

Abstract
Aptamers were discovered more than 25 years ago, yet only one has been approved by the US Food and Drug Administration to date. With some noteworthy advances in their chemical design and the enzymes we use to make them, aptamers and aptamer-based therapeutics have seen a resurgence in interest. New aptamer drugs are being approved for clinical evaluation, and it is certain that we will see increasingly more aptamers and aptamer-like drugs in the future. In this review, we will discuss the production of aptamers with an emphasis on the advances and modifications that enabled early aptamers to succeed in clinical trials as well as those that are likely to be important for future generations of these drugs.
Introduction to Selex and In Vitro Selection
Whether referred to as “SELEX” (Systematic Evolution of Ligands by Exponential Enrichment) or as “in vitro selection” (a demarcation resulting from whether the technique was learned from Tuerk and Gold1 or Ellington and Szostak,2 respectively), the fundamental approach to acquiring novel aptamers remains largely unchanged despite alterations including cell targeting, cell internalization, and even in vivo selections3–7. The core of the selection process, be it for RNA or DNA aptamers, on protein, on cells, or in animals, entails essentially three critical steps: (i) incubation of a library of randomized molecules with a target, (ii) partitioning bound from nonbound species, and (iii) recovering and amplifying the bound species. The process is then repeated until the population is enriched for species with the desired function (Figure 1).
Figure 1.
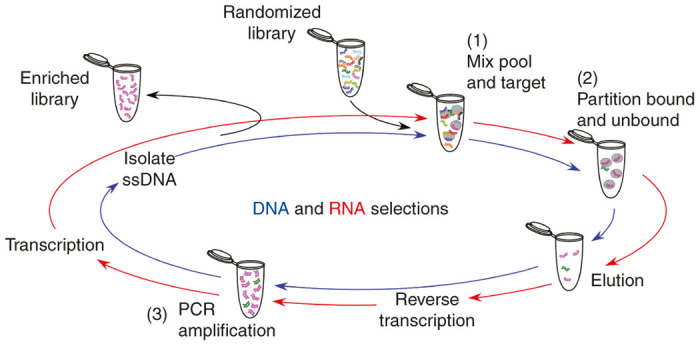
A schematic of the selection process for DNA and RNA aptamer libraries. Starting with randomized library incubated with the target (1), bound species are partitioned and stringently washed (2), followed by elution of desired species. For RNA selections, recovered material must be reverse transcribed, followed by polymerase chain reaction (PCR) amplification (3) and transcription back into RNA to generate the library for the next round. DNA selections however, are ready for PCR amplification after elution (3), but afterwards must be separated from the complement strand before the resulting ssDNA pool can be used for the next round.
For the most part, progress in the aptamer field has not changed this fundamental approach. However, several key advances have significantly changed how aptamers can be identified from the selection, the types of targets that can be probed, and the chemistries that aptamers can incorporate—all of which leads to more expanded uses for aptamers, both in vitro and in vivo. Advances in sequencing technologies and computational analyses now provide greater insight into population dynamics during selections, potentiating the identification of functional molecules from earlier rounds of selection. New polymerases are now available that can yield selection libraries with highly nuclease stabilized sugar-phosphate backbones, diminishing the need for laborious postselection stabilization (see “New RNA Polymerase Variants” below). More excitingly, aptamers with improved binding affinities and kinetics are being generated with modifications on the bases, which render them more protein-like (see “SOMAmers” below). While reviews of aptamers under clinical evaluation abound in the literature,8–12 this review, in addition to summarizing the most recent developments, seeks to contrast the original workflow from aptamer discovery to clinical evaluation, with newer approaches that result in better aptamers and expedited discovery, making way for an expanded US Food and Drug Administration approval pipeline in the future.
Aptamer Production
The enzymatic production of aptamers during the selection process still remains a necessity. However, in order to generate large quantities of material for clinical testing, and for large-scale production, enzymatic synthesis is simply not practical. Thus, a key initial step in the discovery process of any aptamer is the identification of a core, minimal functional sequence, which can be generated synthetically by solid phase synthesis. Aptamer starting libraries are typically between 70 and 90 nucleotides in length including the constant regions. Following selection, candidate aptamers are analyzed by comparing sequence composition and identifying sequence motifs, or by comparing predicted two-dimensional folds using well-established algorithms to identify common structural motifs allowing for minimization. In general, shorter molecules are better for chemical synthesis, as each unnecessary nucleotide (or coupling) both decreases yields and increases manufacturing costs. Indeed, in some clinical aptamers, for example Fovista, this has led the replacement of loop residues with flexible polyethylene glycol (PEG) linkers resulting in a savings of four coupling steps (see below).
For R&D purposes, minimized aptamers 20 to 50 nucleotides in length can be generated in individual labs using “lab scale” DNA or RNA synthesizers (e.g., Expedite 8909, ABI394) or by one of a number of the many oligonucleotide synthesis companies. Depending on oligonucleotide length and the modifications present, synthesis at the 1 μmole scale typically yields ~1 mg of material, which is often sufficient for hundreds of in vitro experiments and enough to perform preclinical analyses in small animal (mouse) models. These machines are capable of scaling to 10–15 µmole, but for larger synthesizes, equipment capable of generating gram scale quantities of aptamers can be readily obtained (e.g., the ÄKTA oligopilot plus 100 can scale from 250 µmole to 4 mmoles). Of course, synthesis ranging from 50 nmoles to 4 mmoles and beyond can also be outsourced to a number of commercial sources that offer large-scale R&D grade materials.
When advancing to clinical evaluation, adherence to current good manufacturing practice must be employed (reviewed in ref. 13). Fortunately, there are now many companies that provide this service with synthesis facilities capable of kilogram scale production. However, while the exact synthesis method for clinically evaluated aptamers, to our knowledge, are not publically available, the synthesis methods all rely on standard solid phase phosphoramidite chemistry.14,15
Limitations of nonmodified aptamers
The advent of chemistries to stabilize aptamers (and nucleic acids) against endogenous, ubiquitous nucleases has been instrumental to the advancement of aptamers that already have, or currently are in the process of undergoing clinical evaluation. Without these modifications, deoxyribonucleic (2’H) and ribonucleic acids (2’OH) (Figure 2a) are highly susceptible to nuclease degradation, with the former displaying serum half-lives of ~1 hour16 and the latter lasting only mere minutes.17 Perhaps just as important, many of these chemistries also render aptamers nonimmunogenic.18–20 This is a significant problem for nonmodified RNA and DNA, which can activate innate responses though interaction with Toll-like receptors, in particular Toll-like receptors 7, 8, 9, and 3 (refs. 21–23). Thus, a number of different strategies and chemical modifications are now available to both enhance the stability of aptamers and render them less immunogenic.
Figure 2.

Modifications utilized to enhance the in vivo stability of aptamers. (a) 2’-modifications can easily be incorporated into aptamers during chemical synthesis and include i. 2’H, ii. 2’OH, iii. 2’ NH2, iv. 2’F, v. 2’OMe and vi. locked nucleic acids. (b) Increased stability can also be garnered though thiolation of the phosphate backbone. Structures shown (from left to right) are for the i. natural phosphodiester, ii. the thiolated phosothioate and iii. phosphorodithioate. (c) 3’ inverted deoxythymidine residue. (d) Non-ribose backbones, which can be incorporated using novel DNA polymerases for the basis for xeno nucleic acids, include i. cyclohexenyl, ii. arabino, iii. α-L-threofuranosy, and iv. 2′-fluoroarabino nucleic acids. (e) Examples of some commercially available functional groups that can be readily attached to the 5’-end during solid phase synthesis and used to facilitate downstream conjugation include i. amine, ii. alkyne, iii. azide, iv. thiol, v. aldehyde and vi. aminooxy.
Backbone modifications
Sugars.
Modifications to the sugars such as 2’-fluoro (2’F) or 2’-amino (2’NH2) ribose groups (Figure 2a) on the pyrimidine residues have been available for incorporation into enzymatically derived nucleic acids for some years using a mutant of T7 RNA polymerase bearing a tyrosine to phenylalanine mutation at position 639 (Y639F; ref. 24). While both are effective at improving serum half-life, 2’F modifications quickly garnered favor over 2’NH2 due to entropic gains in binding caused by reduced backbone flexibility, increased coupling efficiency during solid-phase synthesis, and elimination of extra deprotection steps during 2’NH2 purification.25 The more bulky 2’-O-methyl (2’OMe) modifications (Figure 2a), while incompatible with the Y639F mutant, have been previously used as a postselection modification due to their increased nuclease resistance and higher duplex melting temperature as seen in the clinical examples below. However, more recent advances in polymerase evolution give hope for more prominent use of 2’OMe modifications at the outset of the selection process (see “New RNA Polymerase Variants” below). Another advance in nuclease stabilization is conformationally restricted sugars, or locked nucleic acids, which covalently bridge the 2’ and 4’ ribose positions (Figure 2a).26,27 Despite their enhanced stability, the conformational constraints imposed by locked nucleic acids may relegate such modifications to paired regions and stems, although here again, recent advances in polymerases now allow for their incorporation during the selection process (see “New RNA polymerase variants” below).
Phosphate modifications.
Stabilizing backbone modifications can also be incorporated into aptamers by substituting backbone phosphate linkages (PO) with sulfur-containing phosphorothioate linkages (PS)28 and, more recently, phosphorodiothioate linkages29 (Figure 2b). However, in the case of PS linkages, which are chiral (the PS linkage can exist in the Sp or the Rp configuration), care in placement needs to be exercised as chemical synthesis results in a mixture of enantiomers that could adversely affect function.
Capping modifications
In addition to sugar and backbone modifications, therapeutic aptamers are typically modified in two additional ways. First, the 3’ end of the molecules are usually capped with an inverted dT residue, which provides resistance to serum exonucleases (Figure 2c).16 Second, the molecules are conjugated at the 5’ end to a 40 kDa PEG moiety to slow renal clearance. Indeed, without this moiety, as a result of their small size (10–20 kDa), aptamers are readily cleared by glomerular filtration with a half-life of minutes30,31 similar to that observed for comparatively sized siRNA.32 While this can be slowed by the addition of hydrophobic moieties such as cholesterol or diacyl lipids,30,31,33,34 the addition of PEG has proven to be the most useful and commonly pursued approach, slowing clearance half-lives to 3–50 hours depending on the PEG size.30,31,35–37 The ability to chemically synthesize aptamers affords a significant amount of freedom in the design of conjugation schemes for any chemical moiety. For example, amine, thiol, aminooxy, alkyne, aldehyde, and azide functional groups can all be incorporated into the aptamer during solid phase synthesis (Figure 2e). For small, lipophilic modifications (e.g., cholesterol, palmitate) incorporation can be readily achieved using a phosphoramidite derivative of the molecule. However, for the larger hydrophilic moieties (e.g., PEG 20 or 40 kDa), conjugation is typically achieved post-synthesis via the reaction of a 5’ amine, introduced on the aptamer using a hexylamine linker (Figure 2e; i) and an N-hydroxysuccinimide-derivative of the polymer.
Clinically Evaluated Aptamers: Past and Present
Aptamers evaluated in phase 3 trials
Pegaptanib.
The anti-vascular endothelial growth factor (VEGF) aptamer, pegaptanib (Macugen), originally selected by NeXstar Pharmaceuticals (later taken on by Ophthotech) for the treatment of neovascular (wet) age-related macular degeneration, remains the only US Food and Drug Administration-approved aptamer to date.38
Originally selected from a 2’F pyrimidine RNA library, the lead candidate, clone t44 bound VEGF165 with ~10 pmol/l affinity (Figure 3-t44 and Table 1), though showing no measurable affinity for the VEGF121 isoform.25 For added stability, the 2’OH purines were systematically replaced with 2’OMe purines, and all but two could be converted with binding affinity only rising to 50 pmol/l. The molecule was then further modified with a 3’ inverted dT to inhibit 3’ exonuclease activity and a branched 40 kDa PEG via a 5’ terminal amine to slow clearance (Figure 3-pegaptanib). In its fully modified form, the aptamer is reported to have a serum degradation half-life of 18 hours (Table 1).39 This is even longer in the eye where the molecule is only turned over due to plasma clearance, and, after a single 0.5 mg injection in rhesus monkeys, sees a half-life of 2 days with detectable amounts of intact aptamer for more than 28 days.40 In humans, a half-life of ~10 days has been reported.41 Approved in 2004, Macugen was the first US Food and Drug Administration- approved aptamer and also the first anti-VEGF drug, though it is no longer widely used given its limited isoform recognition compared to its protein competitors Avastin, Lucentis, and Eylea.42–44
Figure 3.
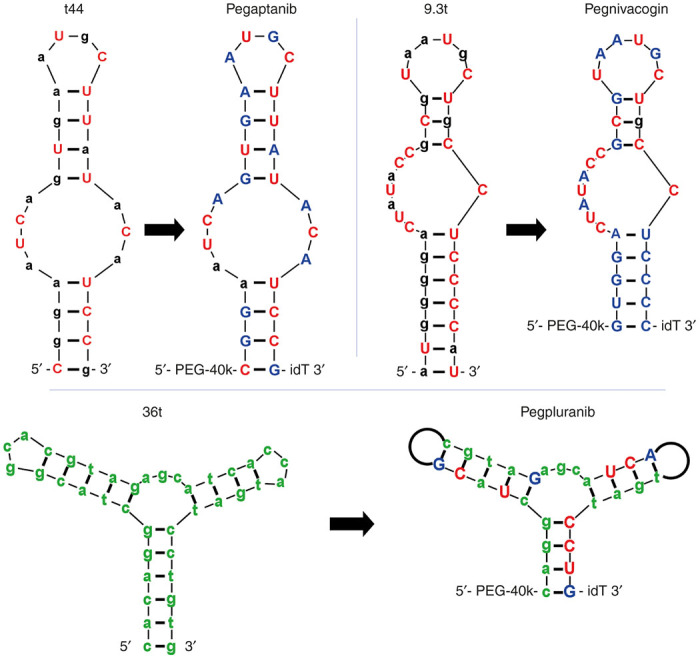
Only the three aptamers have reached Phase III clinical trials to date. Minimized lead molecules as produced from the selection shown next to their counterpart fully stabilized clinical progeny. Lowercase black and green denotes 2’OH and 2’H respectively, uppercase red and blue denotes 2’F and 2’OMe respectively. “idT” represents an inverted deoxythimidine, also known as 3’-3’ dT. PEG-40k represents an amine linked to 40 kDa polyethylene glycol. Black loops on the arms of pegpluranib represents an 18-atom hexaethylene glycol spacer, which replaced the 3 nucleotide loops found in the parent molecule.
Table 1. Summary of characteristics for aptamers discussed in the text.
| Target | Aptamer | 2' Pyrimidine | 2' Purine | 5' mod | 3' Mod | Other | KD (pmol/l) | Stability t1/2 (hours) | Clearance a t1/2 (α, β; hours) | Phase | Status | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Anti-VEGF | t44 | F | OH | -- | -- | -- | 10 | -- | -- | -- | -- | 25 |
| Pegaptanib | F | OMe/OH | PEG 40k | 3'-3' dT | -- | 50 | 18; HP | ~240; H (ivt) | 3 | Approved | 25,39,41 | |
| NX-107 | NH2 | OH | -- | -- | -- | NR | 1.4; MU | -- | -- | -- | 28 | |
| NX-178 | NH2 | OH | PS cap | PS cap | -- | 2,400 | 17; MU | -- | -- | -- | 28 | |
| NX-213 | NH2 | OMe/OH | PS cap | PS cap | -- | 140 | 131; MU | -- | -- | -- | 28 | |
| ARC245 | OMe | OMe | -- | -- | -- | 1,000 | -- | -- | -- | -- | 80 | |
| Anti-FactorIXa | 9.3t | F | OH | -- | 3'-3' dT | -- | 600 | -- | -- | -- | -- | 45 |
| Peg-9.3t | F | OH | PEG 40k | 3'-3' dT | -- | 3,000 | -- | -- | -- | -- | 45 | |
| Pegnivacogin | F | OMe/OH | PEG 40k | 3'-3' dT | -- | 3,000 | -- | >30; H (iv)b ~144; H (sc) | 3 | Terminated | 50,53 | |
| Anti-PDGF | 36t | H | H | -- | 3'-3' dT | -- | 100 | 0.6; RS | -- | -- | -- | 58,59 |
| Pegpleranib | H/F | H/OMe | PEG 40k | 3'-3' dT | -- | 100 | 8; RS | -- | 3 | Ongoing | 59 | |
| SL5 | H | H/OMe | -- | -- | 5-mod dU | 20 | -- | -- | -- | -- | 89 | |
| Anti-VWF | ARC1779 | H/OMe | H/OMe | PEG 20k | 3'-3' dT | 1 PS residue | 2,000 | 63; HPc | ~2; H (iv) | 2 | Terminated | 60 |
| ARC15105 | OMe | OMe | PEG 40k | 3'-3' dT | 1 PS residue | 1,000 | >300; RS >>300; HSd | ~67; CM (iv) ~65; CM (sc) | -- | -- | 63 | |
| Anti-nucleolin | AS1411 | H | H | -- | -- | -- | -- | -- | ~2; H (iv) | 2 | Terminated | 64,68 |
| Anti-TFPI | BAX499 | H/OMe | OMe | PEG 40k | 3'-3' dT | -- | 3,000 | >>72; HSd | -- | 1 | Terminated | 70,72 |
| Anti-hepcidin | NOX-H94 | L-2'OH | L-2'OH | PEG 40k | -- | -- | 650 | >>60; HSd | 23; H (iv) | 2 | Complete | 37,78,79 |
| Anti-CXCL12 | NOX-A12 | L-2'OH | L-2'OH | PEG 40k | -- | -- | 200 | >>60; HSd | 40; H (iv) | 2 | Ongoing | 36,78,79 |
| Anti-CCL2 | NOX-E36 | L-2'OH | L-2'OH | PEG 40k | -- | -- | 500 | >>60; HSd | 50; H (iv) | 2 | Complete | 78,79 |
| Anti-PSMA | ARC591 | F | OH | -- | -- | -- | 1,300–1,800e | ~5; MPf | -- | -- | -- | 94 |
| ARC1725 | OMe/F | H/OMe | -- | 3'-3' dT | 1 PS residue | 4,700e | 20; HP | -- | -- | -- | 94 | |
| anti-IL-6 | SL1025 | H/OMe | H/OMe | -- | 3'-3' dT | 5-mod dU | 200 H | >>48; HSd | -- | -- | -- | 90 |
| SL1023dT | H/OMe | H/OMe | -- | 3'-3' dT | -- | -- | 5.5; HS | -- | -- | -- | 90 | |
| SL1026 | H/OMe | H/OMe | PEG 40k | 3'-3' dT | 5-mod dU | 2,500 CM | -- | 0.4, 160; CM (iv) | -- | -- | 93 |
--, data not available, relevant, or reported.
CM, cynomolgus monkey; H, human; HP, human plasma; HS, human serum; RS, rat serum; i.v., intravenous; s.c., subcutaneous; i.v.t., intravitreal; F, 2’ fluoro; MU, mouse urine; NH2, 2’ amino; OMe, 2’ O-methyl; H; 2’ deoxy; L-OH, L-2’hydroxy; 3’-3’dT, inverted deoxythymidine; PS, phosphorothioate; PSMA, prostate-specific membrane antigen; PDGF, platelet-derived growth factor; VWF, von Willebrand factor; 5-mod dU, deoxyuridine bearing a modification on the 5 position; Approved, clinical trial complete and FDA approved; Terminated, clinical trial terminated; Complete, clinical trial complete; Ongoing, clinical trial ongoing.
Half-lives (t1/2) for clearance are as reported in cited references. t1/2 for the α and β rates were specified if possible.
Indicates median duration of effect instead of t1/2.
Stability was assessed on a non-PEGylated variant.
Denotes insignificant degradation detected at that time point.
Denotes an IC50 for inhibition of NAALDase activity.
Denotes Levy Lab, unpublished results.
It is worth noting that the same authors who reported t44 also performed an earlier VEGF selection with a 2’NH2 pyrimidine RNA library.28 While these molecules did not advance to clinical evaluation, the study did provide important insight regarding how postselection stability enhancements can be used to stabilize aptamers and prepare them accordingly for clinical evaluation. For example, while the lead candidate from this selection, NX-107, displayed a degradation half-life in mouse urine (a nuclease rich surrogate for serum) of ~1.4 hours, the addition of PS caps or a combination of PS caps and 2’OMe purine substitution dramatically increased degradation half-lives to 17 and 131 hours, respectively (Table 1).28
Pegnivacogin.
Similar to Macugen, the Factor IXa aptamer pegnivacogin, which prevents blood coagulation, arose from a selection with a 2’F pyrimidine RNA library performed by Sullenger and colleagues.45 The minimized aptamer, 9.3t (Figure 3-9.3t), displayed a KD of ~600 pmol/l, which rose to 3 nmol/l after conjugation of a 40 kDa PEG on the 5’ end.45 The aptamer, was further stabilized by Regado Biosciences by systematically substituting 2’OH purines and 2’F pyrimidines with corresponding 2’OMe where tolerated (Figure 3-pegnivacogin).46 Pegnivacogin is only half of the Reg1 Anticoagulant System, and is designed to work in conjunction with anivamersen, a complementary (antisense) 2’OMe oligonucleotide, which hybridizes with and inactivates pegnivacogin to quickly reverse the anticoagulation effect, representing a breakthrough in controllable drug design.47 Tested initially in both porcine48 and baboon models,49 the systemically delivered, PEGyated, and backbone modified aptamer demonstrated high in vivo stability with a duration of action of over 30 hours with a 60 mg injection in humans,50 but could be inactivated following i.v. administration of the antisense “antidote” within minutes.51,52 The aptamer could also be delivered subcutaneously to provide sustained release over a period of days.53 The REG1 Anticoagulation System made it as far as phase 3 before a combination of severe allergic reactions, which were observed in 10 of 1,605 patients (including one fatal reaction) and the absence in any difference in the primary endpoint between patients receiving REG1 or the control group, which received bivalirudin, halted the trial.54 While the lack of improved efficacy of REG1 when compared to bivalirudin may be a consequence of the study design,55 more alarming to the aptamer community are safety concerns. However, recent analysis56 of existing samples from the RADAR57 phase 2b study indicated that severe allergic reactions in patients during that trial were correlated to high levels of pre-existing anti-PEG antibody and not caused by the aptamer itself56; good news for the field. Given the widespread use of PEG on the aptamers mentioned in this review (Table 1), the investigation into alternative moieties for avoiding renal clearance may prove important to the field.
Pegpleranib.
The most recent aptamer to make significant progress in clinical trials is Fovista, commonly called pegpleranib (formerly E10030), an aptamer that targets platelet-derived growth factor. Fovista is currently under development by Ophthotech and is undergoing simultaneous phase 2/3 clinical trials. The aptamer, like Macugen, is designed to be delivered via intravitreal injection and is intended to work synergistically with VEGF inhibitors, such as Macugen, Avastin, Lucentis, and Eylea in the treatment of age-related macular degeneration. Unlike Macugen and Reg1, however, Fovista was selected from a ssDNA library with a minimized lead molecule, 36t, displaying an affinity of ~100 pmol/l for the AB and BB isoforms (Figure 3-36t).58 After validation as a ssDNA aptamer, which had a serum degradation half-life of ~0.6 hours, Fovista was back-filled with both 2’F pyrimidines and 2’OMe purines, increasing the serum half-life to ~8 hours while maintaining ~100 pmol/l affinity.59 Additionally, loop regions within the aptamer found to be functionally unimportant were dispensed of in favor of short, PEG linkers, which likely not only added to the stability, but decreased overall cost of production by minimizing the number of nucleotides in the molecule (Figure 3-pegpluranib).59
Though demonstrably effective in their clinical evaluations, one common feature between Macugen, pegnivacogin, and Fovista is that their original selections were all published in the first 12 years of this now 25-year-old field. One is thus left to wonder where new clinical candidates are, and what they look like if they harness the advancements of the last 13 years of progress.
Additional aptamers approved for phase 1/phase 2 clinical evaluation
ARC1779.
Selected to bind the von Willebrand factor (VWF) A1-domain, ARC1179 was originally identified from ssDNA library.60 The minimized aptamer, composed of 39 nucleotides was subsequently modified systematically to improve stability by replacing 26 of the nuclease-labile DNA residues with stable 2’OMe nucleosides, including a single phosphorothioate residue between positions 20 and 21, and by capping at the 3’ end using an inverted dT residue. To increase serum lifetime, the 5’ end of the molecule was synthesized bearing a 5’ amine which was conjugated to a 20 kDa PEG moiety.
ARC1179 displayed high affinity for the VWF A1-domain (KD ~2 nmol/l) and specifically inhibited VWF-mediated platelet aggregation (IC90 ~300 nmol/l). In human trials, following bolus i.v. administration, the aptamer displayed a serum half-life of ~2 hours.35 More importantly, the drug proved safe and well tolerated in phase 1 (ref. 35), and in phase 2 trials was demonstrated to effectively inhibit VWF activity in the treatment of thrombotic thrombocytopenia purpura (ref. 61) as well as cerebral embolism.62 However, when the compound was delivered s.c. it failed to yield plasma concentrations that could effectively control clinical features of thrombocytopenia purpura relegating treatment regimens to bolus primed continuous infusion.
The limitation on route of administration has most recently been addressed by the identification of a second-generation anti-VWF aptamer, ARC15105, composed of 100% 2’OMe RNA that is conjugated to a larger 40 kDa PEG moiety.63 Like ARC1179, ARC15105 binds the A1-domain and inhibits VWF-mediated platelet aggregation (KD ~1 nmol/l; IC90 ~300 nmol/l). Following both i.v. and s.c. in vivo administration in cynomolgus monkeys, the compound displayed half-lives of ~65 hours in serum. The improved PK values are a likely consequence of both improved stability as well as the use of larger, 40 kDa PEG moiety. More impressively, 300 hours following a single 20 mg/kg s.c. dose, VWF activity remained >90% inhibited63 clearly demonstrating the benefit of this dosing method. Future plans for clinical evaluation of ARC15105 are currently unknown.
AS1411.
Originally identified during a screen of antisense oligonucleotide for antiproliferative activity, the aptamer AS1411, formerly AGFO001, is a 26 nucleotide DNA molecule composed exclusively of guanosine and thymidine residues that exists in solution as a G-quadruplex.64 Although the molecule was originally reported to interact with cell surface nucleolin, be taken up by cancer cells, and inhibit cell growth, more recent work has demonstrated a disconnect between cell uptake and nucleolin binding.65 In this work, nucleolin-negative cells were shown to display significant (and sometimes greater) levels of AS1411 uptake via macropinosomes. Interestingly, however, the antiproliferative activity of the molecule still appears to derive from its interaction with nucleolin where it is thought to act as a molecular decoy, competing with bcl-2 mRNA for binding.66 The interaction with nucleolin has also been demonstrated to lead to the induction and enhancement of cellular macropinocytosis,65 yet the mechanism of cytoplasmic access for this molecule remains unclear. Regardless of the exact mechanism of action, numerous biochemical studies have documented the antiproliferative effects on cells in culture, which have led to both phase 1 and phase 2 clinical trials. In a phase 1 trial, AS1411 showed no toxic effects of any type in patients with renal or non-small-cell lung cancer.67 However in a phase 2 study of 35 patients with renal cell carcinoma, only one patient demonstrated a significant response to treatment.68 Similarly, in a phase 2 trial in patients with AML, AS-1411 in combination with cytarabine only demonstrated limited improvement over cytarabine alone.69
BAX499.
Formerly ARC19499, this aptamer was selected to bind recombinant tissue factor pathway inhibitor (TFPI), a negative regulator of both factor VIIa and factor Xa and a causative agent of hemophilia.70 Here, the authors employed an aptamer library composed of 2’H C, 2’OMe A, 2’OMe G, and 2’OMe U residues. Although not specified in the original publication70 or patent,71 the 2’OMe-rich pool, which yields exceptionally nuclease-stable polymers and thus a means to rapidly move to in vivo testing and clinical evaluation, was likely generated using a newer variant of T7 RNA polymerase (discussed in more detail below). Indeed, the patent reveals that no detectible degradation was observed after 72 hours in human serum.72 After minimization to 32 nucleotides, and capping with a 3’-inverted dT and a 5’- 40 kDa PEG, the aptamer was determined to have a KD of ~3 nmol/l and an EC50 of ~2 nmol/l to restore factor Xa activity.70
However, while this aptamer initially showed promise,73–75 administration of the aptamer in humans resulted in increased serum levels of tissue factor pathway inhibitor as a result of increased synthesis and release by endothelial cells and slowed clearance and degradation of the protein.76 The increased levels of tissue factor pathway inhibitor could not be controlled by aptamer administration and development of this molecule was halted.77
Advancements in Aptamer Selection Technology
Mirror image aptamers, Spiegelmers
Generically, postselection incorporation of backbone modifications results in nonimmunogenic, nuclease-stabilized nucleic acids, which display increased serum stability allowing for therapeutic applications. However, this “back filling” approach adds time to the R&D process and is limited because not all positions in an aptamer can be changed to more stable analogs. Indeed, all of the aforementioned clinically evaluated aptamers retain at least one 2’H or 2’OH nucleotides, leaving the aptamers more susceptible to serum nucleases.
A very different, yet effective approach for the generation of highly stable molecules is the use of mirror image aptamers, or so called Spiegelmers (Figure 4). Composed of chains of L-nucleotides, as opposed the natural D-nucleotides, and forming left-handed helices instead of the typical right-handed helices, Spiegelmers display exceptional serum stability as they are not recognized by endogenous nucleases and exhibit essentially no degradation in serum.78 Of course, for this same reason these molecules cannot be made using existing enzymes. As such, Spiegelmers are initially selected using natural D-nucleotides against mirror image targets (i.e., a D-peptide; Figure 4a). The resultant D-aptamers are then synthesized chemically in the inverted stereo configuration as L-aptamers (Spiegelmers), which bind the natural L-peptide or protein target (Figure 4b).
Figure 4.
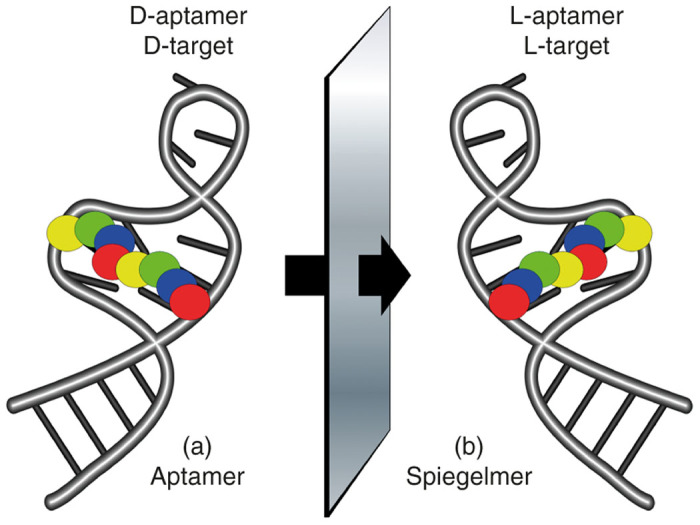
Mirror image aptamers, Spiegelmers, are composed of non-natural L-ribose nucleotides. The molecules are initially selected from natural D-ribose aptamer libraries against a non-natural target, for example a D-peptide (a). Once optimized as a D-aptamer the mirror image L-aptamer (Spiegelmer) is synthesized chemically and intrinsically binds to the natural L-target, such as a naturally occurring protein (b).
To date, three Spieglemers have been described with a 40 kDa PEG at the 5’ end to slow in vivo clearance, which includes the anti-hepcidin lexaptepid pegol (NOX-H94), the anti-CCL2 emapticap pegol (NOX-E36), and the anti-CXCL12 olaptesed pegol (NOX-A12). All three have undergone phase 1 studies where they demonstrated good safety profiles in healthy volunteers. Both NOX-A12 and NOX-H94 have also yielded promising results in phase 2 studies. Additional trials are currently underway with a number of other Spiegelmers in the pipeline (reviewed in ref. 79).
New RNA polymerase variants yield 100% modified RNAs
As noted above, perhaps one of the most significant advancements towards improving the rate of discovering aptamers for clinical evaluation has come from the evolution/creation of novel polymerases that provide a means to incorporate a wider variety of sugar backbone modifications beyond the canonical 2’H and 2’OH. This renders the need for “backfilling”, as was done for pegaptanib, pegnivacogin and pegpleranib, obsolete and streamlines the process flow between selection and approval for clinical trials.
The first published study toward generating 100% modified RNA aptamers was performed by Keefe and colleagues at Archemix and made use of the Y639F variant of T7 RNA polymerase.80 Using optimized buffer conditions, they were able to produce transcripts composed of 2’OMe A, C, U, and G with a trace amount (6%) of 2’OH GTP to maintain transcription yields. Using pools composed of this cocktail, they identified an aptamer to VEGF, ARC245, which, when made synthetically of 100% 2’OMe RNA, maintained function displaying a KD of 1 nmol/l for VEGF165 (ref. 80). This is significantly worse than the KD reported for the 2’F modified t44 (10 pmol/l)25 or the almost fully modified Macugen (50 pmol/l),25 suggesting that while 2’OMe RNA is beneficial for stability, it may hinder affinity. Indeed, the same study noted that similar selections performed using 2’OH GTP instead of 2’OMe GTP yielded aptamers with picomolar binding affinities.80 In addition to its lower affinity, ARC245 also exhibited no measurable affinity for alternate human VEGF isoforms.80
A peek in the patent literature reveals that Archemix subsequently moved to using a different polymerase variant81 combining a Y639L mutation originally identified by Chelliserrykattil and Ellington82 with the H784A mutation originally described by Sousa and co-workers.83 Under optimized conditions, the enzyme was capable of producing increased levels of 100% 2’OMe RNA without the requirement for any 2’OH GTP. Most recently, improved variants of the Chelliserrykattil and Ellington polymerase variant RGVG82 were described by Meyer et al.84 By including a series of stabilizing mutations into the enzyme, they were able to further increase the yields of 100% 2’OMe RNA transcriptions to levels approaching those observed for the wild-type polymerases using unmodified nucleotides.84
Suffice it to say, there are now a number of T7 variants percolating through the community that are capable of synthesizing 100% modified RNAs. For example, Friedman et al.85 recently reported using the Y639L/H784A variant for the selection of aptamers targeting multidrug-resistant Staphylococcus aureus using a combination of 2’F GTP with 2’OMe A, C, and U.
Finally, with regard to polymerases, it is important to note that advances have not just been realized for RNA polymerases but for DNA polymerase as well. For example, Holliger and colleagues have now developed a variety of novel DNA polymerases capable of utilizing a range of nucleotide triphosphate substrates ranging from 2’F, 2’NH2, and 2’OMe ribose, to much more exotic cyclohexenyl, arabino, α-L-threofuranosy, and 2′-fluoroarabino nucleic acids (Figure 2d).86 More importantly, his group has used these to demonstrate the generation of so called xeno nucleic acid aptamers.86 Indeed, xeno nucleic acids may represent the future for aptamers as they have the potential to bring not only added stability and nuclease resistance, but also for novel properties, structures, and folds.
SOMAmers
Although their major focus has been on the development of a diagnostics platform, Somalogic has made significant advancements in the area of utilizing base modifications to give aptamers protein-like functionality. Their slow off-rate modified aptamers (SOMAmers) not only display improved binding affinities and binding kinetics (in particular, slow off-rates) when compared to traditional aptamers, but the inclusion of these modifications in their libraries significantly increased the selection “hit rate”.87,88
The linchpin to this functionality arises from replacing the dT residues within ssDNA aptamer libraries with a dU residue chemically modified at the 5-position of the base (Figure 5). With a variety of mostly hydrophobic modifications available at the 5-position, such as benzyl, napthyl and indole, a number of novel aptamers have been found for previously unselectable targets thanks to these useful modifications.89–91
Figure 5.
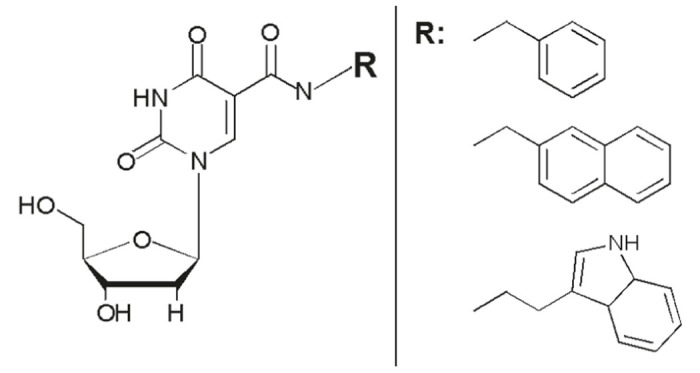
Modified deoxyuridine (dU) residues are at the core of the novel molecules developed by Somalogic. A variety of chemical moieties are attached to the 5-position of dU via a carboxyamide linkage (left). A variety of different modifications (R) have been employed for the selection of SOMAmers including benzyl, napthyl, and indole (right).
Thus, it is perhaps amusing that 10 years after the initial DNA selection against platelet-derived growth factor at NexStar, which resulted in Fovista, Janic and colleagues, now at Somalogic, utilized a benzyl-dU modified DNA library to select for anti-platelet-derived growth factor SOMAmers.89 The resulting SOMAmers were not only backfilled with 2’OMe residues to increase stability, but also subjected to a severe allergic reaction-like study (structure activity relationship) typically used for small molecule compounds to assess how altering the moiety on the modified dU affected function. The resulting molecule, SL5, tolerated 2’OMe stabilization in 4 of the 10 purines with a 20 pmol/l affinity (which the authors admit is approaching the detection limit of the assay), and IC50 for cellular phosphorylation of 50 pmol/l.89
The power of this chemical functionality has been further demonstrated through the discovery of a 32 nucleotide SOMAmer, SL1025, which binds IL-6 with 200 pmol/l binding affinity and exhibits very little nuclease degradation over a 48-hour incubation in human serum.90 Interestingly, the presence of the modified dU moiety was shown to be responsible for a >9-fold increase in the serum stability of these molecules when comparing SL1023 (an all-2’H variant of SL1025) with SL1023dT (a variant of SL1023 where modified dU was replaced with dT) (Table 1).90 A subsequent crystal structure led to the realization that a 16 nucleotide domain within the aptamer formed a G-quartet and bound the target with 270 nmol/l binding affinity. The authors then demonstrated the power of the severe allergic reaction-like approach by determining the appropriate combination of benzyl, phenyl, and napthyl groups within the domain, improving the affinity ~15-fold.92
Most recently, studies with the parent SL1025 have now been conducted in non-human primates.93 Using a rheumatoid arthritis model in cynomolgus monkeys, and SL1026, a variant of SL1025 bearing 40 kDa PEG, animals were dosed at 10 mg/kg q.i.d for 11 days. Treated animals displayed a reduction in IL-6 levels and arthritic symptoms over control. Of additional note, when compared to the clearance rate determined from a single injection of SL1026, the sustained dosing schedule resulted in SL1026 serum concentrations 1,000-fold higher than projected on the sixth day of treatment, indicating that the clearance mechanism for the aptamer may have become saturated. However, most importantly for SL1026 and other SOMAmers, despite the presence of the unnatural base modification on the 5 position of the deoxyuridine residues, SL1026 did not induce any detectable immune response in the monkeys. Though there has been no public movement toward trials to date, this work as well as work targeting platelet-derived growth factor leaves little doubt that we will see SOMAmers approved for clinical trials in the not too distant future.
Future and Conclusions
The advances described herein have facilitated the production of highly stabilized therapeutic aptamers that have advanced to clinical evaluation. Chemical modifications of the sugar-phosphate backbone, whether made synthetically by solid-phase synthesis, or enzymatically through mutant polymerases, have permitted the generation of aptamers that can withstand in vivo nucleases without inducing immunogenicity. Stereo-aptamers, Spiegelmers, bypass the nuclease and immunogenicity problem by making use of non-natural L-nucleotides. SOMAmers, in addition to incorporating modified ribose sugars, are decorated with moieties that impart chemical function beyond what standard RNA or DNA aptamers could ever achieve. Even though the core selection principle remains the same as the first-generation aptamers, these advances have made possible the rapid production of aptamers with significantly improved function and simultaneously reduced effort in postselection optimization.
It is important to note, the aptamers that have been evaluated in clinical trials thus far have primarily been for binding and inhibition of target function. Extrapolating from the progression of antibodies from use as inhibitors to carriers for the targeted delivery of therapeutics, the utilization of aptamers in a similar capacity is likely not far behind. For example, we and others have performed both laboratory as well as preclinical studies investigating the potential to use aptamers for the targeted delivery of cargoes ranging from small molecule drugs and toxins, to proteins, therapeutic oligonucleotides, and nanoparticles (reviewed in ref. 5). Indeed, further exploration of the patent literature reveals work from Archemix that was focused on developing optimized, minimized, and stabilized variants of the aptamer A9 for clinical delivery.94 A9, an aptamer originally reported by Lupold et al., targets the prostate-specific membrane antigen, a protein not just associated with prostate cancer, but found on the neovasculature of most solid tumors.94,95 Starting with a 2’F modified aptamer, the minimized molecule, ARC591, was back filled and stabilized using a combination of 2’OMe, 2’H, and a phosphorothioate linkage to yield a molecule (ARC1725) with a 20-hour degradation half-life in human serum, making the aptamer primed for eventual clinical testing (Table 1). We anticipate that it will not be long before aptamers engineered for delivery of therapeutics debut in clinical trials against prostate-specific membrane antigen and other targets.
With many advances in place, several aptamers in clinical trials, and others in the pipeline, the future of therapeutic aptamers looks extremely hopeful. Where the field progresses from here is uncharted; however, it seems clear the next generation of therapeutic aptamers will be generated more rapidly, encompass more functionality, and require less postselection stabilization than those that have or are currently undergoing clinical testing. Building from the existing advances, it would seem that these aptamers will combine enhanced functionality, as demonstrated by SOMAmers, with polymerases that can incorporate nuclease stabilizing ribose modifications, into the selection. Those advancements, considered with the modern ease of synthesizing oligonucleotides in vast numbers and in large scale, leaves aptamers poised to move toward clinical evaluation and eventual US Food and Drug Administration approval even more quickly.
Acknowledgments
We thank Amy (Amos) Yan for careful reading and editing of this review and Brad Hall for his perspective on state of aptamers in industry.
The authors declare no conflict of interest.
References
- Tuerk, C and Gold, L (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505–510. [DOI] [PubMed] [Google Scholar]
- Ellington, AD and Szostak, JW (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822. [DOI] [PubMed] [Google Scholar]
- Hall, B, Arshad, S, Seo, K, Bowman, C, Corley, M, Jhaveri, SD et al. (2009). In Vitro Selection of RNA Aptamers to a Protein Target by Filter Immobilization. John Wiley & SonsHoboken. pp. 24.3.1–24.3.27. [DOI] [PubMed] [Google Scholar]
- Yan, A and Levy, M (2014). Cell internalization SELEX: in vitro selection for molecules that internalize into cells. Methods Mol Biol 1103: 241–265. [DOI] [PubMed] [Google Scholar]
- Zhou, J and Rossi, JJ (2014). Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol Ther Nucleic Acids 3: e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi, J, Liu, Y, Rabbani, ZN, Yang, Z, Urban, JH, Sullenger, BA et al. (2010). In vivo selection of tumor-targeting RNA motifs. Nat Chem Biol 6: 22–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmostuk, M, Rimpelova, S, Gbelcova, H and Ruml, T (2015). Current approaches in SELEX: an update to aptamer selection technology. Biotechnol Adv 33(6 Pt 2): 1141–1161. [DOI] [PubMed] [Google Scholar]
- Keefe, AD, Pai, S and Ellington, A (2010). Aptamers as therapeutics. Nat Rev Drug Discov 9: 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, X, Castanares, M, Mukherjee, A and Lupold, SE (2011). Nucleic acid aptamers: clinical applications and promising new horizons. Curr Med Chem 18: 4206–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram, P, Kurniawan, H, Byrne, ME and Wower, J (2013). Therapeutic RNA aptamers in clinical trials. Eur J Pharm Sci 48: 259–271. [DOI] [PubMed] [Google Scholar]
- Kong, HY and Byun, J (2013). Nucleic Acid aptamers: new methods for selection, stabilization, and application in biomedical science. Biomol Ther (Seoul) 21: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafuzzaman, M (2014). Aptamers as both drugs and drug-carriers. Biomed Res Int 2014: 697923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia, BG, Rijo, P, Gonçalo, TS and Reis, CP (2015). Good manufacturing practices for medicinal products for human use. J Pharm Bioallied Sci 7: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wincott, FE (2001). Strategies for oligoribonucleotide synthesis according to the phosphoramidite method. Curr Protoc Nucleic Acid Chem Chapter 3: Unit 3.5. [DOI] [PubMed] [Google Scholar]
- Beaucage, SL and Caruthers, MH (2001). Synthetic strategies and parameters involved in the synthesis of oligodeoxyribonucleotides according to the phosphoramidite method. Curr Protoc Nucleic Acid Chem Chapter 3: Unit 3.3. [DOI] [PubMed] [Google Scholar]
- Dass, CR, Saravolac, EG, Li, Y and Sun, LQ (2002). Cellular uptake, distribution, and stability of 10-23 deoxyribozymes. Antisense Nucleic Acid Drug Dev 12: 289–299. [DOI] [PubMed] [Google Scholar]
- Morrissey, DV, Blanchard, K, Shaw, L, Jensen, K, Lockridge, JA, Dickinson, B et al. (2005). Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 41: 1349–1356. [DOI] [PubMed] [Google Scholar]
- Manoharan, M, Akinc, A, Pandey, RK, Qin, J, Hadwiger, P, John, M et al. (2011). Unique gene-silencing and structural properties of 2’-fluoro-modified siRNAs. Angew Chem Int Ed Engl 50: 2284–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekaite, L, Furset, G, Hovig, E and Sioud, M (2007). Gene expression analysis in blood cells in response to unmodified and 2’-modified siRNAs reveals TLR-dependent and independent effects. J Mol Biol 365: 90–108. [DOI] [PubMed] [Google Scholar]
- Judge, AD, Bola, G, Lee, AC and MacLachlan, I (2006). Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther 13: 494–505. [DOI] [PubMed] [Google Scholar]
- Cho, WG, Albuquerque, RJ, Kleinman, ME, Tarallo, V, Greco, A, Nozaki, M et al. (2009). Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc Natl Acad Sci USA 106: 7137–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil, F, Hemmi, H, Hochrein, H, Ampenberger, F, Kirschning, C, Akira, S et al. (2004). Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303: 1526–1529. [DOI] [PubMed] [Google Scholar]
- Kumagai, Y, Takeuchi, O and Akira, S (2008). TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev 60: 795–804. [DOI] [PubMed] [Google Scholar]
- Padilla, R and Sousa, R (1999). Efficient synthesis of nucleic acids heavily modified with non-canonical ribose 2’-groups using a mutantT7 RNA polymerase (RNAP). Nucleic Acids Res 27: 1561–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckman, J, Green, LS, Beeson, J, Waugh, S, Gillette, WL, Henninger, DD et al. (1998). 2’-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J Biol Chem 273: 20556–20567. [DOI] [PubMed] [Google Scholar]
- Barciszewski, J, Medgaard, M, Koch, T, Kurreck, J, and Erdmann, V (2009). Locked nucleic acid aptamers. In: Mayer G (ed.). Nucleic Acid and Peptide Aptamers, vol. 535. Humana PressNew York City. pp. 165–186. [DOI] [PubMed] [Google Scholar]
- Schmidt, KS, Borkowski, S, Kurreck, J, Stephens, AW, Bald, R, Hecht, M et al. (2004). Application of locked nucleic acids to improve aptamer in vivo stability and targeting function. Nucleic Acids Res 32: 5757–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, LS, Jellinek, D, Bell, C, Beebe, LA, Feistner, BD, Gill, SC et al. (1995). Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem Biol 2: 683–695. [DOI] [PubMed] [Google Scholar]
- Yang, X and Gorenstein, DG (2004). Progress in thioaptamer development. Curr Drug Targets 5: 705–715. [DOI] [PubMed] [Google Scholar]
- Healy, JM, Lewis, SD, Kurz, M, Boomer, RM, Thompson, KM, Wilson, C et al. (2004). Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm Res 21: 2234–2246. [DOI] [PubMed] [Google Scholar]
- Watson, SR, Chang, YF, O’Connell, D, Weigand, L, Ringquist, S and Parma, DH (2000). Anti-L-selectin aptamers: binding characteristics, pharmacokinetic parameters, and activity against an intravascular target in vivo. Antisense Nucleic Acid Drug Dev 10: 63–75. [DOI] [PubMed] [Google Scholar]
- Soutschek, J, Akinc, A, Bramlage, B, Charisse, K, Constien, R, Donoghue, M et al. (2004). Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432: 173–178. [DOI] [PubMed] [Google Scholar]
- Rusconi, CP, Roberts, JD, Pitoc, GA, Nimjee, SM, White, RR, Quick, G Jr et al. (2004). Antidote-mediated control of an anticoagulant aptamer in vivo. Nat Biotechnol 22: 1423–1428. [DOI] [PubMed] [Google Scholar]
- Willis, MC, Collins, BD, Zhang, T, Green, LS, Sebesta, DP, Bell, C et al. (1998). Liposome-anchored vascular endothelial growth factor aptamers. Bioconjug Chem 9: 573–582. [DOI] [PubMed] [Google Scholar]
- Gilbert, JC, DeFeo-Fraulini, T, Hutabarat, RM, Horvath, CJ, Merlino, PG, Marsh, HN et al. (2007). First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation 116: 2678–2686. [DOI] [PubMed] [Google Scholar]
- Vater, A, Sahlmann, J, Kröger, N, Zöllner, S, Lioznov, M, Maasch, C et al. (2013). Hematopoietic stem and progenitor cell mobilization in mice and humans by a first-in-class mirror-image oligonucleotide inhibitor of CXCL12. Clin Pharmacol Ther 94: 150–157. [DOI] [PubMed] [Google Scholar]
- Van Eijk, L, Swinkels, DW, John, A, Schwoebel, F, Fliegert, F, Summo, L, et al. (2013). Randomized double-blind placebo-controlled PK/PD study on the effects of a single intravenous dose of the anti-hepcidin Spiegelmer NOX-H94 on serum iron during experimental human endotoxemia. Critical Care 17: P352-P352. [Google Scholar]
- Ng, EW, Shima, DT, Calias, P, Cunningham, ET Jr, Guyer, DR and Adamis, AP (2006). Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat Rev Drug Discov 5: 123–132. [DOI] [PubMed] [Google Scholar]
- Tucker, CE, Chen, LS, Judkins, MB, Farmer, JA, Gill, SC and Drolet, DW (1999). Detection and plasma pharmacokinetics of an anti-vascular endothelial growth factor oligonucleotide-aptamer (NX1838) in rhesus monkeys. J Chromatogr B Biomed Sci Appl 732: 203–212. [DOI] [PubMed] [Google Scholar]
- Drolet, DW, Nelson, J, Tucker, CE, Zack, PM, Nixon, K, Bolin, R et al. (2000). Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm Res 17: 1503–1510. [DOI] [PubMed] [Google Scholar]
- Patel, M, Whitfield, L, Hutmacher, M, Kowalski, K, Burger, P, Dessalegn, B et al. (2006). Population pharmacokinetics/pharmacodynamics (PK/PD) of pegaptanib sodium (Macugen®) in patients with age–related macular degeneration (AMD). Investigative Ophthalmol Visual Sci 47: 2623. [Google Scholar]
- Ferrara, N, Hillan, KJ and Novotny, W (2005). Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun 333: 328–335. [DOI] [PubMed] [Google Scholar]
- Lowe, J, Araujo, J, Yang, J, Reich, M, Oldendorp, A, Shiu, V et al. (2007). Ranibizumab inhibits multiple forms of biologically active vascular endothelial growth factor in vitro and in vivo. Exp Eye Res 85: 425–430. [DOI] [PubMed] [Google Scholar]
- Semeraro, F, Morescalchi, F, Duse, S, Parmeggiani, F, Gambicorti, E and Costagliola, C (2013). Aflibercept in wet AMD: specific role and optimal use. Drug Des Devel Ther 7: 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi, CP, Scardino, E, Layzer, J, Pitoc, GA, Ortel, TL, Monroe, D et al. (2002). RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 419: 90–94. [DOI] [PubMed] [Google Scholar]
- Brooks, D and Rusconi, CP (2012). Method for manufacturing PEGylated oligonucleotides. Patent Application # 20120277419.
- Vavalle, JP and Cohen, MG (2012). The REG1 anticoagulation system: a novel actively controlled factor IX inhibitor using RNA aptamer technology for treatment of acute coronary syndrome. Future Cardiol 8: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimjee, SM, Keys, JR, Pitoc, GA, Quick, G, Rusconi, CP and Sullenger, BA (2006). A novel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol Ther 14: 408–415. [DOI] [PubMed] [Google Scholar]
- Bel, A, Borik, W, Davidson, S, Helies, JM, Stimmer, L, Fremes, S et al. (2016). Inhibition of factor IXa by the pegnivacogin system during cardiopulmonary bypass: a potential substitute for heparin. A study in baboons. Eur J Cardiothorac Surg 49: 682–689. [DOI] [PubMed] [Google Scholar]
- Dyke, CK, Steinhubl, SR, Kleiman, NS, Cannon, RO, Aberle, LG, Lin, M et al. (2006). First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation 114: 2490–2497. [DOI] [PubMed] [Google Scholar]
- Chan, MY, Rusconi, CP, Alexander, JH, Tonkens, RM, Harrington, RA and Becker, RC (2008). A randomized, repeat-dose, pharmacodynamic and safety study of an antidote-controlled factor IXa inhibitor. J Thromb Haemost 6: 789–796. [DOI] [PubMed] [Google Scholar]
- Chan, MY, Cohen, MG, Dyke, CK, Myles, SK, Aberle, LG, Lin, M et al. (2008). Phase 1b randomized study of antidote-controlled modulation of factor IXa activity in patients with stable coronary artery disease. Circulation 117: 2865–2874. [DOI] [PubMed] [Google Scholar]
- Vavalle, JP, Rusconi, CP, Zelenkofske, S, Wargin, WA, Alexander, JH and Becker, RC (2012). A phase 1 ascending dose study of a subcutaneously administered factor IXa inhibitor and its active control agent. J Thromb Haemost 10: 1303–1311. [DOI] [PubMed] [Google Scholar]
- Lincoff, AM, Mehran, R, Povsic, TJ, Zelenkofske, SL, Huang, Z, Armstrong, PW et al. REGULATE-PCI Investigators. (2016). Effect of the REG1 anticoagulation system versus bivalirudin on outcomes after percutaneous coronary intervention (REGULATE-PCI): a randomised clinical trial. Lancet 387: 349–356. [DOI] [PubMed] [Google Scholar]
- Verheugt, FW (2016). An anticoagulant too good to be true for revascularisation. Lancet 387: 314–315. [DOI] [PubMed] [Google Scholar]
- Ganson, NJ, Povsic, TJ, Sullenger, BA, Alexander, JH, Zelenkofske, SL, Sailstad, JM et al. (2015). Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J Allergy Clin Immunol. doi:10.1016/j.jaci.2015.10.034 (in press). [DOI] [PMC free article] [PubMed]
- Povsic, TJ, Vavalle, JP, Aberle, LH, Kasprzak, JD, Cohen, MG, Mehran, R et al. RADAR Investigators. (2013). A phase 2, randomized, partially blinded, active-controlled study assessing the efficacy and safety of variable anticoagulation reversal using the REG1 system in patients with acute coronary syndromes: results of the RADAR trial. Eur Heart J 34: 2481–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, LS, Jellinek, D, Jenison, R, Ostman, A, Heldin, CH and Janjic, N (1996). Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry 35: 14413–14424. [DOI] [PubMed] [Google Scholar]
- Floege, J, Ostendorf, T, Janssen, U, Burg, M, Radeke, HH, Vargeese, C et al. (1999). Novel approach to specific growth factor inhibition in vivo: antagonism of platelet-derived growth factor in glomerulonephritis by aptamers. Am J Pathol 154: 169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener, JL, Daniel Lagassé, HA, Duerschmied, D, Merhi, Y, Tanguay, JF, Hutabarat, R et al. (2009). Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost 7: 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilma-Stohlawetz, P, Gorczyca, ME, Jilma, B, Siller-Matula, J, Gilbert, JC and Knöbl, P (2011). Inhibition of von Willebrand factor by ARC1779 in patients with acute thrombotic thrombocytopenic purpura. Thromb Haemost 105: 545–552. [DOI] [PubMed] [Google Scholar]
- Markus, HS, McCollum, C, Imray, C, Goulder, MA, Gilbert, J and King, A (2011). The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: a randomized trial. Stroke 42: 2149–2153. [DOI] [PubMed] [Google Scholar]
- Siller-Matula, JM, Merhi, Y, Tanguay, JF, Duerschmied, D, Wagner, DD, McGinness, KE et al. (2012). ARC15105 is a potent antagonist of von Willebrand factor mediated platelet activation and adhesion. Arterioscler Thromb Vasc Biol 32: 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates, PJ, Kahlon, JB, Thomas, SD, Trent, JO and Miller, DM (1999). Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J Biol Chem 274: 26369–26377. [DOI] [PubMed] [Google Scholar]
- Reyes-Reyes, EM, Teng, Y and Bates, PJ (2010). A new paradigm for aptamer therapeutic AS1411 action: uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res 70: 8617–8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan, S, Chen, W, Spicer, EK, Courtenay-Luck, N and Fernandes, DJ (2008). The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res 68: 2358–2365. [DOI] [PubMed] [Google Scholar]
- Laber, D, Taft, B, Kloecker, G, Bates, P, Trent, J and Miller, D (2006). Extended phase I study of AS1411 in renal and non-small cell lung cancers. ASCO Annual Meeting Proceedings 24: 13098. [Google Scholar]
- Rosenberg, JE, Bambury, RM, Van Allen, EM, Drabkin, HA, Lara, PN Jr, Harzstark, AL et al. (2014). A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Invest New Drugs 32: 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, R, Stockerl-Goldstein, K, Cooper, M, Devetten, M, Herzig, R, Medeiros, B et al. (2009). Randomized phase II trial of the nucleolin targeting aptamer AS1411 combined with high-dose cytarabine in relapsed/refractory acute myeloid leukemia (AML). ASCO Annual Meeting Proceedings 27: 7019. [Google Scholar]
- Waters, EK, Genga, RM, Schwartz, MC, Nelson, JA, Schaub, RG, Olson, KA et al. (2011). Aptamer ARC19499 mediates a procoagulant hemostatic effect by inhibiting tissue factor pathway inhibitor. Blood 117: 5514–5522. [DOI] [PubMed] [Google Scholar]
- Dockal, M and Scheiflinger, F (2013). Aptamers to tissue factor pathway inhibitor and their use as bleeding disorder therapeutics. Patent Application #WO2012109675A1.
- Schaub, RG, Mcginness, K, Nelson, J, Genga, R, Waters, E, Kurz, JC, Diener, JL (2011). Aptamers to tissue factor pathway inhibitor and their use as bleeding disorder therapeutics. Patent Application # 20110098345.
- Waters, EK, Genga, RM, Thomson, HA, Kurz, JC, Schaub, RG, Scheiflinger, F et al. (2013). Aptamer BAX 499 mediates inhibition of tissue factor pathway inhibitor via interaction with multiple domains of the protein. J Thromb Haemost 11: 1137–1145. [DOI] [PubMed] [Google Scholar]
- Gissel, M, Orfeo, T, Foley, JH and Butenas, S (2012). Effect of BAX499 aptamer on tissue factor pathway inhibitor function and thrombin generation in models of hemophilia. Thromb Res 130: 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorczyca, ME, Nair, SC, Jilma, B, Priya, S, Male, C, Reitter, S et al. (2012). Inhibition of tissue factor pathway inhibitor by the aptamer BAX499 improves clotting of hemophilic blood and plasma. J Thromb Haemost 10: 1581–1590. [DOI] [PubMed] [Google Scholar]
- Dockal, M, Pachlinger, R, Hartmann, R, Knappe, S, Sorensen, B, Wong, WY, et al. (2012). Biological explanation of clinically observed elevation of TFPI plasma levels after treatment with TFPI-antagonistic aptamer BAX 499. Blood 120: 1104. [Google Scholar]
- Dockal, M, Hartmann, R, Knappe, S, Palige, M, Kammlander, W, Kunckova, K, et al. (2012). Effect of increased tissue factor pathway inhibitor (TFPI) levels on factor Xa inhibition and global hemostasis in the presence of TFPI-antagonistic aptamer BAX 499. Blood 120: 2207. [Google Scholar]
- Klussmann, S, Nolte, A, Bald, R, Erdmann, VA and Fürste, JP (1996). Mirror-image RNA that binds D-adenosine. Nat Biotechnol 14: 1112–1115. [DOI] [PubMed] [Google Scholar]
- Vater, A and Klussmann, S (2015). Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer(®) therapeutics. Drug Discov Today 20: 147–155. [DOI] [PubMed] [Google Scholar]
- Burmeister, PE, Lewis, SD, Silva, RF, Preiss, JR, Horwitz, LR, Pendergrast, PS et al. (2005). Direct in vitro selection of a 2’-O-methyl aptamer to VEGF. Chem Biol 12: 25–33. [DOI] [PubMed] [Google Scholar]
- Diener, JL, Dominic, KA, Kristin, T, Chunhua, W and Shuhao, Z (2012). Materials and methods for the generation of fully 2′-modified nucleic acid transcripts. Patent Application # 8105813.
- Chelliserrykattil, J and Ellington, AD (2004). Evolution of a T7 RNA polymerase variant that transcribes 2’-O-methyl RNA. Nat Biotechnol 22: 1155–1160. [DOI] [PubMed] [Google Scholar]
- Padilla, R and Sousa, R (2002). A Y639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucleic Acids Res 30: e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, AJ, Garry, DJ, Hall, B, Byrom, MM, McDonald, HG, Yang, X et al. (2015). Transcription yield of fully 2’-modified RNA can be increased by the addition of thermostabilizing mutations to T7 RNA polymerase mutants. Nucleic Acids Res 43: 7480–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, AD, Kim, D and Liu, R (2015). Highly stable aptamers selected from a 2’-fully modified fGmH RNA library for targeting biomaterials. Biomaterials 36: 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro, VB, Taylor, AI, Cozens, C, Abramov, M, Renders, M, Zhang, S et al. (2012). Synthetic genetic polymers capable of heredity and evolution. Science 336: 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaught, JD, Bock, C, Carter, J, Fitzwater, T, Otis, M, Schneider, D et al. (2010). Expanding the chemistry of DNA for in vitro selection. J Am Chem Soc 132: 4141–4151. [DOI] [PubMed] [Google Scholar]
- Gold, L, Ayers, D, Bertino, J, Bock, C, Bock, A, Brody, EN et al. (2010). Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 5: e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, DR, Gelinas, AD, Zhang, C, Rohloff, JC, Carter, JD, O’Connell, D et al. (2012). Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc Natl Acad Sci USA 109: 19971–19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S, Hirota, M, Waugh, SM, Murakami, I, Suzuki, T, Muraguchi, M et al. (2014). Chemically modified DNA aptamers bind interleukin-6 with high affinity and inhibit signaling by blocking its interaction with interleukin-6 receptor. J Biol Chem 289: 8706–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohloff, JC, Gelinas, AD, Jarvis, TC, Ochsner, UA, Schneider, DJ, Gold, L et al. (2014). Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids 3: e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas, AD, Davies, DR, Edwards, TE, Rohloff, JC, Carter, JD, Zhang, C et al. (2014). Crystal structure of interleukin-6 in complex with a modified nucleic acid ligand. J Biol Chem 289: 8720–8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, M, Murakami, I, Ishikawa, Y, Suzuki, T, Sumida, S, Ibaragi, S et al. (2016). Chemically modified interleukin-6 aptamer inhibits development of collagen-induced arthritis in cynomolgus monkeys. Nucleic Acid Ther 26: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener JL, Killough JR, Wagner-Whyte J, Wilson C and Zhu S (2006). Stabilized aptamers to PSMA and their use as prostate cancer therapeutics. Patent Application # Wo2006096754.
- Lupold, SE, Hicke, BJ, Lin, Y and Coffey, DS (2002). Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 62: 4029–4033. [PubMed] [Google Scholar]