

Significance
Epstein–Barr virus is a human herpesvirus associated with several types of malignancy. We show that spironolactone, a drug used to block mineralocorticoid activity, also has anti-EBV activity and that it acts by inhibiting the function of an essential EBV protein SM, which preferentially increases expression of specific EBV genes. We also show that the mineralocorticoid blocking activity is not the basis for spironolactone’s antiviral activity. Because the SM protein acts at steps of the viral life cycle distinct from those targeted by currently available therapies, this study paves the way for development of novel anti-EBV drugs to address emerging problems of drug resistance and toxicity.
Keywords: Epstein–Barr virus, spironolactone, posttranscriptional regulation, herpesvirus, antiviral
Abstract
Clinically available drugs active against Epstein–Barr virus (EBV) and other human herpesviruses are limited to those targeting viral DNA replication. To identify compounds directed against other steps in the viral life cycle, we searched for drugs active against the EBV SM protein, which is essential for infectious virus production. SM has a highly gene-specific mode of action and preferentially enhances expression of several late lytic cycle EBV genes. Here we demonstrate that spironolactone, a mineralocorticoid receptor antagonist approved for clinical use, inhibits SM function and infectious EBV production. Expression of EBV viral capsid antigen is highly SM dependent, and spironolactone inhibits viral capsid antigen synthesis and capsid formation, blocking EBV virion production at a step subsequent to viral DNA replication. In addition, spironolactone inhibits expression of other SM-dependent genes necessary for infectious virion formation. We further demonstrate that molecules structurally related to spironolactone with similar antimineralocorticoid blocking activity do not inhibit EBV production. These findings pave the way for development of antiherpesvirus drugs with new mechanisms of action directed against SM and homologous essential proteins in other herpesviruses.
Epstein–Barr virus (EBV) is a human lymphotropic herpesvirus associated with several types of lymphoid and epithelial malignancies and lymphoproliferative syndromes (for a review, see ref. 1). EBV infects the majority of humans worldwide and establishes a persistent, lifelong latent infection in memory B cells. Occasional reactivation from latency occurs, and EBV enters a lytic phase of replication, during which a tightly regulated series of lytic genes is expressed, culminating in lysis of the host cell and release of infectious viral progeny. All herpesviruses share these abilities to establish asymptomatic latent infection and reactivate intermittently.
All herpesviruses also express a regulatory protein early in lytic replication that is essential for efficient gene expression and infectious virion production. The EBV member of this family, which includes herpes simplex virus ICP27, cytomegalovirus (CMV) UL69, and Kaposi's sarcoma-associated herpesvirus (KSHV) ORF57, is known as SM (2). SM acts posttranscriptionally to enhance accumulation of many lytic EBV transcripts. SM activity is highly gene-specific and preferentially enhances expression of several late lytic transcripts, including those encoding the major viral capsid antigen (VCA) and gp350, which is required for infection of B lymphocytes (3–5). SM and its homologs in other herpesviruses may also enhance transcription and translation of specific viral genes (6).
Currently, all available antiherpesvirus drugs target viral DNA polymerases and are usually highly effective, but toxicity and development of resistance limits their use (7). To discover potential novel therapeutic compounds, we applied a cell-based screening assay to identify compounds that would specifically target SM function (8). Using this assay, spironolactone (SPR), a drug in clinical use for over 50 y, was identified as inhibiting SM function and EBV virion production. SPR is an aldosterone antagonist that binds to the mineralocorticoid receptor, but comparison with derivatives of SPR demonstrated that mineralocorticoid receptor blocking activity was not the basis of its antiviral activity. Rather, the profile of SPR’s effects on EBV gene expression paralleled that of genetically inactivating SM. SM and its counterparts in other herpesviruses have no cellular homologs and act at a stage of herpesvirus replication distinct from DNA replication (9). Thus, compounds based on SPR have the potential to yield antiviral drugs with a new mechanism of action and limited toxicity.
Results
SPR Inhibits EBV SM Function.
We previously described a screening assay to identify compounds that can inhibit the function of EBV SM protein, which is essential for lytic EBV replication (3, 8). The assay is based on the finding that mRNAs dependent on EBV SM protein for efficient expression confer SM dependence on heterologous genes to which they are fused. The assay uses a reporter cell line stably expressing the SM-responsive KSHV ORF59 gene fused in frame to GFP and in which the fusion product is not efficiently expressed. These also contain a doxycycline-inducible SM gene, and when SM protein is produced, the ORF59-GFP fusion mRNA is expressed and the cells exhibit strong green fluorescence (8). We screened a library of compounds of known activity and structure (10) for their ability to inhibit SM function in this assay and identified SPR as a potential inhibitor of SM function. Treatment of the reporter cells with 10 μM SPR led to inhibition of fluorescence accompanied by almost complete absence of ORF59-GFP RNA and protein accumulation (Fig. 1 A–C). We next asked whether SPR could be affecting SM localization by performing immunofluorescence microscopy for SM in the presence or absence of SPR. As seen in Fig. 1D, SPR did not affect nuclear localization of SM. The specificity of SPR activity was tested by determining the effect of SPR on an analogous construct expressing a CMV UL44-GFP fusion construct that is not SM-dependent or -responsive. SPR did not affect UL44-GFP expression, indicating that its effect is SM-specific (Fig. 1E).
Fig. 1.

Identification of SPR as a drug that inhibits EBV SM function. (A) HEK293 cells were stably transfected with a KSHV ORF59-GFP reporter plasmid and transduced with a lentivirus expressing inducible EBV SM protein. Fluorescence microscopy was performed on reporter cells in the presence or absence of SPR. Cells expressed GFP protein only when SM was expressed (+SM) but were GFP-negative when treated with 10 μM SPR (S). (B) SPR inhibits SM-dependent ORF59-GFP RNA accumulation. The expression of ORF59-GFP fusion RNA was measured by qRT-PCR 24 h after 10 μM SPR treatment. (C) SPR inhibits ORF59-GFP protein accumulation. Immunoblotting with anti-GFP or actin antibody was performed on lysates from cells treated as shown. (D) The reporter cell line was induced to express SM and treated with either vehicle (–S) or treated with SPR (+S). Cells were fixed 30 h after induction and stained with either DAPI (blue) or anti-SM antibodies (red). (E) 293 cells were transfected with CMV UL44-GFP and empty vector (–) or SM (+) and treated with either vehicle or 10 μM SPR as shown.
Antiviral Activity of SPR Is Not Due to Mineralocorticoid Receptor Blocking Activity.
SPR acts to block aldosterone activity by binding to the mineralocorticoid receptor and acting as a receptor antagonist (11). To ask whether SM inhibition by SPR was related to its mineralocorticoid blocking activity, we compared its effect on SM with that of eplerenone (EPR), a chemically similar congener also used clinically as an aldosterone antagonist (12). As shown in Fig. 2, SPR displayed a dose-dependent SM inhibitory effect in the SM functional assay, whereas EPR had no effect. These results therefore indicate that mineralocorticoid receptor blockade is not sufficient for SM inhibition and that it is not likely to be related to the effect of SPR on SM activity.
Fig. 2.

Inhibitory effect of SPR on SM activation of gene expression is independent of mineralocorticoid receptor antagonist activity. The ORF59-GFP reporter cell line was either mock-treated (–) or treated with dox (+) to induce SM expression, which is required for GFP expression. The induced cells were also treated with either DMSO vehicle (0) or various concentrations of SPR (S) or the mineralocorticoid receptor antagonist EPR (E), and cells were examined for GFP expression by fluorescence microscopy 24 h after treatment. Drug concentrations are shown below the panel.
SPR Exhibits Potent Structure-Dependent Antiviral Activity Against EBV.
To determine whether SPR inhibition of SM activity translated into antiviral activity, we measured its effect on infectious EBV production. An epithelial gastric carcinoma cell line (AGS) infected with a GFP-expressing EBV (13) was used to test the effect of SPR and SPR-related compounds on EBV production. AGS cells were induced to permit lytic EBV replication and treated with either vehicle (DMSO) or SPR. Release of infectious EBV was then measured by incubating the supernatant from the induced AGS cells with uninfected 293 cells. EBV-infected cells express GFP, allowing them to be accurately counted by flow cytometry as previously described (5, 14). SPR led to a dose-related reduction in infectious EBV titer in the supernatant (Fig. 3A). The IC50 of SPR (2.1 μM) was similar to that of acyclovir (3.4 μM) in this assay (Fig. S1). The antiviral effect of EPR and two metabolites of SPR that also have mineralocorticoid antagonist activity (11), canrenone (CAN) and 7α-thiomethyl SPR (TMS), was compared with that of SPR. Interestingly, CAN displayed antiviral activity, albeit less than that of SPR, whereas TMS and EPR did not (Fig. 3B). Comparison of their structures reveals that these molecules differ only at the modifications to the number 7 carbon of the parent molecule (Fig. 3C). The residues at this position therefore appear to be critical for anti-SM– and EBV-directed antiviral activity but not for mineralocorticoid activity. All four compounds were also tested in the SM reporter assay, and SPR was highly active, with CAN displaying slight activity and EPR and TMS having no detectable activity (Fig. S2). To confirm the aldosterone blocking activity of the four compounds, a cell-based reporter assay for aldosterone function was performed and verified that all four compounds, although varying in potency, had maximal activity at concentrations of 10 μM and above (Fig. S3).
Fig. 3.

Antiviral activity of SPR in epithelial cells. (A) EBV lytic replication was induced with TPA (+ ind.) in an GFP-EBV–infected gastric carcinoma cell line (AGS), which was then treated with SPR or EPR at various concentrations as shown. Virion production was measured by passage to 293 cells and quantitated by flow cytometry. EBV titer expressed as the number of GFP-transducing units per mL is shown on the y axis. (B) Antiviral activities of CAN, TMS, and EPR (E) were compared with SPR as in A. Cells were either untreated (–) or treated (+) to induce lytic replication (ind). (C) Structures of SPR, CAN, TMS, and EPR are shown. The thioacetyl group at C-7 of SPR is circled.
Fig. S1.

Comparison of SPR and acyclovir antiviral activity in AGS cells. AGS cells were induced to produce infectious GFP-positive EBV and used to infect 293 cells. SPR or acyclovir was added to the culture medium at concentrations shown at the time of infection. Filtered cell supernatants were collected at 5 d postinduction and were used to infect 293T cells, and the numbers of green fluorescent-positive cells representing infectious viral particles were quantitated by flow cytometry. The 50% inhibitory concentrations (IC50) for SPR and ACV were calculated using GraphPad PRISM version 6.0 software.
Fig. S2.

Comparison of SPR and congeners in SM-inhibition reporter assay. The ORF59-GFP reporter cell line was either mock treated (–) or treated with dox (+) to induce SM expression, which is required for GFP expression. The induced cells were also treated with either DMSO vehicle (0) or various concentrations of SPR (S) or the mineralocorticoid receptor antagonists CAN, TMS, or EPR, and cells were examined for GFP expression by fluorescence microscopy 24 h after treatment. Phase contrast images for each treatment are shown above each panel, and drug concentrations are shown below.
Fig. S3.

Mineralocorticoid antagonist assay of SPR and congeners. Aldosterone blocking assay was performed by addition of aldosterone and serial dilutions of each antagonist (SPR, EPR, CAN, and TMS) to cells carrying a mineralocorticoid receptor-driven luciferase gene. Luciferase activity was measured 24 h after drug treatment and expressed as relative light units (RLUs). The concentration of each drug is shown on the x axis.
SPR Inhibits Virus Production at a Stage Subsequent to EBV DNA Replication but Before Virus Release.
To investigate the mechanism of SPR’s antiviral action, we measured the effect of SPR on EBV DNA replication and early (E) and immediate-early (IE) gene expression, which occur before DNA replication (1). EBV-infected AGS cells were induced to replicate and were treated with SPR or vehicle. DNA replication was measured by qPCR of DNA harvested from induced cells, and EBV protein expression was assessed by Western blotting of cell lysates (Fig. 4 A and C). Despite SPR having the expected inhibitory effect on infectious EBV production, SPR did not affect the increase in intracellular EBV copy number that occurs with lytic replication (Fig. 4 A and B). SPR also had no effect on IE or E gene expression, including SM expression (Fig. 4C). Many E genes are required for EBV lytic DNA replication, whereas EBV late gene expression is highly dependent on EBV DNA replication (1). Preservation of IE and E gene expression and DNA replication in the presence of SPR therefore indicated that SPR was inhibiting the expression of one or more late genes required for infectious EBV production. To determine whether defective, noninfectious EBV particles were being released after SPR treatment, we measured the effect of SPR on release of extracellular EBV DNA by qPCR. As shown in Fig. 4D, SPR, but not EPR, prevented release of EBV DNA into the supernatant. To determine whether SPR had similar effects in EBV-infected B lymphocytes, we measured E and IE lytic protein expression in EBV-positive P3HR1-ZHT lymphoma cells treated with SPR (Fig. S4). SPR did not affect E or IE gene expression in lymphoma cells. However, as in epithelial cells, SPR inhibited release of extracellular EBV DNA into the supernatant (Fig. S4A). These data therefore indicate that because EBV DNA is replicated normally in the presence of SPR, subsequent stages of viral packaging, assembly, or egress may be inhibited by SPR.
Fig. 4.

SPR inhibits virus production at a lytic replicative stage after DNA replication. (A) Antiviral effect of SPR is not due to inhibition of viral DNA replication. EBV-positive AGS gastric carcinoma cells were transduced with a lentivirus expressing EBV transactivator ZTA protein under control of a doxycycline-inducible promoter to generate a cell line in which EBV replicates robustly when treated with doxycycline (AGSiZ). The AGSiZ cell line was either mock treated (–) or treated (+) with doxycycline to induce lytic DNA replication (ind.) and also treated with SPR or EPR at concentrations shown. DNA copy number was measured by qPCR of cell pellets 70 h postinduction. RQ, relative quantity of intracellular viral DNA. (B) SPR but not EPR specifically inhibits infectious virus production. Serial dilutions of virus-containing supernatant were used to infect 293 cells, and the number of GFP-positive cells representing infectious viral particles was measured by flow cytometry. (C) SPR does not affect expression of IE or E EBV proteins. Protein cell lysates from cells treated with SPR (S) or EPR (E) were immunoblotted using anti-SM, EA-D, RTA, ZTA, or actin antibodies at 24 and 48 h after induction of replication. (D) SPR but not EPR inhibits production of extracellular EBV. Extracellular EBV DNA was measured by DNA qPCR in cell supernatants harvested 5 d after SPR (S) or EPR (E) treatment. RQ, relative quantity of DNA.
Fig. S4.

Antiviral activity of SPR in lymphoma cells. P3HR1-ZHT cells were treated with tamoxifen (Tam) to induce EBV lytic replication (ind.) and also treated with various concentrations of SPR (S) as shown. (A) SPR inhibits EBV extracellular virus production in a dose-dependent manner. Cell supernatants were harvested 5 d after induction of replication, and extracelluar EBV DNA was measured by qPCR. RQ of DNA (RQ) is shown on the y axis. (B) SPR does not affect IE or E EBV protein expression. Protein cell lysates from cells treated with SPR were immunoblotted using anti-EAD, anti-SM, and anti-ZTA antibodies. Z-HT is the ZTA-hydroxytamoxifen receptor fusion protein made from the transduced gene in P3RH1-ZHT cells.
SPR Prevents EBV Capsid Formation.
Because SPR prevented infectious virus production despite having no effect on DNA replication, we examined the effect of SPR on virion assembly and egress. Electron microscopy was performed on EBV-infected AGS cells after induction of EBV replication and SPR treatment to examine the potential effect of SPR on virion formation. As shown in Fig. 5, SPR completely abolished EBV capsid formation. Whereas intranuclear EBV capsids at various stages of formation were readily detectable in EBV-infected AGS cells (32% of induced cells), no capsids were found in SPR-treated cells (0%). Viral DNA replication was again unaffected by drug treatment (Fig. S5). These data therefore suggested that SPR inhibits expression of one or more proteins required for EBV capsid formation but did not rule out additional effects on other late genes.
Fig. 5.

SPR inhibits EBV capsid formation. AGSiZ cells were induced to permit EBV replication with doxycycline and treated with 10 μM SPR (+S) or vehicle alone (–S). The cells were harvested, fixed at 4 d postinduction, and analyzed by electron microscopy. Viral particles were seen in vehicle-treated cells (–S) as shown in magnifications of 1,500× and 5,000× . No viral particles were detectable in the SPR-treated samples (+S).
Fig. S5.
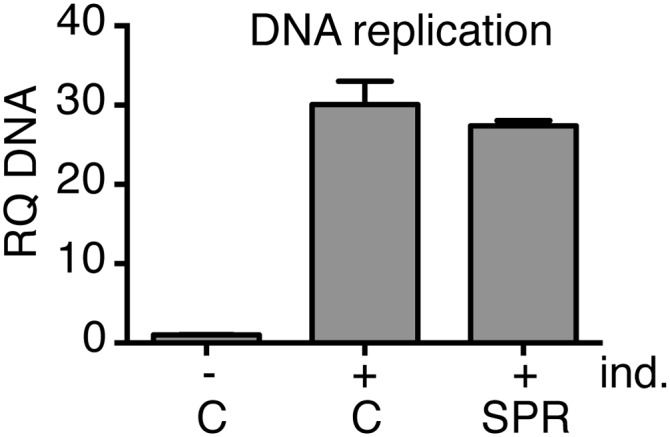
Lack of SPR effect on EBV DNA replication. Aliquots of AGSiZ cells used for electron microscopy were centrifuged, and DNA was prepared from cell pellets, followed by qPCR to measure EBV DNA copy number. Cells were treated with dox to permit lytic EBV replication (+ind.) and treated with either vehicle (C) or 10 μM SPR (SPR) as shown. Experiments were performed in triplicate. RQ, relative quantity of DNA.
SPR Inhibits Expression of an Essential SM-Dependent Late Lytic Capsid Gene.
We have recently shown that SM preferentially enhances expression of several late lytic genes but is not required for efficient expression of IE or E genes (9). To investigate the effect of SPR on EBV late gene expression, we compared the effect of SPR on EBV protein expression from several EBV genes of different temporal classes, by immunofluorescence microscopy. As expected, SPR had no effect on expression of IE gene Z or E genes EA or SM (Fig. 6). However, SPR did strongly inhibit expression of VCA, which is highly dependent on SM for efficient expression (4, 9). Although the percentage of BMRF1 (EA-D)-, ZTA-, and SM-expressing cells was unaffected or actually increased slightly with SPR treatment, the percentage of VCA-positive cells decreased by 73% (P < 0.0001; Fig. 6B). To confirm these results and examine SPR effects on additional SM-dependent genes, qPCR for several EBV lytic genes was performed using RNA from SPR-treated AGS cells. As shown in Fig. S6A, the effect of SPR on late lytic genes was gene-specific, inhibiting expression of SM-dependent mRNAs, such as BDLF1, BLLF1 (gp350), and BcLF1 (VCA), but having little effect on BFRF3 or BALF4, which are not SM-dependent (9). The effect of SPR on these genes in EBV-infected lymphoma cells was similar to that observed in AGS epithelial cells (Fig. S6B). We also compared the effect of SPR on SM-responsive and SM-unresponsive transcripts in cotransfection assays (Fig. S7) and demonstrated that SPR inhibits only SM-responsive gene expression.
Fig. 6.

SPR inhibits EBV capsid protein expression. AGSiZ cells were either mock-treated (–D), treated with doxycycline (+D) to induce replication, or induced and also treated with SPR (+D+S) for 48 h. The cells were fixed and stained with BMRF1, SM, ZTA, and VCA antibodies and analyzed by immunofluorescence microscopy. DAPI-stained nuclei (blue), GFP-positive EBV-infected cells (green), and specific antigen expression (red) are shown. (A) Representative images for expression of each protein are shown. (B) Percentage of cells positive for each protein is shown.
Fig. S6.

Gene-specific effects of SPR on EBV RNA expression during lytic replication. (A) AGSiZ cells were either mock-treated (–D), treated with doxycycline (+D) to induce replication, or induced and also treated with SPR (+D+S). RNA was harvested at 48 h postinduction and drug treatment. qPCR was performed to quantitate the effect of SPR on SM-dependent RNAs (BDLF1, BcLF1, BLLF1, and BILF2) or SM-independent RNAs (BMRF1, SM, BFRF3, BALF4). RQ, relative quantity of RNA. (B) P3HR1 cells were either mock-treated (–T), treated with tamoxifen (+T) to induce replication, or induced and also treated with SPR (+T+S), and RNAs were measured as in A.
Fig. S7.

SPR effects on SM function in cotransfection assays. 293 cells were transfected with SM-responsive EBV genes (BILF2, BDLF1) or SM-independent genes (EBER2, ZTA) cloned in an expression vector with either empty vector (C) or SM. SM-transfected cells were treated with either vehicle (SM) or SPR (SM +S). RNA was harvested 28 h posttransfection, and gene expression was measured by RNA qPCR. RQ, relative quantity of RNA.
Concordance Between the Effects of SPR Treatment and SM Deletion on the EBV Transcriptome.
We have comprehensively characterized the specificity of SM effects on the EBV transcriptome by comparing the transcriptome of SMKO EBV-infected cells with that of cells infected with wt EBV and SM-rescued SMKO EBV (9). The effect of SM is highly gene-dependent and primarily enhances expression of several essential late genes, including VCA and gp350 (4, 5, 9). It was therefore possible to conduct a similar global comparison between the effects of SPR on WT EBV-infected cells and the effect of genetically knocking out SM function. We treated AGS (epithelial cells) and P3HR1-ZHT (lymphoma cells) with SPR or vehicle and harvested RNA from the infected cells 48 h after induction of lytic EBV replication. The gene expression patterns in the presence and absence of SPR were then determined by high-throughput sequencing and transcriptomic analysis in which read counts for each EBV gene were determined. The results were then computed as binary ratios between gene expression in the absence or presence of SPR and gene expression in the presence or absence of SM. As shown in Fig. 7, many of the genes highly inhibited by SPR in both cell types were also highly SM-dependent. To examine the overall correlation between the effects of SM deletion and SPR, an unsupervised clustering analysis of the differential expression data was performed. Although the effect of SPR was not identical in each cell line, there was a high degree of concordance between the effect of SPR and the effect of inactivating SM (Fig. S8), with a Pearson’s correlation coefficient of 0.68 and 0.63 between the effect of SM knockout and SPR treatment in P3HR1 and AGS cells, respectively. These data therefore indicate that a major aspect of the antiviral effect of SPR is mediated by its inhibitory effect on SM and its target-specific mode of action.
Fig. 7.

Comparison of the effect of SPR treatment to SM knockout on EBV gene expression in epithelial and lymphoid cells. (A) SMKO EBV-infected 293 cells were transfected with ZTA to induce replication or transfected with both ZTA and SM to rescue SM-dependent gene expression. RNA was prepared from cells 48 h postreplication induction. High-throughput RNA sequencing was performed to determine the effects of SPR or SM knockout on the lytic EBV transcriptome. Differential expression of SM-dependent genes is shown as the ratio of RNA expression in SMKO EBV transfected with Z+SM versus SMKO Z alone. Genes whose expression is most highly dependent on SM are marked in blue. (B) Epithelial AGSiZ cells were treated with doxycycline to induce replication. The differential expression of SPR-sensitive genes during EBV replication is shown as the ratio of RNA in vehicle-treated (–S) versus that in SPR-treated cells (+S). The most SPR-sensitive genes therefore have the highest ratios of RNAs. (C) Comparison of SPR-treated versus untreated EBV-infected P3HR1-ZHT lymphoma cells was performed as in B except that cells were treated with tamoxifen to induce lytic replication. +, values greater than limit of y axis; *, genes in the region that were deleted in B95.8 and P3HR1 genomes.
Fig. S8.

Clustering analysis of SM-dependent and SPR-sensitive genes. Differential expression of EBV lytic genes in the presence and absence of SM was compared with the effect of SPR in both epithelial (AGS) and B lymphocyte cells (P3). Differential expression between SM-expressing vs. SM KO cells, untreated (C) vs. SPR-treated AGS, and untreated (C) vs. SPR-treated P3HR1 cells, respectively, is shown as log2 fold changes. Red coloring indicates increased expression, and blue coloring indicates lower expression compared with SMKO or SPR-treated cells.
Discussion
In this study, we describe the ability of SPR to inhibit EBV SM protein function, decreasing accumulation of SM target RNAs and thereby preventing production of infectious EBV particles. This property of SPR is not based on its mineralocorticoid receptor blocking activity, as congeners with antialdosterone activity did not possess anti-SM or antiviral activity. The activity of SPR was structure-dependent, with specific substitutions at the 7 position of SPR altering anti-SM and anti-EBV activity. Consistent with its anti-SM activity, the effect of SPR on EBV gene expression was highly concordant with the effect of mutationally deleting SM from the EBV genome. The most highly SM-dependent and SPR-sensitive genes are late lytic genes, and most encode either tegument proteins or glycoproteins (4, 5, 9). In addition, two genes that are essential for encapsidation before tegument incorporation and virion envelopment, the major VCA and the minor capsid gene (BDLF1) product, are also highly SM-dependent (9). Inhibition of VCA expression thus represents the most proximate block in EBV production due to SPR, and SPR completely prevented capsid formation despite adequate EBV DNA replication.
The mechanism(s) of action of SM protein remains incompletely characterized. SM binds EBV RNA and affects RNA stability (15–19). Although it preferentially enhances accumulation of some EBV mRNAs, SM action likely depends on inherent characteristics of inefficiently expressed RNAs, such as stability or nuclear exportability (8, 9). Further, the possibility that SM exerts transcriptional effects on one or more of its target genes has not been excluded. ORF57, the KSHV homolog of SM, may also act transcriptionally as well as posttranscriptionally (6, 20). SPR may directly interact with SM or cellular partners of SM to block function. Even if SPR does not directly affect the ability of SM to interact with target RNAs, it may still act to interfere with formation of RNA-binding protein complexes recruited by SM. SPR could also act indirectly by inhibiting expression of cellular proteins required for SM function at the transcriptional level. SPR has been shown to exert inhibitory effects on several transcription factors in mononuclear cells independently of mineralocorticoid receptor antagonism (21). Establishing the exact mechanism(s) by which SPR inhibits SM function will therefore require further investigation of SM’s mechanism of action.
Substitutions at C-7 of SPR appear to be critical for antiviral activity. The fact that CAN and SPR retain activity but TMS does not suggests that the molecules may be acting as an electrophilic trap. The thioacetate at C-7 in SPR may serve as an acyl donor, or it may be eliminated to the γ,δ-unsaturated form, producing CAN in vivo. Thus, the target nucleophile would react to produce an acylated variant or undergo a Michael-addition to produce a CAN-alkylated adduct. The thiomethylether in TMS cannot transfer an acyl group, and elimination is less likely based on the pKa of the methanethiolate. Either rationale would explain the lack of activity with TMS.
Although SPR has been used clinically for decades as a diuretic and to treat heart failure, hyperaldosteronism, and edematous states, several side effects limit its use (22, 23). These include effects on reproductive and other metabolic functions. It should be noted that the IC50 of SPR (∼2.1 μM) for blocking infectious EBV production is lower than that of acyclovir (3.4 μM) in the same assay (Fig. S1). Although the steady-state levels of SPR achieved in vivo are likely to be lower than the concentrations used in vitro in our study (24), the metabolite CAN has a much longer half-life in vivo (24) and may potentiate the activity of the parent compound. Nevertheless, because the mineralocorticoid antagonist activity of SPR does not seem to be related to its anti-EBV activity, SPR provides an ideal lead compound for developing antiherpesvirus drugs without SPR’s known side effects. Resistance to currently available antiherpesvirus drugs such as acyclovir and ganciclovir is an emerging problem in the treatment of immunocompromised patients (25). Cross-resistance to other agents active against viral DNA polymerase, such as foscarnet and cidofovir, not infrequently complicates therapy (25). In addition, these agents have significant myelosuppressive or nephrotoxic toxicities. Drugs directed against other viral proteins such as SM therefore have the potential to significantly extend the antiviral armamentarium. Use of combination therapy with agents directed against different steps in the viral life cycle also has the potential to limit the emergence of drug resistance. Each human herpesvirus expresses an SM homolog that is essential for virus production (3, 26–28). Although they generally do not cross-complement each other in rescuing viral replication (3, 26, 29), they are structurally and functionally similar. SPR and its derivatives therefore have the potential to be active against other human herpesviruses. Further, the screening assay that we have used to specifically identify SM inhibitors can be easily adapted to screen for agents active against SM homologs from other herpesviruses (8).
In summary, we have shown that SPR specifically acts to inhibit EBV SM protein function, which is required for expression of several late lytic EBV genes and infectious EBV production. The effect of SPR parallels the effect of knocking out SM function, which is critical for expression of EBV VCA and therefore results in an inability of the virus to make viral capsids. In addition to preventing capsid formation, SPR inhibits expression of other SM-dependent EBV genes important for infectivity, including gp350, thereby inhibiting EBV at several steps in virion production. SPR may be used as the template for development of antiherpesvirus drugs that act at stages of herpesvirus replication distinct from that of all other currently available drugs.
Materials and Methods
Cell Lines.
HEK293 cells stably carrying an ORF59-GFP fusion gene that also inducibly expresses SM-dependent GFP were maintained as previously described (8). P3HR1-ZHT is an EBV-positive lymphoma cell line that contains the EBV BZLF1 gene fused to the hormone-binding domain of the 4-hydroxytamoxifen (4HT) receptor that allows robust induction of EBV replication with 4HT treatment (30). The gastric carcinoma cell line AGS was infected with GFP-expressing EBV Akata BX1 (31). AGS Akata BX1 cells stably express EBV transactivator protein BZLF1 under control of a doxycycline inducible promoter (AGSiZ). See Supporting Information for details of AGSiZ cell line construction using lentivirus transduction (32).
Induction Reagents and Antiviral Compound Screening.
EBV lytic replication in AGS Akata BX1, AGSiZ, and P3HR1-ZHT cells was performed by addition of 12-O-tetradecanoylphorbol-13-acetate (TPA), doxycycline, or 4HT, respectively. HEK293 reporter cells carrying ORF59-GFP and inducible SM were induced to express SM with 1 μg/mL doxycycline. SPR (Sigma, S3378), CAN (TOCRIS Bioscience, 3281), TMS (TRC Toronto, T353500), and EPR (Sigma, E6657) were used at various concentrations from 0 to 20 μM as indicated. Screening was performed using ORF59-GFP-iSM cells, an assay developed and validated for high throughput screening of SM inhibitors as previously described (8).
Quantification of Infectious Virion Production and Viral DNA Replication.
EBV lytic replication in AGS and AGSiZ cells was induced by TPA or doxycycline treatment, respectively, and also treated with either DMSO control or with SPR, CAN, TMS, or EPR for 5 d. Filtered cell supernatants were used to infect 293T cells, and the numbers of GFP-positive 293T cells representing infectious viral particles were quantitated by flow cytometry as described previously (14, 33). For P3HR1-ZHT cells, cell supernatants were harvested at 5 d after induction of replication, and EBV DNA released in the supernatant was measured by qPCR. To measure DNA replication, cell pellets were harvested at 70 h after treatment as described above, and EBV DNA copy number was measured by qPCR as described previously (5, 14).
Electron Microscopy.
AGSiZ cells were treated with doxycycline to induce lytic replication and also treated with SPR. Cells were harvested at 4 d posttreatment and examined by electron microscopy. At least 150 cells were examined from three different preparations.
RNA Sequencing and Data Analysis.
cDNA libraries were prepared from poly(A) RNA and were sequenced on a HiSeq2000 instrument with 50 cycle single end reads. Sequenced reads obtained from EBV-infected AGS and P3HR1 cells were aligned to the EBV Akata (GenBank accession no. KC207813.1) and P3HR1 genomes (GenBank accession no. DQ279927.1). Differential gene expression was measured using USeq’s Defined Region Differential Seq application as described previously (14). Heat maps showing clustering of SM-dependent and SPR-sensitive genes and log2 transformed fold changes were clustered using the R package pheatmap (cran.r-project.org/web/packages/pheatmap/index.html). The distance method was “euclidean” and the clustering method was “complete.”
Additional methods are included in Supporting Information.
Cell Culture and Reagents
AGS/Akata BX1 and AGSiZ cells were grown in F-12 media with 500 μg/mL G-418 or 0.5 μg/mL puromycin plus 500 μg/mL G-418, respectively. HEK293 and P3HR1-ZHT cells were grown in DMEM and RPMI, respectively. All media were supplemented with 10% (vol/vol) charcoal-stripped FBS and 1% GlutaMAX (ThermoFisher). EBV lytic replication in AGS Akata BX1, AGSiZ, and P3HR1-ZHT cells was induced by addition of 50 ng/mL 12-O-tetradecanoylphorbol-13-acetate (Sigma, P8139), 0.05 μg/mL doxycycline (Sigma, D9891), and 100 nM 4HT (Sigma, H6278), respectively.
RNA Isolation and qPCR
RNA was isolated from cell lines cultured for 48 h after induction of EBV lytic replication and drug treatment. Biological triplicates of each experiment were performed, and qPCR was performed with gene-specific primers using 50 ng of total RNA as previously described (9). RT-PCR was performed in triplicate, and the relative quantity (RQ) for each gene was calculated using cellular GAPDH as an endogenous control in all reactions. The primers used to amplify specific EBV transcripts have been previously reported (9).
Generation of AGSiZ
A lentivirus expressing doxycycline-inducible BZLF1 was constructed by amplification of the coding sequence for B95.8 EBV BZLF1 and cloned into the pTRIPZ/GFP vector (32). Lentivirus stocks were made in 293T cells by transfecting the BZLF1 lentiviral vector together with the helper plasmid pMd2.G and packaging construct pCMVdeltaR8.74 exactly as described earlier (8). Lentiviral transductions of AGS Akata BX1 cells stably infected with EBV were performed in six-well plates by spin inoculation. Briefly, 250,000 cells were plated in six-well plates and the next day were infected with 1.0 mL of medium containing 500 μL of the lentiviral supernatant by centrifugation at 2,000 × g for 60 min at room temperature. At 48 h after infection, fresh F-12 medium with 500 μg/mL G418 and 1.0 μg/mL puromycin was added, and cells were selected for 2 wk. Individual clones were tested for BZLF1 expression and EBV DNA replication by Western blot and qPCR, respectively, after the addition of doxycycline. After screening, cells were maintained in F-12 medium containing 10% charcoal-stripped FBS (Sigma) with 500 μg/mL G418 and 0.5 μg/mL puromycin.
Drug Screening
The screen was performed by treating ORF59-GFP-iSM cells with 1.0 μg/mL doxycycline to induce SM expression and SM-dependent GFP expression (8). Cells were simultaneously incubated with small molecule compounds from the Spectrum Collection at the drug discovery core laboratory at the University of Utah. The screen was performed in 96-well clear-bottom plates (BD Falcon, cat. no. 353219) by seeding 35,000 cells per well in 100 μL medium without any antibiotics. The next day, cells were treated with doxycycline and compounds from the Spectrum library (10) at 10 μM with 0.1% DMSO using a Tecan EVO100/MCA96 Liquid Handler.
The Spectrum library (www.msdiscovery.com/spectrum.html) has over 2,000 compounds and was used in our initial screening assay. Cells were incubated at 37 °C in a CO2 incubator, and cells were visually examined under fluorescence microscopy to detect suppression of GFP expression at 30 h after treatment.
Electron Microscopy
AGSiZ cells were treated with 0.05 μg/mL doxycycline to induce lytic replication and harvested 4 d after induction. AGSiZ cell pellets were fixed with 2.5% glutaraldehyde and 1% paraformaldehyde in cacodylate buffer overnight at 4 °C. After fixation, cells were postfixed with 2% osmium tetroxide for 1 h at 25 °C, washed with cacodylate buffer, and stained en bloc with aqueous-saturated uranyl acetate. After washing, the cells were dehydrated in graded ethanol followed by acetone and infiltrated and embedded in epon-araldite resin. Ultrathin sections at 70-nm thickness were obtained using a Leica UC6 Ultratome and were collected on 200-mesh copper grids. Sections were counterstained by incubation with aqueous-saturated uranyl acetate for 10 min followed by staining with lead citrate for 5 min (34). Sections were viewed on a JEOL (JEM-1400) transmission electron microscope, operated at 120 kV, and images were acquired on a Gatan 2Kx2K digital camera.
Immunofluorescence Microscopy and Immunoblotting
HEK 293 reporter cells were treated with 1 μg/mL doxycycline to induce SM and SM-dependent GFP expression and also treated with DMSO, SPR, or EPR at various concentrations (5 μM to 20 μM) as described in each experiment. Cells were examined for GFP expression at 24 h after doxycycline induction and drug treatment and were photographed with a fluorescence microscope. EBV protein expression in AGSiZ cells was measured 24 and 48 h after induction of EBV lytic replication and SPR treatment. For Western blots, whole-cell lysates were prepared after induction of EBV replication and drug treatments and were analyzed by SDS/PAGE as described previously (15). Briefly, immunoblotting was performed with an anti-SM polyclonal (1:500), EAD (1:250), ZTA (1:100), RTA (Argene, 11–008, 1:100), and actin (Sigma A-5441 1:5,000) monoclonal or an anti-GFP polyclonal (1:1,000) antibody using a horseradish peroxidase-conjugated secondary antibody (anti-mouse or anti-rabbit) followed by incubation with Bio-Rad chemiluminescence detection reagent.
For immunofluorescence, cells were fixed and stained with antibodies as previously described (5, 35). AGSiZ cells were plated on coverslips in six-well plates and treated with 0.05 μg/mL doxycycline to induce EBV replication and also treated with either DMSO or SPR for 48 h. Cells were washed with PBS and fixed in 4% paraformaldehyde and stored at 4 °C until staining. Cells were permeabilized with 0.1% Triton X-100, blocked with 20% goat serum, and incubated with respective antibodies. The following antibodies and dilutions were used: SM polyclonal (1:500), ZTA (Argene, 11–007, 1:100), BMRF1 (Capricorn EBV-018–48180, 1:250), and VCA (Capricorn EBV-018–48184, 1:250) monoclonal antibodies. The cells were washed and incubated with Alexa Fluor 594 conjugated with either goat anti-rabbit IgG or anti-mouse IgG. Nuclei were stained with ProLong Gold antifade reagent with DAPI (Molecular Probes, P36935), and mounted slides were visualized and imaged using an Axio Imager M2 microscope (Zeiss).
Quantification of Virion Production and Viral DNA Replication
For virus production, AGS Akata BX1 and AGS Akata BX1iZ cells were plated in six-well plates, and lytic replication was induced by treatment with TPA (50 ng/mL) and doxycycline (0.05 μg/mL), respectively. These cells were also treated with either DMSO as a control or treated with increasing concentrations of SPR, CAN, TMS, or EPR for 5 d. Cell supernatants were harvested, filtered through 0.8-μm cellulose acetate filters, and serial dilutions of supernatants were used to infect 293T cells by spin inoculation. The infected cells were incubated at 37 °C, and the numbers of GFP-positive 293T cells representing infectious EBV particles were quantitated by flow cytometry at 2 d postinfection as previously described (5, 14).
For virus production in lymphoma cells, P3HR1-ZHT cells were treated with tamoxifen to induce EBV lytic replication and also treated with SPR for 5 d. Cell supernatants were harvested at 5 d after induction of replication and EBV DNA was purified. Briefly, 1.0 mL cleared, cell supernatants were centrifuged at 16,000 × g for 2 h at 4 °C, and viral particles were dissolved in 25 μL PBS, treated with 2.5 U of RQ1DNase (Promega, M6101) for 30 min followed by heat inactivation of DNase at 65 °C for 10 min. The viral particles were treated with 1 mg/mL proteinase K for 30 min at 55 °C. DNA was purified by phenol chloroform extraction, followed by G-50 columns. A fraction of DNA was used to measure the EBV copy number by qPCR using EBV BMRF1 primers.
For DNA copy number quantification, cell pellets were harvested at 70 h postinduction as described above, and DNA was isolated with a blood DNeasy kit (Qiagen). EBV DNA copy number was measured by qPCR using 50 ng total DNA with EBV BMRF1 primers, and RQ was calculated using cellular GAPDH primers as an internal control.
Cotransfection Assays and SMKO Cell Transfections
HEK293 or 293 SMKO EBV-infected cells were grown in DMEM supplemented with 10% FBS and 1% GlutaMAX supplement (Life Technologies). Transfections were performed with TransIT293 reagent (Mirus) according to the manufacturer’s protocol. SM, BDLF1, BILF2, and ZTA expression vectors have been previously described (9). The EBV EcoJ fragment was cloned into pcDNA3 for expression of the EBER2 transcript. We added 5 μM SPR or DMSO in triplicate to six-well plates transfected with 0.5 μg of the gene of interest and 0.5 μg of either SM or empty pcDNA3 vector plasmid (Invitrogen) and harvested it at 28 h. All transfections were performed in triplicate.
Human Mineralocorticoid Receptor Reporter Assay
The Human Mineralocorticoid Receptor Reporter Assay System (Indigo Biosciences IB00501-32) was used according to the manufacturer’s protocol with fourfold antagonist dilutions (2.5 nM to 40.96 μM) and an aldosterone concentration of 333 pM. Luciferase expression was measured using a Turner Biosystems 96-well plate luminometer.
Acknowledgments
We thank Linda Nikolova of the University of Utah School of Medicine EM core facility, Tim Mosbruger of the Huntsman Cancer Institute Bioinformatics Shared Resource, and Ryan Looper of the Department of Chemistry for their assistance with electron microscopy, RNAseq data analysis, and structural analysis, respectively. This work was funded by Public Health Service Grant R01 81133 (to S.S.) from the National Cancer Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1523686113/-/DCSupplemental.
References
- 1.Longnecker RM, Kieff E, Cohen JI. Epstein-Barr virus. In: Knipe DM, Howley PM, editors. Fields Virology. 6th Ed. Vol 2. Wolters Kluwer/Lippincott Williams and Wilkins; Philadelphia: 2013. pp. 1898–1959. [Google Scholar]
- 2.Swaminathan S, Kenney S. 2009. The Epstein-Barr virus lytic lifecycle. DNA Tumor Viruses, eds Damania B, Pipas J (Springer, New York)
- 3.Gruffat H, et al. Epstein-Barr virus mRNA export factor EB2 is essential for production of infectious virus. J Virol. 2002;76(19):9635–9644. doi: 10.1128/JVI.76.19.9635-9644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batisse J, Manet E, Middeldorp J, Sergeant A, Gruffat H. Epstein-Barr virus mRNA export factor EB2 is essential for intranuclear capsid assembly and production of gp350. J Virol. 2005;79(22):14102–14111. doi: 10.1128/JVI.79.22.14102-14111.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han Z, et al. Multiple roles of Epstein-Barr virus SM protein in lytic replication. J Virol. 2007;81(8):4058–4069. doi: 10.1128/JVI.02665-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmeri D, Spadavecchia S, Carroll KD, Lukac DM. Promoter- and cell-specific transcriptional transactivation by the Kaposi’s sarcoma-associated herpesvirus ORF57/Mta protein. J Virol. 2007;81(24):13299–13314. doi: 10.1128/JVI.00732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coen DM, Richman DD. Antiviral agents. In: Knipe DM, Howley PM, editors. Fields Virology. 6th Ed. Vol 1. Wolters Kluwer/Lippincott Williams and Wilkins; Philadelphia: 2013. pp. 338–373. [Google Scholar]
- 8.Verma D, Kim EA, Swaminathan S. Cell-based screening assay for antiviral compounds targeting the ability of herpesvirus posttranscriptional regulatory proteins to stabilize viral mRNAs. J Virol. 2013;87(19):10742–10751. doi: 10.1128/JVI.01644-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson J, Verma D, Li D, Mosbruger T, Swaminathan S. Identification and characterization of the physiological gene targets of the essential lytic replicative EBV SM protein. J Virol. 2016;90(3):1206–1221. doi: 10.1128/JVI.02393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kocisko DA, et al. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77(19):10288–10294. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szasz G, Budvari-Barany Z. Pharmaceutical Chemistry of Antihypertensive Agents. Boca Raton, FL: CRC Press; 1990. p. 288. [Google Scholar]
- 12.Kolkhof P, Borden SA. Molecular pharmacology of the mineralocorticoid receptor: Prospects for novel therapeutics. Mol Cell Endocrinol. 2012;350(2):310–317. doi: 10.1016/j.mce.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 13.Wang HB, et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat Commun. 2015;6:6240. doi: 10.1038/ncomms7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li DJ, Verma D, Mosbruger T, Swaminathan S. CTCF and Rad21 act as host cell restriction factors for Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog. 2014;10(1):e1003880. doi: 10.1371/journal.ppat.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruvolo V, Wang E, Boyle S, Swaminathan S. The Epstein-Barr virus nuclear protein SM is both a post-transcriptional inhibitor and activator of gene expression. Proc Natl Acad Sci USA. 1998;95(15):8852–8857. doi: 10.1073/pnas.95.15.8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruvolo V, Gupta AK, Swaminathan S. Epstein-Barr virus SM protein interacts with mRNA in vivo and mediates a gene-specific increase in cytoplasmic mRNA. J Virol. 2001;75(13):6033–6041. doi: 10.1128/JVI.75.13.6033-6041.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han Z, Verma D, Hilscher C, Dittmer DP, Swaminathan S. General and target-specific RNA binding properties of Epstein-Barr virus SM posttranscriptional regulatory protein. J Virol. 2009;83(22):11635–11644. doi: 10.1128/JVI.01483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiriart E, et al. A region of the Epstein-Barr virus (EBV) mRNA export factor EB2 containing an arginine-rich motif mediates direct binding to RNA. J Biol Chem. 2003;278(39):37790–37798. doi: 10.1074/jbc.M305925200. [DOI] [PubMed] [Google Scholar]
- 19.Hiriart E, et al. A novel nuclear export signal and a REF interaction domain both promote mRNA export by the Epstein-Barr virus EB2 protein. J Biol Chem. 2003;278(1):335–342. doi: 10.1074/jbc.M208656200. [DOI] [PubMed] [Google Scholar]
- 20.Malik P, Blackbourn DJ, Cheng MF, Hayward GS, Clements JB. Functional co-operation between the Kaposi’s sarcoma-associated herpesvirus ORF57 and ORF50 regulatory proteins. J Gen Virol. 2004;85(Pt 8):2155–2166. doi: 10.1099/vir.0.79784-0. [DOI] [PubMed] [Google Scholar]
- 21.Sønder SU, Mikkelsen M, Rieneck K, Hedegaard CJ, Bendtzen K. Effects of spironolactone on human blood mononuclear cells: Mineralocorticoid receptor independent effects on gene expression and late apoptosis induction. Br J Pharmacol. 2006;148(1):46–53. doi: 10.1038/sj.bjp.0706700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lainscak M, et al. Safety profile of mineralocorticoid receptor antagonists: Spironolactone and eplerenone. Int J Cardiol. 2015;200:25–29. doi: 10.1016/j.ijcard.2015.05.127. [DOI] [PubMed] [Google Scholar]
- 23.Meng Q, et al. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J Virol. 2010;84(9):4534–4542. doi: 10.1128/JVI.02487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardiner P, et al. Spironolactone metabolism: Steady-state serum levels of the sulfur-containing metabolites. J Clin Pharmacol. 1989;29(4):342–347. doi: 10.1002/j.1552-4604.1989.tb03339.x. [DOI] [PubMed] [Google Scholar]
- 25.Hanson KE, Swaminathan S. Cytomegalovirus antiviral drug resistance: Future prospects for prevention, detection and management. Future Microbiol. 2015;10(10):1545–1548. doi: 10.2217/fmb.15.82. [DOI] [PubMed] [Google Scholar]
- 26.Han Z, Swaminathan S. Kaposi’s sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J Virol. 2006;80(11):5251–5260. doi: 10.1128/JVI.02570-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toth Z, Stamminger T. The human cytomegalovirus regulatory protein UL69 and its effect on mRNA export. Front Biosci. 2008;13:2939–2949. doi: 10.2741/2899. [DOI] [PubMed] [Google Scholar]
- 28.Sandri-Goldin RM. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 2011;6(11):1261–1277. doi: 10.2217/fmb.11.119. [DOI] [PubMed] [Google Scholar]
- 29.Boyer JL, Swaminathan S, Silverstein SJ. The Epstein-Barr virus SM protein is functionally similar to ICP27 from herpes simplex virus in viral infections. J Virol. 2002;76(18):9420–9433. doi: 10.1128/JVI.76.18.9420-9433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma D, Ling C, Johannsen E, Nagaraja T, Swaminathan S. Negative autoregulation of Epstein-Barr virus (EBV) replicative gene expression by EBV SM protein. J Virol. 2009;83(16):8041–8050. doi: 10.1128/JVI.00382-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol. 2000;74(14):6324–6332. doi: 10.1128/jvi.74.14.6324-6332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linnstaedt SD, Gottwein E, Skalsky RL, Luftig MA, Cullen BR. Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus. J Virol. 2010;84(22):11670–11678. doi: 10.1128/JVI.01248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li DJ, Verma D, Swaminathan S. Binding of cellular export factor REF/Aly by Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF57 protein is not required for efficient KSHV lytic replication. J Virol. 2012;86(18):9866–9874. doi: 10.1128/JVI.01190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruvolo V, et al. Functional analysis of Epstein-Barr virus SM protein: Identification of amino acids essential for structure, transactivation, splicing inhibition, and virion production. J Virol. 2004;78(1):340–352. doi: 10.1128/JVI.78.1.340-352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]