

Abstract
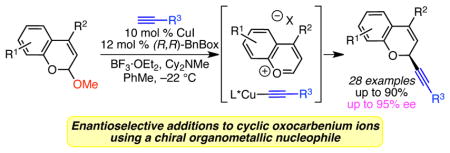
We have developed highly enantioselective, copper-catalyzed alkynylations of benzopyranyl acetals. By using a copper(I) catalyst equipped with a chiral bis(oxazoline) ligand, high yields and enantioselectivities are achieved in the alkynylation of widely available, racemic isochroman and chromene acetals to deliver α-chiral oxygen heterocycles. This method demonstrates that chiral organometallic nucleophiles can be successfully used in enantioselective additions to oxocarbenium ions.
Graphical Abstract
1. INTRODUCTION
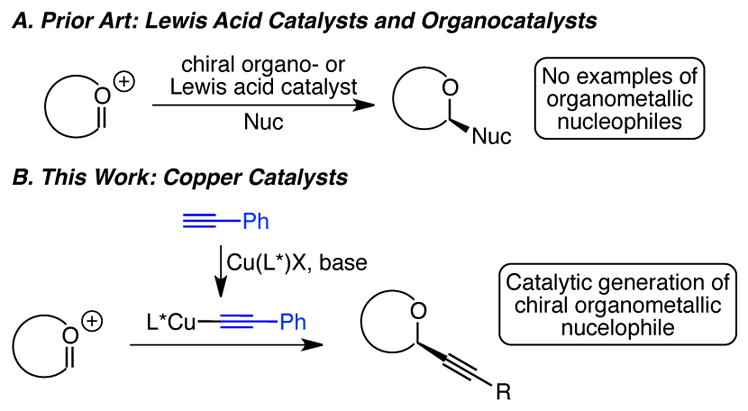
Controlling enantioselectivity in additions to oxocarbenium ions represents a long-standing challenge in asymmetric catalysis. In terms of intermolecular additions to cyclic oxocarbenium ions, few methods have been developed to confront this problem, despite the power of such a transformation to deliver α-chiral oxygen heterocycles, an important class of biologically active compounds.1,2,3,4 The challenge – and opportunity – of controlling enantioselectivity in additions to these electrophiles stems in part from the fact that oxocarbenium ions lack a Lewis basic site (except for the counter-anion, as discussed below). This fact distinguishes oxocarbenium ions from other carbonyl substrates and precludes the well-established strategy of using a chiral Lewis acid catalyst to control enantioselectivity in additions to these special C=X electrophiles. Furthermore, the high reactivity of oxocarbenium ion intermediates can make decomposition reactions competitive with desired addition pathways.
Recognizing these challenges, a select number of enantioselective additions to cyclic oxocarbenium ion intermediates have been developed. The majority relies on either organocatalysts or Lewis acid catalysts (Scheme 1A). In the first report of an enantioselective addition involving a cyclic oxocarbenium ion, Braun described a single example of allylation of dihydropyranyl acetal catalyzed by a chiral titanium(IV) Lewis acid.5 These additions are proposed to involve SN2 additions to diastereomeric titanium-bound acetals, which equilibrate via an oxocarbenium ion. For substrates that form more stable oxocarbenium ions, two distinct strategies have been used to control enantioselectivity. In a seminal report, Jacobsen developed conditions for the catalytic generation of a chiral oxocarbenium electrophile by using chiral thiourea catalysts in concert with 1-chloroisochroman substrates.6 The Jacobsen group has now also demonstrated that chiral thiourea catalysts can also control enantioselectivity in both intra- and intermolecular cyclizations of pyrilium ion intermediates.7 Subsequently, Terada and Floreancig showed that phosphoric acid catalysts can also be used to catalytically generate chiral oxocarbenium ion electrophiles, which undergo enantioselective attack by hydride or allyl nucleophiles, respectively.8 In a distinct strategy, Schaus has demonstrated the complementary approach of catalytic generation of a chiral nucleophile via tartarate-derived diol-catalysis of vinyl and aryl boronate esters.9 Rueping, Lou and Liu, and Cozzi have also shown that chiral enamine nucleophiles, catalytically generated from amine catalysts and aldehydes, add to oxocarbenium ions with high enantioselectivities.10 These methods are powerful in delivering specific classes of nucleophiles (allyl, vinyl, aryl, enolate equivalents, and hydride) to cyclic oxocarbenium ion intermediates and indeed demonstrate that catalytic asymmetric additions to oxocarbenium ions are feasible.
Scheme 1.
Enantioselective Additions to Cyclic Oxocarbenium Ion Intermediates
Given the success of using catalytically generated chiral nucleophiles for highly enantioselective additions to cyclic oxocarbenium ions, we envisioned that the use of chiral organometallic nucleophiles, generated in situ using a chiral metal catalyst, would provide an alternative strategy for enantioselective additions to oxocarbenium ion intermediates. In particular, inspired by zinc- and copper-catalyzed alkynylations of aldehydes,11 ketones,12 imines and iminium ions,13 we have focused on the addition of alkynes. Alkynes are a class of nucleophiles not addressed by organo- or Lewis acid-catalyzed methods, and provide a powerful functional group handle for elaboration of the α-chiral oxygen heterocycle products. Herein, we report our development of a copper(I)-catalyzed alkynylation of benzopyranyl acetals, which represents the first example of enantioselective addition of an organometallic nucleophile to a prochiral cyclic oxocarbenium ion (Scheme 1B).14 Using a copper catalyst equipped with a bis(oxazoline) ligand, we have achieved high yields and enantioselectivities in the alkynylation of both isochroman and chromene substrates.
2. RESULTS AND DISCUSSION
2.1. Substrate Synthesis
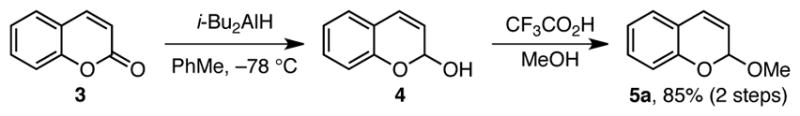
One advantage of using enantioselective additions to oxocarbenium ions to generate α-chiral oxygen heterocycles is the wide availability of the requisite acetal precursors. Isochroman acetals are readily prepared in one step via oxidation of the isochroman precursor (eq 1).6 Reduction of chromenones delivers chromene acetals (Scheme 2).9 These acetal substrates are stable for months to years when stored neat at −15 °C.
Scheme 2.
Synthesis of Chromene Acetals
 |
(1) |
2.2 Alkynylations with Achiral Catalysts
Our first challenge in developing a metal-catalyzed alkynylation of oxocarbenium ion intermediates was to identify conditions to generate the requisite oxocarbenium ion that would be compatible with a metal acetylide intermediate. Specifically, we were concerned that the Lewis acid used to ionize an acetal substrate may quench the metal acetylide. However, Downey had demonstrated that zinc-catalyzed alkynylations of aldehydes can be performed, and even accelerated, in the presence of trimethylsilyl triflate (TMSOTf), suggesting that zinc acetylides are compatible with TMSOTf.15
Encouraged by Downey’s report, we began by investigating the use of achiral zinc(II) catalysts in the alkynylation of benzopyranyl acetals. In the presence of either catalytic ZnBr2 or CuI, as well as trimethylsilyl triflate (TMSOTf) and Et3N, both isochroman and chromene acetals indeed undergo alkynylation in good yields (Scheme 3). Although a small amount of trimethylsilyl acetylene byproducts are formed, only a slight excess of alkyne (1.0–1.3 equiv) is required to achieve high yields. Alkynes with a broad range of substituents, including aryl, primary and secondary alkyl, trimethylsilyl, and protected aminomethyl, can be used in this transformation. These results demonstrate that organometallic nucleophiles, catalytically generated in situ, indeed undergo efficient additions to oxocarbenium ion intermediates.
Scheme 3. Oxocarbenium Ion Alkynylation with Achiral Catalystsa.
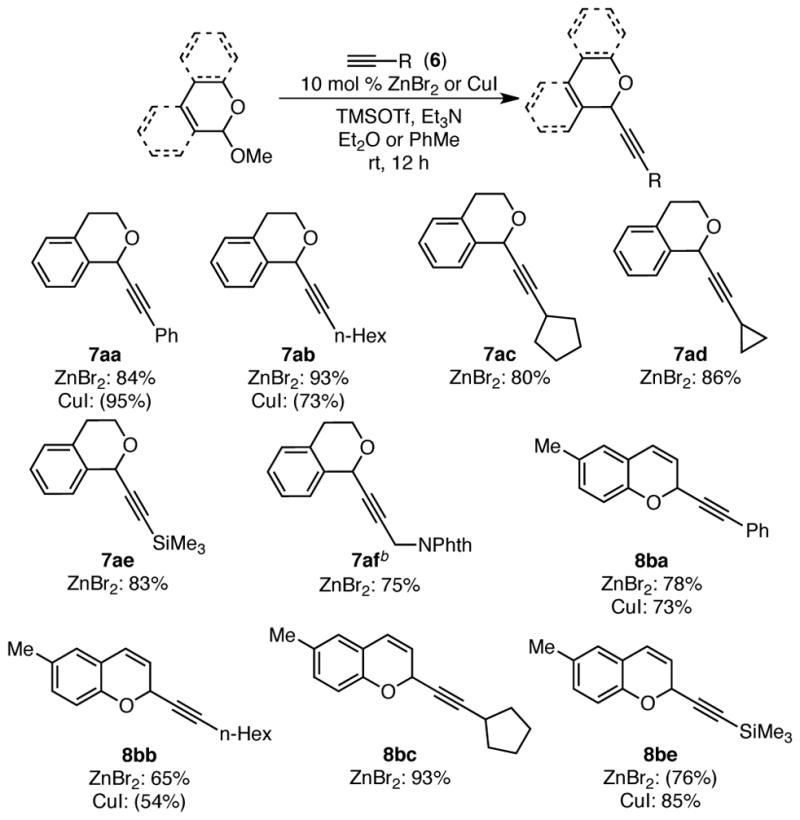
a Conditions: Acetal 2a or 5b (1.0 equiv), ZnBr2 or CuI (10 mol %), alkyne (1.0–1.3 equiv), Et3N (1.0–1.3 equiv), TMSOTf (1.1–1.2 equiv), Et2O or PhMe, rt, 12 h, unless otherwise noted. See Supporting Information for specific conditions. Yields in parentheses determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard. b CH2Cl2 used as solvent.
2.3 Enantioselective Alkynylations
As reported in our initial communication in this area, the copper-catalyzed alkynylation of isochroman acetals is rendered enantioselective by the addition of a bis(oxazoline) ligand.14 In particular, by using a copper(I) catalysts generated from Cu(MeCN)4(PF6) and BnBox, high yields and enantioselectivities were achieved with a range of isochroman acetals and aryl-substituted alkynes (Scheme 4). Notably, use of a non-coordinating counter-anion in the copper pre-catalyst was critical; CuI led to low enantioselectivities. Further, despite the promising reactivity of ZnBr2 to form racemic products, we have yet to identify a chiral zinc catalyst capable of delivering high reactivity or enantioselectivity.
Scheme 4. Enantioselective Alkynylation of Isochroman Acetalsa.
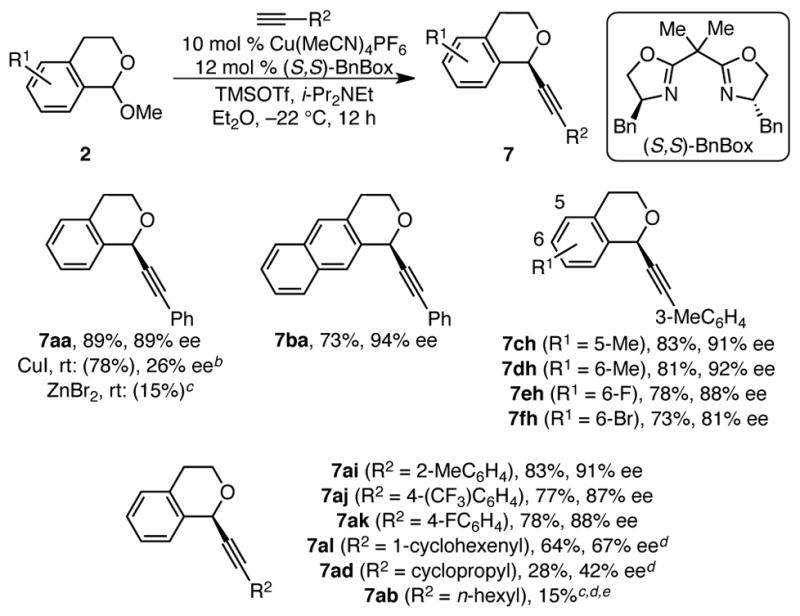
a Conditions: Acetal 2 (0.30 mmol, 1.0 equiv), [Cu(MeCN)4]PF6 (0.030 mmol, 10 mol %), (S,S)-BnBox (0.036 mmol, 12 mol %), alkyne (0.34 mmol, 1.1 equiv), i-Pr2NEt (0.396 mmol, 1.3 equiv), TMSOTf (0.365 mmol, 1.2 equiv, Et2O, −22 °C, 12 h, unless otherwise noted. Average isolated yields (±3%) and ee’s (±2%) from duplicate experiments, unless otherwise noted. Yields in parentheses determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard. Ee’s determined by HPLC analysis using a chiral stationary phase. b TMSOTf (1.1 equiv), i-Pr2Net (1.2 equiv). c ee not determined. d 20 mol % [Cu], 23 mol % BnBox, PhMe, 0 °C. e 0.1 mmol scale, single experiment.
Having established that enantioselective alkynylation of oxocarbenium ion intermediates provides an efficient route to enantioenriched α-chiral isochromans, we then sought to demonstrate the generality of using catalytically generated, chiral organometallic nucleophiles in enantioselective additions to oxocarbenium ions. Herein, we describe our application of this strategy to the preparation of enantioenriched α-alkynyl chromenes. This work demonstrates that our alkynylation strategy is effective in providing high enantioselectivity in reactions of both benzylic and aromatic oxocarbenium ions.
We began by studying the reaction of phenyl acetylene and chromene acetal 5a. Despite the similarities between the benzylic cation of isochroman oxocarbenium ions and the aromatic cation of chromene oxocarbenium ions, we quickly discovered that they react differently in these alkynylations. In our previous optimization of isochroman acetal 2, we had found that use of Cu(I) catalysts with weakly coordinating counterions was crucial for high enantioselectivity. In particular, Cu(MeCN)4PF6 had proven best. However, under similar conditions to those optimal for the alkynylation of isochroman acetal 2, low enantioselectivity (40% ee) of α-alkynyl chromene 8aa was observed (Table 1, entry 1). In examining the effect of the Cu counter-ion, we were surprised to find that catalysts derived from CuI provided much greater enantioselectivity (60% ee) than Cu salts with other counter-ions (entries 1–5). This result is in direct contrast to the alkynylation of isochroman acetals, in which CuI provided some of the lowest enantioselectivities.
Table 1.
Identification of Catalysta
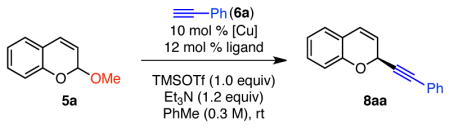
| ||||
|---|---|---|---|---|
| entry | [Cu] | ligand | yield (%)b | ee (%)c |
| 1 | Cu(MeCN)4PF6 | BnBox | 49 | 40 |
| 2 | Cu(MeCN)4BF4 | BnBox | 58 | 42 |
| 3 | Cu(OAc)2 | BnBox | 60 | 21 |
| 4 | CuBr | BnBox | 55 | 20 |
| 5 | CuI | BnBox | 87 | 60 |
| 6 | CuI | PhBox | 63 | 26 |
| 7 | CuI | i-PrBox | 57 | 48 |
| 8 | CuI | t-BuBox | 59 | 26 |
| 9 | CuI | L1 | 75 | 54 |
| 10 | CuI | L2 | 82 | 63 |
| 11 | CuI | L3 | 70 | 61 |
| 12 | CuI | L4 | 76 | 61 |
|
| ||||
Conditions: Acetal 5a (0.12 mmol, 1.0 equiv), [Cu] (0.012 mmol, 10 mol %), L* (0.014 mmol, 12 mol %), HCCPh (6a, 0.15 mmol, 1.2 equiv), Et3N (0.15 mmol, 1.2 equiv), TMSOTf (0.12 mmol, 1.0 equiv), PhMe (0.31 M), 0 °C, 15 h.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard.
Determined by HPLC analysis using a chiral stationary phase.
Despite this difference, BnBox remained the best ligand. Our efforts to improve the enantioselectivity by identifying an alternative ligand were unsuccessful; we investigated a variety of other chiral ligand scaffolds, but none provided higher enantioselectivity than BnBox under these reaction conditions. Other bis(oxazoline) ligands also resulted in lower enantioselectivities (entries 6–8). Curious about the potential importance of an aryl ring in the ligand, we investigated bis(oxazoline) ligands with substituted benzyl substituents, including those with greater steric bulk (L1, entry 9), increased electron-donating ability (L2 and L4, entries 10 and 12), and extended π-faces (L3 and L4, entries 11 and 12). Although p-methoxybenzyl-substituted L2 resulted in slightly higher enantioselectivity (63% ee), no significant improvements were observed with these ligands. Because BnBox is commercially available and easier to synthesize than L2, we pursued further optimization with BnBox.
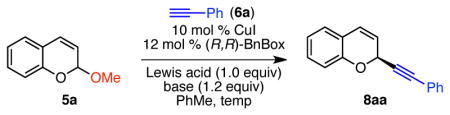
Having identified CuI/BnBox as the best catalyst system, we undertook a systematic evaluation of the other reaction variables. By lowering the reaction temperature to 0 °C, chromene 8aa was formed in 71% yield and 63% ee (Table 2, entry 1). Lowering the temperature more did not result in further increases in enantioselectivity. However, the overall reaction concentration influenced the enantioselectivity. By reducing the [5a] to 0.08 M, 73% ee of 8aa was achieved (Table 2, entry 2). Under these more dilute conditions, optimization of the base and Lewis acid revealed that the use of dicyclohexyl methyl amine (Cy2NMe) and BF3·OEt2 resulted in even higher enantioselectivity (entries 3–4). At a reaction temperature to −22 °C, chromene 8aa was formed in 83% ee, but at the cost of yield (entry 5). Increasing the equivalents of BF3·OEt2 led to synthetically useful yields of chromene 8aa in equivalent enantioselectivity (entry 6). Under these optimal conditions, chromene 8aa was formed in 74% yield and 83% ee.
Table 2.
Optimization of Reaction Conditionsa
| ||||||
|---|---|---|---|---|---|---|
| entry | [5] (M) | base | Lewis acid | temp (°C) | yield (%)b | ee (%)c |
| 1 | 0.31 | Et3N | TMSOTf | 0 | (71) | 63 |
| 2 | 0.08 | Et3N | TMSOTf | 0 | 56 | 73 |
| 3 | 0.08 | Cy2NMe | TMSOTf | 0 | 45 | 77 |
| 4 | 0.08 | Cy2NMe | BF3·OEt2 | 0 | 40 | 80 |
| 5 | 0.08 | Cy2NMe | BF3·OEt2 | –22 | 31 | 83 |
| 6d | 0.08 | Cy2NMe | BF3·OEt2 | –22 | (74) | 83 |
Conditions: Acetal 5a (0.12 mmol, 1.0 equiv), CuI (0.14 mmol, 10 mol %), BnBox (0.014 mmol, 12 mol %), HCCPh (6a, 0.15 mmol, 1.2 equiv), base (0.15 mmol, 1.2 equiv), Lewis acid (0.12 mmol, 1.0 equiv), PhMe, unless otherwise noted.
Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard. Numbers in parentheses are isolated yields.
Determined by HPLC analysis using a chiral stationary phase.
1.75 equiv BF3·OEt2 was used.
Under these optimized conditions, a variety of chromene acetal substrates underwent alkynylation in high yields and enantioselectivities (Scheme 5). In particular, alkynylation of chromene acetals substituted with electron-donating groups resulted in high enantioselectivies (8ba, 8ca). 4-Aryl chromene products were also formed in high ee’s (8ea–8ia). Notably, a number of biologically active chromene natural products contain this 4-aryl substituent.16 In this 4-aryl-substituted series, the importance of electronic effects is clear; substrates with more electron-donating 4-aryl substituents result in higher enantioselectivities (discussed in detail below). The highest ee’s are observed for chromene acetals with both an electron-donating R1 and a 4-phenyl substituent (8ja, 8ka). In contrast with the beneficial effect of 4-aryl substitution, 3-phenyl chromene acetal underwent reaction with phenyl acetylene in only 30% ee, and 4-methyl chromene acetal decomposed under the reaction conditions (not shown). For convenience, we set up our reactions in an inert-atmosphere glovebox. However, these reactions can also be set up on the bench-top with little change in the yield or enantioselectivity (see 8ea).
Scheme 5. Scope of Chromene Acetala.
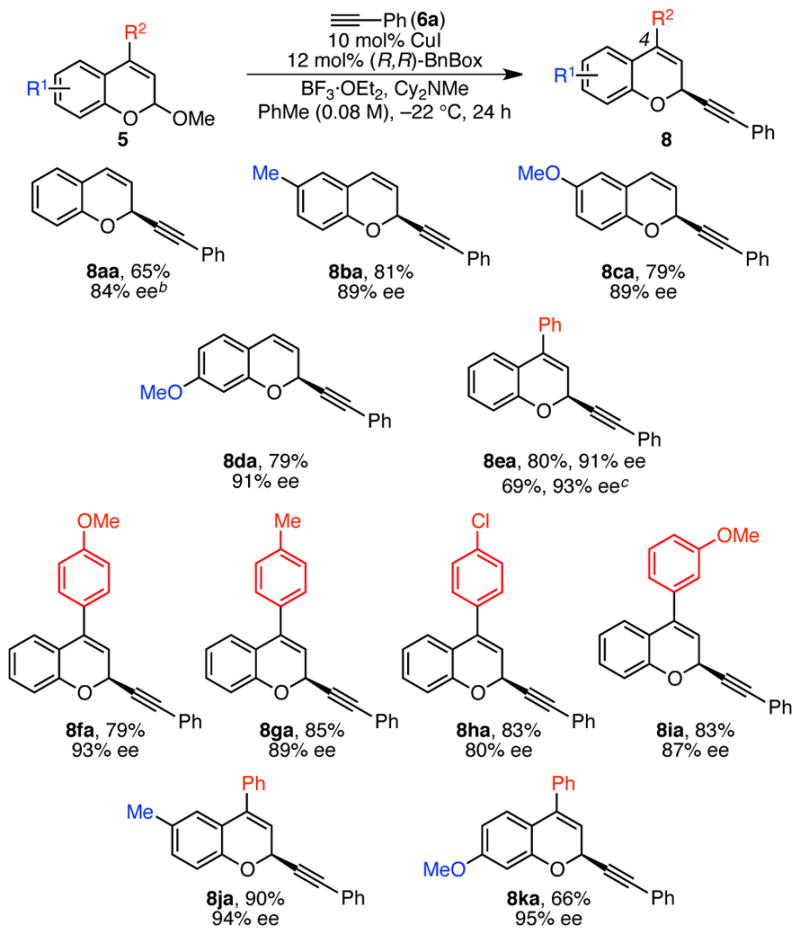
a Conditions: Acetal 5 (0.25 mmol, 1.0 equiv), CuI (0.025 mmol, 10 mol %), (R,R)-BnBox (0.030 mmol, 12 mol %), HCCPh (6a, 0.31 mmol, 1.2 equiv), Cy2NMe (0.31 mmol, 1.2 equiv), BF3·OEt2 (0.44 mmol, 1.8 equiv), PhMe (0.08 M), 24 h, unless otherwise noted. Average yields (±7%) and ee’s (±1%) of isolated products of duplicate reactions. Ee determined by HPLC analysis using a chiral stationary phase. b HCCPh (0.38 mmol, 1.5 equiv), BF3·OEt2 (0.36 mmol, 1.5 equiv). c Reaction set up outside glovebox, HCCPh (0.38 mmol, 1.5 equiv).
Wide scope was also observed with respect to the alkyne (Scheme 6). Both electron-rich and electron-poor aryl-substituted alkynes were effective. In addition, a wide range of functional groups was well tolerated, including ether (8em, 8fx), chloride (8eo, 8et, 8ft), bromide (8es), fluoride (8eu, 8fu), trifluoromethyl (8ej, 8fj), nitrile (8eq, 8ev), and ester (8fw) groups. However, reactions of alkynes with vinyl or aliphatic substituents result in lower yields and enantioselectivities. For example, cyclohexene 8el is formed in only 49% yield and 70% ee, and the analogous reaction of cyclopropylacetylene provides product in only 43% ee (not shown). Although we do not currently understand this trend, it mirrors observations with isochroman acetal substrates. Although alkyl-substituted alkynes undergo reaction in the presence of achiral Zn and Cu catalysts, they fail when chiral Cu(BnBox) catalysts are employed.
Scheme 6. Scope of Alkynea.

a Conditions: Acetal 5 (0.25 mmol, 1.0 equiv), CuI (0.025 mmol, 10 mol %), (R,R)-BnBox (0.030 mmol, 12 mol %), alkyne 6 (0.31 mmol, 1.2 equiv), Cy2NMe (0.31 mmol, 1.2 equiv), BF3·OEt2 (0.44 mmol, 1.8 equiv), PhMe (0.08 M), 24 h, unless otherwise noted. Average yields (±3%) and ee’s (±1%) of isolated products of duplicate reactions. Ee determined by HPLC analysis using a chiral stationary phase.
Reduction of these 2-alkynyl chromene products readily delivers 2-alkyl chromans with high levels of stereochemical fidelity. For example, hydrogenation of alkyne 8aa, prepared in 83% ee using (R,R)-BnBox as ligand, resulted in 2-phenethylchroman (9) in 87% yield and 82% ee (eq 2). Comparison of the optical rotation of chroman 9 to reported values confirmed that the absolute configuration of alkyne 8aa is S.9 In addition, the absolute configuration of products 8ea and 8eo were also determined to be S by crystallography.17 These absolute configurations confirm that the copper acetylide adds to the re face of the oxocarbenium ion when (R,R)-BnBox is used.
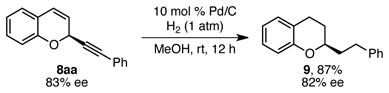 |
(2) |
2.4 Mechanistic Hypothesis and Model for Enantioinduction
We propose that these alkynylations of both isochroman and chromene acetals proceed via a catalytic cycle as shown in Scheme 7 (illustrated with chromene acetal 5a). Combination of BnBox and Cu(MeCN)4PF6 or CuI leads to formation of copper(I) species 10. Consistent with this proposal, a crystal structure of [(S,S)-BnBox]CuI shows bidentate coordination of BnBox to a trigonal planar copper(I) center (Figure 2).17 Importantly, consistent and high enantioselectivitives are only observed when the copper salt and ligand are stirred for at least 30 min at room temperature prior to the addition of other reagents, suggesting that the ligation event is slow. Addition of alkyne and base likely lead to formation of chiral copper acetylide 11. Simultaneously, acetal 5a undergoes Lewis acid-promoted ionization to deliver oxocarbenium ion 12. Nucleophilic attack of copper acetylide 11 onto oxocarbenium ion 12 would then form the new C–C bond and stereogenic center.18 Subsequent release of product 8aa frees catalyst 10 to re-enter the catalytic cycle.
Scheme 7.
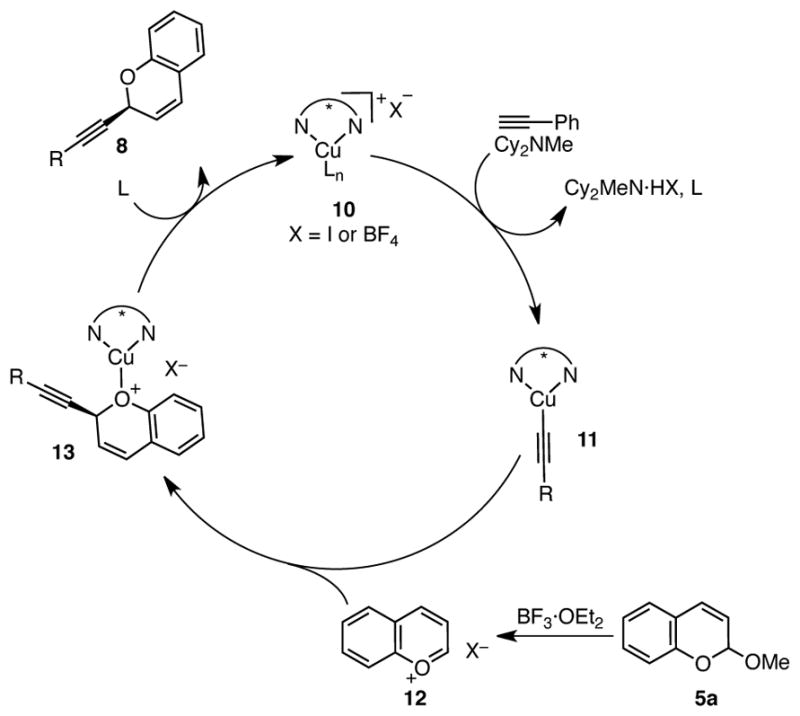
Proposed Catalytic Cycle
Figure 2.
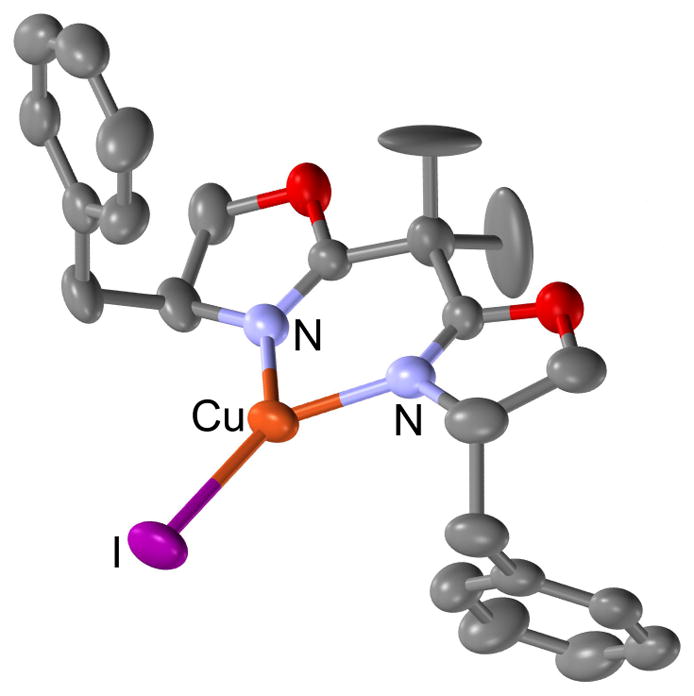
Molecular diagram of [(S,S)-BnBox]CuI with ellipsoids at 30% probability. H-atoms omitted for clarity.
As discussed above, there is a strong correlation between the stability of the oxocarbenium ion intermediate and the enantioselectivity. As shown by the Hammett correlation between σ+ values of substituents on the chromene acetal and the enantiomeric ratio of the products (Figure 1),19 higher enantioselectivities are observed for substrates with electron-donating substituents, which stabilize oxocarbenium ion 12. In general, electron-donating substituents on isochroman substrates also lead to higher enantioselectivies, but the Hammett correlation is less conclusive, suggesting other factors also affect enantioselectivity in this case.17 These trends are consistent with the C–C bond formation being enantiodetermining; a more stable oxocarbenium ion intermediate will undergo a later transition state in the addition of copper acetylide 11 to oxocarbenium ion 12. A later transition state will have a shorter C–C distance in the nascent bond, resulting in greater interaction of the oxocarbenium ion with the chiral catalyst.
Figure 1.
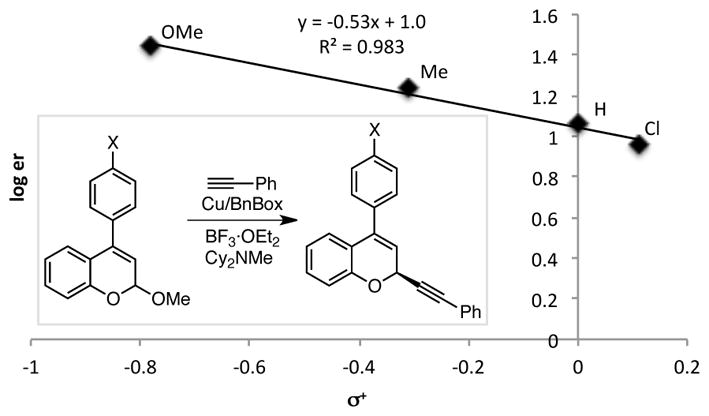
Hammett Plot of Substituent Effects of 4-Aryl-Substituted Chromene Acetals vs. Enantioselectivity
Focusing on C–C bond formation as the probable enantiodetermining step, our current model for enantioinduction is largely based on minimization of steric interactions between the oxocarbenium ion and the benzyl substituents of the catalyst. We assume that copper acetylide 11 adopts a pseudotetrahedral geometry at copper in the C–C bond-forming transition state. We also propose that the copper acetylide approaches the oxocarbenium ion from a Bürgi–Dunitz-like angle. Within these constraints, addition of the copper acetylide to the Re face of the oxocarbenium ion would be disfavored by a significant steric interaction between the benzene of the oxocarbenium ion and the benzyl substitutent of the catalyst (15, Figure 3). This destabilizing interaction is absent in attack of the Si face of the oxocarbenium ion (14). This model correctly predicts the observed major enantiomer in the alkynylation of isochroman acetals using (S,S)-BnBox as the ligand. With respect to chromene oxocarbenium ions, steric hindrance between the benzene of the oxocarbenium ion and the benzyl of the catalyst disfavors addition to the Re face (17), which is consistent with the observed major enantiomer when (S,S)-BnBox is used. However, in this case, maintenance of a Bürgi–Dunitz-like approach rotates the benzene of the oxocarbenium ion away from the benzyl group of the catalyst, leading to somewhat less steric hindrance (15 vs. 17), potentially explaining why chromene acetal 5a undergoes alkynylation in lower enantioselectivities than isochroman acetals 2 under identical conditions (see Table 1, entry 1). As noted above, 4-aryl chromene acetals generally undergo alkynylation in higher enantioselectivities. This effect of 4-aryl substituents may be due to a later transition state in the C–C bond formation due to stabilization of the oxocarbenium ion intermediate via conjugation to the aryl ring. It may also occur partially due to a steric interaction between the 4-aryl substituent and the benzyl group of the catalyst (17). This model is also consistent with the formation of racemic product in the alkynylation of benzaldehyde dimethyl acetal (20, eq 3). In this case, the oxocarbenium ion likely adopts an E configuration, instead of the Z configuration enforced for cyclic oxocarbenium ions. Little difference would then be expected between additions to the Re vs. Si face of the oxocarbenium ion (Figure 3C).
Figure 3.
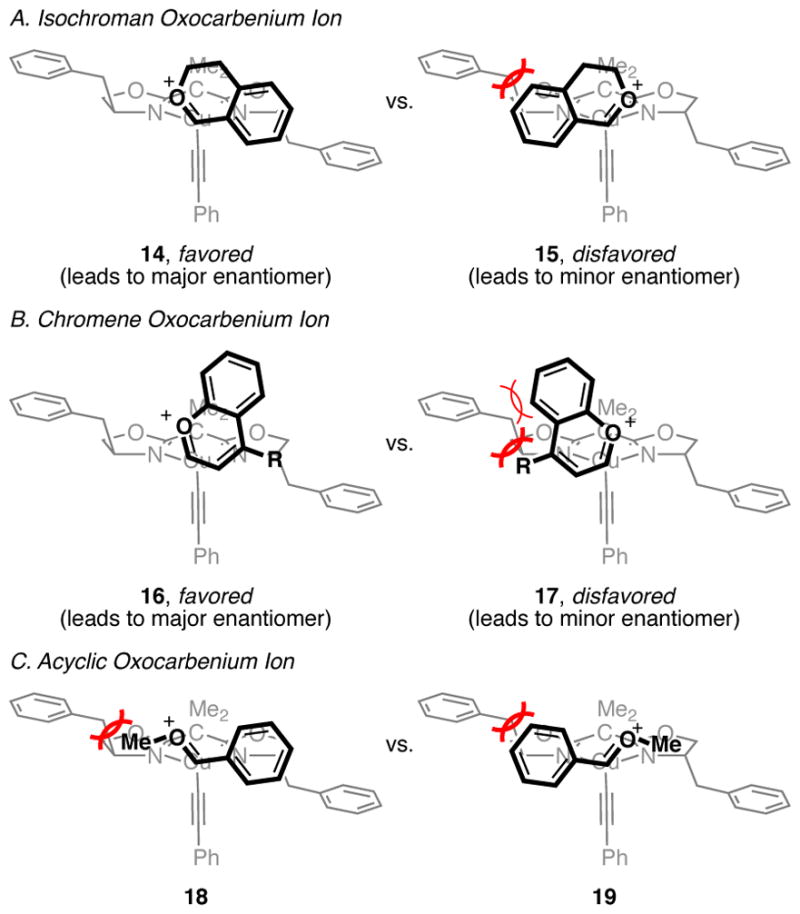
Putative stereochemical rationale. Shown with (S,S)-BnBox ligand.
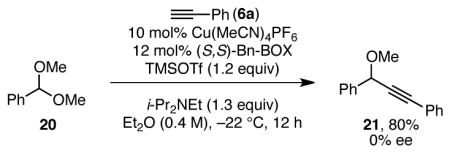 |
(3) |
Although this stereochemical model is satisfying in its rationalization of the observed major enantiomers and its simplicity, there are several results it does not explain. Notably, this model is predicated on minimization of steric hindrance, but ligand substituents larger than benzyl result in lower enantioselectivities. For example, in the alkynylation of chromene acetals, PhBox, i-PrBox, and t-BuBox give 26, 48 and 26% ee, respectively, under conditions where BnBox provides 60% ee (see Table 1, entries 5–8). Similar trends are observed with isochroman acetals. Further, the identity of the Lewis acid, base and copper counter-anion affect enantioselectivity. However, the optimal Lewis acid, base and copper counter-anion differ for the two acetal classes, hindering the development of a straightforward explanation of their effects. These observations suggest that the mechanism and particularly the enantiodetermining transition state are more complicated than our current understanding. Experiments are underway to increase the sophistication of our mechanistic understanding of this highly enantioselective transformation.
CONCLUSIONS
We have developed an efficient, enantioselective, copper-catalyzed alkynylation of benzopyranyl acetals. This method enables formation of highly enantioenriched α-chiral oxygen heterocycles from widely available, racemic isochroman and chromene acetal substrates. This reaction relies on the use of a copper/BnBox catalyst and demonstrates that chiral organometallic nucleophiles can be used in highly enantioselective additions to cyclic oxocarbenium ions. Ongoing efforts in our laboratory are directed towards establishing the generality of using chiral organometallic nucleophiles in enantioselective additions to oxocarbenium ions and towards developing a sophisticated understanding of the mechanism of this class of reactions.
EXPERIMENTAL SECTION
General Information
Reactions were performed either in a N2-atmosphere glovebox or in round-bottomed flasks. Flasks were fitted with rubber septa, and reactions were conducted under a positive pressure of N2. Syringes were used to transfer air- and moisture-sensitive liquids. Flash chromatography was performed on silica gel 60 (40–63μm, 60Å). Thin layer chromatography (TLC) was performed on glass plates coated with silica gel 60 with F254 indicator. Commercial reagents were purchased and used as received with the following exceptions: toluene, CH2Cl2, and Et2O were dried by passing through drying columns.20 Toluene was then degassed by sparging with N2 and stored over activated 4Å MS in a N2-atmosphere glovebox. Et3N and Cy2NMe were distilled from CaH2. MeOH was distilled from CaH2. BF3·OEt2 was purchased in sure sealed bottles and used as such. CDCl3 was stored over oven-dried potassium carbonate. Alkynes were degassed before use by either freeze-pump-thaw cycles or sparging with N2. Proton nuclear magnetic resonance (1H NMR) spectra and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on 400 MHz and 600 Mz spectrometers. Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CHCl3 = δ 7.28) and ((CD3)2CO = δ 2.05). Chemical shifts for carbon are reported in parts per million downfield from tetramethylsilane and are referenced to the carbon resonance of the solvent (CDCl3 = δ 77.07) and (CD3)2CO = δ 28.94). Data are represented as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants in Hertz (Hz), integration. Infrared (IR) spectra were obtained using FTIR spectrophotometers with material loaded onto a NaCl plate. Optical rotations were measured using a 2.5 mL cell with a 0.1 dm path length. BOX ligands were prepared as described in the literature.21 Alkynes 6j,22 6q,23 6r,24 6s,25 6v,26 6w,27 and 6x28 were prepared as described in the literature.
General Procedure for Preparation of Chromene Acetal Substrates
2-Methoxy-2H-chromene (5a)
This procedure was adapted from literature.9 To a flame-dried, 250-mL round-bottomed flask was added coumarin (6.0 g, 41.1 mmol, 1.0 equiv) and CH2Cl2 (60 mL). The solution was cooled to −78 °C and DIBAL-H (1.2 M in PhMe, 36.0 mL, 43.1 mmol, 1.05 equiv) was added via syringe over 15 min. The solution was then stirred for an additional 2 h at −78 °C and then allowed to warm to 0 °C and stirred for 15 min. The solution was then diluted with EtOAc (200 mL) and quenched with H2O (200 mL). The resulting mixture was vigorously stirred for 1 h and then filtered through Celite. The aqueous layer was extracted with EtOAc (2 × 200 mL). The combined organic layers were washed with sat. NaCl (200 mL), dried (Na2SO4), filtered and concentrated. The hemiacetal was used in the subsequent step without further purification.
The crude hemiacetal was dissolved in MeOH (50 mL). Trifluoroacetic acid (95.4 μL, 1.2 mmol, 3 mol %) was added, and the solution was stirred for 4 h at room temperature. K2CO3 (228 mg, 1.65 mmol, 0.04 equiv) was added. The mixture was filtered, and the filtrate was concentrated. The resulting residue was purified by silica gel chromatography (2–4% Et2O/hexanes with 5% Et3N; Rf = 0.44 to afford 5a (5.65 g, 85%) as pale yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.24 – 7.20 (m, 1H), 7.15 – 7.13 (m, 1H), 7.00 – 6.94 (m, 2H), 6.74 (d, J = 9.6 Hz, 1H), 5.87 (dd, J = 9.7, 3.8 Hz, 1H), 5.60 (d, J = 3.8 Hz, 1H), 3.50 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 151.3, 129.4, 127.1, 126.7, 121.6, 120.7, 119.7, 116.6, 95.9, 55.1; FTIR (NaCl, thin film) 2912, 2830, 1642, 1606, 1488, 1457, 1403, 1205 cm−1; LRMS (EI+) [M]+ calculated for C10H10O2: 162.07, found: 162.1.
2-Methoxy-6-methyl-2H-chromene (5b)
Prepared via the general procedure described above on a 31.0 mmol scale. The crude product was purified by silica gel chromatography (3–4% Et2O/hexanes with 5% Et3N; Rf = 0.5) to give 5b (5.1 g, 93%) as colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.03– 7.01 (m, 1H), 6.96 – 6.94 (m, 1H), 6.90 – 6.88 (m, 1H), 6.70 (d, J = 9.6 Hz, 1H), 5.85 (dd, J = 9.6, 3.8 Hz, 1H), 5.56 (d, J = 3.8 Hz, 1H), 3.48 (s, 3H), 2.28 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 149.1, 130.8, 130.0, 127.4, 126.7, 120.4, 119.7, 116.3, 95.8, 54.9, 20.6; FTIR (NaCl, thin film) 2914, 1641, 1493, 1083, 1023 cm−1; LRMS (EI+) [M]+ calculated for C11H12O2: 176.08, found: 176.1.
2,6-Dimethoxy-2H-chromene (5c)
Prepared via the general procedure described above on a 12.5 mmol scale. 6-Methoxy coumarin preparation method was adapted from literature.29 The crude product was purified by silica gel chromatography (10% Et2O/hexanes with 5% Et3N; Rf = 0.4) to give 5c (1.96 g, 82%) as colorless oil: 1H NMR (400 MHz, CDCl3) δ 6.93 (d, J = 8.8 Hz, 1H), 6.79 (dd, J = 8.8, 3.0 Hz, 1H), 6.70 – 6.68 (m, 2H), 5.90 (dd, J = 9.7, 3.8 Hz, 1H), 5.53 (d, J = 3.8 Hz, 1H), 3.77 (s, 3H), 3.48 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 154.2, 145.2, 126.6, 121.2, 120.4, 117.2, 115.3, 111.5, 95.8, 55.8, 55.1; FTIR (NaCl, thin film) 2932, 2831, 1611, 1604, 1578, 1492, 1263, 1207 cm−1; LRMS (EI+) [M]+ calculated for C11H12O3: 192.08, found: 192.1.
2,7-Dimethoxy-2H-chromene (5d)
Prepared via the general procedure described above on a 12.5 mmol scale. 7-Methoxy coumarin was prepared as reported in the literature.29 The crude product was purified by silica gel chromatography (10% Et2O/hexanes with 5% Et3N; Rf = 0.42) to give 5d (1.46g, 61%) as colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 8.3 Hz, 1H), 6.69 (d, J = 9.7 Hz, 1H), 6.59 – 6.48 (m, 2H), 5.73 (dd, J = 9.6, 3.7 Hz, 1H), 5.58 (d, J = 3.7 Hz, 1H), 3.80 (s, 3H), 3.49 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.8, 152.6, 127.8, 126.4, 116.9, 114.1, 107.8, 102.09, 96.2, 55.4, 54.9; FTIR (NaCl, thin film) 2933, 2830, 1641, 1615, 1569, 1506, 1274 cm−1; LRMS (EI+) [M]+ calculated for C11H12O3: 192.08, found: 192.1.
2-Methoxy-4-phenyl-2H-chromene (5e)
Prepared via the general procedure described above on a 4.86 mmol scale. 4-Phenyl coumarin was prepared as reported in the literature.30 The crude product was purified by silica gel chromatography (3–4% Et2O/hexanes with 5% Et3N; Rf = 0.45) to give 5e (856 mg, 74%) as a white solid (mp 81–83 °C): 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.37 (m, 5H), 7.28 – 7.24 (m, 1H), 7.14 (dd, J = 7.8, 1.6 Hz, 1H), 7.10 – 7.08 (m, 1H), 6.95 – 6.91 (m, 1H), 5.86 (d, J = 4.2 Hz, 1H), 5.66 (d, J = 4.1 Hz, 1H), 3.54 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 151.5, 138.8, 137.6, 129.5, 128.8, 128.4, 128.1, 126.3, 121.7, 121.4, 117.9, 117.0, 95.8, 55.2; FTIR (NaCl, thin film) 2928, 2827, 1637, 1636, 1604, 1483, 1483, 1452, 1219, 1045 cm−1; LRMS (EI+) [M]+ calculated for C16H14O2: 238.1, found: 238.1.
2-Methoxy-4-(4-methoxyphenyl)-2H-chromene (5f)
Prepared via the general procedure described above on a 3.98 mmol scale. 4-(4-Methoxyphenyl) coumarin was prepared as reported in the literature.30 The crude product was purified by silica gel chromatography (7–8% Et2O/hexanes with 5% Et3N; Rf = 0.33) to give 5f (810 mg, 76%) as a white solid (mp 79–82 °C): 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.31 (m, 2H), 7.26 – 7.22 (m, 1H), 7.17 – 7.14 (m, 1H), 7.08 – 7.06 (m, 1H), 6.95 – 6.90 (m, 3H), 5.81 (d, J = 4.2 Hz, 1H), 5.62 (d, J = 4.1 Hz, 1H), 3.85 (s, 3H), 3.53 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 159.6, 151.6, 138.3, 130.1, 129.9, 129.4, 126.3, 121.9, 121.4, 117.3, 117.1, 113.7, 95.9, 55.3, 55.2; FTIR (NaCl, thin film) 2930, 2833, 1696, 1636, 1606, 1573, 1612, 1452, 1248, 1095, cm−1; LRMS (EI+) [M]+ calculated for C17H16O3: 268.1, found: 268.1.
2-Methoxy-4-(p-tolyl)-2H-chromene (5g)
Prepared via the general procedure described above on a 3.26 mmol scale. 4-(p-Tolyl) coumarin was prepared as reported in the literature.30 The crude product was purified by silica gel chromatography (3–4% Et2O/hexanes with 5% Et3N; Rf = 0.45) to give 5g (517 mg, 63%) as a white solid (mp 85–88 °C): 1H NMR (400 MHz, CDCl3) δ 7.33 – 7.21 (m, 5H), 7.16 – 7.14 (m, 1H), 7.09 – 7.07 (m, 1H), 6.94 – 6.90 (m, 1H), 5.86 (d, J = 4.2 Hz, 1H), 5.66 (d, J = 4.1 Hz, 1H), 3.55 (s, 3H), 2.42 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 151.5, 138.7, 137.9, 134.6, 129.4, 129.1, 128.7, 126.4, 121.8, 121.4, 117.5, 117.1, 95.8, 55.2, 21.2; FTIR (NaCl, thin film) 2923, 2827, 1639, 1603, 1558, 1484, 1452, 1220, 1095, 1045, cm−1; LRMS (EI+) [M]+ calculated for C17H16O2: 252.2, found: 252.1.
4-(4-Chlorophenyl)-2-methoxy-2H-chromene (5h)
Prepared via the general procedure described above on a 1.75 mmol scale. 4-(4-Chlorophenyl) coumarin was prepared as reported in the literature.31 The crude product was purified by silica gel chromatography (5% Et2O/hexanes with 5% Et3N; Rf = 0.44) to give 5h (286 mg, 60%) as a white solid (mp 99–102 °C): 1H NMR (400 MHz, CDCl3) 7.41 – 7.33 (m, 4H), 7.25 – 7.29 (m, 1H), 7.08 – 7.10 (m, 2H) 6.96 – 6.92 (m, 1H), 5.84 (d, J = 4.2 Hz, 1H), 5.64 (d, J = 4.1 Hz, 1H), 3.54 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 146.3, 132.6, 130.8, 128.9, 125.02, 124.5, 123.4, 120.8, 116.3, 116.2, 113.0, 112.01, 90.5, 50.1; FTIR (NaCl, thin film) 2926, 2827, 1652, 1636, 1558, 1483, 1455, 1219, 1088, 1045, cm−1; LRMS (EI+) [M]+ calculated for C16H13ClO2: 272.1, found: 272.1.
2-Methoxy-4-(3-methoxyphenyl)-2H-chromene (5i)
Prepared via the general procedure described above on a 2.78 mmol scale. 4-(3-Methoxyphenyl) coumarin was prepared as reported in the literature.30 The crude product was purified by silica gel chromatography (6–7% Et2O/hexanes with 5% Et3N; Rf = 0.34) to give 5i (469 mg, 63%) as a white solid (mp 87–90 °C): 1H NMR (400 MHz, CDCl3) δ 7.34 – 7.23 (m, 2H), 7.17 – 7.15 (m, 1H), 7.09 – 7.07 (m, 1H), 6.99 – 6.90 (m, 4H), 5.87 (d, J = 4.1 Hz, 1H), 5.65 (d, J = 4.1 Hz, 1H), 3.82 (s, 3H), 3.54 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 159.5, 151.4, 139.03, 138.8, 129.5, 129.4, 126.3, 121.70, 121.5, 121.3, 117.8, 117.1, 114.1, 113.9, 95.8, 55.3, 55.2; FTIR (NaCl, thin film) 2931, 2831, 1636, 1604, 1577, 1483, 1453, 1218, 1097, 1045, cm−1; LRMS (EI+) [M]+ calculated for C17H16O3: 268.1, found: 268.1.
2-Methoxy-6-methyl-4-phenyl-2H-chromene (5j)
Prepared via the general procedure described above on a 2.39 mmol scale. 6-Methyl-4-phenyl coumarin was prepared as reported in the literature.32 The crude product was purified by silica gel chromatography (4% Et2O/hexanes with 5% Et3N; Rf = 0.45) to give 5j (500 mg, 83%) as a white solid (mp 89–92 °C): 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.40 (m, 5H), 7.09 – 7.07 (m, 1H), 7.02 – 7.00 (m, 1H), 6.96 – 6.94 (m, 1H), 5.87 (d, J = 4.2 Hz, 1H), 5.64 (d, J = 4.2 Hz, 1H), 3.55 (s, 3H), 2.25 (s, 3H); 13C NMR (101 MHz, CDCl3), δ 149.2, 138.9, 137.8, 130.7, 130.1, 128.9, 128.4, 128.08, 126.5, 121.5, 118.03, 116.8, 95.7, 55.2, 20.8; FTIR (NaCl, thin film) 2923, 2827, 1637, 1489, 1445, 1227, 1093, 1045 cm−1; LRMS (EI+) [M]+ calculated for C17H16O2: 252.1, found: 252.1.
2,7-Dimethoxy-4-phenyl-2H-chromene (5k)
Prepared via the general procedure described above on a 1.50 mmol scale., 7-Methoxy-4-phenyl coumarin was prepared as reported in the literature.32 The crude product was purified by silica gel chromatography (7–8% Et2O/hexanes with 5% Et3N; Rf = 0.4) to give 5k (210 mg, 52%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.44 – 7.42 (m, 5H), 7.07 (d, J = 8.6 Hz, 1H), 6.68 (d, J = 2.5 Hz, 1H), 6.52 (dd, J = 8.6, 2.6 Hz, 1H), 5.74 (d, J = 4.1 Hz, 1H), 5.67 (d, J = 4.1 Hz, 1H), 3.84 (s, 3H), 3.57 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 160.8, 152.9, 138.7, 137.8, 128.8, 128.3, 128.1, 127.2, 115.2, 115.11, 107.6, 102.4, 96.3, 55.4, 55.1; FTIR (NaCl, thin film) 2925, 2830, 1612, 1567, 1504, 1444, 1157, 1043 cm−1; LRMS (EI+) [M]+ calculated for C17H16O3: 268.1, found: 268.1.
General Procedure for the Enantioselective, Copper-Catalyzed Alkynylation of Chromene Acetals
In a N2-atmosphere glovebox, CuI (4.8 mg, 0.025 mmol, 10 mol %) was weighed into a 10-mL round-bottomed flask. (+)-2,2′-Isopropylidene[(4R)-4-benzyl-2-oxazoline] (BnBox, 11.0 mg, 0.030 mmol, 12 mol %) and toluene (3.2 mL, 0.08 M) were added. The round-bottomed flask was sealed with a septum. The mixture was stirred for 60 minutes at room temperature. Then the alkyne (0.305 mmol, 1.2 equiv), dicyclohexylmethyl amine (65.5 μL, 0.305 mmol, 1.2 equiv) and chromene acetal (0.254 mmol, 1.0 equiv) were added. The flask was again sealed with a septum, removed from the glovebox, and cooled to −22 °C. After 10 min, BF3·OEt2 (55.0 μL, 0.444 mmol, 1.75 equiv) was added via syringe, and the reaction mixture was stirred for 24 h at −22 °C. The reaction mixture was quenched MeOH (3.0 ml), allowed to warm to room temperature, diluted with Et2O (10 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (10 mL). The filtrate was concentrated and purified by silica gel chromatography.
(S)-2-(Phenylethynyl)-2H-chromene (8aa)
Chromene 8aa was prepared according to the General Procedure described above, except that 1.5 equiv of alkyne and 1.45 equiv BF3·OEt2 was used. The crude material was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.40) to give 8aa (run 1: 39.3 mg, 67%; run 2: 36.9 mg, 63%) as a colorless oil. The enantiomeric excess was determined to be 84% (run 1: 84% ee; run 2: 83% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 10.9 min, tR(minor) = 10.30 min. [α]D24 = −110.1° (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.41 (m, 2H), 7.34 – 7.26 (m, 3H), 7.20 – 7.14 (m, 1H), 7.07 –7.03 (m, 1H), 6.96 – 6.90 (m, 2H), 6.53 (d, J = 9.5 Hz, 1H), 5.87 – 5.81 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 152.4, 131.9, 129.5, 128.7, 128.2, 126.8, 124.6, 122.15, 122.10, 121.8, 121.4, 116.5, 86.0, 85.7, 65.0; HRMS (EI+) [M]+ calculated for C17H12O: 232.0888, found: 232.0895. The spectral data for this compound matches that reported in the literature.14
(S)-6-Methyl-2-(phenylethynyl)-2H-chromene (8ba)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (2–3% Et2O/hexanes, Rf = 0.5) to give 8ba (run 1: 52.8 mg, 84%; run 2: 48 mg, 77%) as a colorless oil. The enantiomeric excess was determined to be 89% (run 1: 89% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 11.00 min, tR(minor) = 10.24 min. [α]D24 = −223.5° (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.45 – 7.43 (m, 2H), 7.35 – 7.27 (m, 3H), 6.98 (dd, J = 8.2, 1.7 Hz, 1H), 6.88 – 6.87 (m, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.52 – 6.49 (m, 1H), 5.86 (dd, J = 9.5, 4.0 Hz, 1H), 5.79 (dd, J = 4.0, 1.6 Hz, 1H), 2.29 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 150.2, 131.9, 131.1, 129.9, 128.6, 128.2, 127.2, 124.8, 122.2, 122.1, 121.2, 116.2, 86.1, 85.5, 65.0, 20.6; FTIR (NaCl, thin film) 2918, 2830, 2214, 1725, 1665, 1632, 1586, 1487, 1442, 1206, 1022 cm−1; HRMS (EI+) [M]+ calculated for C18H14O: 246.1044, found: 246.1048.
(S)-6-Methoxy-2-(phenylethynyl)-2H-chromene (8ca)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (5% Et2O/hexanes, Rf = 0.38) to give 8ca (run 1: 51 mg, 77%; run 2: 54 mg, 81%) as a colorless oil. After the column fractions were concentrated, HPLC and NMR analysis were immediately performed on compound 8ca. When stored neat at room temperature, compound 8ca begins to decompose within minutes, but can be stored in solution in CHCl3 below −5 °C for days. The enantiomeric excess was determined to be 89% (run 1: 89% ee; run 2: 88% ee) by chiral HPLC analysis (CHIRALPAK IC, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 9.21 min, tR(minor) = 10.71 min. [α]D24 = −173.1° (c 1.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.40 (m, 2H), 7.29 – 7.26 (m, 3H), 6.85 (d, J = 8.7 Hz, 1H), 6.72 (dd, J = 8.8, 3.0 Hz, 1H), 6.61 (d, J = 3.0 Hz, 1H), 6.49 (d, J = 9.6, 1H), 5.89 (dd, J = 9.6, 4.1 Hz, 1H), 5.74 – 5.73 (m, 1H), 3.77 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 154.5, 146.3, 131.9, 128.6, 128.2, 124.8, 123.1, 122.2, 122.1, 117.1, 114.6, 111.8, 86.0, 85.6, 65.0, 55.7; FTIR (NaCl, thin film) 2935, 2832, 2216, 1635, 1609, 1577, 1489, 1450, 1269, 1199 cm−1; HRMS (EI+) [M]+ calculated for C18H14O 2: 262.0993, found: 262.0988.
(S)-7-Methoxy-2-(phenylethynyl)-2H-chromene (8da)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.4) to give 8da (run 1: 56.3 mg, 85%; run 2: 48 mg, 72%) as light yellow oil. After the column fractions were concentrated, HPLC and NMR analysis were immediately performed on compound 8da. When stored neat at room temperature, compound 8da begins to decompose within minutes, but can be stored in solution in CHCl3 below −5 °C for days. The enantiomeric excess was determined to be 91% (run 1: 90% ee; run 2: 91% ee) by chiral HPLC analysis (CHIRALPAK IC, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 10.14 min, tR(minor) = 9.53 min. [α]D24 = −110.8° (c 1.2, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.45 (d, J = 7.2 Hz, 2H), 7.34 – 7.28 (m, 3H), 6.97 (d, J = 8.1 Hz, 1H), 6.52 – 6.48 (m, 3H), 5.80 – 5.79 (m, 1H), 5.73 (dd, J = 9.6, 4.0 Hz, 1H), 3.81 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 160.9, 153.7, 131.9, 128.6, 128.2, 127.5, 124.3, 122.2, 119.1, 114.7, 107.7, 102.3, 86.2, 85.5, 65.2, 55.3; FTIR (NaCl, thin film) 2932, 2830, 2213, 1635, 1612, 1550, 1481, 1269 cm−1; HRMS (EI+) [M]+ calculated for C18H14O2: 262.0993, found: 262.0985.
(S)-4-Phenyl-2-(phenylethynyl)-2H-chromene (8ea)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.40) to give 8ea (run 1: 60.0 mg, 77%; run 2: 64.0 mg, 82%) as a white solid (mp 111–114 °C). The enantiomeric excess was determined to be 91% (run 1: 91% ee; run 2: 90% ee) by chiral HPLC analysis (CHIRALPAK IC, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 7.66 min, tR(minor) = 7.30 min. [α]D24 = −103.7° (c 1.6, CHCl3); 1H NMR (600 MHz (CD3)2CO) δ 7.48-7.33 (m, 10H), 7.25 (t, J = 7.8, Hz, 1H), 7.04 (d, J= 7.6 Hz, 1H), 7.00 (d, 1 J = 8.1 Hz, 1H), 6.95 (t, J = 7.5 Hz 1H), 5.98 (d, J = 4.7 Hz, 1H), 5.95 (d, J = 4.6, 1H); 13C NMR (151 MHz, (CD3)2CO) δ 153.1, 137.6, 136.7, 131.6, 129.6, 128.8, 128.6, 128.54, 128.51, 128.1, 125.8, 122.7, 122.1, 121.6, 120.4, 116.9, 86.2, 85.1, 64.5; FTIR (NaCl/thin film) 2922, 2850, 2215, 1629, 1601, 1573, 1481, 1451, 1214, 1110 cm−1; HRMS (EI+) [M]+ calculated for C23H16O: 308.1201, found: 308.1191. X-ray quality crystals were obtained from an Et2O/hexanes mixture cooled to −18 °C. The crystal structure demonstrated that the absolute configuration is S (Figure 1)
Product 8ea was also prepared in a reaction set up outside a N2-atmosphere glovebox. In a flame-dried, 10-mL round-bottomed flask, CuI (4.8 mg, 0.025 mmol, 10 mol %) and (+)-2,2′-isopropylidene[(4R)-4-benzyl-2-oxazoline] (BnBox, 11.0 mg, 0.0305 mmol, 12 mol %) were combined. The flask was sealed with a septum. The flask was evacuated and refilled with N2 three times before PhMe (3.18 mL, 0.08 M) was added. The solution was stirred for 60 min at room temperature. Then phenyl acetylene (38.9 mg, 0.381 mmol, 1.5 equiv), dicyclohexylmethyl amine (65.5 μL, 0.305 mmol, 1.2 equiv) and chromene acetal 5e (60.5 mg, 0.254 mmol, 1.0 equiv) were added. The flask was cooled to −22 °C. After 10 min, BF3·OEt2 (55.0 μL, 0.444 mmol, 1.75 equiv) was added via syringe, and the reaction mixture was stirred for 24 h at −22 °C. MeOH (3.0 mL) was then added. After warming to room temperature, the mixture was diluted with Et2O (10 mL) and filtered through a short plug of silica gel, which was then washed with Et2O (10 mL). The filtrate was concentrated. The crude product was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.40) to give 8ea (run 1: 54.3 mg, 69%; run 2: 53.0 mg, 68%) as a white solid. The enantiomeric excess was determined to be 93% (run 1: 93% ee; run 2: 93% ee) by chiral HPLC analysis (CHIRALPAK IC, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 7.10 min, tR(minor) = 6.77 min. The spectral data for this compound matches that reported above.
(S)-4-(4-Methoxyphenyl)-2-(phenylethynyl)-2H-chromene (8fa)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (5% Et2O/hexanes, Rf = 0.4) to give 8fa (run 1: 70 mg, 81%; run 2: 65.8 mg, 77%) as a white solid (mp 94–97 °C). The enantiomeric excess was determined to be 93% (run 1: 93% ee; run 2: 92% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 8.67 min, tR(minor) = 7.38 min. [α]D24 = −140.5° (c 1.0, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.45 – 7.33 (m, 7H), 7.29 – 7.25 (m, 1H), 7.10 (dd, J = 7.7, 1.7 Hz, 1H), 7.05 – 6.96 (m, 4H), 6.00 – 5.94 (m, 2H), 3.85 (s, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 159.8, 153.2, 136.2, 131.6, 129.8, 129.7, 129.5, 128.8, 128.5, 125.9, 122.9, 122.2, 121.6, 119.5, 116.9, 113.8, 86.3, 84.9, 64.5, 54.7; FTIR (NaCl, thin film) 2929, 2832, 2216, 1608, 1571, 1481, 1450, 1346, 1247, 1213 cm−1; HRMS (EI+) [M]+ calculated for C24H18O2: 338.1306, found: 338.1300.
(S)-2-(Phenylethynyl)-4-(p-tolyl)-2H-chromene (8ga)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.5) to give 8ga (run 1: 70 mg, 86%; run 2: 67.7 mg, 83%) as a colorless oil. The enantiomeric excess was determined to be 89% (run 1: 89% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 10.45 min, tR(minor) = 7.32 min. [α]D24 = −38.3° (c 1.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.44 (m, 2H), 7.34 – 7.28 (m, 5H), 7.25 – 7.21 (m, 3H), 7.11 (dd, J = 7.7, 1.6 Hz, 1H), 7.05 – 7.03 (m, 1H), 6.95 – 6.91 (m, 1H), 5.88 – 5.85 (m, 2H), 2.43 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 153.1, 137.9, 137.1, 134.7, 131.9, 129.5, 129.1, 128.7, 128.6, 128.2, 126.1, 122.9, 122.2, 121.6, 119.6, 116.9, 86.1, 85.7, 65.1, 21.2; FTIR (NaCl, thin film) 2920, 2826, 2230, 1683, 1635, 1601,1481, 1456, 1213 cm−1; HRMS (EI+) [M]+ calculated for C24H18O: 322.1357, found: 322.1360.
(S)-4-(4-Chlorophenyl)-2-(phenylethynyl)-2H-chromene (8ha)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.45) to give 8ha (run 1: 75.4 mg, 87%; run 2: 68.7 mg, 79%) as a colorless oil. The enantiomeric excess was determined to be 80% (run 1: 80% ee; run 2: 80% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 8.62 min, tR(minor) = 6.47 min. [α]D24 = −31.0° (c 2.0, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.54 – 7.51 (m, 2H), 7.48 – 7.33 (m, 7H), 7.30 – 7.26 (m, 1H), 7.06 – 6.94 (m, 3H), 6.04 (d, J = 4.7 Hz, 1H), 5.96 (d, J = 4.7 Hz, 1H); 13C NMR (101 MHz, (CD3)2CO) δ 153.1, 136.3, 135.5, 133.5, 131.6, 130.3, 129.8, 128.9, 128.6, 128.5, 125.7, 122.3, 122.1, 121.8, 120.9, 117.0, 86.0, 85.2, 64.4; FTIR (NaCl, thin film) 2924, 2840, 2216, 2235, 1635, 1658, 1506, 1488, 1481, 1213, 1110, 1088 cm−1; HRMS (EI+) [M]+ calculated for C23H15OCl: 342.0811, found: 342.0804.
(S)-4-(3-Methoxyphenyl)-2-(phenylethynyl)-2H-chromene (8ia)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (5% Et2O/hexanes, Rf = 0.4) to give 8ia (run 1: 68.5 mg, 80%; run 2: 73.5 mg, 86%) as a white solid (mp 97–100 °C). The enantiomeric excess was determined to be 87% (run 1: 87% ee; run 2: 86% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 8.00 min, tR(minor) = 7.16 min. [α]D24 = −123° (c 1.2, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.43 – 7.33 (m, 6H), 7.28 – 7.23 (m, 1H), 7.09 (dd, J = 7.7, 1.6 Hz, 1H), 7.00 – 6.94 (m, 5H), 6.01 (d, J = 4.6 Hz, 1H), 5.95 (d, J = 4.6 Hz, 1H), 3.83 (s, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 159.9, 153.1, 138.9, 136.5, 131.6, 129.6, 129.5, 128.9, 128.5, 125.9, 122.6, 122.1, 121.7, 120.8, 120.3, 116.9, 114.0, 113.7, 86.2, 85.1, 64.5, 54.7; FTIR (NaCl, thin film) 2929, 2843, 2217, 1597, 1481, 1451, 1211 cm−1; HRMS (EI+) [M]+ calculated for C24H18O 2: 338.1306, found: 338.1310.
(S)-6-Methyl-4-phenyl-2-(phenylethynyl)-2H-chromene (8ja)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.6) to give 8ja (run 1: 73 mg, 89%; run 2: 73.4 mg, 90%) as a white solid (mp 152–154 °C). The enantiomeric excess was determined to be 94% (run 1: 93% ee; run 2: 95% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 7.38 min, tR(minor) = 6.21 min. [α]D24 = −80.6° (c 1.6, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.48 – 7.34 (m, 10H), 7.06 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.85 (s, 1H), 5.96 (d, J = 4.6 Hz, 1H), 5.90 (d, J = 4.6 Hz, 1H), 2.19 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 150.9, 137.8, 137.3, 131.9, 131.0, 130.09, 128.8, 128.6, 128.4, 128.2 128.03, 126.4, 122.6, 122.2, 120.2, 116.7, 86.1, 85.7, 65.1, 20.8; FTIR (NaCl, thin film) 2922, 2830, 2217, 1683, 1635, 1601, 1481, 1456, 1213 cm−1; HRMS (EI+) [M]+ calculated for C24H18O: 322.1357, found: 322.1363.
(S)-7-Methoxy-4-phenyl-2-(phenylethynyl)-2H-chromene (8ka)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography using N2 to pressurize the column (3–5% Et2O/hexanes, Rf = 0.42) to give 8ka (run 1: 59 mg, 69%; run 2: 54 mg, 63%) as a colorless oil. After the column fractions were concentrated, HPLC and NMR analysis were immediately performed on compound 8ka. When stored neat at room temperature, compound 8ka begins to decompose within minutes, but can be stored in solution in CHCl3 under a N2 atmosphere below −5 °C for at least 12 hours. The enantiomeric excess was determined to be 95% (run 1: 95% ee; run 2: 94% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 8.35 min, tR(minor) = 7.11 min. [α]D24 = −66.2° (c 1.6, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.44 – 7.36 (m, 7H), 7.33 – 7.27 (m, 3H), 6.98 (d, J = 8.6 Hz, 1H), 6.61 – 6.60 (m, 1H), 6.47 (dd, J = 8.6, 2.5 Hz, 1H), 5.84 (d, J = 4.2 Hz, 1H), 5.73 (d, J = 4.2 Hz, 1H), 3.80 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 160.8, 154.5, 137.9, 137.1, 132.0, 128.7, 128.6, 128.3, 128.2, 128.0, 127.0, 122.2, 117.2, 116.1, 107.7, 102.5, 86.1, 85.6, 65.4, 55.4; FTIR (NaCl, thin film) 2930, 2835, 2215, 1630, 1602, 1480, 1450, 1348, 1213 cm−1; HRMS (EI+) [M]+ calculated for C24H18O2: 338.1306, found: 338.1298.
(S)-2-((4-Methoxyphenyl)ethynyl)-4-phenyl-2H-chromene (8em)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (7% Et2O/hexanes, Rf = 0.35) to give 8em (run 1: 61.5 mg, 72%; run 2: 65.8 mg, 77%) as a colorless oil. The enantiomeric excess was determined to be 86% (run 1: 85% ee; run 2: 86% ee) by chiral HPLC analysis (CHIRALPAK IC, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 8.69 min, tR(minor) = 8.18 min. [α]D24 = −72.9° (c 1.4, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.51 - 7.34 (m, 7H), 7.28 - 7.24 (m, 1H), 7.06 (dd, J= 7.7 Hz, 1.7 Hz, 1H), 7.01-6.90 (m, 4H), 5.99 (d, J = 4.6 Hz, 1H), 5.94 (d, J = 4.6 Hz, 1H), 3.82 (s, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 160.2, 153.2, 137.6, 136.5, 133.2, 129.6, 128.6, 128.5, 128.1, 125.8, 122.8, 121.6, 120.6, 116.9, 114.1, 114.0, 85.2, 84.7, 64.6, 54.8; FTIR (NaCl, thin film) 2928, 2836, 2216, 1604, 1570, 1480, 1451, 1249 cm−1; HRMS (EI+) [M]+ calculated for C24H18O2: 338.1306, found: 338.1315.
(S)-4-Phenyl-2-(p-tolylethynyl)-2H-chromene (8en)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.5) to give 8en (run 1: 64 mg, 78%; run 2: 58.8 mg, 72%) as a white solid (mp 147–149 °C). The enantiomeric excess was determined to be 89% (run 1: 89% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 10.89 min, tR(minor) = 7.23 min. [α]D24 = −117.0° (c 0.8, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.47 - 7.38 (m, 5H), 7.30 (d, J =7.8 Hz, 2H), 7.25 (t, J= 7.7 Hz, 1H), 7.17 (d, J = 7.8 Hz, 2H), 7.04 (d, J = 7.7 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 6.94 (t, J = 7.6 Hz, 1H), 5.97 (d, J = 4.6 Hz, 1H), 5.93 (d, J = 4.6 Hz, 1H), 2.31 (s, 3H); 13C NMR (151 MHz, (CD3)2CO) δ 153.2, 139.0, 137.7, 136.6, 131.6, 129.6, 129.2, 128.6, 128.5, 128.1, 125.8, 122.8, 121.6, 120.5, 119.2, 116.9, 85.6, 85.3, 64.6, 20.5; FTIR (NaCl/thin film) 2916, 2848, 2214, 1635, 1508, 1450, 1453, 1212 cm−1; HRMS (EI+) [M]+ calculated for C24H18O: 322.1357, found: 322.1365.
(S)-2-((4-Chlorophenyl)ethynyl)-4-phenyl-2H-chromene (8eo)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (6% Et2O/hexanes, Rf = 0.5) to give 8eo (run 1: 63.4 mg, 73%; run 2: 65.3 mg, 75%) as a light yellow solid (mp 142–145 °C). The enantiomeric excess was determined to be 89% (run 1: 89% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 7.24 min, tR(minor) = 6.46 min. [α]D24 = −134.2° (c 0.8, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.51-7.40 (m, 9H), 7.30-7.25 (m, 1H), 7.07 – 6.95 (m, 3H), 5.97–6.00 (m, 2H); 13C NMR (101 MHz, (CD3)2CO) δ 153.0, 137.5, 136.7, 134.4, 133.3, 129.7, 128.8, 128.57, 128.56, 128.2, 125.8, 122.7, 121.7, 120.9, 120.1, 116.9, 87.3, 83.8, 64.4; FTIR (NaCl, thin film) 2921, 2820, 2240, 1659, 1631, 1481, 1452, 1214 cm−1; HRMS (EI+) [M]+ calculated for C23H15OCl: 342.0811, found: 342.0808. X-ray quality crystals were obtained from an Et2O/hexanes mixture cooled to −18 °C. The enantiomeric excess of these crystals was determined to be >99% by chiral HPLC analysis. The crystal structure demonstrated that the absolute configuration is S (Figure 2).
(S)-4-Phenyl-2-((4-(trifluoromethyl)phenyl)ethynyl)-2H-chromene (8ej)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.55) to give 8ej (run 1: 72.3 mg, 76%; run 2: 69.2 mg, 73%) as a light yellow solid (mp 123–126 °C). The enantiomeric excess was determined to be 83% (run 1: 82% ee; run 2: 83% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 8.30 min, tR(minor) = 6.98 min. [α]D24 = −81.2° (c 1.6, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.71 (d, J = 8.1 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 7.48 – 7.40 (m, 5H), 7.26 (t, J = 7.6 Hz, 1H), 7.05 (d, J = 6.0 Hz, 1H), 7.02 (d, J = 8.1 Hz, 1H), 6.97 (t, J = 7.6 Hz, 1H), 6.05 – 6.00 (m, 2H); 13C NMR (151 MHz, (CD3)2CO) δ 153.0, 137.5, 137.0, 129.9 (q, JC–F = 31.7 Hz), 129.7,129.2 128.4, 128.2,127.8, 126.3, 125.9, 125.4 (q, JC–F = 3.0 Hz), 124.0 (q, JC–F = 271.8 Hz), 122.7, 121.8, 119.9, 116.9, 88.9, 83.6, 64.4; FTIR (NaCl, thin film) 2925, 2820, 2232, 1615, 1481, 1452 cm−1; HRMS (EI+) [M]+ calculated for C24H15OF3: 376.1075, found: 376.1070.
(S)-4-((4-Phenyl-2H-chromen-2-yl)ethynyl)benzonitrile (8eq)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (9% Et2O/hexanes, Rf = 0.3) to give 8eq (run 1: 62.2 mg, 74%; run 2: 63.8 mg, 76%) as light yellow oil. The enantiomeric excess was determined to be 85% (run 1: 85% ee; run 2: 85% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 13.24 min, tR(minor) = 11.74 min. [α]D24 = −106.7° (c 0.8, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.77 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 8.1 Hz, 2H), 7.48 – 7.40 (m, 5H), 7.27 (t, J = 7.7 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.07 – 5.99 (m, 2H); 13C NMR (151 MHz, (CD3)2CO) δ 152.9, 137.5, 137.1, 132.4, 132.3, 129.7, 128.6, 128.5, 128.2, 126.8, 125.9, 122.7, 121.8, 119.7, 117.9, 116.9, 112.2, 90.3, 83.5, 64.4; FTIR (NaCl, thin film) 2924, 2853, 2228, 2235, 1717, 1683, 1652, 1603, 1558, 1480, 1213 cm−1; HRMS (EI+) [M]+ calculated for C24H15ON: 333.1153, found: 333.1148.
(S)-2-((3,5-Dimethylphenyl)ethynyl)-4-phenyl-2H-chromene (8er)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (2–3% Et2O/hexanes, Rf = 0.57) to give 8er (run 1: 68 mg, 80%; run 2: 73.3 mg, 86%) as a colorless oil. The enantiomeric excess was determined to be 91% (run 1: 90% ee; run 2: 92% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 10.59 min, tR(minor) = 7.63 min. [α]D24 = −214.5° (c 0.4, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.51 – 7.41 (m, 5H), 7.29 – 7.25 (m, 1H), 7.07 – 6.94 (m, 6H), 5.99 (d, J = 4.7 Hz, 1H), 5.96 (d, J = 4.7 Hz, 1H), 2.26 (s, 6H); 13C NMR (101 MHz, (CD3)2CO) δ 153.1, 138.1, 137.6, 136.5, 130.5, 129.6, 129.2, 128.6, 128.5, 128.1, 125.8, 122.7, 121.8, 121.6, 120.4, 116.9, 85.49, 85.48, 64.5, 20.1; FTIR (NaCl, thin film) 2917, 2820, 2212, 1637, 1599, 1481, 1452, 1214 cm−1; HRMS (EI+) [M]+ calculated for C25H20O: 336.1514, found: 336.1509.
(S)-4-Phenyl-2-(m-tolylethynyl)-2H-chromene (8eh)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (3% Et2O/hexanes, Rf = 0.5) to give 8eh (run 1: 70.3 mg, 86%; run 2: 65 mg, 80%) as a white solid (mp 98–102°C). The enantiomeric excess was determined to be 90% (run 1: 90% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IC, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 7.35 min, tR(minor) = 6.78 min. [α]D24 = −126.6° (c 1.2, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.51 – 7.41 (m, 5H), 7.30 – 7.20 (m, 5H), 7.07 – 6.95 (m, 3H), 6.00 (d, J = 4.7 Hz, 1H), 5.97 (d, J = 4.7 Hz, 1H), 2.31 (s, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 153.1, 138.2, 137.6, 136.6, 132.1, 129.7, 129.6, 128.7, 128.58, 128.56, 128.4, 128.1, 125.8, 122.7, 122.0, 121.6, 120.4, 116.9, 85.8, 85.2, 64.5, 20.1; FTIR (NaCl, thin film) 2920, 2823, 2216, 1601, 1481, 1451, 1341, 1294, 1214 cm−1; HRMS (EI+) [M]+ calculated for C24H18O: 322.1357, found: 322.1352.
(S)-2-((3-Bromophenyl)ethynyl)-4-phenyl-2H-chromene (8es)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.5) to give 8es (run 1: 72.4 mg, 74%; run 2: 74 mg, 76%) as a yellow oil. The enantiomeric excess was determined to be 87% (run 1: 87% ee; run 2: 86% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 7.03 min, tR(minor) = 6.43 min. [α]D24 = −129° (c 1.0, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.58 – 7.56 (m, 2H), 7.48 – 7.39 (m, 6H), 7.34 – 7.24 (m, 2H), 7.05 – 6.94 (m, 3H), 6.00 – 5.96 (m, 2H); 13C NMR (101 MHz, (CD3)2CO) δ 153.0, 137.5, 136.8, 134.1, 132.0, 130.6, 130.5, 129.7, 128.59, 128.57, 128.2, 125.9, 124.3, 122.7, 121.8, 121.7, 120.0, 116.9, 87.7, 83.40, 64.4; FTIR (NaCl, thin film) 2919, 2849, 2214, 1589, 1554, 1480, 1349, 1213, 1110 cm−1; HRMS (EI+) [M]+ calculated for C23H15O Br: 386.0306, found: 386.0302.
(S)-2-((3-Chlorophenyl)ethynyl)-4-phenyl-2H-chromene (8et)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.4) to give 8et (run 1: 70.3 mg, 81%; run 2: 73 mg, 84%) as a yellow oil. The enantiomeric excess was determined to be 89% (run 1: 90% ee; run 2: 88% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 8.58 min, tR(minor) = 7.32 min. [α]D24 = −71.2° (c 0.8, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.48 – 7.36 (m, 9H), 7.26 (t, J = 7.7 Hz, 1H), 7.05 (d, J = 7.7 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 5.98 (m, 2H); 13C NMR (151 MHz, (CD3)2CO) δ 153.0, 137.5, 136.9, 133.8, 131.2, 130.3, 130.2, 129.7, 129.08, 128.59, 128.55, 128.2, 125.8, 124.09, 122.7, 121.7, 120.1, 116.9, 87.3, 83.8, 64.4; FTIR (NaCl, thin film) 2922, 2832, 2215, 1683, 1652, 1558, 1540, 1506, 1521, 1457, 1436 cm−1; HRMS (EI+) [M]+ calculated for C23H15OCl: 342.0811, found: 342.0821.
(S)-2-((3-Fluorophenyl) ethynyl)-4-phenyl-2H-chromene (8eu)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.4) to give 8eu (run 1: 65 mg, 79%; run 2: 60.2 mg, 73%) as a colorless oil. The enantiomeric excess was determined to be 93% (run 1: 93% ee; run 2: 92% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 2% i-PrOH/hexane, λ= 220 nm); tR(major) = 6.64 min, tR(minor) = 6.12 min. [α]D24 = −180.0° (c 0.4, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.48 – 7.38 (m, 6H), 7.27 – 7.24 (m, 2H), 7.18 – 7.15 (m, 2H), 7.05 (d, J = 7.6 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 5.99 – 5.96 (m, 2H); 13C NMR (151 MHz, (CD3)2CO) δ 162.3 (d, JC–F = 246.1 Hz), 153.1, 137.5, 136.8, 130.6 (d, JC–F = 7.6 Hz), 129.7, 128.6, 128.5, 128.1, 127.8 (d, JC–F = 3.02 Hz), 125.8, 124.1 (d, JC–F = 10.5 Hz), 122.7, 121.7, 120.1, 118.1(d, JC–F = 22.6 Hz), 116.9, 116.1(d, JC–F = 21.1 Hz), 87.3, 83.7(d, JC–F = 3.02 Hz), 64.4; FTIR (NaCl, thin film) 2916, 2848, 2224, 1601, 1581, 1481, 1452, 1264 cm−1; HRMS (EI+) [M]+ calculated for C23H15OF : 326.1107, found: 326.1106.
(S)-3-((4-Phenyl-2H-chromen-2-yl) ethynyl) benzonitrile (8ev)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (8% Et2O/hexanes, Rf = 0.33) to give 8ev (run 1: 65.8 mg, 78%; run 2: 63.2 mg, 75%) as a colorless oil. The enantiomeric excess was determined to be 88% (run 1: 87% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 12.66 min, tR(minor) = 11.59 min). [α]D24 = −140.8° (c 1.2, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.81 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 7.9 Hz, 1H), 7.60 (t, J = 7.8 Hz, 1H), 7.48 – 7.40 (m, 5H), 7.28 – 7.25 (m, 1H), 7.05 (d, J = 7.5 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.00 – 5.98 (m, 2H); 13C NMR (151 MHz, (CD3)2CO) δ 153.0, 137.5, 137.0, 135.9, 134.8, 132.2, 129.8, 129.7, 128.59, 128.55, 128.2, 125.9, 123.6, 122.7, 121.8, 119.8, 117.5, 116.9, 112.9, 88.5, 82.9, 64.4; FTIR (NaCl, thin film) 2924, 2853, 2226, 2232, 1600, 1572, 1451, 1293 cm−1; HRMS (EI+) [M]+ calculated for C24H15ON: 333.1153, found: 333.1149.
(S)-2-(Cyclohex-1-en-1-ylethynyl)-4-phenyl-2H-chromene (8el)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (2–3% Et2O/hexanes, Rf = 0.5) to give 8el (run 1: 40.1 mg, 51%; run 2: 36.2 mg, 46%) as a colorless oil. The enantiomeric excess was determined to be 70% (run 1: 70% ee; run 2: 70% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 6.74 min, tR(minor) = 6.15 min. [α]D24 = −82.0° (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3), δ 7.42 – 7.35 (m, 5H), 7.21 – 7.17 (m, 1H), 7.04 – 7.02 (m, 1H), 6.99 – 6.98 (m, 1H), 6.90 – 6.87 (m, 1H), 6.15 – 6.13 (m, 1H), 5.81 (d, J = 4.1 Hz, 1H), 5.76 (d, J = 4.1 Hz, 1H), 2.28 – 2.08 (m, 4H), 1.69 – 1.47 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 153.2, 137.7, 136.9, 136.5, 129.5, 128.8, 128.4, 128.0, 126.0, 122.9, 121.5, 120.6, 119.8, 116.9, 87.8, 83.2, 65.2, 29.0, 25.7, 22.2, 21.4; FTIR (NaCl, thin film) 2925, 2855, 2214, 1630, 1602, 1480, 1450, 1348, 1213 cm−1; HRMS (EI+) [M]+ calculated for C23H20O: 312.1514, found: 312.1522.
(S)-Ethyl 4-((4-(4-methoxyphenyl)-2H-chromen-2-yl)ethynyl)benzoate (8fw)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (12% Et2O/hexanes, Rf = 0.33) to give 8fw (run 1: 88 mg, 85%; run 2: 89.8 mg, 86%) as a colorless oil. The enantiomeric excess was determined to be 89% (run 1: 88% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IA, 0.8 mL/min, 3% i-PrOH/hexane, λ=254 nm); tR(major) = 17.69 min, tR(minor) = 14.33 min. [α]D24 = −105° (c 0.8, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 8.03 – 8.00 (m, 2H), 7.58 – 7.55 (m, 2H), 7.39 – 7.37 (m, 2H), 7.32 – 7.28 (m, 1H), 7.11 (dd, J = 7.7, 1.6 Hz, 1H), 7.08 – 6.99 (m, 4H), 6.03 – 5.98 (m, 2H), 4.36 (q, J = 7.1 Hz, 2H), 3.89 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 165.1, 159.8, 153.1, 136.4, 131.7, 130.5, 129.7, 129.63, 129.60, 129.3, 126.6, 125.9, 122.9, 121.7, 119.1, 116.9, 113.8, 89.3, 84.1, 64.4, 60.8, 54.7, 13.6; FTIR (NaCl, thin film) 2931, 2835, 2232, 1716, 1678, 1606, 1572, 1511, 1481 cm−1; HRMS (EI+) [M]+ calculated for C27H22O4: 410.1518, found: 410.1527.
(S)-4-(4-Methoxyphenyl)-2-((4-(trifluoromethyl)phenyl)ethynyl)-2H-chromene (8fj)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (6% Et2O/hexanes, Rf = 0.33) to give 8fj (run 1: 76.4 mg, 74%; run 2: 81.3 mg, 79%) as a colorless oil. The enantiomeric excess was determined to be 87% (run 1: 87% ee; run 2: 87% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 11.72 min, tR(minor) = 10.55 min. [α]D24 = − 19.2 ° (c 1.0, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.72 (d, J = 8.3 Hz, 2H), 7.63 (d, J = 8.2 Hz, 2H), 7.36 – 7.31 (m, 2H), 7.29 – 7.24 (m, 1H), 7.09 (dd, J = 1.6, 7.7 Hz, 1H), 7.05 – 6.95 (m, 4H), 6.02 – 5.90 (m, 2H), 3.85 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 159.6, 153.1, 137.0, 132.2, 130.3 (q, JC–F = 33.2 Hz), 129.9, 129.86, 129.6, 126.2, 126.1 125.1(q, JC–F = 3.02 Hz), 123.8 (q, JC–F = 271.8 Hz), 123.0, 121.8 118.7, 116.9, 113.9, 88.6, 84.2, 64.9, 55.4; FTIR (NaCl, thin film) 2929, 2834, 2226, 1608, 1570, 1511, 1481, 1451, 1323, 1290, 1248 cm−1; HRMS (EI+) [M]+ calculated for C25H17O2F3: 406.1180, found: 406.117.
(S)-4-(4-Methoxyphenyl)-2-((3-methoxyphenyl)ethynyl)-2H-chromene (8fx)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (8% Et2O/hexanes, Rf = 0.3) to give 8fx (run 1: 80.7 mg, 86%; run 2: 77.0 mg, 82%) as a colorless oil. The enantiomeric excess was determined to be 90% (run 1: 90% ee; run 2: 89% ee) by chiral HPLC analysis (CHIRALPAK IA, 0.8 mL/min, 2% i-PrOH/hexane, λ=254 nm); tR(major) = 16.46 min, tR(minor) = 13.94 min. [α]D24 = −56.8 ° (c 1.0, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.37 – 7.33 (m, 2H), 7.30 – 7.242 (m, 2H), 7.09 (dd, J = 7.7, 1.6 Hz, 1H), 7.05 – 6.95 (m, 7H), 5.96 – 5.93 (m, 2H), 3.87 (s, 3H), 3.80 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 159.5, 159.2, 153.2, 136.7, 130.0, 129.9 129.5, 129.3, 126.1, 124.5, 123.2, 123.06, 121.7, 119.3, 116.9, 116.6, 115.4, 113.8, 85.9, 85.6, 65.1, 55.36, 55.31; FTIR (NaCl, thin film) 2928, 2832, 2228, 2221, 1604, 1573, 1511, 1480 cm−1; HRMS (EI+) [M]+ calculated for C25H20O3: 368.1412, found: 368.1404.
(S)-2-((3-Chlorophenyl)ethynyl)-4-(4-methoxyphenyl)-2H-chromene (8ft)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.5) to give 8ft (run 1: 75.7 mg, 80%; run 2: 70.8 mg, 75%) as a light yellow solid (mp 112–115 °C). The enantiomeric excess was determined to be 91% (run 1: 91% ee; run 2: 90% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 11.23 min, tR(minor) = 10.17 min. [α]D24 = −70.8° (c 1.2, CHCl3); 1H NMR (400 MHz, (CD3)2CO) δ 7.45 – 7.33 (m, 6H), 7.30 – 7.25 (m, 1H), 7.10 (dd, J = 7.7, 1.7 Hz, 1H), 7.06 – 6.96 (m, 4H), 6.00 – 5.94 (m, 2H), 3.86 (s, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 159.8, 153.1, 136.4, 133.8, 131.1, 130.3, 130.1, 129.7, 129.62, 129.61, 129.1, 125.9, 124.1, 122.9, 121.7, 119.1, 116.9, 113.9, 87.8, 83.3, 64.3, 54.7; FTIR (NaCl, thin film) 2930, 2832, 2216, 2235, 1608, 1627, 1529, 1560, 1480, 1214 cm−1; HRMS (EI+) [M+] calculated for C24H17O2 Cl: 372.0917, found: 372.0913.
(S)-2-((3-Fluorophenyl)ethynyl)-4-(4-methoxyphenyl)-2H-chromene (8fu)
Prepared via the General Procedure. The crude material was purified by silica gel chromatography (4% Et2O/hexanes, Rf = 0.4) to give 8fu (run 1: 77.6 mg, 86%; run 2: 81.3 mg, 90%) as a colorless oil. The enantiomeric excess was determined to be 91% (run 1: 91% ee; run 2: 91% ee) by chiral HPLC analysis (CHIRALPAK IB, 0.8 mL/min, 1% i-PrOH/hexane, λ=254 nm); tR(major) = 11.07 min, tR(minor) = 9.84 min. [α]D24 = −115.7° (c 1.4, CHCl3); 1H NMR (600 MHz, (CD3)2CO) δ 7.45 – 7.41 (m, 1H), 7.36 (d, J = 8.2 Hz, 2H), 7.29 – 7.26 (m, 2H), 7.20 – 7.18 (m, 2H), 7.12 (d, J = 7.6 Hz, 1H), 7.05 – 6.98 (m, 4H), 5.98 – 5.95 (m, 2H), 3.88 (s, 3H); 13C NMR (151MHz, (CD3)2CO) δ 162.3 (d, JC–F = 247.6 Hz), 159.9, 153.1, 136.4, 130.6 (d, JC–F = 9.06 Hz), 129.7, 129.6, 129.5, 127.8 (d, JC–F = 3.0 Hz), 125.9, 124.2 (d, JC–F = 10.57 Hz), 122.9, 121.7, 119.2, 118.1 (d, JC–F = 24.16 Hz), 116.8, 116.07 (d, JC–F = 21.1 Hz), 113.9, 87.5, 83.6 (d, JC–F = 3.0 Hz), 64.5, 54.8; FTIR (NaCl, thin film) 2930, 2835, 2222, 1667, 1580, 1510, 1481, 1463, 1450, 1247, 1213 cm−1; HRMS (EI+) [M]+ HRMS (EI+) [M+] calculated for C24H17O2F: 356.1212, found: 356.1203.
(R)-2-Phenethylchroman (9)
Alkyne 8aa (25.0 mg, 0.107 mmol, 83% ee, prepared from acetal 5a and phenylacetylene using (R,R)-BnBox as ligand) and MeOH (2.5 mL) were combined in a flame-dried, 10-mL round-bottomed flask fitted with a 3-way adapter with a T-bore stopcock. Via this adapter, the reaction vessel was connected to a N2/vacuum manifold and to a H2-filled balloon. The flask was evacuated and refilled with nitrogen three times. 10% Pd/C (3.0 mg, 0.0028 mmol, 0.026 equiv) was added, and the flask was again evacuated and refilled with nitrogen three times. The flask was then evacuated and refilled with H2 five times. The reaction mixture was stirred under H2 (1 atm) for 12 h. After consumption of alkyne 8aa as determined by TLC analysis, the mixture was filtered through a short pad of celite, which was then washed with Et2O (10 mL). The filtrate was concentrated, and the crude material was purified by silica gel chromatography (1–3% Et2O/hexanes, Rf = 0.4) to give 9 (22.3 mg, 87%) as colorless oil. The enantiomeric excess was determined to be 82% by chiral HPLC analysis (CHIRALPAK IA, 0.8 mL/min, 0.5% i-PrOH/hexane, λ=254 nm); tR(major) = 7.72 min, tR(minor) = 8.6 min. [α]D24 = +43.1 ° (c 0.8, CHCl3). The sign of observed rotation is opposite to that of (S)-9 reported in literature,9 allowing assignment of the absolute configuration of 9 as R via our synthesis. The absolute configuration of alkyne 8aa is thus assigned as S. 1H NMR (600 MHz, CDCl3) δ 7.32 – 7.29 (m, 4H), 7.22 – 7.20 (m, 1H), 7.12 (t, J = 7.8 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 6.87 – 6.84 (m, 2H), 4.01 – 3.99 (m, 1H), 2.95– 2.76 (m, 4H), 2.13 – 2.07 (m, 1H), 2.02 – 2.00 (m, 1H), 1.95 – 1.89 (m, 1H), 1.83 – 1.76 (m, 1H); 13C NMR (151 MHz, CDCl3) δ 155.0, 141.9, 129.5, 128.6, 128.4, 127.2, 125.8, 122.1, 120.0, 116.8, 74.8, 37.1, 31.5, 27.5, 24.8; HRMS (EI+) [M]+ calculated for C17H18O: 238.1357, found: 238.1353.
1-(Oct-1-yn-1-yl)isochroman (7ab)
In a N2-atmosphere glovebox, ZnBr2 (9.0 mg, 0.04 mmol, 10 mol %) was weighed into a 1-dram vial. 1-Octyne (6b, 57.3 mg, 0.52 mmol, 1.3 equiv) and Et2O (1.0 mL, 0.4 M) were added. Then triethyl amine (72.5 μL, 0.52 mmol, 1.3 equiv) and isochroman acetal 2a (65.7 mg, 0.4 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (87.5 μL, 0.48 mmol, 1.2 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (1–2 % Et2O/hexanes, Rf = 0.50) to give product 7ab (90.1 mg, 93%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 1H), 7.22 – 7.21 (m, 2H), 7.15–7.13 (m, 1H), 5.55 (s, 1H), 4.27–4.24 (m, 1H), 3.98–3.95 (m, 1H), 2.90–2.87 (m, 2H), 2.27–2.24 (m, 2H), 1.58–1.50 (m, 2H), 1.41–1.26 (m, 6H), 0.91–0.88 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 135.6, 132.6, 128.8, 127.0, 126.2, 125.9, 86.7, 79.0, 67.2, 62.6, 31.3, 28.58, 28.53, 28.1, 22.5, 18.8, 14.06; HRMS LIFDI [M]+ calculated for C17H22O: 242.1671, found: 242.1694. The spectra matches with that reported in the literature.14
1-(Cyclopentylethynyl)isochroman (7ac)
In a N2-atmosphere glovebox, ZnBr2 (9.0 mg, 0.04 mmol, 10 mol %) was weighed into a 1-dram vial. Cyclopentylacetylene (6c, 90%, 54.4 mg, 0.52 mmol, 1.3 equiv) and Et2O (1.0 mL, 0.4 M) were added. Then triethyl amine (72.5 μL, 0.52 mmol, 1.3 equiv) and isochroman acetal 2a (65.7 mg, 0.4 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (87.5 μL, 0.48 mmol, 1.2 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (3–4% Et2O/hexanes, Rf = 0.40) to give product 7ac (72.4 mg, 80%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.31–7.28 (m, 1H), 7.23 – 7.20 (m, 2H), 7.13–7.11 (m, 1H), 5.55 (s, 1H), 4.28–4.22 (m, 1H), 3.97–3.92 (m, 1H), 2.91–2.86 (m, 2H), 2.70–2.66 (m, 1H), 1.95–1.91 (m, 2H), 1.73–1.55 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 136.3, 133.2, 129.3, 127.5, 126.7, 126.5, 91.3, 79.04, 67.9, 63.3, 34.3, 30.7, 28.6, 25.5; FTIR (NaCl, thin film) 2959, 2868, 2160, 1729, 1491, 1451, 1289, 1085 cm−1; HRMS LIFDI [M]+ calculated for C16H18O: 226.1358, found: 226.1332.
1-(Cyclopropylethynyl)isochroman (7ad)
In a N2-atmosphere glovebox, ZnBr2 (9.0 mg, 0.04 mmol, 10 mol %) was weighed into a 1-dram vial. Cyclopropylacetylene (6d, 34.4 mg, 0.52 mmol, 1.3 equiv) and Et2O (1.0 mL, 0.4 M) were added. Then triethyl amine (72.5 μL, 0.52 mmol, 1.3 equiv) and isochroman acetal 2a (65.7 mg, 0.4 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (87.5 μL, 0.48 mmol, 1.2 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (1–2% Et2O/hexanes, Rf = 0.4) to give product 7ad (68.2 mg, 86%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.29–7.27 (m, 1H), 7.22 – 7.20 (m, 2H), 7.13–7.10 (m, 1H), 5.51 (s, 1H), 4.27–4.21 (m, 1H), 3.97–3.92 (m, 1H), 2.88 (t, J= 5.7 Hz, 2H), 1.32–1.27 (m, 1H), 0.81–0.73 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 135.4, 132.6, 128.8, 127.1, 126.2, 125.9, 89.6, 74.2, 67.2, 62.6, 28.05, 8.3, 8.2; HRMS LIFDI [M]+ calculated for C14H14O: 198.1044, found: 198.1045. The spectra matches with that reported in the literature.14
(Isochroman-1-ylethynyl)trimethylsilane (7ae)
In a N2-atmosphere glovebox, ZnBr2 (9.0 mg, 0.04 mmol, 10 mol %) was weighed into a 1-dram vial. Trimethyl silylacetylene (6e, 51.1 mg, 0.52 mmol, 1.3 equiv) and Et2O (1.0 mL, 0.4 M) were added. Then triethyl amine (72.5 μL, 0.52 mmol, 1.3 equiv) and isochroman acetal 2a (65.7 mg, 0.4 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (87.5 μL, 0.48 mmol, 1.2 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (1% Et2O/hexanes, Rf = 0.50) to give product 7ae (76.2 mg, 83%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.30–7.28 (m, 1H), 7.24 – 7.22 (m, 2H), 7.14–7.10 (m, 1H), 5.55 (s, 1H), 4.29–4.23 (m, 1H), 3.99–3.94 (m, 1H), 2.89–2.91 (m, 2H), 0.20 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 134.6, 132.7, 128.8, 127.2, 126.3, 126.0, 104.1, 90.4, 67.4, 62.9, 27.9, 0.14; FTIR (NaCl, thin film) 2961, 2899, 2166, 1652, 1558, 1492, 1452, 1426, 1426, 1249, 1093 cm−1; HRMS LIFDI [M]+ calculated for C14H18OSi: 230.1127, found: 230.1106.
2-(3-(Isochroman-1-yl)prop-2-yn-1-yl)isoindoline-1,3-dione (7af)
In a N2-atmosphere glovebox, ZnBr2 (9.0 mg, 0.04 mmol, 10 mol %) was weighed into a 1-dram vial. Propargyl phthalimide (6f, 96.3 mg, 0.52 mmol, 1.3 equiv) and CH2Cl2 (1.0 mL, 0.4 M) were added. Then triethyl amine (72.5 μL, 0.52 mmol, 1.3 equiv) and isochroman acetal 2a (65.7 mg, 0.4 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (87.5 μL, 0.48 mmol, 1.2 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (40% Et2O/hexanes, Rf = 0.30) to give product 7af (95.2 mg, 75%) as a white solid (mp 158–162 °C): 1H NMR (400 MHz, CDCl3) δ 7.90–7.88 (m, 2H), 7.76–7.74 (m, 2H), 7.26–7.20 (m, 3H), 7.12–7.11 (m, 1H), 5.55 (s, 1H), 4.53 (s, 2H), 4.24–4.18 (m, 1H), 3.99–3.93 (m, 1H), 2.90–2.79 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 167.0, 134.3, 134.1, 132.7, 132.03, 128.9, 127.2, 126.3, 126.0, 123.5, 82.1, 79.2, 66.6, 62.4, 27.8, 27.3; FTIR (NaCl, thin film) 2927, 2240, 1771, 1719, 1611, 1491, 1467, 1426, 1452, 1391, 1344, 1190, 1116 cm−1; HRMS LIFDI [M]+ calculated for C20H15NO3: 317.1052, found: 317.1064.
6-Methyl-2-(oct-1-yn-1-yl)-2H-chromene (8bb)
In a N2-atmosphere glovebox, ZnBr2 (6.4 mg, 0.028 mmol, 10 mol %) was weighed into a 1-dram vial. 1-Octyne (6b, 34.4 mg, 0.312 mmol, 1.1 equiv) and toluene (916 μL, 0.31 M) were added. Then triethyl amine (50.0 μL, 0.355 mmol, 1.25 equiv) and chromene acetal 5b (50.0 mg, 0.284 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (56.9 μL, 0.312 mmol, 1.1 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (1–2 % Et2O/hexanes, Rf = 0.40) to give 8bb (46.9 mg, 65%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 6.96 (dd, J = 8.12, 1.8 Hz, 1H), 6.85 – 6.84 (m, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.43 (dd, J = 9.6, 1.1 Hz, 1H), 5.76 (dd, J = 9.6, 3.8 Hz, 1H), 5.54 – 5.52 (m, 1H), 2.28 (s, 3H), 2.23 – 2.19 (m, 2H), 1.51 – 1.46 (m, 2H) 1.38 – 1.23 (m, 6H), 0.89 (t, J = 6.9, 3H); 13C NMR (101 MHz, (CD3)2CO) δ 150.3, 130.6, 129.5, 127.1, 123.7, 123.3, 121.6, 115.9, 85.9, 77.7, 64.3, 31.1, 28.2, 28.1, 22.3, 19.6, 18.1, 13.4; FTIR (NaCl, thin film) 3077, 2990, 2812, 2338, 2311, 2281, 1900, 1719, 1623, 1547, 1271, 1088 cm−1; HRMS (EI+) [M]+ calculated for C18H22O: 254.1671, found: 254.1686.
2-(Cyclopentylethynyl)-6-methyl-2H-chromene (8bc)
In a N2-atmosphere glovebox, ZnBr2 (3.2 mg, 0.014 mmol, 10 mol %) was weighed into a 1-dram vial. Cyclopentylacetylene (6c, 90%, 18.5 μL, 0.142 mmol, , 1.0 equiv) and toluene (460 μL, 0.31 M) were added. Then triethyl amine (24.8 μL, 0.177 mmol, 1.25 equiv) and chromene acetal 5b (25.0 mg, 0.142 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (28.5 μL, 0.156 mmol, 1.1 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (1–2 % Et2O/hexanes, Rf = 0.40) to give 8bc (31.4 mg, 93%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 6.96 (dd, J = 8.12, 1.8 Hz, 1H), 6.84–6.83 (m, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.42 (dd, J = 9.6, 1.5 Hz, 1H), 5.74 (dd, J = 9.6, 3.7 Hz, 1H), 5.56 – 5.54 (m, 1H), 2.68 – 2.60 (m, 1H), 2.27 (s, 3H), 1.95 – 1.88 (m, 2H), 1.75– 1.66 (m, 2H) 1.65 – 1.47 (m, 4H); 13C NMR (101 MHz, (CD3)2CO) δ 150.4, 130.5, 129.5, 127.1, 123.7, 123.4, 121.5, 115.9, 90.0, 77.2, 64.4, 33.42, 33.41, 24.5, 19.6; FTIR (NaCl, thin film) 3077, 2958, 2868, 2311, 2222, 1652, 1558, 1489, 1209, 1125, 1024 cm−1; HRMS (EI+) [M]+ calculated for C17H18O: 238.1358, found: 238.1381.
Trimethyl((6-methyl-2H-chromen-2-yl)ethynyl)silane (8be)
In a N2-atmosphere glovebox, CuI (5.4 mg, 0.028 mmol, 10 mol %) was weighed into a 1-dram vial. Trimethylsilylacetylene (6e, 40.0 μL, 0.283 mmol, 1.0 equiv) and toluene (912 μL, 0.31 M) were added. Then triethyl amine (50.0 μL, 0.354 mmol, 1.25 equiv) and chromene acetal 5b (50.0 mg, 0.283 mmol, 1.0 equiv) were added. The vial was sealed and removed from the glovebox. After 10 min, TMSOTf (56.8 μL, 0.312 mmol, 1.1 equiv) was added via syringe, and the reaction mixture was stirred for 12 h at rt. The reaction mixture was quenched MeOH (1.0 ml), diluted with Et2O (5.0 mL), and filtered through a short plug of silica gel, which was then washed with Et2O (5.0 mL). The filtrate was concentrated and purified by silica gel chromatography (1 % Et2O/hexanes, Rf = 0.5) to give 8be (58.5 mg, 85%) as a white solid (mp 72–75 °C): 1H NMR (400 MHz, CDCl3) δ 6.96 (dd, J = 8.12, 1.9 Hz, 1H), 6.84 – 6.83 (m, 2H), 6.79 (d, J = 8.1 Hz, 1H), 5.74 (dd, J = 9.6, 1.6 Hz, 1H), 5.57–5.55 (m, 1H), 2.28 (s, 3H), 0.18 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 150.2, 131.0, 129.9, 127.2, 124.7, 122.2, 121.1, 116.1, 102.1, 90.8, 65.1, 20.5, −0.21; FTIR (NaCl, thin film) 2966, 2896, 2167, 1684, 1652, 1489, 1250, 1206, 1026 cm−1; HRMS (EI+) [M]+ calculated for C15H18OSi: 242.1127, found: 242.1148.
Supplementary Material
Acknowledgments
Acknowledgement is gratefully made to the National Science Foundation (CAREER CHE 1151364) and the University of Delaware Research Fund for support of this research. NMR and other data were acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF CHE 0421224, CHE 1229234, and CHE 0840401; NIH P20 GM103541 and S10 RR02682).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Spectra of new compounds, X-ray crystal structures of 8ea, 8eo, and [(S,S)-BnBox]CuI. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For examples of bioactive benzopyrans, see: Albrecht U, Lalk M, Langer P. Bioorg Med Chem. 2005;13:1531. doi: 10.1016/j.bmc.2004.12.031.TenBrink RE, Bergh CL, Duncan JN, Harris DW, Huff RM, Lahti RA, Lawson CF, Lutzke BS, Martin IJ, Rees SA, Schlachter SK, Sih JC, Smith MW. J Med Chem. 1996;39:2435. doi: 10.1021/jm960084f.Xu J, Kjer J, Sendker J, Wray V, Guan H, Edrada R, Muller WEG, Bayer M, Lin W, Wu J, Proksch P. Bioorg Med Chem. 2009;17:7362. doi: 10.1016/j.bmc.2009.08.031.Bauer DJ, Selway JWT, Batchelor JF, Tisdale M, Caldwell IC, Young DAB. Nature. 1981;292:369. doi: 10.1038/292369a0.Lu ZY, Lin ZJ, Wang WL, Du L, Zhu TJ, Fang YC, Gu QQ, Zhu WM. J Nat Prod. 2008;71:543. doi: 10.1021/np0704708.Sawadjoon S, Kittakoop P, Kirtikara K, Vichai V, Tanticharoen M, Thebtaranonth Y. J Org Chem. 2002;67:5470. doi: 10.1021/jo020045d.Gauthier S, Caron B, Cloutier J, Dory YL, Favre A, Larouche D, Mailhot J, Ouellet C, Schwerdtfeger A, Leblanc G, Martel C, Simard J, Merand Y, Belanger A, Labrie C, Labrie F. J Med Chem. 1997;40:2117. doi: 10.1021/jm970095o.
- 2.For routes towards racemic α-substituted benzopyrans, see: Correia CA, Li CJ. Heterocycles. 2010;82:555.Zhang Y, Li CJ. J Am Chem Soc. 2006;128:4242. doi: 10.1021/ja060050p.Hayashi M, Inubushi A, Mukaiyama T. Bull Chem Soc Jpn. 1988;61:4037.Mitchell TA, Bode JW. J Am Chem Soc. 2009;131:18057. doi: 10.1021/ja906514s.Vieira AS, Fiorante PF, Hough TLS, Ferreira FP, Ludtke DS, Stefani HA. Org Lett. 2008;10:5215. doi: 10.1021/ol8022177.Vo CVT, Mitchell TA, Bode JW. J Am Chem Soc. 2011;133:14082. doi: 10.1021/ja205174c.Chen W, Xie Z, Zheng H, Lou H, Liu L. Org Lett. 2014;16:5988. doi: 10.1021/ol503004a.Yu Y, Yang W, Rominger F, Hashmi ASK. Angew Chem Int Ed. 2013;52:7586. doi: 10.1002/anie.201302402.
- 3.For enantioselective additions to acyclic oxocarbenium ions (or acetals), see: Evans D, Thomson R. J Am Chem Soc. 2005;127:10506. doi: 10.1021/ja053386s.Corić I, Vellalath S, List B. J Am Chem Soc. 2010;132:8536. doi: 10.1021/ja102753d.Corić I, List B. Nature. 2012;483:315. doi: 10.1038/nature10932.Corić I, Müller S, List B. J Am Chem Soc. 2010;132:17370. doi: 10.1021/ja108642s.Umebayashi N, Hamashima Y, Hashizume D, Sodeoka M. Angew Chem Int Ed. 2008;47:4196. doi: 10.1002/anie.200705344.Terada M, Tanaka H, Sorimachi K. J Am Chem Soc. 2009;131:3430. doi: 10.1021/ja8090643.Kobayashi S, Arai K, Yamakawa T, Chen YJ, Salter MM, Yamashita Y. Adv Synth Catal. 2011;353:1927.
- 4.For an alternative enantioselective strategies towards α-chiral oxygen heterocycles, see: Hoveyda A, Hird A, Kacprzynski M. Chem Commun. 2004:1779. doi: 10.1039/b401123f.Ravindra B, Das BG, Ghorai P. Org Lett. 2014;16:5580. doi: 10.1021/ol502614n.Biddle M, Lin M, Scheidt K. J Am Chem Soc. 2007;129:3830. doi: 10.1021/ja070394v.Chen J, Chen J, Lang F, Zhang X, Cun L, Zhu J, Deng J, Liao J. J Am Chem Soc. 2010;132:4552. doi: 10.1021/ja1005477.Dittmer C, Raabe G, Hintermann L. Eur J Org Chem. 2007;35:5886.Korenaga T, Hayashi K, Akaki Y, Maenishi R, Sakai T. Org Lett. 2011;13:2022. doi: 10.1021/ol2004148.Trost B, Toste F. J Am Chem Soc. 1998;120:9074.Uozumi Y, Kato K, Hayashi T. J Am Chem Soc. 1997;119:5063.Wang HF, Cui HF, Chai Z, Li P, Zheng CW, Yang YQ, Zhao G. Chem Eur J. 2009;15:13299. doi: 10.1002/chem.200902303.Wang L, Liu X, Dong Z, Fu X, Feng X. Angew Chem Int Ed. 2008;47:8670. doi: 10.1002/anie.200803326.Zhang H, Lin S, Jacobsen EN. J Am Chem Soc. 2014;136:16485. doi: 10.1021/ja510113s.
- 5.Braun M, Kotter W. Angew Chem Int Ed. 2004;43:514. doi: 10.1002/anie.200352128. [DOI] [PubMed] [Google Scholar]
- 6.Reisman SE, Doyle AG, Jacobsen EN. J Am Chem Soc. 2008;130:7198. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Witten MR, Jacobsen EN. Angew Chem Int Ed. 2014;53:5912. doi: 10.1002/anie.201402834. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burns NZ, Witten MR, Jacobsen EN. J Am Chem Soc. 2011;133:14578. doi: 10.1021/ja206997e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Terada M, Li F, Toda Y. Angew Chem Int Ed. 2014;53:235. doi: 10.1002/anie.201307371. [DOI] [PubMed] [Google Scholar]; (b) Terada M, Yamanaka T, Toda Y. Chem Eur J. 2013;19:13658. doi: 10.1002/chem.201302486. [DOI] [PubMed] [Google Scholar]; (c) Cui Y, Villafane LA, Clausen DJ, Floreancig PE. Tetrahedron. 2013;69:7618. doi: 10.1016/j.tet.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moquist PN, Kodama T, Schaus SE. Angew Chem Int Ed. 2010;49:7096. doi: 10.1002/anie.201003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Hsiao CC, Liao HH, Sugiono E, Atodiresei I, Rueping M. Chem Eur J. 2013;19:9775. doi: 10.1002/chem.201300766. [DOI] [PubMed] [Google Scholar]; (b) Rueping M, Volla CMR, Atodiresei I. Org Lett. 2012;14:4642. doi: 10.1021/ol302084q. [DOI] [PubMed] [Google Scholar]; (c) Meng Z, Sun S, Yuan H, Lou H, Liu L. Angew Chem Int Ed. 2014;53:543. doi: 10.1002/anie.201308701. [DOI] [PubMed] [Google Scholar]; (d) Benfatti F, Benedetto E, Cozzi PG. Chem Asian J. 2010;5:2047. doi: 10.1002/asia.201000160. [DOI] [PubMed] [Google Scholar]
- 11.(a) Anand NK, Carreira EM. J Am Chem Soc. 2001;123:9687. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]; (b) Asano Y, Hara K, Ito H, Sawamura M. Org Lett. 2007;9:3901. doi: 10.1021/ol701534n. [DOI] [PubMed] [Google Scholar]; (c) Asano Y, Ito H, Hara K, Sawamura M. Organometallics. 2008;27:5984. [Google Scholar]; (d) Boyall D, Frantz DE, Carreira EM. Org Lett. 2002;4:2605. doi: 10.1021/ol026282k. [DOI] [PubMed] [Google Scholar]; (e) Frantz DE, Fässler R, Carreira EM. J Am Chem Soc. 2000;122:1806. [Google Scholar]; (f) Ito JI, Asai R, Nishiyama H. Org Lett. 2010;12:3860. doi: 10.1021/ol1015338. [DOI] [PubMed] [Google Scholar]; (g) Koyuncu H, Dogan Ö. Org Lett. 2007;9:3477. doi: 10.1021/ol701535y. [DOI] [PubMed] [Google Scholar]; (h) Takita R, Yakura K, Ohshima T, Shibasaki M. J Am Chem Soc. 2005;127:13760. doi: 10.1021/ja053946n. [DOI] [PubMed] [Google Scholar]; (i) Trost BM, Chan VS, Yamamoto D. J Am Chem Soc. 2010;132:5186. doi: 10.1021/ja910656b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Yang F, Xi P, Yang L, Lan J, Xie R, You J. J Org Chem. 2007;72:5457. doi: 10.1021/jo0707535. [DOI] [PubMed] [Google Scholar]
- 12.(a) Chen C, Hong L, Xu ZQ, Liu L, Wang R. Org Lett. 2006;8:2277. doi: 10.1021/ol060526+. [DOI] [PubMed] [Google Scholar]; (b) Zhou S, Chen CR, Gau HM. Org Lett. 2010;12:48. doi: 10.1021/ol902454n. [DOI] [PubMed] [Google Scholar]
- 13.(a) Ahamed M, Todd MH. Eur J Org Chem. 2010;31:5935. [Google Scholar]; (b) Aschwanden P, Stephenson CRJ, Carreira EM. Org Lett. 2012;8:2437. doi: 10.1021/ol060876w. [DOI] [PubMed] [Google Scholar]; (c) Black DA, Beveridge RE, Arndtsen BA. J Org Chem. 2008;73:1906. doi: 10.1021/jo702293h. [DOI] [PubMed] [Google Scholar]; (d) Muncipinto G, Moquist P, Schreiber S, Schaus S. Angew Chem Int Ed. 2011;50:8172. doi: 10.1002/anie.201103271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Taylor AM, Schreiber SL. Org Lett. 2006;8:143. doi: 10.1021/ol0526165. [DOI] [PubMed] [Google Scholar]
- 14.For our initial work on isochroman acetals, see: Maity P, Srinivas HD, Watson MP. J Am Chem Soc. 2011;133:17142. doi: 10.1021/ja207585p.
- 15.Downey C, Mahoney B, Lipari V. J Org Chem. 2009;74:2904. doi: 10.1021/jo900102w. [DOI] [PubMed] [Google Scholar]
- 16.(a) Maloney DJ, Deng JZ, Starck SR, Gao Z, Hecht SM. J Am Chem Soc. 2005;127:4140. doi: 10.1021/ja042727j. [DOI] [PubMed] [Google Scholar]; (b) Deng JZ, Starck SR, Li S, Hecht SM. J Nat Prod. 2005;68:1625. doi: 10.1021/np058064g. [DOI] [PubMed] [Google Scholar]; (c) Foo LY, Porter LJ. J Chem Soc, Perkin Trans. 1983;1:1535. [Google Scholar]; (d) Williamson G, Manach C. Am J Clin Nutr. 2005;81:243S. doi: 10.1093/ajcn/81.1.243S. [DOI] [PubMed] [Google Scholar]
- 17.CCDC-973165 (8ea), CCDC-943166 (8eo), and CCDC-973164 ([(S,S)-BnBox]CuI) contain the supplementary crystallographic data for this paper These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif
- 18.π-Complexation of the oxocarbenium ion to Cu may proceed C–C bond formation. For examples of Cu-arene and Cu-olefin complexes, see: Wang X-S, Zhao H, Li Y-H, Xiong R-G, You X-Z. Top Catal. 2005;35:43.Badiei YM, Warren TH, Chiang KP, Holland PL. Inorg Synth. 2010;35:50.Pérez-Galán P, Delpont N, Herrero-Gómez E, Maseras F, Echavarren AM. Chem Eur J. 2010;16:5324. doi: 10.1002/chem.200903507.
- 19.(a) Hansch C, Leo A, Taft R. Chem Rev. 1991;91:165. [Google Scholar]; (b) Ritchie CD, Sager WF. Prog Phys Org Chem. 1964;2:323. [Google Scholar]
- 20.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 21.Matt PVL-JGC, Minidis ABE, Pfaltz A, Macko L, Neudurger M, Zehnder M, Rüegger H, Pregosin PS. Helv Chim Acta. 1995;78:265. [Google Scholar]
- 22.Shimada TMK, Shinohara A, Han JW, Hayashi T. J Am Chem Soc. 2002;124:1584. doi: 10.1021/ja017617g. [DOI] [PubMed] [Google Scholar]
- 23.Hirsch KAWSR, Moore JS. J Am Chem Soc. 1997;119:10401. [Google Scholar]
- 24.Barett AGMHBT, Love AC, Tedeschi L. Org Lett. 2004;6:835. doi: 10.1021/ol049915z. [DOI] [PubMed] [Google Scholar]
- 25.Cañeque TCAM, Alvarez-Builla J, Pérez-Moreno J, Clays K, Marcelo G, Mendicuti F, Castaño O, Andrés JL, Vaquero JJ. Eur J Org Chem. 2010;33:6323. [Google Scholar]
- 26.Kempson JGJ, Das J, Moquin RV, Spergel SH, Watterson SH, Langevine CM, Dyckman AJ, Pattoli M, Burke JR, Yang X, Gillooly KM, McIntyre KW, Chen L, Dodd JH, Mckinnon M, Barrish JC, Pitts WJ. Bioorg Med Chem Lett. 2009;19:2646. doi: 10.1016/j.bmcl.2009.03.159. [DOI] [PubMed] [Google Scholar]
- 27.Schaate ARP, Preuße T, Lohmeier SJ, Godt A, Behrens P. Chem Eur J. 2011;17:9320. doi: 10.1002/chem.201101015. [DOI] [PubMed] [Google Scholar]
- 28.Montalbetti CSM, Bonnefis F, Genêt JP. Tetrahedron Lett. 1995;36:5891. [Google Scholar]
- 29.Zubia ELFR, Guillermo MM, Collado IG. Tetrahedron. 1992;48:4239. [Google Scholar]
- 30.Tang Z, Hu Q. Adv Synth Catal. 2004;346:1635. [Google Scholar]
- 31.Yamamoto Y, Kirai N. Org Lett. 2008;10:5513. doi: 10.1021/ol802239n. [DOI] [PubMed] [Google Scholar]
- 32.Li YQZ, Wang H, Fu X, Duan C. J Org Chem. 2012;77:2053. doi: 10.1021/jo202577m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.