in skeletal muscle (19). However, caffeine directly stimulates SR Ca2+ release, bypassing the voltage sensor in the EC coupling machinery; hence, a direct physiological role of S100A1 in EC coupling has not been demonstrated in skeletal muscle. We therefore sought to evaluate the effect of S100A1 on the electrically evoked Ca2+ transient in skeletal muscle fibers using a ratiometric Ca2+ indicator widely used to monitor changes in myoplasmic Ca2+ concentration. After loading cultured FDB muscle fibers by exposure to Indo-1 AM in the bathing medium (see “Experimental Procedures”), individual fibers were stimulated with a 1-ms pulse from a field electrode, and the muscle fibers were assayed for changes in fluorescence emission intensity using whole cell epifluorescent microscopy. Fig. 2A shows the average response of KO and WT fibers to single AP stimulation (WT, n = 9 fibers; KO, n = 12 fibers) initiated at t = 0 ms. KO fibers exhibited no significant differences in resting Indo-1 ratios (measured without stimulation) from WT controls (WT = 1.56 ± 0.04, KO = 1.57 ± 0.08, p = 0.37) (Fig. 2B). However, KO fibers demonstrated a significantly decreased peak amplitude of the Indo-1 ratio following single AP stimulation, as quantified in Fig. 2C (WT =2.91 ±0.05, KO =2.53 ± 0.07, p = .0005).
FIGURE 2. S100A1−/− muscle fibers exhibit normal resting [Ca2+ ]i Indo-1 ratio but a decreased peak amplitude of the Indo-1 Ca2+ transient following single AP stimulation.

A, average Ca2+ transient from WT (black trace) and KO (gray trace) fibers. Isolated fibers were stimulated with field electrodes at time 0, and emission ratio was assayed. B, bar plot summarizing resting Indo-1 ratio averages of WT and KO fibers. No significant differences in resting Ca2+ concentration were detected (p =0.3656). WT =1.56 ±0.04, n = 9; KO = 1.57 = 0.08, n = 12. C, bar plot representation of peak Indo-1 ratio reached in A. WT = 2.91 ± 0.05, n = 9; KO = 2.53 ± 0.07, n = 12. *, data were significantly different at p = 0.0005.
To increase the temporal resolution of the assayed Ca2+ transient and to improve the signal to noise ratio of these Ca2+ signals, we used high speed confocal microscopy (50 μs/line) as monitored by the non-ratiometric fluorescent indicator Fluo-4 AM. The difference in the peak amplitude of the Ca2+ transient seen in Fig. 2A is preserved using ΔF/Fo signals from these Fluo-4 recordings (Fig. 3A). As quantified in Fig. 3B, KO fibers demonstrate a 31.6% decrease in peak ΔF/Fo when compared with WT controls following single AP stimulation (WT = 5.780 ± 0.377, n =26. KO = 3.953 ± 0.274, n = 25. p = 0.0003). Because resting [Ca2+]i is the same in WT and KO FDB fibers (Fig. 2B), and as ΔF/Fo records correct for differences in dye loading, these values represent differences in the Ca2+ transients between WT and KO fibers. The above results demonstrate that S100A1 positively modulates SR Ca2+ release following electrical stimulation in murine FDB fibers, implicating a physiological role of S100A1 in skeletal muscle EC coupling.
FIGURE 3. S100A1−/−muscle fibers exhibit decreased peak amplitude of the Fluo-4 Ca2+transient.

A, average transient responses to single AP stimulation from similar assay to Fig. 2A, performed with non-ratiometric Fluo-4 AM loaded fibers and assayed with high speed confocal line scan microscopy (50 μs/line) for increased temporal resolution. Data presented as ΔF/Fo. B, bar plot summarizing differences in peak amplitude of fluorescent transient. WT =5.78 ± 0.38, n =26; KO =3.95 ±0.27, n =25. *, data were significantly different at p = 0.0003.
Adenoviral Expression of S100A1 Rescues the Ca2+ Signaling Phenotype Seen in KO Fibers
We next attempted to rescue this depressed Ca2+ signaling phenotype in S100A1 KO fibers through adenoviral expression of S100A1. In this experiment, KO fibers were infected with an adenovirus containing S100A1 and GFP cDNA (Ad-S100A1) or simply GFP cDNA (Ad-GFP) alone. WT fibers were infected with Ad-GFP alone. 48 h following infection GFP fluorescence could be seen clearly in ~95% of all WT and KO fibers (representative fiber in Fig. 1E). Fibers were then loaded with Indo-1 AM, and those fibers that were strongly positive for GFP fluorescence and passed our selection criteria (see “Experimental Procedures”) were then evaluated for their responses to single AP stimulation (Fig. 4A). Fluo-4 was not used for this experiment as the emission spectrum significantly overlaps with that of GFP. KO fibers infected with Ad-GFP, without S100A1, showed a significantly reduced peak amplitude of the Ca2+ transient when compared with WT fibers infected with the same virus, as expected from our previous findings (WT-GFP =2.09 ± 0.07, n = 5; KO-GFP = 1.81 ± .07, n = 5, p = 0.021) (Fig. 4B). KO fibers infected with Ad-S100A1 showed nearly complete rescue to the WT phenotype, demonstrating a significantly increased peak amplitude of the Ca2+ transient when compared with KO fibers infected with Ad-GFP (KO-A1 = 2.03 ± 0.06, n = 6; KO-GFP = 1.81 ± 0.07, n = 5, p = 0.035) (Fig. 4B).
FIGURE 4. Adenoviral expression of S100A1 restores the peak amplitude of the Indo-1 Ca2+transient in S100A1 KO muscle fibers.

A, average Indo-1 Ca2+ transient of WT fibers infected with Ad-GFP alone (black trace), KO fibers infected with Ad-GFP alone (gray trace), and KO fibers infected with Ad-S100A1-GFP (red trace). Traces were smoothed following signal averaging to increase signal to noise ratio. B, quantification of peak amplitude of Indo-1 ratio reached for 3 groups depicted in A. WT-GFP = 2.09 ± 0.07, n = 5; KO-GFP = 1.81 ± 0.07 n = 5; KO-A1 = 2.03 ± 0.06. *, data were significantly different at p < 0.05.
S100A1+/+ Muscle Fibers Exhibit a Prolonged Rising Phase of the Ca2+ Transient Following Single Action Potential Stimulation
The duration of RyR1-mediated Ca2+ release from the SR following electrical excitation can be roughly monitored by evaluating the duration of the rising phase of the Ca2+ transient. Here, we quantify the rising phase of the Ca2+ transient as the time elapsed from 10 to 90% of the peak ΔF/Fo signal. It was observed that the rising phase following single AP stimulation occurs in ~1–2 ms in murine FDB muscle fibers. Using high speed confocal microscopy with a sampling rate of 50 μs/line, we were able to discern differences in the duration of Ca2+ release between WT and S100A1−/− fibers, as depicted in Fig. 5. The rising phase from 10 to 90% of the Ca2+ transient was prolonged by 38.6% in KO fibers compared with controls, as quantified in Fig. 5B (WT = 1.284 ± 0.06 ms, n = 19; KO = 1.78 ± 0.11 ms, n = 19, p = 0.0003). The time to half-peak amplitude was also increased in KO fibers to 1.48 ms, compared with 0.98 ms for controls, a 33.8% increase in duration. Taken together (Figs. 2–5), these results suggest that S100A1 positively modulates the Ca2+ transient at least in part by augmenting the kinetics of RyR-mediated Ca2+ release following electrical stimulation in fast twitch skeletal muscle.
FIGURE 5. S100A1−/− muscle fibers exhibit prolonged rising phase of the Fluo-4 Ca2+ transient following single AP stimulation.

A, average rising phase of Ca2+ transient in Fluo-4 loaded WT (black trace) and KO (gray trace) fibers. Data presented as ΔF/Fo normalized for peak amplitude of Ca2+ transient. B, bar plot summarizing temporal differences seen in the rising phase of the Ca2+ transient, quantified as the time duration from 10 to 90% peak ΔF/Fo. WT = 1.28 ± .06 ms, n = 19; KO = 1.78 ± .11 ms, n = 19. * data were significantly different at p = 0.0003.
S100A1 Competes with CaM for Binding to the Intact RyR1
Because S100A1 and RyR colocalize to the same region in both skeletal and cardiac muscle and because S100A1 and RyR2 coimmunoprecipitate (10), the question of whether S100A1 binds directly to RyR1 was explored further. It is not uncommon for S100 proteins to bind similar structural motifs as CaM (46, 47), and in RyR the well characterized cytosolic CaM binding domain (3616–3643) (48–51) has a region that fits very closely to the S100 consensus binding sequence (52). To directly test if S100A1 binds to the CaM-binding site in intact RyR1, competition experiments were conducted in which intact SR vesicles were mixed with CaM-linked agarose beads in the presence of increasing concentrations of either S100A1 or CaM. As visualized in Fig. 6, both S100A1 and free CaM fully compete RyR1 away from CaM beads in a dose-dependent manner with similar affinities at both high (2 mM) and low (100 nM) Ca2+ concentrations. Therefore, S100A1 binds to the myo-plasmic CaM binding domain on intact RyR1 and can fully displace CaM bound to RyR1 at physiological Ca2+ concentrations. CaM binds tightly to only one site on the intact RyR1 in skeletal muscle (48–51, 53). Although it remains possible that S100A1 binds to other regions of RyR1 as reported previously (20, 54), it is clear from these studies that S100A1 competes with CaM for binding this site on RyR1. Because both cardiac and skeletal isoforms of RyR contain this same CaM binding domain, the local structure of this site is most likely similar, and the mechanism by which S100A1 modulates EC coupling may be conserved between both muscle types.
FIGURE 6. S100A1 competes with CaM for binding to intact RyR1 in isolated SR vesicles.

A, S100A1 competes RyR1 away from agarose-linked CaM beads at high Ca2+ concentrations, as visualized by a decreased RyR1 band intensity in an anti-RyR Western blot. Each bar graph is the average of at least three images. At the bottom are representative bands from a single blot. Free CaM, a positive control, also competes RyR away from CaM-linked beads in the same protein concentration range. B, S100A1 and CaM compete RyR1 from CaM beads at low (100 nM) Ca2+ concentrations similar to those established for resting skeletal muscle (65).
S100A1 Binds a Peptide from the CaM-binding Site of RyR
In general, S100 proteins bind most tightly to amphipathic helices containing the consensus sequence (K/R)(L/I)XWXX(I/ L)L. The beginning of the CaM-binding site, 3616KKAVWH-KLLSKQ (termed RyRP12), fits closely to the S100 consensus sequence with the only deviation being a very minor difference of having an alanine residue in the second position (i.e. versus a Leu or Ile). This peptide is fully conserved between cardiac and skeletal muscle isoforms of RyR, and no other sequence in the entirety of the RyR molecule fits this S100 consensus binding sequence as closely as that of the RyRP12 peptide. By using tryptophan fluorescence, we determined the KD value of the S100A1 protein/RyR peptide to be 8.1 ± 1.1 μM at saturating [Ca2+] levels (Fig. 7). This is over 2-fold tighter than the binding of S100A1 to the canonical S100 binding sequence TRTK12, carried out under identical conditions (data not shown) (4). An identical tryptophan fluorescence experiment conducted with CaM and the KD was found to be ~5 μM (data not shown). Thus because the peptide binding affinities are similar between the two proteins, it is reasonable to expect that both S100A1 and CaM bind to this same sequence in RyR as found in the competition assay (Fig. 6). Another peptide derived from RyR (residues 3788–3800) that was previously identified as a putative S100A1 interaction sequence (20) was also tested for binding to S100A1; however, it was completely insoluble, and thus no binding data could be obtained.
FIGURE 7. The RyRP12 peptide binds S100A1 in the presence of Ca2+.
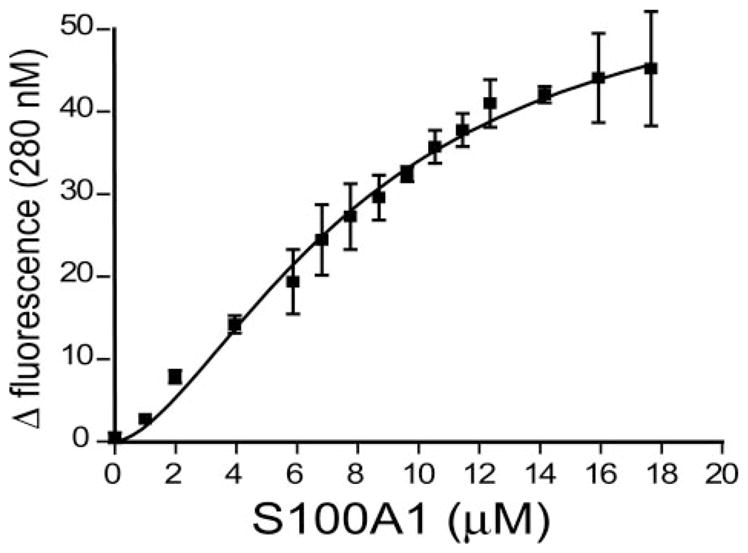
Increasing amounts of purified S100A1 protein were added to a cuvette containing 20 μM of the RyRP12 peptide, 25 mM NaCl, 10 mM CaCl2, 15 mM Tris, pH 7.2, and 15 mM DTT. Tryptophan fluorescence was excited at 280 nm, and the change in fluorescence at 350 nm was measured, giving a binding curve between S100A1 and RyR-12 with a KD of 8.1 ± 1.1 μM.
S100A1-RyRP12 Residue Assignments and S100A1 Secondary Structure
To assess whether S100A1 binds this RyRP12 peptide in a Ca2+-dependent manner, unlabeled RyRP12 peptide was titrated into either 15N-labeled apoS100A1 or Ca2+-loaded S100A1 and a fast HSQC was collected. In the absence of Ca2+, no changes to the HSQC spectra were observed; however, in the presence of Ca2+ a large population of the S100A1 resonances shifted. After the protein was fully loaded with the RyRP12 peptide, no further shifts were seen, even when severalfold higher peptide concentrations were added to the mixture. In an effort to see both if binding the RyRP12 peptide changed the global shape of the protein, and where on S100A1 the target peptide bound, the 1H, 13C, and 15N resonances of RyRP12-Ca2+-S100A1 were assigned sequence-specifically. Because several 1H-15N chemical shift perturbations were slowly exchanging peaks, it was necessary to re-assign the resonances a priori using two-dimensional, three-dimensional, and four-dimensional NMR experiments (supplemental Table 1). Namely, a three-dimensional 15N-edited CBCA(CO)NH, a three-dimensional HNCACB, and a three-dimensional HNCA were used to correlate the C+ and C+ resonance to their corresponding backbone amides. These resonance assignments together with data from three-dimensional H(CCO)NH and three-dimensional C(CO)NH NMR experiments were used to extend the assignments to include most side chain 1H and 13C resonances (>95%). A three-dimensional HNCO experiment was sufficient to assign nearly all of the backbone carbonyl resonances in RyRP12-Ca2+-S100A1 (≥94%). A very small number of ambiguous assignments remained from the three-dimensional data sets described above; however, data from the three-dimensional 15N-edited NOESY, four-dimensional 15N, 13C-edited NOESY and the four-dimensional 13C, 13C-edited NOESY were sufficient to resolve all of these ambiguities as well as to confirm assignments made using the triple resonance suite of NMR experiments. S100A1 is a homodimer of 93 residues per subunit, and 90 of the expected 93 correlations in the two-dimensional 1H-15N HSQC spectrum were observed and assigned (Fig. 8). The three missing residues (Leu-45, Ala-51, and Gln-72) are exchange broadened and could not be observed. The HSQC spectrum shows all seven pairs of side chain amide protons, corresponding to glutamines and aspar-agines, that are connected with horizontal lines in the HSQC spectrum. The 1H, 13C, and 15N sequence-specific assignments described here were deposited in the BioMagResBank under accession number 15296.
FIGURE 8. Two-dimensional 1H-15N HSQC spectrum of the RyRP12-bound Ca2+-S100A1 at 600-MHz (1H) with the resonance assignments included.

Backbone 1H-15N correlations are labeled sequence-specifically, and correlations connected by horizontal lines correspond to glutamine and asparagine side chain NH2 groups.
Chemical shift indexes (27, 55), amide exchange rates, J-coupling data (3JNH-Hα), and NOE data (56) were used to determine the secondary structure of RyRP12-Ca2+-S100A1 (supplemental Fig. 1). Briefly, the structure is consistent with two helix-loop-helix EF-hand calcium binding domains with four α-helices (H1, residues 3–19; H2, residues 30–39; H3, residues 51–61, and H4, residues 71–89) and two short β-strands (β1, residues Lys-27 to Ser-29; β2, residues Glu-68 to Asp-70). NOE correlations between residues Lys-27 and Asp-70, Leu-28 and Val-69, and Ser-29 and Glu-68 were observed, indicating that these residues align as a small antiparallel β-sheet as in other S100 proteins. There is virtually no difference in the overall secondary structure of the peptide-bound S100A1 protein as compared with Ca2+-bound S100A1. This suggests that all of the HSQC perturbations are caused by the peptide binding event and not as a result of large peptide-induced conformational change(s).
RyRP12 Binds the Hydrophobic Binding Pocket of S100A1
Large chemical shifts between two states of the same protein reflect major changes in the molecular environment of individual residues, thus providing a means of mapping the target protein-binding site to a specific area on the protein surface. The overall structure of S100A1 does not dramatically change upon peptide binding, and so it was relatively straightforward to map the areas of greatest chemical shift perturbations out onto the solution structure of Ca2+-S100A1. Significant chemical shift perturbations (>120 Hz) in HN and N were observed for residues in the hinge region (Phe-44, Leu-45, and Val-47), helix 3 (Ala-51, Val-54, and Ile-57), and helix 4 (Glu-73, Val-75, Ala-79, Ala-80, Leu-81, Thr-82, Ala-84, Cys-85, Phe-89, and Trp-90), with those residues of RyRP12-Ca2+-S100A1 that show the greatest chemical shift changes being located on helix 4 (Fig. 9, A–C). These regions are close to each other in the three-dimensional space and reveal a contiguous binding site of RyRP12 peptide to Ca2+-S100A1. Of note, the population of residues involved in this binding site is very similar to that involved in S100A1 binding to the CapZ peptide, TRTK-12, which also binds in the hydrophobic pocket of both S100A1 and S100B (2, 6).
FIGURE 9. The RyRP12 peptide-binding site on Ca2+-S100A1 as defined by NMR chemical shift perturbations.

A, HN and N chemical shift perturbations between Ca2+-S100A1 and RyRP12-Ca2+-S100A1. B, chemical shift changes mapped on the Ca2+-S100A1 structure. The helices of the first subunit are colored blue, and the second subunit are colored red, and the loops are colored gray. Regions of large chemical shift, defined as Δδ HN Δδ N > 120 Hz are colored in yellow. These residues are Phe-44, Leu-45, Val-47, Ala-51, Val-54, Ile-57, Glu-73, Val-75, Ala-79, Ala-80, Leu-81, Thr-82, Ala-84, Cys-85, Phe-89, and Trp-90. C, surface representation of Ca2+-S100A1, with the subunits colored blue and red and large chemical shifts colored in yellow, showing a contiguous exposed region of S100A1, located in the hydrophobic cleft where RyRP12 binds.
DISCUSSION
S100A1 is a known enhancer of cardiac contractility and exhibits strong potential as a gene therapeutic agent for treatment of cardiomyopathy. This potential is attributable in large part to the role of S100A1 in increasing the gain of EC coupling and potentiating Ca2+ cycling, both of which are compromised in myopathic conditions (for review see Ref. 57). However, no direct evaluation of the physiological contribution of S100A1 to EC coupling in skeletal muscle has been described.
Here we demonstrate for the first time using S100A1 KO muscle fibers that endogenous S100A1 plays a physiological role in skeletal muscle EC coupling. The well characterized ratiometric Ca2+ indicator dye Indo-1 was used to qualitatively compare resting [Ca2+]i between KO and WT muscle fibers. Comparable resting ratios in KO and WT fibers indicated no alterations in the steady state regulation of myoplasmic Ca2+ content in S100A1-deficient muscle. However, following AP stimulation, depressed peak ratios of the Indo-1 fluorescence signal in KO fibers revealed a decrease in myoplasmic [Ca2+] following activation of the SR Ca2+ release channel. Restoration of S100A1 through adenoviral expression in KO fibers rescued this peak ratio of the Ca2+ transient. This suggests that S100A1 specifically mediates this Ca2 +signaling phenotype, and validates that our initial findings were not a result of compensatory mechanisms in S100A1-deficient mice. We next sought to more accurately assay the temporal properties of the Ca2 + transient utilizing high temporal resolution analysis (50 μs/line confocal microscopy) with the non-ratiometric indicator Fluo-4. With this system we were able to confirm our Indo-1 findings of alterations in the level of Ca2+ release from the SR following AP stimulation, as well as identify differences in the temporal properties of the developing Ca2+ transient. S100A1 KO fibers demonstrated an increase of 34% in the time to the half-maximal peak of the Ca2+ transient and an increase in duration of 39% between 10 and 90% of the peak Ca2+ transient. These results are suggestive of a decreased activation state of the SR Ca2+ release channel and a sluggishness of Ca2+ release following AP stimulation in S100A1-deficient muscle.
Supporting this concept, Treves et al. (20) demonstrated that S100A1 increases the open probability and activation state of isolated RyR in lipid bilayers. Furthermore, Most et al. (10) showed significant increases in the caffeine-evoked twitch force following addition of exogenous S100A1. Taken together, these studies with our own present a picture where S100A1 increases the activation state of the RyR following initial activation from electrical (AP) or chemical (caffeine) stimuli. This stimulation then triggers a faster rate of Ca2+ release and an increase in the global Ca2+ transient, leading to greater force generation and increased contraction.
In our experimental setting we analyzed EC coupling in isolated FDB fast twitch muscle fibers. Fast twitch muscle expresses lower levels of S100A1 than either slow twitch or cardiac myocytes (5, 58). Despite this lower cellular expression, multiple studies have demonstrated S100A1 modulation of Ca2+ release mechanisms at nanomolar concentrations, which falls well within proposed endogenous levels for fast twitch muscle (19, 20). Additionally, as Most et al. (10) saw similar increases in force generation in both slow and fast twitch muscle with exogenous S100A1, it is likely that S100A1 plays a similar physiologic role in both skeletal muscle types. Furthermore, S100A1 interaction with RyR has been demonstrated in both skeletal and cardiac muscle (10, 12, 20). Our RyRP12 peptide is conserved between RyR1 in skeletal muscle, RyR2 in cardiac muscle, and RyR3 found primarily in embryonic muscle and the brain. Therefore, we propose that S100A1 increases the amount and rate of Ca2+ release following RyR activation because of a conserved molecular mechanism driven by S100A1 binding to this specific region of RyR in all striated muscle.
Although previous studies have indicated that RyR1 and S100A1 interact directly (10, 20), this is the first study to (a) locate an S100A1 binding site on the intact RyR and (b) explore intermolecular interactions of this binding site on a residue by residue basis. Although this binding site seems likely to be physiologically relevant, other binding sites within RyR could also be S100A1 targets. This notion was supported by Treves et al. (20), who demonstrated S100A1 binding to a different domain of RyR. However, no other regions of the skeletal or cardiac RyR isoform display as close a match to the S100 consensus binding sequence as RyRP12. In general, amphipathic sequences containing positive charges and anchored by tryptophan have the highest affinity for S100 molecules (47). To this end, we have found that the permutation of either tryptophan or the first leucine to aspartate completely abolishes the RyRP12-S100A1 interaction.
The RyRP12 peptide is contained within the CaM-binding site (RyR-(3616–3643)) in skeletal muscle (48, 50). In vitro studies using SR vesicles and purified RyR1 incorporated in lipid bilayers show that in its Ca2+-free form apoCaM is a weak agonist of RyR1 (59). In further support of its partial activating role, addition of apoCaM to permeabilized frog skeletal muscle fibers has been shown to increase the frequency of spontaneous Ca2+ sparks (60), which are local, discrete elevations in intra-cellular Ca2+ concentration because of RyR opening (61). An increase in the frequency of Ca2+ sparks is an indicator of a more activated state of RyR. In contrast to apoCaM, at higher Ca2+ concentrations Ca2+-CaM acts as an inhibitor of RyR activation and opening (58). It has been demonstrated that upon binding Ca2+, Ca2+-CaM shifts in its binding site, allowing the N terminus to interact with a second, potentially inhibitory site on an adjacent RyR monomer. This noncontiguous binding has been proposed to account for the inhibitory effect of Ca2+-CaM on RyR1 (62, 63), as Ca2+-CaM may inhibit SR Ca2+ release by stabilizing intersubunit interactions. Conversely, Rodney et al. (52) have proposed that apoCaM binding this intersubunit site of RyR1 may sensitize the RyR channel to activation by disrupting these homotetramer subunit interactions. This would increase the open probability of RyR, thereby increasing Ca2+ release upon depolarization.
Based on our results and these previous findings, we propose that S100A1 may act in a similar fashion as apoCaM to increase the RyR activation state and therefore SR Ca2+ release in striated muscle. Because S100A1 and CaM have similar binding affinities for RyR1 in SR vesicles, both proteins most likely endogenously compete for this binding site (Fig. 6). It is important to note that although in its apo-form S100A1 did not interact with the RyRP12 peptide in NMR experiments, S100A1 did compete with CaM for binding to RyR1 in SR vesicles under low Ca2+ conditions (100 nM Ca2+, Fig. 6). One possible explanation for this discrepancy is that in the NMR experimental setting there is zero Ca2+ in the test solution, and therefore apoS100A1 is unable to bind Ca2+ and interact with its target RyRP12 peptide. However, S100 proteins have been shown to strongly increase their affinity for Ca2+ in the presence of their target protein (64). Therefore, it is reasonable to expect that in the presence of RyR1 a significant portion of S100A1 may be Ca2+-bound at 100 nM Ca2+, and therefore able to interact with RyR1 and occupy the CaM binding domain. A second explanation for the contrast in binding affinities of S100A1 for RyRP12 and the intact RyR1, which is not necessarily mutually exclusive from the first, is that the RyRP12 peptide contains only 12 of the 28 amino acids of the CaM binding domain, and therefore may have a lower affinity for S100A1 then the intact RyR1. Based on these findings, we propose that Ca2+-bound S100A1 may be interacting with RyR1 at resting physiological Ca2+ concentrations, sensitizing the channel to activation, which leads to faster and increased Ca2+ release upon electrical stimulation.
As an alternative mechanism to explain our results, S100A1 may augment SR Ca2+ release by preventing Ca2+-CaM-mediated inhibition of RyR1 following activation of the channel. According to this hypothesis, in WT muscle fibers as local [Ca2+] increases, S100A1 competes with CaM binding to RyR1 and therefore alleviates Ca2+-CaM-mediated inhibition of Ca2+ release. Conversely, in S100A1-deficient mice, Ca2+-CaM can bind without competition to this domain of RyR1, and therefore maximal SR Ca2+ release is inhibited. Although CaM modulation of RyR1 has been extensively studied in vitro, further studies are needed to evaluate the interplay between endogenous CaM, S100A1, and RyR1 in vivo. The local expression of these two proteins surrounding RyR1 in resting and excited muscle fibers and their responses to increases in myo-plasmic [Ca2+] in vivo may determine the balance of RyR1 activation/inhibition proposed in these models.
Previous studies on mice show that the addition of S100A1 elicits its most profound effect in compromised myopathic conditions (15). Additionally, S100A1 knock-out mice only display an obvious impaired cardiac phenotype when the heart is stressed (13). Mirroring these observations, preliminary data from our lab suggest that addition of exogenous S100A1 protein produces a much more pronounced effect on the Ca2+ transient of isolated FDB fibers with depressed Ca2+ signaling phenotypes, compared with those with more typical transient responses. It is therefore tempting to speculate that S100A1 function, while at least partially redundant in healthy myocytes, plays an important role in maintaining the function and health of compromised muscle fibers. These findings both may explain the lack of an overt phenotype in S100A1 KO mice and provide a basis for further studies looking at S100A1 function in skeletal muscle fatigue or myopathic conditions. Our results support a potential therapeutic role for S100A1.
Here we demonstrate for the first time a role of S100A1 in physiological EC coupling in skeletal muscle. We also identify a novel S100A1-binding site within RyR that is conserved in both cardiac and skeletal muscle. We propose that S100A1 interacts with this binding site in a similar fashion to CaM, increasing the open probability of RyR. This leads to an enhancement of global Ca2+ release following electrical or ligand-induced excitation. In turn, this translates to increased contractility and force generation in striated muscle.
Supplementary Material
Acknowledgments
The NMR spectrometers used in these studies were purchased in part with funds from National Institutes of Health Shared Instrumentation Grants S10 RR10441, S10 R15741, and S10 RR16812 (to D. J. W.) and National Science Foundation Grant DBI 0115795 (to D. J. W.).
Footnotes
This work was supported by National Institutes of Health NIAMS Research Grant RO1 AR055099 (to M. F. S.) and National Institutes of Health Grants GM58888 and CA107331 (to D. J. W.).
The abbreviations used are: RyR, ryanodine receptor; DTT, dithiothreitol; SR, sarcoplasmic reticulum; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; KO, knock-out; WT, wild type; Ad, adeno-virus; GFP, green fluorescent protein; EC, excitation-contraction; MEM, minimal essential medium; CaM, calmodulin; TES, 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid; FDB, flexor digito-rum brevis; AP, action potential.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Fig. 1.
References
- 1.Moore BW. Biochem Biophys Res Commun. 1965;19:739–744. doi: 10.1016/0006-291x(65)90320-7. [DOI] [PubMed] [Google Scholar]
- 2.Landar A, Rustandi RR, Weber DJ, Zimmer DB. Biochemistry. 1998;37:17429–17438. doi: 10.1021/bi9817921. [DOI] [PubMed] [Google Scholar]
- 3.Donato R. Int J Biochem Cell Biol. 2001;33:637–668. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 4.Zimmer DB, Cornwall EH, Landar A, Song W. Brain Res Bull. 1995;37:417–429. doi: 10.1016/0361-9230(95)00040-2. [DOI] [PubMed] [Google Scholar]
- 5.Haimoto H, Kato K. J Neurochem. 1987;48:917–923. doi: 10.1111/j.1471-4159.1987.tb05604.x. [DOI] [PubMed] [Google Scholar]
- 6.Wright NT, Varney KM, Ellis KC, Markowitz J, Gitti RK, Zimmer DB, Weber DJ. J Mol Biol. 2005;353:410–426. doi: 10.1016/j.jmb.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 7.Rustandi RR, Baldisseri DM, Inman KG, Nizner P, Hamilton SM, Landar A, Zimmer DB, Weber DJ. Biochemistry. 2002;41:788–796. doi: 10.1021/bi0118308. [DOI] [PubMed] [Google Scholar]
- 8.Inman KG, Yang R, Rustandi RR, Miller KE, Baldisseri DM, Weber DJ. J Mol Biol. 2002;324:1003–1014. doi: 10.1016/s0022-2836(02)01152-x. [DOI] [PubMed] [Google Scholar]
- 9.Weber DJ, Rustandi RR, Carrier F, Zimmer DB. In: The Molecular Basis of Calcium Action in Biology and Medicine. Pochet R, editor. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2000. pp. 521–539. [Google Scholar]
- 10.Most P, Remppis A, Pleger ST, Loffler E, Ehlermann P, Bernotat J, Kleuss C, Heierhorst J, Ruiz P, Witt H, Karczewski P, Mao L, Rockman HA, Duncan SJ, Katus HA, Koch WJ. J Biol Chem. 2003;278:33809–33817. doi: 10.1074/jbc.M301788200. [DOI] [PubMed] [Google Scholar]
- 11.Kiewitz R, Acklin C, Schafer BW, Maco B, Uhrik B, Wuytack F, Erne P, Heizmann CW. Biochem Biophys Res Commun. 2003;306:550–557. doi: 10.1016/s0006-291x(03)00987-2. [DOI] [PubMed] [Google Scholar]
- 12.Most P, Bernotat J, Ehlermann P, Pleger ST, Reppel M, Borries M, Niroomand F, Pieske B, Janssen PM, Eschenhagen T, Karczewski P, Smith GL, Koch WJ, Katus HA, Remppis A. Proc Natl Acad Sci U S A. 2001;98:13889–13894. doi: 10.1073/pnas.241393598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Most P, Seifert H, Gao E, Funakoshi H, Volkers M, Heierhorst J, Remppis A, Pleger ST, DeGeorge BR, Jr, Eckhart AD, Feldman AM, Koch WJ. Circulation. 2006;114:1258–1268. doi: 10.1161/CIRCULATIONAHA.106.622415. [DOI] [PubMed] [Google Scholar]
- 14.Remppis A, Greten T, Schafer BW, Hunziker P, Erne P, Katus HA, Heizmann CW. Biochim Biophys Acta. 1996;1313:253–257. doi: 10.1016/0167-4889(96)00097-3. [DOI] [PubMed] [Google Scholar]
- 15.Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, Loffler E, Janssen PM, Eckhart AD, Martini J, Williams ML, Katus HA, Remppis A, Koch WJ. J Clin Investig. 2004;114:1550–1563. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pleger ST, Remppis A, Heidt B, Volkers M, Chuprun JK, Kuhn M, Zhou RH, Gao E, Szabo G, Weichenhan D, Muller OJ, Eckhart AD, Katus HA, Koch WJ, Most P. Mol Ther. 2005;12:1120–1129. doi: 10.1016/j.ymthe.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Pleger ST, Most P, Heidt B, Voelkers M, Hata JA, Katus HA, Remppis A, Koch WJ. Eur J Med Red. 2006;10:418–422. [PubMed] [Google Scholar]
- 18.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, Gao E, Dasgupta A, Rengo G, Remppis A, Katus HA, Eckhart AD, Rabinowitz JE, Koch WJ. Circulation. 2007;115:2506–2515. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 19.Most P, Remppis A, Weber C, Bernotat J, Ehlermann P, Pleger ST, Kirsch W, Weber M, Uttenweiler D, Smith GL, Katus HA, Fink RH. J Biol Chem. 2003;278:26356–26364. doi: 10.1074/jbc.M303338200. [DOI] [PubMed] [Google Scholar]
- 20.Treves S, Scutari E, Robert M, Groh S, Ottolia M, Prestipino G, Ronjat M, Zorzato F. Biochemistry. 1997;36:11496–11503. doi: 10.1021/bi970160w. [DOI] [PubMed] [Google Scholar]
- 21.Matise MP, Auerbach W, Joyner AL. Gene Targeting: A Practical Approach. 2. Oxford University Press; Oxford, UK: 2000. [Google Scholar]
- 22.Liu Y, Kranias EG, Schneider MF. Am J Physiol. 1997;273:C1915–C1924. doi: 10.1152/ajpcell.1997.273.6.C1915. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Cseresnyés Z, Randall WR, Schneider MF. J Cell Biol. 2001;155:27–39. doi: 10.1083/jcb.200103020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacLennan DH. J Biol Chem. 1970;245:4508–4518. [PubMed] [Google Scholar]
- 25.Rustandi RR, Baldisseri DM, Drohat AC, Weber DJ. Protein Sci. 1999;8:1743–1751. doi: 10.1110/ps.8.9.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 27.Spera S, Bax A. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 28.Baldisseri DM, Rustandi RR, Zhang Z, Tang C, Bair CL, Landar A, Landar A, Zimmer DB, Weber DJ. J Biomol NMR. 1999;14:91–92. doi: 10.1023/a:1008301518346. [DOI] [PubMed] [Google Scholar]
- 29.Vuister GW, Bax A. J Biomol NMR. 1992;2:401–405. doi: 10.1007/BF01874818. [DOI] [PubMed] [Google Scholar]
- 30.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 31.Spera S, Ikura M, Bax A. J Biomol NMR. 1991;1:155–165. doi: 10.1007/BF01877227. [DOI] [PubMed] [Google Scholar]
- 32.Edison AS, Abildgaard F, Westler WM, Mooberry ES, Markley JL. Methods Enzymol. 1994;239:3–79. doi: 10.1016/s0076-6879(94)39003-7. [DOI] [PubMed] [Google Scholar]
- 33.Live DH, Davis DG, Agosta WC, Cowburn D. J Am Chem Soc. 1984;106:1939–1941. [Google Scholar]
- 34.Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 35.Ikura M, Bax A, Clore GM, Gronenborn AM. J Am Chem Soc. 1990;112:9020–9022. [Google Scholar]
- 36.Kuboniwa H, Grzesiek S, Delaglio F, Bax A. J Biomol NMR. 1994;4:871–878. doi: 10.1007/BF00398416. [DOI] [PubMed] [Google Scholar]
- 37.Grzesiek S, Anglister J, Bax A. J Magn Reson B. 1993;101:114–119. [Google Scholar]
- 38.Carlomagno T, Luy B, Glaser SJ. J Magn Reson. 1996;126:110–119. doi: 10.1006/jmre.1997.1157. [DOI] [PubMed] [Google Scholar]
- 39.Wittekind M, Mueller L. J Magn Reson B. 1993;101:205–210. [Google Scholar]
- 40.Kay LE, Clore GM, Bax A, Gronenborn AM. Science. 1990;249:411–414. doi: 10.1126/science.2377896. [DOI] [PubMed] [Google Scholar]
- 41.Muhandiram DR, Guang YX, Kay LE. J Biomol NMR. 1993;3:463–470. [Google Scholar]
- 42.Du SJ, Cole JJ, Tenis N, Gao XM, Kontgen F, Kemp BE, Heierhorst J. Mol Cell Biol. 2002;22:2821–2829. doi: 10.1128/MCB.22.8.2821-2829.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maco B, Mandinova A, Durrenberger MB, Schafer BW, Uhrik B, Heizmann CW. Physiol Res. 2001;50:567–574. [PubMed] [Google Scholar]
- 44.Kettlewell S, Most P, Currie S, Koch WJ, Smith GL. J Mol Cell Cardiol. 2005;39:900–910. doi: 10.1016/j.yjmcc.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 45.DiFranco M, Capote J, Vergara JL. J Membr Biol. 2005;208:141–153. doi: 10.1007/s00232-005-0825-9. [DOI] [PubMed] [Google Scholar]
- 46.Rhoads AR, Friedberg F. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 47.Wilder PT, Lin J, Bair CL, Charpentier TH, Yang D, Liriano M, Varney KM, Lee A, Oppenheim AB, Adhya S, Carrier F, Weber DJ. Biochim Biophys Acta. 2006;1763:1284–1297. doi: 10.1016/j.bbamcr.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 48.Moore CP, Rodney G, Zhang JZ, Santacruz-Toloza L, Strasburg G, Hamilton SL. Biochemistry. 1999;38:8532–8537. doi: 10.1021/bi9907431. [DOI] [PubMed] [Google Scholar]
- 49.Rodney GG, Moore CP, Williams BY, Zhang JZ, Krol J, Pedersen SE, Hamilton SL. J Biol Chem. 2001;276:2069–2074. doi: 10.1074/jbc.M008891200. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi N, Xin C, Meissner G. J Biol Chem. 2001;276:22579–22585. doi: 10.1074/jbc.M102729200. [DOI] [PubMed] [Google Scholar]
- 51.O’Connell KM, Yamaguchi N, Meissner G, Dirksen RT. J Gen Physiol. 2002;120:337–347. doi: 10.1085/jgp.20028617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ivanenkov VV, Jamieson GA, Jr, Gruenstein E, Dimlich RV. J Biol Chem. 1995;270:14651–14658. doi: 10.1074/jbc.270.24.14651. [DOI] [PubMed] [Google Scholar]
- 53.Rodney GG, Wilson GM, Schneider MF. J Biol Chem. 2005;280:11713–11722. doi: 10.1074/jbc.M408189200. [DOI] [PubMed] [Google Scholar]
- 54.Menegazzi P, Larini F, Treves S, Guerrini R, Quadroni M, Zorzato F. Biochemistry. 1995;333:9078–9084. doi: 10.1021/bi00197a008. [DOI] [PubMed] [Google Scholar]
- 55.Wishart DS, Sykes BD. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 56.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons, Inc; New York: 1986. pp. 117–129. [Google Scholar]
- 57.Pleger ST, Most P, Katus HA. Cardiovasc Res. 2007;75:1–2. doi: 10.1016/j.cardiores.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 58.Zimmer DB. Cell Motil Cytoskeleton. 1991;20:325–337. doi: 10.1002/cm.970200408. [DOI] [PubMed] [Google Scholar]
- 59.Tripathy A, Xu L, Mann G, Meissner G. Biophys J. 1995;69:106–119. doi: 10.1016/S0006-3495(95)79880-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodney GG, Schneider MF. Biophys J. 2003;85:921–932. doi: 10.1016/S0006-3495(03)74531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng H, Lederer WJ, Cannell MB. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 62.Xiong LW, Newman RA, Rodney GG, Thomas O, Zhang JZ, Persechini A, Shea MA, Hamilton SL. J Biol Chem. 2002;277:40862–40870. doi: 10.1074/jbc.M206763200. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H, Zhang JZ, Danila CI, Hamilton SL. J Biol Chem. 2003;278:8348–8355. doi: 10.1074/jbc.M209565200. [DOI] [PubMed] [Google Scholar]
- 64.Markowitz J, Rustandi RR, Varney KM, Wilder PT, Udan R, Wu SL, Horrocks WD, Weber DJ. Biochemistry. 2005;44:7305–7314. doi: 10.1021/bi050321t. [DOI] [PubMed] [Google Scholar]
- 65.Westerblad H, Allen DG. J Gen Physiol. 1991;98:615–635. doi: 10.1085/jgp.98.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.