

Our appreciation of the different molecular subtypes [1] and the complex genomic architecture of myeloma [2] has been recently enhanced by whole-exome and whole-genome DNA sequencing, indicating that myeloma cells typically harbor dozens of non-synonymous mutations [3, 4]. Although myeloma is associated with recurrent mutations in a number of genes, including NRAS, KRAS, FAM46C, DIS3 and IRF4 [3], there is no evidence for hallmark “unifying” mutations that have been found in other blood cancers, such as BRAFV600E in hairy cell leukemia [5] and MYD88L265P in Waldenström macroglobulinemia [6]. The newly discovered myeloma alleles have raised the bar for preclinical mouse models of myeloma that can be used for interrogating pathways of tumor development and responses to new therapies. To rise to the challenge, more accurate, cost-effective and scalable models than what is currently available should be devised. Additionally, next-generation mouse models of myeloma should be able to distinguish myeloma drivers from innocent bystander mutations, validate molecular targets for experimental myeloma interventions, elucidate mechanisms of acquired drug resistance, and determine whether a myeloma gene operates in the tumor cells, the tumor microenvironment (TME), or both.
Encouraged by the push in the mouse cancer genetics community to supplement the time-consuming “conventional” or “germline” transgenic mouse models of cancer with more flexible and less expensive “non-germline” models [7], we have recently begun with the development of non-germline models for the in vivo validation of candidate myeloma genes. The cornerstone of the new method is adoptive B cell transfer. Our first approach relied on Myc-transgenic cells. Although it provided proof-of-principle for the utility of adoptive B-cell transfer and showed that the TME is the critical source of IL-6 for neoplastic plasma cell (PC) development [8], the requirement to prime the host mice with an intraperitoneal (IP) injection of pristane was a severe limitation. Treatment with pristane results in peritoneal plasmacytoma, an insufficient model for human myeloma. Here we demonstrate that this limitation can be overcome by employing a different type of B cell for the adoptive transfer: BCL2+IL6+. This cell is prone to malignant transformation by virtue of classic oncogene collaboration; i.e., a survival-enhancing BCL2 transgene [9] and a pro-inflammatory IL6 transgene [10]. BCL2+IL6+ B cells can be readily isolated from secondary lymphoid tissues of double-transgenic mice (e.g., spleen, mesenteric lymph node), genetically modified in vitro by viral gene transduction, and transferred to syngeneic BALB/c hosts primed with whole-body irradiation to facilitate the homeostatic expansion of the incoming B cells. The stable engraftment of these cells yields myeloma-like plasma cell neoplasms (PCNs) in immunocompetent hosts that have not been treated with IP pristane (Figure 1A).
Figure 1. Adoptive transfer of BCL2+IL6+ B cells gives rise to plasma cell neoplasms (PCN) in host mice.
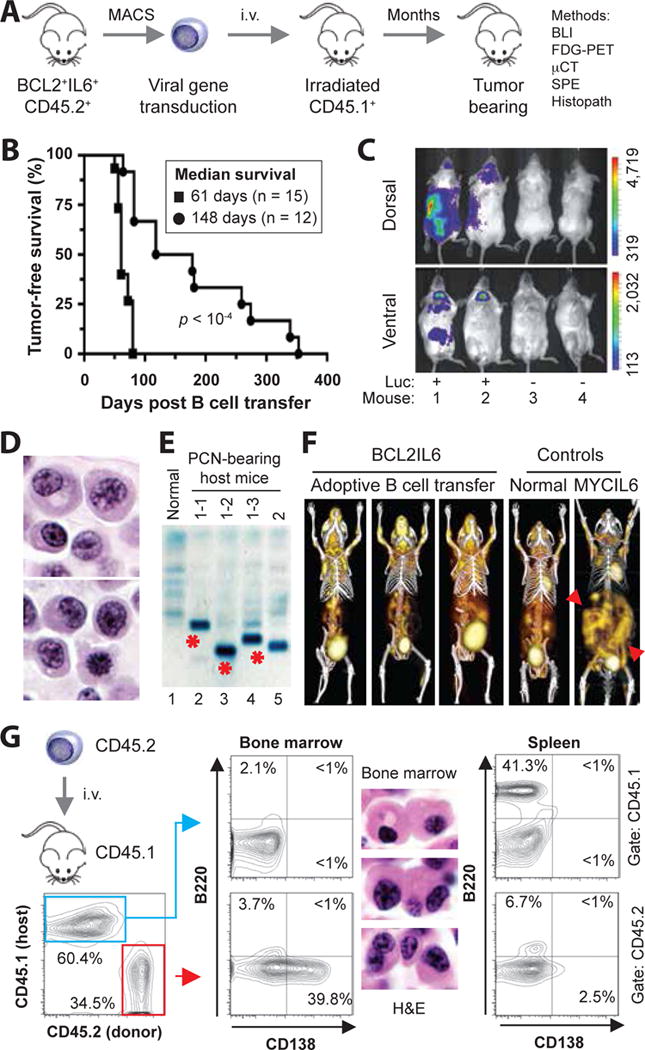
(A) Schematic overview of tumor induction and analysis. The experimental approach relied on the adoptive transfer of B220+ B cells obtained from spleens of 5–8 weeks old, double-transgenic BCL2+IL6+ mice congenic for CD45.2+. Donor B cells were isolated with the assistance of the MACS® B220 mouse B-cell kit from Miltenyi, were in some cases genetically modified in vitro by retroviral reporter gene (luciferase) transduction, and were transferred to normal (not transgenic) CD45.1+ host mice that had been conditioned with whole-body irradiation a few hours prior to the cell transfer. Neoplasms arising in B cell-reconstituted hosts were analyzed using a variety of immunological, imaging and histopathological methods, the results of which are presented in panels B to G of this figure and Figure 2. Note that one donor mouse suffices to reconstitute 20–30 hosts, which mimic the clonal heterogeneity of human myeloma [4] by developing distinct tumors. See panel E for an example. Donor B cells at the time of harvest can be operationally defined as premalignant. However, their malignant potential is high, as all BCL2+IL6+ mice spontaneously develop striking hypergammaglobulinemia by ~3 months of age (Supplemental Figure 6) and PC tumors by ~5 months of age (Supplemental Figure 7).
(B) Line graphs indicating tumor-free survival of B-cell reconstituted host mice pre-treated with either a sublethal dose (4.5 Gy, black circles) or lethal dose (11 Gy, black squares) of whole-body irradiation. The latter was administered as split dose (4.5 Gy and 6.5 Gy, delivered 4 hrs apart) and required hematopoietic stem cell rescue using bone marrow transplantation (3 × 106 cells) for survival of mice. Median tumor-free survival of sublethally irradiated hosts (148 days; n=12) was significantly longer than lethally irradiated hosts (61 days; n = 15) based on Mantel-Cox log-rank analysis (p < 10−4). In both studies, 2 × 106 donor B cells were transferred. B cells were stimulated for about 48 hrs in vitro using 12.5 ng/ml mIL-4 and 10 μg/ml LPS (both from Sigma, St. Louis, MO).
(C) Bioluminescence image of 4 B cell-reconstituted, tumor-bearing CD45.1+ recipients that had received on day -76 either luciferase-expressing CD45.2+B220+ B cells (Luc+) or B cells not transduced with luciferase-encoding cDNA (Luc−). The result indicates that tumor precursors may be genetically modified in future studies by retroviral transduction of over-expressed candidate myeloma driver genes or shRNAs knocking down those genes.
(D) High-power view (100×) of H&E-stained PC tumor tissue section demonstrating typical features of malignant PCs, such as large amounts of cytoplasm, eccentric nucleus with marginated chromatin and paranuclear hof.
(E) Serum protein electropherogram demonstrating pronounced M-spikes (monoclonal Ig) in 4 PCN-bearing CD45.1+ mice reconstituted with B cells from the same donor mouse (lanes 2–4) or a different donor mouse (lane 5). Normal serum (lane 1) was used as control. The different electrophoretic mobility of the paraproteins in lanes 2–4 (red asterisks) strongly suggests that the underlying PC tumors are unique; i.e., different tumor precursors from the donor B cell pool underwent tumor progression and attained clonal dominance. Additional changes of serum proteins in tumor-bearing mice included elevated levels of cyto- and chemokines (e.g., IL-5, IL-13, IL-17, MCP-1 and TNFα) relative to tumor-bearing IL6MYC mice [8] and BCL2+IL6+ mice (Supplemental Tables 1 and 2, Supplemental Figure 8).
(F) Three dimensional renderings of fused FDG-PET/CT images obtained from three adoptively transferred mice that carried BCL2+IL6+ PC tumors (images 1–3), an age-matched normal C mouse used as control (image 4) and a PC tumor-bearing MYCIL6-transgenic mouse (image 5) that exhibited pronounced extraosseous disease in abdominal lymph nodes (red arrowhead pointing down) and spleen (red arrowhead pointing up). All images were normalized to the same maximal standard uptake value (SUVmax) to facilitate comparison of PET lesions. High FDG uptake and PET signal strength not associated with tumor burden is apparent in tissues with physiologically high levels of glucose utilization (e.g., brain, heart) and in the FDG excretion pathway, particularly the urinary bladder.
(G) Flow cytometric histograms of a representative CD45.2+ myeloma-like tumor that resides in a CD45.1+ host. Forward scatter- and side scatter-gated bone marrow cells in the contour plot to the left were first distinguished as donor derived (CD45.2+, 34.5%) or host derived (CD45.1+, 60.4%) and then evaluated for the expression of B220 (B cell marker) and CD138 (syndecan-1, plasma cell marker). Unlike host cells, which included a small number of B220+CD138− B cells (2.1%) but essentially no B220+CD138+ plasmablasts (<1%) or B220−CD138+ PCs (<1%; center plot, top) in the bone marrow, donor cells contained a sizable fraction of PCs (39.8%; center plot, bottom). These cells exhibited the hallmark cytological features of myeloma in H&E-stained tissue sections (original magnification 100×). The spleen of the same mouse harbored a large number of host-derived B cells (41.3%; right plot, top), as one might have expected in a situation in which the infiltration with donor-derived B cells (6.7%) and plasma cells (2.5%) is just beginning. The low percentage of malignant PCs in the spleen underlines the bone-seeking, myeloma-like homing pattern of the adoptive-transfer tumors presented in panel F.
To evaluate the potential of BCL2+IL6+ B cells to give rise to myeloma-like tumors in genetically compatible hosts, B220+CD45.2+ splenocytes were obtained from BCL2+IL6+ mice and adoptively transferred, via retro-orbital IV injection, to CD45.1+ hosts pre-treated with a lethal dose of whole-body irradiation (Figure 1B, squares). Tumor development was fully penetrant (100% tumor incidence) and tumor onset was short (61 days median, 50–80 days range) in all B cell-reconstituted mice (n=15). All tumors that arose from B cells that had been retrovirally transduced in vitro using a luciferase (Luc)-encoding cDNA gene exhibited strong reporter gene activity upon bioluminescence imaging in vivo (Figure 1C, Supplemental Figure 1). Disappointingly, however, serum paraproteins were not detected (Supplemental Figure 2A) and the tumors were classified as diffuse large B cell lymphoma (DLBCL) based on histopathologic criteria (Supplemental Figure 2B).
To assess the possibility that the tumor pattern changes under conditions of less severe suppression of the host immune system, the tumor induction study was repeated using sub-lethally irradiated mice without bone marrow rescue (n=12). Tumor onset was slowed (148 days median) and less predictable (64 days minimum, 353 days maximum) compared to lethally irradiated hosts, but tumor development was again complete (100% tumor incidence, Figure 1B, circles). Importantly, histopathological investigations demonstrated that the majority of tumors (9 of 12, 75%) consisted of homogeneous sheets of abnormal PCs, consistent with the diagnosis of PCN (Figure 1D). Two mice harbored DLBCL with plasmacytic differentiation (not shown) and one mouse contained a coexisting PCN and DLBCL (Supplemental Figure 3). The blood serum of PCN-bearing mice harbored abundant paraproteins (M-spikes), hallmarks of monoclonal expansions of immunoglobulin-producing PCs (Figure 1E), which secreted predominantly IgG isotypes (Supplemental Figure 4) and were associated with myeloma-like cast nephropathy in all cases (Supplemental Figure 5). These findings demonstrated that host priming (lethal vs. sub-lethal irradiation) is key for determining tumor onset (early vs. late) and tumor pattern (lymphoma vs. PC tumor) in the BCL2+IL6+ adoptive transfer model.
Because our chief interest concerns improved preclinical modeling of myeloma, the follow-up investigations concentrated on the PCN-bearing mice. To evaluate tumor burden, tumor dissemination pattern and extent of extra-medullary disease, 18F-FDG-PET imaging was used [11]. Extra-medullary tumor load was modest (Figure 1F) relative to mice harboring IL-6/MYC-driven PCNs for which comparable PET data were available [11]. In keeping with the large M-spikes presented in Figure 1E, the PET results indicated that the malignant PCs exhibit a myeloma-like preponderance for hematopoietic bone marrow. To confirm that with an independent, quantitative method, we performed a flow-cytometric analysis of femural bone marrow samples, taking advantage of specific antibodies to CD45.2 and CD45.1, respectively, to unambiguously distinguish donor and host cells (Figure 1G, left). In 5 of 5 cases that featured dense plasmacytic infiltrates in the femur, the tumors were donor-derived, mature CD45.2+CD138+B220− PCs (Figure 1G, center). The low abundance of CD45.2+ B cells and PCs in spleen (Figure 1G, right) and lymph nodes (not shown) lent further support to the contention that the BCL2+IL6+ tumors recapitulate the homing pattern of human myeloma.
To assess whether tumor manifestation in the bone marrow was accompanied by myeloma-like bone disease, we performed radiological and histochemical studies on the skeleton of PCN-bearing mice. Quantitative bone analysis using Carestream planar radiography provided preliminary evidence for bone loss (not shown), but a more sensitive approach was desired. To that end, we employed high-resolution CT, which revealed skull and spine as apparent hotspots of bone disease. The skull demonstrated separations and erosions of the coronal, sagittal, and lambdoidal cranial sutures (Figure 2A), reminiscent of changes seen in the Bcl-XL/iMyc model of myeloma [12]. CT imaging of the spine, using lumbar vertebrae L2-4 of tumor-bearing mice as indicator bones, demonstrated osteolytic perforations of the cortical bone (Figure 2B, left). Agreeing with that, quantitative CT measurements of bone mass and secondary spongiosa showed a reduction in trabecular bone volume (Figure 2B, top right) and trabecular thickness (Figure 2B, center right), and a concomitant increase in trabecular space and separation (Figure 2B, bottom right). These changes were confirmed in 2D sections of L2-fexposure time, 80 kV voltage and 504, demonstrating discontinuities in the cortical ring (osteolytic lesions) and overall loss of cortical and trabecular bone (Figure 2C, left). Additional evidence for the kind of highly active medullary disease that leads to generalized and focal osteopenia in patients with myeloma was provided by: BoneJ Thickness-generated graphical output data on trabecular bone mass (Figure 2C, right); frequent detection of hind limb paralysis in tumor-bearing mice (Figure 2D and Supplemental Movie); and striking increases in TRAP+ (tartrate-resistant acid phosphatase) osteoclasts at the border of diseased bone and extensive sheets of malignant PCs (Figure 2E) that completely effaced the normal hematopoietic elements of the marrow cavity.
Figure 2. Myeloma-like bone disease.
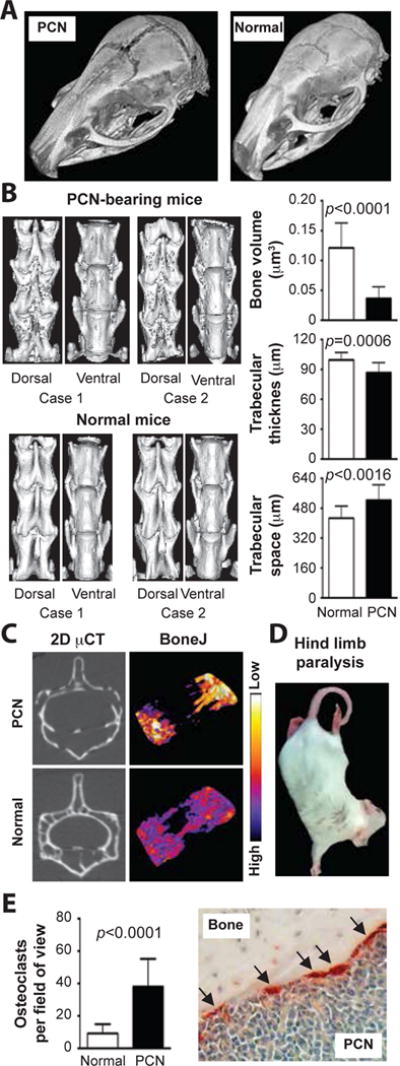
(A) Ex vivo computed tomography (CT) images of the skull presented as 3-dimensional renderings. The widening of the sagittal and parietal cranial sutures of the PCN-bearing mouse is evident. Bone surface roughness analysis revealed that the bone loss involved the cranial bone plates (not shown). CT was performed on formalin-fixed cadavers using an Inveon (Siemens Knoxville, TN) preclinical PET/CT/SPECT scanner (Inveon Acquisition Workplace version 1.5). Acquisition parameters included 360 rotation, 720 projections, 755 milliseconds exposure time, 80 kV voltage and 500 μA current. Scan projections were reconstructed to a pixel size of 20 μm2.
(B) CT analysis of lumbar spine. Shown to the left are high-resolution 3D images of lumbar vertebrae 2–4 (L2–4) from two representative, tumor-bearing CD45.2+ mice (top panel; a total of 8 diseased mice were analyzed) compared to two normal mice (bottom; a total of 5 mice were analyzed). Corresponding false color images of quantitative bone surface roughness analyses are presented in Supplemental Figure 9. Shown to the right are diagrams of CT analytical data obtained with the help of the BoneJ (v. 1.3.14) plugin of ImageJ (v. 1.49m). Trabecular bone was evaluated in a region spanning 0.6 mm below the cranial growth plate and 0.6 mm above the caudal growth plate. Regions of interest (ROI) were identified manually, but thresholding of bone from non-bone relied on software algorithms of BoneJ. BoneJ’s Volume Fraction tool was used to determine the bone volume (top). The Thickness and Spacing tool was used to assess trabecular thickness (center) and inter-trabecular space (bottom). Mean values and standard deviations were plotted and statistically compared using the Mann-Whitney test. The same bone parameters from age-matched normal mice treated with 4.5 Gy whole-body irradiation (data not shown) were indistinguishable from those in normal mice (left untreated) included in the diagrams. This indicated that the osteopenia found in tumor-bearing mice was caused by neoplastic growth, not by irradiation.
(C) CT analysis of individual vertebrae. Shown to the left are 2D grey-scale images of tomographic slices of L2 from a representative tumor-bearing and normal mouse. Corresponding false-color images of trabecular bone thickness (BoneJ) are presented to the right. Blue and white indicates strong and weak bone, respectively.
(D) Hind limb paralysis in tumor-bearing mice. See the Supplemental Video for examples of different disease stages, ranging from subtle hind limb weakness to complete paralysis.
(E) Elevated numbers of TRAP+ osteoclasts in PCN-bearing mice. TRAP (tartrate resistant alkaline phoshatase) activity in osteoclasts (arrows) was detected using an acid phosphatase leukocyte kit from Sigma. Osteoclasts were enumerated in five high-power microscopy fields of each bone specimen. Five specimens per mouse were evaluated. Six tumor-bearing mice were compared to 6 normal controls. Mean values and SEM of the two datasets were plotted.
Advantages of the new myeloma model described above include the possibility to (1) generate – de novo, and in a predictable, economic manner – “waves” of primary PCNs derived from the same donor B-cell pool, (2) employ in vivo imaging to evaluate myeloma bone disease (CT) and tumor development (BLI, PET) in live mice – in a reproducible, objective, sequential and, if desired, lesion-specific fashion, (3) elucidate pleiotropic myeloma drivers, such as Bruton tyrosine kinase [13], specifically in the “seed” or “soil” of myeloma – as recently demonstrated for IL-6 in plasmacytoma [8], (4) study the mechanisms of and new treatments for myeloma bone disease, and (5) last but not least, take advantage of allotype-specific antibodies to CD45 to precisely identify rare CD45.2+ tumor cells in a CD45.1+ environment. If the latter will be carried out in CD45.2+ tumor-bearing CD45.1+ mice that have achieved complete drug-induced remissions, it may afford an extraordinary opportunity to shed light on long-standing, thorny questions in myeloma research; e.g., the nature and anatomical location of stem cell-like myeloma cells, the biological underpinnings of minimal residual disease, and the early events in tumor relapse. In sum, the findings reported here not only confirm two recent, insightful, independent studies on adoptive B-cell transfer mouse models of myeloma [14, 15], they also provide a blueprint for the biological validation of candidate myeloma drivers as we go forward.
Supplementary Material
Acknowledgments
This work was supported in part by: NIH Hematology Training Grant T32 HL007344 to VT; NIH Prefdoctoral Training Grant T32 AI007485 to TRR; NCI Core Grant P30CA086862 in support of the Holden Comprehensive Cancer Center; Senior Research Awards from the Multiple Myeloma Research Foundation and International Waldenström’s Macroglobulinemia Foundation to SJ; and R01CA151354 from the NCI to SJ. C.CD45.1 congenic mice and the retroviral transduction system were kindly provided, respectively, by Drs. Lyse A. Norian and Dawn E. Quelle (both UI). Special thanks to the UI Flow Cytometry, Central Microscopy Research, and Comparative Pathology facilities for expert assistance.
Footnotes
Conflict-of-interest
The authors declare no competing financial interests.
Supplementary information is available at Leukemia’s website.
References
- 1.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–348. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 3.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer cell. 2014;25(1):91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. The New England journal of medicine. 2011;364(24):2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. The New England journal of medicine. 2012;367(9):826–833. doi: 10.1056/NEJMoa1200710. [DOI] [PubMed] [Google Scholar]
- 7.Heyer J, Kwong LN, Lowe SW, Chin L. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10(7):470–480. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosean TR, Tompkins VS, Olivier AK, Sompallae R, Norian LA, Morse HC, 3rd, et al. The tumor microenvironment is the main source of IL-6 for plasma cell tumor development in mice. Leukemia. 2014 doi: 10.1038/leu.2014.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva S, Kovalchuk AL, Kim JS, Klein G, Janz S. BCL2 accelerates inflammation-induced BALB/c plasmacytomas and promotes novel tumors with coexisting T(12;15) and T(6;15) translocations. Cancer research. 2003;63(24):8656–8663. [PubMed] [Google Scholar]
- 10.Kovalchuk AL, Kim JS, Park SS, Coleman AE, Ward JM, Morse HC, 3rd, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci USA. 2002;99(3):1509–1514. doi: 10.1073/pnas.022643999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duncan K, Rosean TR, Tompkins VS, Olivier A, Sompallae R, Zhan F, et al. (18)F-FDG-PET/CT imaging in an IL-6- and MYC-driven mouse model of human multiple myeloma affords objective evaluation of plasma cell tumor progression and therapeutic response to the proteasome inhibitor ixazomib. Blood Cancer J. 2013;3:e165. doi: 10.1038/bcj.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, et al. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011;17(23):7313–7323. doi: 10.1158/1078-0432.CCR-11-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tai YT, Chang BY, Kong SY, Fulciniti M, Yang G, Calle Y, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120(9):1877–1887. doi: 10.1182/blood-2011-12-396853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Zheng M, Gali R, Tian Z, Topal Gorgun G, Munshi NC, et al. A novel rapid-onset high-penetrance plasmacytoma mouse model driven by deregulation of cMYC cooperating with KRAS12V in BALB/c mice. Blood Cancer J. 2013;3:e156. doi: 10.1038/bcj.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dechow T, Steidle S, Gotze KS, Rudelius M, Behnke K, Pechloff K, et al. GP130 activation induces myeloma and collaborates with MYC. The Journal of clinical investigation. 2014;124(12):5263–5274. doi: 10.1172/JCI69094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.