

Abstract
A revolution is occurring in ecological and evolutionary genetics, driven by the development of techniques such as Restriction-site-Associated DNA sequencing (RADseq) that allow relatively low-cost discovery and genotyping of thousands of genetic markers for any species, including non-model species. Here we provide an overview of the diverse RADseq techniques that have been developed and highlight some of the research questions these powerful methods can be used to answer. We discuss how technical differences among the many variant methods lead to trade-offs in experimental design and analysis, and describe general considerations for designing a RADseq study.
Introduction
The development of Restriction site-Associated DNA Sequencing (RADseq) was deemed among the most significant scientific breakthroughs within the last decade1. RADseq has fueled studies in ecological, evolutionary, and conservation genomics by harnessing the massive throughput of next-generation sequencing to uncover hundreds or thousands of polymorphic genetic markers across the genome in a single, simple, and cost-effective experiment2,3. Like other reduced-representation sequencing approaches, RADseq targets a subset of the genome, thus providing advantages over whole-genome sequencing including greater depth of coverage per locus (and therefore improved confidence in genotype calls) and sequencing of greater numbers of samples for a given budget. Unlike many other methods for generating genome-wide data, RADseq does not require any prior genomic information for the taxa being studied. Consequently, RADseq has become the most widely used genomic approach for high-throughput SNP discovery and genotyping in ecological and evolutionary studies of non-model organisms.
The widespread adoption of RADseq has sparked further innovation of the core RADseq technique (BOX 1). Numerous variations promise to increase flexibility (e.g., in the number of loci assayed) and decrease cost and effort in ecological and evolutionary genomics studies. However, technical differences among the methods lead to important considerations for all steps of genomic studies, from costs of library preparation and sequencing, to the types of bias and error inherent in the resulting data, to the types of scientific questions that can be addressed. A comprehensive review of RADseq methods is thus critically needed to aid researchers in choosing an approach and avoiding erroneous scientific conclusions from RADseq data, a problem that has plagued other new marker types in the past4–6.
Box 1. Common RADseq-related techniques.
I. Sequence adjacent to single restriction enzyme cut sites
Original RAD10,64 digests genomic DNA with one restriction enzyme, followed by mechanical shearing to reduce fragments to the appropriate length for sequencing, which (unlike other methods) creates variance in fragment sizes at each locus.
2bRAD15,65 uses type IIB restriction enzymes, which cleave DNA upstream and downstream of the recognition site, resulting in short fragments of uniform length (33–36bp).
II. Sequence fragments flanked by two restriction enzyme cut sites
a. Single enzyme, indirect size selection
Genotyping by Sequencing (GBS)66 uses a common-cutter enzyme and PCR preferentially amplifies short fragments.
Sequence-based Genotyping (SBG)67 uses a rare-cutter and one or two common-cutters and PCR preferentially amplifies short fragments
b. Double enzyme, indirect size selection
Complexity Reduction of Polymorphic Sequences (CRoPS)68 uses two enzymes and a proprietary library preparation kit (originally developed for 454 pyrosequencing).
c. Single enzyme, direct size selection
Reduced Representation Libraries (RRL)11,69 are unique in using a blunt-end common-cutter enzyme, followed by a size selection step and a proprietary Illumina library preparation kit.
Multiplexed shotgun genotyping (MSG)55 uses one common-cutter enzyme and a size selection step.
ezRAD16 uses one or more common-cutter enzymes, and a proprietary kit for Illumina library preparation.
d. Double enzyme, direct size selection
Double-digest RAD (ddRAD)14 uses two restriction enzymes, with adaptors specific to each enzyme, and size selection by automated gel cut.
Variations on the above techniques include using methylation-sensitive enzymes70; adding more restriction enzymes to existing protocols to further reduce the set of loci67,71; adding a second digestion to eliminate adaptor dimers18; adapting RADseq techniques to other sequencing platforms such as Ion Torrent71–73; and other minor technical modifications56,74.
Figure within BOX 1.
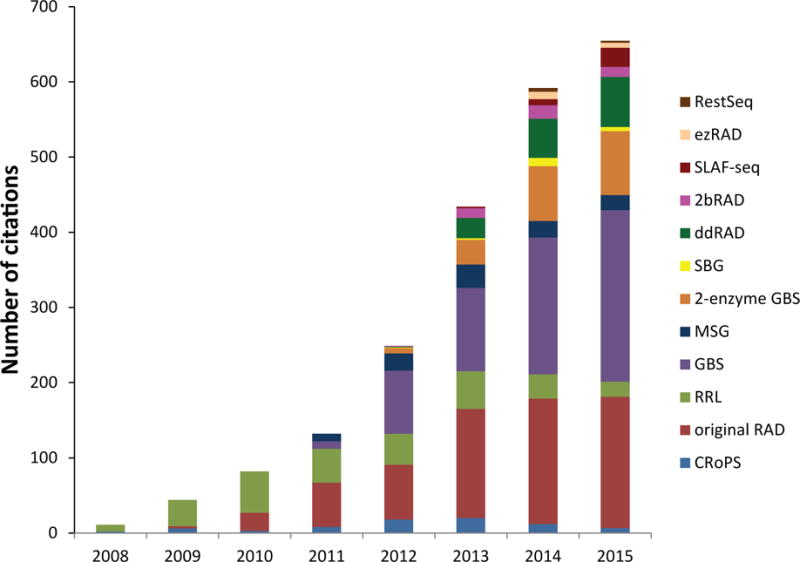
Numbers of articles citing the original papers describing each RADseq protocol over time. Data for 2015 are extrapolated using numbers of articles cited from January through September 2015. Protocols are arranged by order of first appearance in the literature. Data generated using Web of Science.
The core feature of RADseq techniques is the use of restriction enzymes to obtain DNA sequence at a genome-wide set of loci. Restriction enzymes have long been utilized to sample loci across the genome and generate information on population-level variation7,8, including genome-wide surveys for genetic variation in humans9. Whereas these previous techniques focused on polymorphisms within restriction cut sites or used Sanger sequencing, RADseq uses next-generation sequencing to generate sequence data adjacent to a large number of restriction cut sites10–12. RADseq loci can occur in all areas of the genome (i.e. coding and non-coding regions), and individuals within or between closely related species generally share most loci due to conservation of cut sites. All RADseq methods are broadly applicable across a wide range of taxa and scientific questions (BOX 2). However, some techniques have been used more widely in certain systems, largely due to historical contingencies rather than relative suitability of the various approaches to different species (e.g. CRoPS, GBS, and RRL have been used primarily in agricultural species13).
Box 2. Ecological and evolutionary insights from RADseq data.
Exemplar studies illustrate ecological, evolutionary, and conservation related questions that can be answered using RADseq.
Inbreeding and genomic diversity
A study investigating heterozygosity-fitness correlations in seals found that genome-wide heterozygosity estimated using RADseq had a nearly fivefold higher correlation with fitness than did 27 microsatellite loci52. RADseq genomic diversity estimates were also used to characterize the influence of social structure on autosome vs. sex chromosome diversity in Tonkean macaque monkeys75.
Effective population size (Ne)
Thousands of SNPs generated using RADseq were used to estimate Ne in salmon and smelt from western North America76,77.
Population structure, phylogeography, and conservation units
RADseq was used to develop a population-informative SNP panel to monitor stock composition in salmon and delineate population units to harvest as discrete rather than mixed stocks76,78; see also Emerson et al. 201079, Combosch & Vollmer 201580, Gaither et al. 201581.
Introgression
Hohenlohe et al.20 used RADseq to identify 3180 species-diagnostic SNPs and calculate admixture between a native and an invasive trout species; see also Eaton & Ree 201382, Ford et al. 201583.
Genomics of adaptation
A study using GWAS and FST outlier tests of RADseq data from two butterfly species in four parallel hybrid zones found that genomic regions harboring genes controlling color pattern were the most divergent between the two species49; see also Hohenlohe et al. 201025, Chutimanitsakun et al. 201184, Ruegg et al. 201450.
Phylogenomics
RADseq data generated a highly resolved tree for 16 species of Lake Victoria cichlid fish, whereas previous analyses using AFLP, microsatellites, or a handful of sequence-based markers failed to resolve species level relationships for these species85.
Note that we use the term RADseq here to refer to any of several related methods that rely on restriction enzymes to determine the set of loci that will be sequenced in a reduced-representation library. The term “RAD” was originally used to describe one particular method10, but has subsequently been used to describe a range of methods14–16. “Genotyping-by-sequencing” (GBS) has also been used to describe these methods17, but we use the term “RADseq” here because the methods we review are united by their use of restriction enzymes, which is captured in the RADseq acronym. Although we focus primarily on RADseq applications to ecological and evolutionary genetics in natural populations (BOX 2), much of our discussion is also relevant to other RADseq applications, such as trait-mapping in agricultural species13.
The RADseq family of methods
RADseq techniques share several basic steps (FIG. 1). All methods start with relatively high molecular weight genomic DNA18 and begin by digesting it with one or more restriction enzymes. All methods add specific sequencing adapters, or double-stranded oligonucleotides, that are required by all next-generation sequencing platforms. Adapters added during RADseq protocols may contain barcodes – short unique sequences generally 6–12bp – that are used to identify individual samples that are sequenced together (multiplexed) in a single library. Depending on the enzyme(s) used, RADseq protocols also reduce and/or select sizes of DNA fragments optimal for next-generation sequencing.
Figure 1.
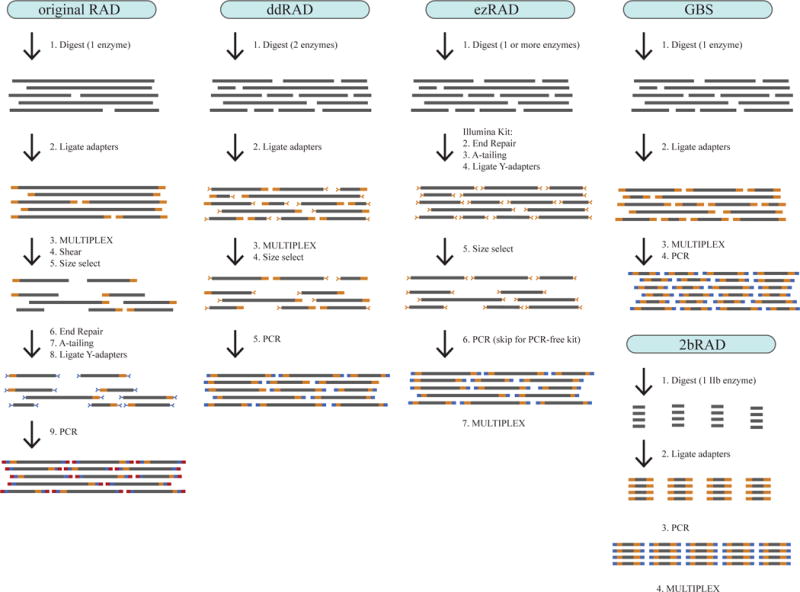
Step-by-step illustration of five RADseq library prep protocols. All protocols begin by digesting relatively high quality genomic DNA with one or more restriction enzymes. For most protocols, the sequencing adapters (oligos) are added in two stages, with one set of oligos added during a ligation step early in the protocol, and a second set of oligos incorporated during a final PCR step. The second set of oligos extends the length of the total fragment to produce the entire Illumina adapter sequences. In contrast, the original RADseq adds adapters in three stages. For Illumina sequencing, the adapters on either end of each DNA fragment must differ, and therefore some protocols (e.g. original RADseq, ddRAD, ezRAD) use “Y-adapters” that are structured to ensure that only fragments with different adapters on either end are PCR-amplified (illustrated here as Y-shaped adapters). Other protocols (e.g. GBS) simply rely on the fact that fragments without the correct adaptors will not be sequenced. To generate fragments of an ideal length for sequencing, most methods use common-cutter enzymes (e.g. 4–6bp cutters) to generate a wide range of fragment sizes, followed by a direct size selection (gel-cutting or magnetic beads, e.g. ddRAD, ezRAD) or an indirect size selection (as a consequence of PCR amplification or sequencing efficiency, e.g. GBS). In contrast, the original RADseq uses a mechanical shearing step to produce fragments of an appropriate size, and incorporates a size selection step only to increase Illumina sequencing efficiency and remove adapter dimers. 2bRAD uses IIB restriction enzymes to produce small fragments of equal size across all loci (33–36bp).
RADseq methods differ in the order and details of enzyme digestion, adapter ligation, barcoding, and size selection, as well as type of sequence data that can be produced at each locus. These differences can be used to place techniques into major groups (BOX 1). Below we discuss important variations among methods at each step and some of the consequences for library preparation, the resulting data, and subsequent bioinformatic analyses.
Starting genomic DNA
RADseq techniques have been optimized based on starting material comprised of high molecular weight genomic DNA, and thus these techniques may perform poorly with highly degraded genomic DNA18. For example, in methods without enzyme-specific adaptors (e.g., ezRAD, CRoPS), smaller fragments of starting genomic DNA not adjacent to cut sites may end up in the sequencing library, thus wasting sequencing effort on non-RAD loci. The original RADseq technique10 also requires higher-molecular-weight DNA than other methods, because the mechanical shearing step is most consistent and efficient with relatively large fragments remaining after enzyme digestion (see below).
In general, more starting DNA is often beneficial, as it may reduce the number of PCR cycles required and thus reduce the problem of PCR duplicates (see below). Some of the protocols originally recommended fairly large amounts of DNA (up to 1 μg per sample for original RAD19 or 5.5 μg for RRL11); however, most RADseq methods are somewhat flexible in the total amount of DNA required per sample, and can often be implemented with 50–100 ng of DNA per sample. One exception would be when using a PCR-free library preparation method that requires larger amounts of starting DNA, as in one implementation of ezRAD16.
Restriction enzyme digestion
RADseq protocols differ in the number of restriction enzymes used and the frequency with which these enzymes cut the genome, with “common-cutters” defined as restriction enzymes that cut more frequently than “rare-cutters,” generally a result of the length of their recognition sequence. Techniques also fall into two major groups depending on how the set of loci sequenced relates to the distribution of enzyme cut sites across the genome. The original RADseq protocol and 2bRAD aim to produce sequence data at all cut sites for the restriction enzyme. In contrast, all other techniques depend on sequencing of genomic fragments produced by two enzyme cut sites separated by a specified genomic distance (typically 300–600bp apart, with the distance determined by direct or indirect size selection; see below). These cut sites may be from the same or different enzymes, depending on whether the method uses one or two enzymes. For each method, common-cutter or rare-cutter enzymes can be used to tailor the number of loci produced. For example, for the original RADseq protocol, a very rough estimate is that an 8-cutter will cut every 4ˆ8 = 65,536bp, while a 6-cutter will cut every 4ˆ6 = 4,096bp; this calculation can be adjusted to account for the GC content of the recognition sequence and the genome under study (Davey et al. 2011).
Adapter ligation
RADseq techniques differ in how adapters are constructed and ligated to DNA fragments, and also how they are designed to ensure that only the target genomic DNA fragments (i.e. those adjacent to restriction cut sites) are sequenced. In some cases, adapters are designed to ligate only at the characteristic single-stranded sticky end that is left at restriction cut sites after digestion. Many Illumina sequencing-based RADseq protocols also use “Y-adapters” that are structured to ensure that only fragments with the adapter combinations required for sequencing are PCR amplified (FIG 1). Some techniques adopt proprietary library preparation kits for adapter ligation (e.g. ezRAD, CRoPS, RRL), which may increase the reliability as well as the cost of reagents for library construction. Using adapters from proprietary kits can also lead to lower specificity in ligation, because these adapters do not ligate to the sticky ends, and therefore sequence data could be generated from fragments of degraded DNA not adjacent to restriction cut sites16.
Size selection
For most protocols, the restriction digest reduces genomic DNA to a wide range of fragment lengths, and then a size selection step is used to isolate fragments of ideal lengths for sequencing. This leads to key distinctions among RADseq protocols (BOX 1): for all the methods that sequence DNA fragments flanked by two cut sites, the set of loci to be genotyped is further reduced by this size selection, because each potential locus has a characteristic fragment size determined by the distance between cut sites. In these techniques, size selection is done either indirectly, as a consequence of PCR amplification or sequencing efficiency (e.g. GBS, CRoPS), or directly, using manual or automated gel cutting techniques or magnetic beads (e.g. RRL, MSG, ezRAD, ddRAD). For these methods, consistency of size selection across libraries is critical for producing data on a comparable set of loci across samples; inconsistency can lead to different sets of loci appearing in different libraries, resulting in wasted sequencing effort and high levels of missing genotypes.
In contrast, original RADseq and 2bRAD do not use size selection to reduce the set of loci to be sequenced; instead, all loci adjacent to restriction cut sites are targeted by these two methods. The original RADseq follows digestion by a single enzyme with a mechanical shearing step to produce fragments appropriate for Illumina sequencing. This means that each sequenced fragment has a cut site on one end and a randomly sheared end on the other, and a distribution of fragment sizes is produced at each locus. As a result, the size selection step does not further reduce the set of loci, but is used only to optimize Illumina sequencing efficiency and remove adapter dimers. The 2bRAD method is unique among the RADseq protocols in that it uses IIB restriction enzymes to produce short fragments that are of equal size across all loci (33–36 bp).
Barcoding
Use of barcodes built into the adapters allows multiplexing of individual samples early in library preparation for some of the protocols (this is sometimes called “pooling,” but should not be confused with pooling of individuals into one barcode; BOX 3). During library preparation, as soon as barcoded adapters are ligated to each sample, the samples can be multiplexed, which can greatly reduce the time and expense of subsequent steps in studies with large numbers of samples. Multiplexing of samples early in the library preparation requires the use of “in-line” barcodes, which are short sequences (typically 6–12bp) immediately adjacent to the genomic DNA. Adapters from proprietary kits do not have in-line barcodes, and therefore custom-made adapters are required for in-line barcoding. Many techniques can also be used with combinatorial barcoding, in which DNA fragments from each sample are identified by a unique combination of two different identifiers, typically one in-line barcode, and one Illumina index (6–8bp located near the middle of the adapter) added at the PCR stage to the opposite end of the DNA fragment (e.g. Peterson et al.14). An alternative combinatorial barcoding strategy would be to use two Illumina indexes, one on each side of the DNA fragment. However, this strategy would not allow multiplexing of samples early in the library preparation. Another alternative would be to use in-line barcodes on both sides of the DNA fragment; however, this strategy would be redundant because all Illumina libraries have at least one index, and would also waste sequencing effort on a redundant inline barcode. Combinatorial barcoding decreases the total number of adapters required to distinguish individual samples, so for instance a set of 24 barcoded adapters and 16 indexes can uniquely identify 384 samples in a sequencing lane.
Box 3. Pooling.
Pooling of samples without individual barcoding during RADseq library prep can allow estimation of population allele frequencies at reduced cost86–88. However, several sources of error are unique or magnified for pooled sequencing. Unequal representation of DNA from individual samples could lead to inaccurate allele frequency estimates89,90, and PCR duplicates will amplify this problem88. In addition, identification of allele dropout, paralogs, mapping errors, and hidden population structure is more difficult or impossible for pooled data86,88. Similarly, distinguishing sequencing error from low-frequency alleles is more difficult for pooled data.
Errors caused by unequal representation of individual samples in pooled sequencing libraries can be substantially reduced by using large per-pool sample sizes and depth of coverage, and removal of PCR duplicates88,91,92. The prevalence of PCR duplicates can be reduced by using a small number of PCR cycles, which should be feasible for pooled sequencing with a large starting amount of genomic DNA. Generating and comparing sequence data for replicate pools for each population can also help identify and correct for unequal representation of individual samples88. However, this does not mitigate problems with identifying paralogs or allele drop-out.
Researchers should also be aware of restrictions in analyses that can be conducted with pooled sequence data. Analyses requiring individual genotypes are not possible with this type of data, such as assignment tests (e.g. Bayesian clustering analyses with STRUCTURE93), relatedness tests, or estimates of inbreeding coefficients. Several approaches for inferring population history or detecting selection depend on accurate estimates of linkage disequilibrium (LD)94,95, and while there is limited power to estimate LD with the unphased data that results from individually-barcoded RADseq data, it is not possible at all with pooled data. More fundamentally, pooling assumes that all samples in a pool are from a single well-mixed population, and cryptic population structure will be obscured if multiple groups are unknowingly combined within a pool.
Type of sequence data
Most RADseq techniques currently use Illumina sequencing. Illumina machines offer a range of sequence read lengths (currently 50 to 300bp, and likely to increase further) and also the option of either single-end sequencing, which produces one “forward” read per DNA fragment, or paired-end sequencing, which produces one “forward” read and one “reverse” read per fragment. These options can be applied to all RADseq libraries, although paired-end sequencing would not be beneficial for 2bRAD, which produces very short fragments (33–36bp). For all other methods, forward reads begin from the restriction enzyme cut site, and longer reads typically capture more genomic sequence. For all methods that target loci flanked by two cut sites, reverse reads begin at the second cut site, and therefore these reads will align at identical locations in the genome for each locus. In contrast, paired-end sequencing using the original RADseq protocol produces a very different type of data. While the forward reads begin at the cut site, the reverse reads start from the randomly sheared end, typically 400–700bp away. Therefore the reverse reads at any given locus are staggered20, and these data can be used to assemble long contigs, for example as long as 1kb if library fragments are tailored to be this length19,21. These RAD contigs allow for better identification of paralogs22, provide more sequence for BLAST searching of functionally important loci20, and could provide haplotype data for genealogical or phylogenetic analysis. Longer contig sequences also allow for the design of PCR primers or sequence capture probes to target loci of interest for further study23,24.
For all methods, read pairs produced by paired-end sequencing may overlap depending on read length and fragment size range, so that if fragments are less than 200–300bp long (e.g., some fragments produced using GBS with a common-cutter enzyme), increasing read lengths or using paired-end sequencing may not gain any genomic sequence information. However, overlapping read pairs may be used to improve genotyping accuracy by increasing depth of coverage toward the ends of the reads, which tend to have higher rates of sequencing error.
Bioinformatic analyses
Post-sequencing analyses will generally share several basic steps for data generated using all RADseq methods. Initial analyses will include de-multiplexing and trimming of barcodes (if present), filtering reads based on the presence of the expected restriction enzyme cut site and sequence quality, and possibly trimming if quality declines toward the end of reads. For some RADseq methods, PCR duplicates can be removed during initial analyses to improve downstream genotyping accuracy (see below). If a reference genome is available, loci can then be identified by alignment of sequence reads to the reference. Alternatively, loci can be assembled de novo by clustering similar sequence reads together and assuming that variation among reads at a locus represents either sequencing error or allelic variation. After locus discovery, long contigs can be generated for paired-end data generated using the original RADseq (see above). Genotyping can be conducted using maximum likelihood25 or Bayesian approaches26,27; maximum likelihood methods may require higher depth of coverage than Bayesian methods, particularly if Bayesian approaches make use of population-level allele frequencies to set prior probabilities on genotypes.
Several programs designed specifically for analyzing RADseq data are available (e.g., Stacks28, pyRAD29, UNEAK30, in addition to other publicly available scripts and pipelines). Stacks contains a number of flexible modules to conduct all parts of the analysis, from quality filtering to locus identification (either reference-aligned or de novo) to genotyping and calculating population genetic statistics. pyRAD, designed specifically for phylogenetic applications, conducts quality filtering and de novo locus identification and genotyping, with the advantage that it can handle insertion-deletion variation among alleles and may thus be better suited to studies with a broader taxonomic scale. UNEAK is part of the TASSEL pipeline for association mapping with GBS data31 and applies a network-based SNP detection algorithm, but is somewhat less flexible than other software in aspects such as read trimming and parameters for de novo locus identification. RADseq data can also be analyzed using more generic software tools for quality filtering, alignment to a reference genome, and genotyping. Following genotyping, further filtering is typically recommended to remove loci and/or individual samples with large proportions of missing data. The appropriate level of filtering at this stage depends on the study goals and the subsequent analyses to be conducted, as these vary in their sensitivity to missing data and sample size of individuals and loci. Several recent publications have highlighted how the details of RADseq data analysis, particularly the parameters used in de novo locus identification, can profoundly affect analytical results32–34. Some of this work provides explicit recommendations for how to apply bioinformatic tools to RADseq data. Overall, it is critical for researchers to vary the parameters used in all steps of the analysis, from quality filtering to locus identification and genotyping, to critically evaluate the sensitivity of the results and optimize the analysis depending on the study goals.
Sources of Error and Bias
RADseq methods share some sources of sequencing and genotyping errors with all next-generation sequencing methods35. However, there are also several unique potential sources of error and bias in RADseq methods, the impact of which can vary across library preparation protocols and statistical analyses.
Allele dropout and null alleles
Allele dropout manifests in RADseq when a polymorphism occurs at a restriction enzyme recognition site, resulting in a failure to cut the genomic DNA at that location. Alleles lacking the complete recognition site will not be sequenced and are therefore “null alleles.” If a SNP occurs within a null allele, failure to sequence the allele could cause genotyping errors, with individuals heterozygous for the null allele appearing as homozygotes. The absence of a restriction cut site could also drive allele dropout for loci at neighboring cut sites, because the post-digestion fragment lengths may fall outside the selected size range for methods that use size selection to reduce the set of loci (FIG. 3A).
The frequency of allele dropout will increase with the cumulative length of the restriction enzyme recognition sites, simply due to an increase in the probability of mutations in longer sequences36. Simulation studies also indicate that allele dropout will increase with overall levels of polymorphism in the study system, and will have a greater impact on data generated by ddRAD than original-RAD because loci depend on the presence of two cut sites rather than one36,37.
Genotyping errors caused by allele dropout can bias population genetic statistics through underestimation of genomic diversity, overestimation of FST, and an increase in false positives and negatives in FST outlier tests36,37. However, there is evidence that the impact of these biases may be limited unless effective population sizes are large (Ne>105)36. FST biases can be largely compensated by removing loci with null alleles from the dataset. In theory, loci with null alleles should be identifiable by high variance in depth of coverage across individual samples, as some individuals will lack one or both copies at the locus. However, many other factors cause variance in depth of coverage (see below), so it is not always a reliable indicator of null alleles. Nevertheless, loci with a high prevalence of null alleles will be removed by many standard filtering practices that retain only loci that are successfully genotyped across some minimum percentage of individual samples. Although removal of loci with null alleles should largely compensate for biased FST estimates, it may do little to compensate for biased diversity estimates. Loci with null alleles are expected to occur more frequently in genomic regions with higher mutation rates and/or levels of standing genetic diversity, and thus the absence of these loci from the dataset will tend to lead to systematic underestimation of overall genomic diversity37.
PCR duplicates and genotyping errors
Most next-generation sequencing library preparation protocols have a PCR step during which clonal DNA fragments (“PCR duplicates”) are generated from the original genomic DNA fragments (“parent fragments”)38,39. During PCR, stochastic processes can cause one allele to amplify more than the other at a given locus in an individual sample. This potential skew can lead to downstream genotyping errors because heterozygotes can appear as homozygotes, or alleles containing PCR errors can appear as true alleles (FIG 3B). Studies report that PCR duplicates can occur at high frequencies in RADseq data (e.g. 20–60% of reads20,38,39). In theory, PCR should not systematically favor one allele over another at a given locus, and therefore parameters estimated from a large number of loci are unlikely to be substantially biased. However, analyses requiring high genotyping accuracy at individual loci, such as outlier tests or parentage assignments, could produce erroneous results if PCR duplicates are present.
For sequence data generated using most next-generation sequencing protocols, PCR duplicates can be identified and removed bioinformatically to improve genotyping accuracy. This is possible in protocols that have a mechanical or random enzymatic fragmentation step, so that PCR duplicates can be identified as fragments that start and end at identical positions in the genome. This method can also be used to identify PCR duplicates in sequence data generated using original RADseq with paired-end sequencing, because of the mechanical shearing step (FIG. 3B). In some circumstances (when the distance between forward and reverse reads is very short or local coverage is very high), this filter will remove fragments that are not duplicates but that, by chance, have the same start and end points. However, this should occur rarely and should be conservative with respect to genotyping accuracy. This method cannot be used to identify PCR duplicates in any other RADseq protocols, because all fragments for a given locus will have identical start and stop positions for these protocols2.
Another recently developed method shows promise for identifying PCR duplicates through the use of degenerate base regions within the sequencing adapters to tag parent fragments prior to PCR39–41. This method could be incorporated into any protocol that uses custom-designed adaptors. An alternative method for dealing with PCR duplicates is to eliminate the PCR step of library prep altogether, as in ezRAD with Illumina PCR-free kits16. However, PCR-free kits are currently much more expensive and require much more genomic DNA (1μg) than other RADseq protocols.
Variance in depth of coverage among loci
Whereas PCR duplicates and allele dropout can cause genotyping errors as a result of preferential sequencing of certain alleles within RADseq loci, several other phenomena can cause preferential sequencing of certain loci over other loci. These phenomena should not cause genotyping errors, but will require greater overall sequencing effort to obtain sufficient depth for the loci that sequence less commonly. One well-known phenomenon is preferential amplification of fragments based on GC content during PCR2,42–44, and this bias should equally affect all RADseq methods that include a PCR step. Another is the preferential amplification of shorter fragments over longer fragments. This issue will affect all RADseq methods that sequence fragments flanked by two cut sites (BOX 1), because each locus has a characteristic fragment length. This issue will not affect 2bRAD because all loci are uniform in length, and will not affect the original RADseq because each locus is represented by a variety of fragment lengths (see above).
Another phenomenon influencing variance in depth of coverage among loci is driven by the mechanical shearing step in the original RAD. Fragments <10kb shear with lower efficiency, and therefore loci originating from shorter restriction fragments will yield fewer reads than loci originating from longer fragments42. However, this phenomenon should have less influence on the majority of original RADseq studies, which typically use rare-cutters that digest genomic DNA to fragments >10kb.
When coverage varies widely among loci, obtaining sufficient numbers of reads to accurately genotype the low-coverage loci will require an increase in the average depth of coverage across all loci. To accomplish this, the number of individuals multiplexed per sequence lane must be decreased, and this will increase the cost of the research project or decrease the number of individual samples that can be analyzed. Alternatively, low-coverage loci could simply be removed from the dataset if sufficient data can be obtained from high-coverage markers, and this is commonly done in practice.
How to design a RADseq study
Designing a RADseq study for a particular application requires several major considerations regarding the most appropriate RADseq method, sampling and sequencing strategies, budget, and other methodological details. Trade-offs among selected methods are summarized in Table 1.
TABLE 1.
Summary of trade-offs among five RADseq methods.
| Original RAD | 2bRAD | GBS | ddRAD | ezRAD | |
|---|---|---|---|---|---|
| Options for tailoring number of loci | Change restriction enzyme | Change restriction enzyme | Change restriction enzyme | Change restriction enzyme or size selection window | Change restriction enzyme or size selection window |
| Number of loci per 1 Mb of genome size* | 30–500 | 50–1000 | 5–40 | 0.3–200 | 10–800 |
| Length of single-end loci | ≤1kb if building contigs; otherwise ≤300bp** | 33–36bp | <300bp** | ≤300bp** | ≤300bp** |
| Cost per barcoded/indexed sample | Low | Low | Low | Low | High |
| Effort per barcoded/indexed sample | Medium | Low | Low | Low | High |
| Uses proprietary kit? | No | No | No | No | Yes |
| Can identify PCR duplicates? | with paired-end sequencing | No | with degenerate barcodes | with degenerate barcodes | No |
| Specialized equipment needed | Sonicator | None | None | Pippin Prep*** | Pippin Prep*** |
| Suitability for large or complex genomes**** | good | poor | moderate | good | good |
| Suitability for de novo locus identification (no reference genome)***** | good | poor | moderate | moderate | moderate |
| Available from commercial companies (in 2015) | Yes | No | Yes | Yes | No |
Estimated as follows: original RAD, assuming either a 6-cutter or 8-cutter; 2bRAD, assuming type IIB enzymes with recognition sites containing 5–7 specific nucleotides; GBS, values from Elshire et al.66; ddRAD, from Table 1 in Peterson et al14 and allowing for up to double the size range; ezRAD, values from Toonen et al16 for species with reference genomes.
Based on current limits in sequencing technology
Can alternatively be used with standard gel equipment
Based on ability to reduce total number of loci and lengths of loci
Based on lengths of loci to distinguish paralogs and duplicate sequence
Number of loci
The number of loci identified and genotyped by RADseq methods depends on the genome size, the frequency of the restriction cut sites in the genome, and the number of cut sites targeted for sequencing. Computational tools are available to estimate the number of loci expected for each protocol43,45. RADseq methods that target all cut sites (original RAD and 2bRAD) or use common-cutter enzymes (GBS) without a direct size selection step generally provide more loci, but the number can be adjusted by the choice of enzyme. In contrast, protocols involving an explicit size-selection step (e.g. ddRAD, ezRAD) can not only adjust the number of loci by choice of enzyme(s), but also by changing the size range selected, and thus they typically have more flexibility to provide a smaller number of loci. Alternatively, another way to reduce the number of loci in any RADseq protocol is to design probes for a subset of RADseq loci and use these to capture and sequence selected loci (i.e., “RAD Capture” or “Rapture,” unpublished data, see Further Information).
The optimal number of loci depends on the goals of the study. Studies focused on estimating neutral or genome-wide processes, such as phylogenetic relationships, geographic population structure, gene flow, introgression, or individual inbreeding (identity by descent) often require only several hundred to a few thousand SNP-containing RADseq loci to adequately sample the genome20,46–48. In contrast, studies seeking to characterize functionally important regions across the entire genome, such as those exhibiting signatures of selection, require a larger set of markers (e.g., up to tens or even hundreds of thousands of RADseq loci)25,49,50. In mapping studies the optimal number of RADseq loci depends on the expected extent of linkage disequilibrium along chromosomes and recombination patterns. For instance, a laboratory F2 cross or very recently admixed population would require fewer loci, although statistical power may be increased with large numbers of progeny and more markers. For association mapping in an outbred population, many more markers would be required. Quantifying diversity patterns along chromosomal stretches (e.g. runs of homozygosity) to estimate recent and historical effective population size and inbreeding also requires tens of thousands of loci48,51,52.
Some biological factors may also increase the number of loci that should be targeted. Bottlenecked or small populations with low genomic variation may require sequencing more loci to accurately quantify levels of variation. Genomes with a history of whole-genome or gene duplication, or genomes with high levels of transposable elements or other repeat sequence, may also require large numbers of loci to compensate for stringent filtering (removal) of problematic loci.
Type of sequence reads
Longer sequence reads and/or paired-end sequencing reads provide many advantages, including improved locus identification, discrimination of paralogous or repetitive sequence, and BLAST searching for functionally important loci. For most RADseq protocols, sequence length is limited primarily by sequencing technology (e.g. typically up to 150bp reads with Illumina, but up to 300bp in some cases). Many research questions can be sufficiently addressed with relatively short reads (e.g. 100bp) and single-end sequencing. However, as described above, longer RADseq loci can be obtained by assembling contigs from paired-end sequence reads with the original RAD (up to 1kb20), and this method can be particularly advantageous for complex genomes in the absence of a reference genome. 2bRAD produces the shortest reads of all methods (33–36bp), so is not recommended for de novo locus identification or in the case of large and complex genomes (e.g. the human genome53).
Prior genomic resources
Prior reference sequence can provide numerous advantages for RADseq studies. A reference genome sequence, a poorly assembled set of genomic scaffolds, or even a set of previously identified RAD loci can greatly improve the ability to filter paralogous or repetitive sequences, identify insertion-deletion variation, and remove non-target DNA sequence (e.g. bacterial contamination)54. A well-assembled reference genome provides further advantages. For instance, mapping studies can use information on physical positions of loci to infer haplotypes across larger chromosomal regions covering multiple loci55. The GBS and MSG methods have been used in this way for trait mapping in model species, where chromosomal blocks of parental ancestry are relatively large. Population genomic studies can use a reference genome assembly to conduct sliding window analyses and increase statistical power to detect genomic regions of interest, such as regions under divergent selection between populations25,50. In the absence of a reference genome, long contigs generated with the original RAD should provide the greatest ability to distinguish paralogous or repetitive sequences (see above)19–21.
Depth of sequencing coverage
Libraries from all RADseq methods can be sequenced to produce different depths of coverage, and the ideal depth for individually barcoded samples varies widely across studies. At one extreme, laboratory mapping studies with a well-assembled reference genome may be most efficient with very low coverage (<1×). Much higher coverage is required (e.g. 20–30×) for confident de novo locus discovery and genotyping in diploids, and even higher coverage would be required in polyploid taxa. Alternatively, in some cases individuals may be pooled into single barcodes (BOX 3), with much lower coverage per individual because individual genotypes are not assigned.
Budget
Often the major expense in producing RADseq data is the sequencing itself. The total sequencing effort is divided among the number of loci, the number of samples and populations, and the desired coverage per locus per individual. However, the different protocols can also differ considerably in the expense of library preparation, and in the way in which library preparation costs scale with the number of samples. For instance, while the original RADseq protocol has a relatively large number of steps, samples are multiplexed early in the protocol and the subsequent steps are conducted on mixtures of up to 96 or more barcoded samples, so the marginal cost of increasing samples is minimized both in terms of time and money (FIG 2). In contrast, the cost of ezRAD scales roughly linearly with samples because multiplexing does not occur until the end, so this method may be most appropriate for small numbers of samples or pools of samples16. Some RADseq protocols also require an initial financial investment in specialized barcoded adapters, although a single set of such oligonucleotides is often sufficient for a large number of libraries. In addition, some RAD protocols may require the purchase of specialized laboratory equipment. The original RAD requires the use of a DNA sonicator, and RADseq protocols that use a direct size selection (e.g. ddRAD, ezRAD) can increase precision and consistency of size selection, and decrease the possibility of cross-contamination, by using a Pippin Prep14 (Sage Science, Beverly, MA).
Figure 2.
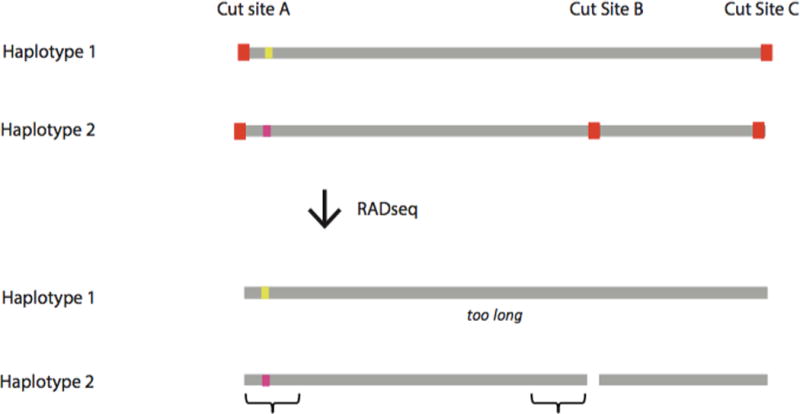
Sources of error and bias in RADseq data. (a) Example of allele dropout for a RADseq protocol that uses size selection to reduce the number of loci to be sequenced. Gray lines represent chromosomes within one individual, red squares represent restriction cut sites, colored squares represent heterozygous SNPs, and brackets represent genomic regions that are sequenced. Mutation in Restriction Cut Site B for Haplotype 1 makes the post-digestion fragment containing the SNP too long to be retained during size selection for Haplotype 1, eliminating the possibility of sequencing of any loci on that fragment, and causing the individual to appear homozygous at the heterozygous SNP. (b) See Figure 1 of Andrews et al. 201438. Example of fragments produced after PCR for one heterozygous locus for different RADseq protocols, and the reads retained after bioinformatic analyses. PCR duplicates are shown with the same symbol (circle, square, asterisk or triangle) as the parent fragment from the original template DNA. By chance, some alleles will amplify more than others during PCR. For all protocols, PCR duplicates will be identical in sequence composition and length to the original template molecule. For the original RADseq, this feature (i.e., identical length) can be used to identify and remove PCR duplicates bioinformatically, because original template molecules for a given locus will not be identical in length. For alternative RADseq methods, this feature cannot be used to identify PCR duplicates, because all original template molecules for a given locus are identical in length. High frequencies of PCR duplicates can cause heterozygotes to appear as homozygotes or can cause PCR errors to appear as true diversity.
Comparability of data
A final consideration when designing a RADseq study is the consistency of data across sequencing runs and across laboratories. Inconsistency in size selection could produce variation among libraries for methods that use size selection to reduce the set of loci. The consistency of different size selection techniques (automated or manual gel extraction vs. bead-based selection) has not been rigorously quantified, but magnetic beads are likely much less consistent56. Methods that target every cut site (original RAD, 2bRAD) are generally expected to be more consistent across libraries (but see Sources of Error). There can be some consistency in the loci genotyped even across methods, depending on the choice of restriction enzymes. For instance, the loci sequenced using SbfI and EcoRI in a ddRAD protocol should be a subset of those sequenced using SbfI with original RAD.
Complementary approaches
Although RADseq has many benefits as a tool for SNP genotyping and discovery, it is not the best method of choice for every ecological and evolutionary study. Two major alternative reduced representation approaches that take advantage of next generation sequencing are transcriptome sequencing (RNAseq)57 and targeted (probe-based) capture58 (BOX 4). Whole genome re-sequencing and whole genome pooled sequencing are other alternatives that provide much more genomic information than reduced representation techniques59–61. However, despite the increasing feasibility of whole-genome re-sequencing for population studies, many ecological and evolutionary questions stand to gain little from such an increase in genome-wide data. For example, a RADseq study using several thousands of markers to detect selection based on allele frequency or linkage disequilibrium is more likely to be limited by the number of individuals sampled than the density of markers.
Box 4. Alternatives to RADseq.
Two major alternative reduced representation next-generation sequencing methods are transcriptome sequencing (RNAseq) and targeted (probe-based) capture.
Transcriptome sequencing (RNAseq): sequencing transcribed regions of the genome using RNA as a starting point in library preparation.
Advantages
RNAseq can be used to quickly sequence thousands of functional genomic regions in virtually any species with limited or no genomic resources96. Most transcripts can be annotated against existing genome databases97, providing a much stronger functional context when compared to anonymous RADseq loci.
Disadvantages
RNAseq provides limited opportunity to dynamically scale sequencing effort based on question or experimental design. Individual transcripts may differ by several orders of magnitude in relative abundances98, complicating genotyping99 and increasing sequencing costs. Functional annotation may be limited in taxonomic groups with poor database representation. RNAseq requires high quality samples, which can limit its feasibility for many studies.
Targeted (Probe-based) capture: sequencing of pre-selected genomic regions using a DNA probe to isolate regions of interest
Advantages
Targeted capture is highly scalable, able to sequence a single locus100 or hundreds of thousands of loci101,102. Technical performance is typically very high103, with low variance in sequencing coverage across regions and individuals36,42,104. Capture can be applied across moderate to deep evolutionary timescales105–107 and on degraded DNA samples, making it popular for phylogenetic34,108,109 and ancient DNA studies110–115.
Disadvantages
Primary limitations for capture are the availability of genomic resources for designing probes, and generally higher cost than RADseq or RNAseq58.
Alternative genomic approaches can also be used to complement RADseq for more comprehensive or flexible investigation in a particular system. For instance, the development of de novo reference genomes for non-model species is becoming increasingly feasible as sequencing and assembly technologies continue to improve62,63, and such a reference provides numerous advantages for analysis of RADseq data from population-level sampling25,49,50,54. Transcriptome sequencing can also complement RADSeq data by targeting coding (and presumably functional) sequence, whereas RADseq interrogates both coding and non-coding loci. RADseq can also be used as the first step in a larger study to focus on significant loci. For instance, RADseq can provide a genome-wide scan to identify candidate loci of interest, and sequence data at these loci can then be used to design probes for sequence capture. Subsequent targeted sequencing could then be conducted on a large number of samples at greatly reduced cost per sample, and with poorer quality DNA.
Conclusions
RADseq techniques have enormous power and versatility for SNP discovery and genotyping in ecological and evolutionary genomics, but researchers should employ careful consideration in choosing and applying these methods. Numerous RADseq protocols have been developed that differ not only in the technical details and cost of the library prep, but also in the types of data produced and the sources of genotyping error and bias. Therefore different protocols will be better suited to different study systems, budgets, and research questions. Despite rapid changes in sequencing technology and costs, we anticipate that reduced representation sequencing approaches like RADseq will continue to be a critical tool for genomic studies of natural populations into the foreseeable future. When implemented appropriately, RADseq approaches provide efficient, flexible, and cost-effective avenues to unleash the power of next-generation sequencing technologies for gaining new insights into ecological, evolutionary, and conservation-related questions.
KEY POINTS.
RADseq has fueled studies in ecological, evolutionary, and conservation genomics by using next-generation sequencing to uncover hundreds or thousands of polymorphic loci across the genome in a single, simple, and cost-effective experiment. RADseq does not require any prior genomic information for the taxa being studied, and is therefore particularly advantageous for studies of non-model organisms.
Numerous technical variations on RADseq have been developed, promising to increase flexibility and decrease cost and effort in genomics studies. Differences among the methods lead to important considerations for all steps of genomic studies, from costs of library preparation and sequencing, to the types of bias and error inherent in the resulting data, to the types of scientific questions that can be addressed.
Allele dropout, PCR duplicates, and variance in depth of coverage among loci are important sources of error and bias in RADseq studies, and the prevalence of these phenomena will vary across RADseq methods.
Other important considerations when designing a RADseq study include the number, length, and coverage of loci needed to address the research question; availability of prior genomic resources; budget; and consistency of data across sequencing runs and laboratories.
There is no single best or most flexible RADseq method. Researchers must weigh the trade-offs of the different methods, and choose the approach best suited to their study goals.
Acknowledgments
We thank Michelle Gaither, Emma Carroll, Andre Moura, Ryan Bracewell, and Matt Jones for helpful discussions. PAH received support from NIH grant P30 GM103324 and NSF grant 1316549. JMG is supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD073439) and the National Institute of General Medical Sciences (R01GM098536) of the National Institutes of Health. GL was supported by grants from U.S. National Science Foundation (DEB-0742181 and DEB-1067613) and NASA-(NNX14AB84G).
GLOSSARY
- Allele dropout
failure of an allele present in a sample to be detected by sequencing
- Adapters
double-stranded oligos that must be ligated to DNA fragments prior to next-generation sequencing. Illumina adapters contain regions that anneal to the flow cell, an “index” sequence that act as a barcode to identify individual samples, and primer binding sites for bridge amplification and sequencing of the DNA fragment and indexes
- Barcode (also called “inline barcode”)
a short unique sequence (typically 6–12bp) used to identify individual samples. Inline barcodes occur on the end of the adapter that is immediately adjacent to the genomic DNA fragment after adapter ligation. The barcode is sequenced immediately prior to sequencing of the DNA fragment, and thus the barcode sequence will appear at the beginning of sequence reads
- Combinatorial barcoding
using two different barcoding methods, usually a standard Illumina index and an inline barcode. This method can reduce the number of adapters that must be purchased, thus reducing library prep cost
- Contig
a group of overlapping sequence reads assembled to form a longer sequence
- Depth of coverage
the number of sequence reads for a given locus or nucleotide site
- Filtering
removing unwanted sequence reads from a dataset due to low sequence quality, low depth of coverage, evidence for paralogy, or other reasons
- Illumina index
a unique 6bp or 8bp sequence incorporated into Illumina adapters that functions as a barcode to identify individual samples
- Sequencing library
DNA prepared for next-generation sequencing. The DNA must be an appropriate length for sequencing and must have sequencing adapters ligated
- Next-generation sequencing (“Massively parallel sequencing”)
Technologies first emerging around 2005 that sequence millions of DNA molecules simultaneously
- Null allele
An allele present in a sample that fails to be identified by genotyping. The presence of a null allele leads to “allele dropout.”
- Paired-end sequencing
Illumina sequencing of both ends of each DNA fragment
- Paralog
sequence originating through duplication within the genome
- Pooling
combining multiple individual samples into a DNA library with only one unique identifier (e.g. one barcode or one index)
- Reduced-representation library
DNA library comprised of a subset of loci, rather than the entire genome
- Restriction site Associated DNA (RADseq)
a method for sequencing thousands of genetic loci adjacent to restriction cut sites across the genome using massively parallel (“next generation”) sequencing. Sometimes referred to as “Genotyping by Sequencing.”
- Single-end sequencing
Illumina sequencing of only one end of each DNA fragment
- Sticky end (also called “DNA overhang”)
the string of single-stranded DNA remaining on the end of a DNA fragment that has been digested with a restriction enzyme. Also called a “DNA overhang.” Some restriction enzymes produce “blunt ends” (double-stranded ends) rather than sticky ends
Biographies
Kimberly R. Andrews is a postdoctoral research fellow in Lisette Waits’ group (Department of Fish & Wildlife Sciences) collaborating with Paul Hohenlohe’s group (Department of Biological Sciences) at the University of Idaho. Her work focuses on investigating the ecological and evolutionary factors driving dispersal, gene flow, adaptation, and speciation, and applying this knowledge to conservation and management issues. She received her PhD from the University of Hawai‘i at Mānoa and has conducted postdoctoral research at the University of Hawai‘i and as a Marie Curie Research Fellow at Durham University.
Jeffrey Good is an assistant professor in the Division of Biological Sciences at the University of Montana, Missoula, USA. His laboratory combines population, comparative, and functional genomics to understand the genetic basis of speciation and adaptation in model and non-model systems.
Paul A. Hohenlohe is an Assistant Professor in the Institute for Bioinformatics and Evolutionary Studies, and the Departments of Biological Sciences and Statistical Science at the University of Idaho. His research group focuses on evolutionary and conservation genomics in a wide variety of organisms, using RADseq and other tools. He received his Ph.D. from the University of Washington, worked as a conservation biologist and conducted postdoctoral research at Oregon State University and the University of Oregon before joining UI in 2011.
Footnotes
FURTHER INFORMATION
RAD Capture (Rapture) O.A. Ali, S.M. O’Rourke, S.J. Amish, M.H. Meek, G. Luikart, C. Jeffres, M.R. Miller. RAD Capture (Rapture): Flexible and efficient sequence-based genotyping. BioRxiv preprint doi: http://dx.doi.org/10.1101/028837. In review. (2015). Extends RADseq with the addition of a sequence-capture step to target a subset of RAD loci, and also presents a substantially revised new version of the original RADseq protocol.
LITERATURE CITED
- 1.Science. Breakthrough of the Year. Areas to Watch. Science 330. 2010;6011:1608–1609. doi: 10.1126/science.330.6011.1608-c. [DOI: 1610.1126/science.1330.6011.1608-c] [DOI] [PubMed] [Google Scholar]
- 2.Davey JW, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics. 2011;12:499–510. doi: 10.1038/nrg3012. Reviews methods for genomic marker discovery and genotyping using next-generation sequencing methods. [DOI] [PubMed] [Google Scholar]
- 3.Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: From genotyping to genome typing. Nature Reviews Genetics. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- 4.Hedges SB, Schweitzer MH. Detecting dinosaur DNA. Science. 1995;268:1191–1192. doi: 10.1126/science.7761839. [DOI] [PubMed] [Google Scholar]
- 5.Pérez T, Albornoz J, Domínguez A. An evaluation of RAPD fragment reproducibility and nature. Molecular Ecology. 1998;7:1347–1357. doi: 10.1046/j.1365-294x.1998.00484.x. [DOI] [PubMed] [Google Scholar]
- 6.Taberlet P, Waits LP, Luikart G. Noninvasive genetic sampling: look before you leap. Trends in Ecology & Evolution. 1999;14:323–327. doi: 10.1016/s0169-5347(99)01637-7. [DOI] [PubMed] [Google Scholar]
- 7.Avise JC, Lansman RA, Shade RO. Use of restriction endonucleases to measure mitochondrial DNA sequence relatedness in natural populations. I. Population structure and evolution in the genus. Peromyscus Genetics. 1979;92:279–295. doi: 10.1093/genetics/92.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown WM. Polymorphism in mitochondrial DNA of humans as revealed by restricion endonuclease analysis. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:3605–3609. doi: 10.1073/pnas.77.6.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altshuler D, et al. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407 doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- 10.Baird NA, et al. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. Plos One. 2008;3 doi: 10.1371/journal.pone.0003376. doi: e337610.1371/journal.pone.0003376. Introduces one of the most widely-used RADseq methods, which we call “original RAD.”. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Tassell CP, et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nature Methods. 2008;5:247–252. doi: 10.1038/nmeth.1185. [DOI] [PubMed] [Google Scholar]
- 12.Wiedmann RT, Smith TPL, Nonneman DJ. SNP discovery in swine by reduced representation and high throughput pyrosequencing. BMC Genetics. 2008;9 doi: 10.1186/1471-2156-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poland JA, Rife TW. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome. 2012;5:92–102. doi: 10.3835/plantgenome2012.05.0005. [DOI] [Google Scholar]
- 14.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. DoubleDigest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. Plos One. 2012;7 doi: 10.1371/journal.pone.0037135. Introduces “double digest RAD (ddRAD),” one of the most widely-used RADseq methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Meyer E, McKay JK, Matz MV. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nature Methods. 2012;9:808–812. doi: 10.1038/nmeth.2023. [DOI] [PubMed] [Google Scholar]
- 16.Toonen RJ, et al. ezRAD: a simplified method for genomic genotyping in non-model organisms. PeerJ. 2013;1:e203–e203. doi: 10.7717/peerj.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narum SR, Buerkle CA, Davey JW, Miller MR, Hohenlohe PA. Genotyping-by-sequencing in ecological and conservation genomics. Molecular Ecology. 2013;22:2841–2847. doi: 10.1111/mec.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham C, et al. Impacts of degraded DNA on restriction enzyme associated DNA sequencing (RADSeq) Molecular Ecology Resources. 2015 doi: 10.1111/1755-0998.12404. [DOI] [PubMed] [Google Scholar]
- 19.Etter PD, Preston JL, Bassham S, Cresko WA, Johnson EA. Local De Novo Assembly of RAD Paired-End Contigs Using Short Sequencing Reads. Plos One. 2011;6 doi: 10.1371/journal.pone.0018561. Introduces a method for generating long contigs from paired-end RADseq data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohenlohe PA, et al. Genomic patterns of introgression in rainbow and westslope cutthroat trout illuminated by overlapping paired-end RAD sequencing. Molecular Ecology. 2013;22:3002–3013. doi: 10.1111/mec.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willing EM, Hoffmann M, Klein JD, Weigel D, Dreyer C. Paired-end RAD-seq for de novo assembly and marker design without available reference. Bioinformatics. 2011;27:2187–2193. doi: 10.1093/bioinformatics/btr346. [DOI] [PubMed] [Google Scholar]
- 22.Waples RK, Seeb LW, Seeb JE. Linkage mapping with paralogs exposes regions of residual tetrasomic inheritance in chum salmon (Oncorhynchus keta) Molecular Ecology Resources. 2015 doi: 10.1111/1755-0998.12394. [DOI] [PubMed] [Google Scholar]
- 23.Amish SJ, et al. RAD sequencing yields a high success rate for westslope cutthroat and rainbow trout species-diagnostic SNP assays. Molecular Ecology Resources. 2012;12:653–660. doi: 10.1111/j.1755-0998.2012.03157.x. [DOI] [PubMed] [Google Scholar]
- 24.Ali OA, et al. RAD Capture (Rapture): Flexible and efficient sequence-based genotyping. BioRxiv. 2015 doi: 10.1534/genetics.115.183665. preprint http://dx.doi.org/10.1101/028837. In review. [DOI] [PMC free article] [PubMed]
- 25.Hohenlohe PA, et al. Population Genomics of Parallel Adaptation in Threespine Stickleback using Sequenced RAD Tags. Plos Genetics. 2010;6 doi: 10.1371/journal.pgen.1000862. doi: e100086210.1371/journal.pgen.1000862. An early application of RADseq for population genomics, identifies loci under selection in multiple, independently derived freshwater stickleback populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nielsen R, Korneliussen T, Albrechtsen A, Li Y, Wang J. SNP Calling, Genotype Calling, and Sample Allele Frequency Estimation from New-Generation Sequencing Data. Plos One. 2012;7 doi: 10.1371/journal.pone.0037558. Introduces Bayesian methods for SNP-calling using the sample allele frequency spectra estimated from next-generation sequencing data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fumagalli M, et al. Quantifying population genetic differentiation from next-generation sequencing data. Genetics. 2013;195:979–992. doi: 10.1534/genetics.113.154740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Molecular Ecology. 2013;22:3124–3140. doi: 10.1111/mec.12354. Introduces Stacks, a widely used software package for locus discovery, genotyping, and population genomic analysis using RADseq data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eaton DAR. PyRAD: assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics. 2014;30:1844–1849. doi: 10.1093/bioinformatics/btu121. [DOI] [PubMed] [Google Scholar]
- 30.Lu F, et al. Switchgrass Genomic Diversity, Ploidy, and Evolution: Novel Insights from a Network-Based SNP Discovery Protocol. Plos Genetics. 2013;9 doi: 10.1371/journal.pgen.1003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradbury PJ, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 32.Ilut DC, Nydam ML, Hare MP. Defining Loci in Restriction-Based Reduced Representation Genomic Data from Nonmodel Species: Sources of Bias and Diagnostics for Optimal Clustering. Biomed Research International. 2014 doi: 10.1155/2014/675158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mastretta-Yanes A, et al. Gene Duplication, Population Genomics, and Species-Level Differentiation within a Tropical Mountain Shrub. Genome Biology and Evolution. 2014;6:2611–2624. doi: 10.1093/gbe/evu205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leaché AD, et al. Phylogenomics of Phrynosomatid Lizards: Conflicting Signals from Sequence Capture versus Restriction Site Associated DNA Sequencing. Genome Biology and Evolution. 2015;7:706–719. doi: 10.1093/gbe/evv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 36.Gautier M, et al. The effect of RAD allele dropout on the estimation of genetic variation within and between populations. Molecular Ecology. 2013;22:3165–3178. doi: 10.1111/mec.12089. Uses computer simulations to investigate the influence of allele dropout on population genomic statistics for RADseq data. [DOI] [PubMed] [Google Scholar]
- 37.Arnold B, Corbett-Detig RB, Hartl D, Bomblies K. RADseq underestimates diversity and introduces genealogical biases due to nonrandom haplotype sampling. Molecular Ecology. 2013;22:3179–3190. doi: 10.1111/mec.12276. [DOI] [PubMed] [Google Scholar]
- 38.Andrews KR, et al. Trade-offs and utility of alternative RADseq methods: Reply to Puritz et al. 2014. Molecular Ecology. 2014;23:5943–5946. doi: 10.1111/mec.12964. [DOI] [PubMed] [Google Scholar]
- 39.Schweyen H, Rozenberg A, Leese F. Detection and Removal of PCR Duplicates in Population Genomic ddRAD Studies by Addition of a Degenerate Base Region (DBR) in Sequencing Adapters. Biological Bulletin. 2014;227:146–160. doi: 10.1086/BBLv227n2p146. [DOI] [PubMed] [Google Scholar]
- 40.Casbon JA, Osborne RJ, Brenner S, Lichtenstein CP. A method for counting PCR template molecules with application to next-generation sequencing. Nucleic Acids Research. 2011;39 doi: 10.1093/nar/gkr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tin MMY, Rheindt FE, Cros E, Mikheyev AS. Degenerate adaptor sequences for detecting PCR duplicates in reduced representation sequencing data improve genotype calling accuracy. Molecular Ecology Resources. 2014 doi: 10.1111/1755-0998.12314. [DOI] [PubMed] [Google Scholar]
- 42.Davey JW, et al. Special features of RAD Sequencing data: implications for genotyping. Molecular Ecology. 2013;22:3151–3164. doi: 10.1111/mec.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DaCosta JM, Sorenson MD, Amplification Biases. Consistent Recovery of Loci in a Double-Digest RAD-seq Protocol. Plos One. 2014;9 doi: 10.1371/journal.pone.0106713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benjamini Y, Speed TP. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Research. 2012;40 doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lepais O, Weir JT. SimRAD: an R package for simulation-based prediction of the number of loci expected in RADseq and similar genotyping by sequencing approaches. Molecular Ecology Resources. 2014;14:1314–1321. doi: 10.1111/1755-0998.12273. [DOI] [PubMed] [Google Scholar]
- 46.Cariou M, Duret L, Charlat S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecology and Evolution. 2013;3:846–852. doi: 10.1002/ece3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruaud A, et al. Empirical Assessment of RAD Sequencing for Interspecific Phylogeny. Molecular Biology and Evolution. 2014;31:1272–1274. doi: 10.1093/molbev/msu063. [DOI] [PubMed] [Google Scholar]
- 48.Kardos M, Luikart G, Allendorf FW. Measuring individual inbreeding in the age of genomics: marker-based measures are better than pedigrees. Heredity. 2015;115:63–72. doi: 10.1038/hdy.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nadeau NJ, et al. Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Research. 2014;24:1316–1333. doi: 10.1101/gr.169292.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruegg K, Anderson EC, Boone J, Pouls J, Smith TB. A role for migration-linked genes and genomic islands in divergence of a songbird. Molecular Ecology. 2014;23:4757–4769. doi: 10.1111/mec.12842. [DOI] [PubMed] [Google Scholar]
- 51.Kirin M, et al. Genomic Runs of Homozygosity Record Population History and Consanguinity. Plos One. 2010;5 doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffman JI, et al. High-throughput sequencing reveals inbreeding depression in a natural population. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:3775–3780. doi: 10.1073/pnas.1318945111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmieri N, Schloetterer C. Mapping Accuracy of Short Reads from Massively Parallel Sequencing and the Implications for Quantitative Expression Profiling. Plos One. 2009;4 doi: 10.1371/journal.pone.0006323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hand BK, et al. Genomics and introgression: Discovery and mapping of thousands of species-diagnostic SNPs using RAD sequencing. Current Zoology. 2015;61:146–154. [Google Scholar]
- 55.Andolfatto P, et al. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Research. 2011;21:610–617. doi: 10.1101/gr.115402.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heffelfinger C, et al. Flexible and scalable genotyping-by-sequencing strategies for population studies. Bmc Genomics. 2014;15 doi: 10.1186/1471-2164-15-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones M, Good J. Targeted capture in evolutionary and ecological genomics. Molecular Ecology. 2015 doi: 10.1111/mec.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellegren H, et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature. 2012;491:756–760. doi: 10.1038/nature11584. [DOI] [PubMed] [Google Scholar]
- 60.Kardos M, et al. Whole genome resequencing uncovers molecular signatures of natural and sexual selection in wild bighorn sheep. Molecular Ecology. 2015 doi: 10.1111/mec.13415. [DOI] [PubMed] [Google Scholar]
- 61.Schlötterer C, Tobler R, Kofler R, Nolte V. Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nature Reviews Genetics. 2014;15:749–763. doi: 10.1038/nrg3803. [DOI] [PubMed] [Google Scholar]
- 62.Huddleston J, et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Research. 2014;24:688–696. doi: 10.1101/gr.168450.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Putnam N, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. arXiv. 2015;1502:05331. doi: 10.1101/gr.193474.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research. 2007;17:240–248. doi: 10.1101/gr.5681207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo Y, et al. An improved 2b-RAD approach (I2b-RAD) offering genotyping tested by a rice (Oryza sativa L.) F2 population. Bmc Genomics. 2014;15 doi: 10.1186/1471-2164-15-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elshire RJ, et al. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. Plos One. 2011;6 doi: 10.1371/journal.pone.0019379. Introduces “Genotyping by Sequencing (GBS),” one of the most widely-used RADseq methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Truong HT, et al. Sequence-Based Genotyping for Marker Discovery and Co-Dominant Scoring in Germplasm and Populations. Plos One. 2012;7 doi: 10.1371/journal.pone.0037565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Orsouw NJ, et al. Complexity Reduction of Polymorphic Sequences (CRoPS (TM)): A Novel Approach for Large-Scale Polymorphism Discovery in Complex Genomes. Plos One. 2007;2 doi: 10.1371/journal.pone.0001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greminger MP, et al. Generation of SNP datasets for orangutan population genomics using improved reduced-representation sequencing and direct comparisons of SNP calling algorithms. BMC Genomics. 2014;15 doi: 10.1186/1471-2164-15-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schield DR, et al. EpiRADseq: scalable analysis of genomewide patterns of methylation using next-generation sequencing. Methods in Ecology and Evolution. 2015 doi: 10.1111/2041-1210X.12435. [DOI] [Google Scholar]
- 71.Stolle E, Moritz RFA. RESTseq – Efficient Benchtop Population Genomics with RESTriction Fragment SEQuencing. Plos One. 2013;8 doi: 10.1371/journal.pone.0063960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pukk L, et al. Less is more: extreme genome complexity reduction with ddRAD using Ion Torrent semiconductor technology. Molecular Ecology Resources. 15:1145–1152. doi: 10.1111/1755-0998.12392. [DOI] [PubMed] [Google Scholar]
- 73.Recknagel H, Jacobs A, Herzyk P, Elmer KR. Double-digest RAD sequencing using Ion Proton semiconductor platform (ddRADseq-ion) with nonmodel organisms. Molecular Ecology Resources. 2015 doi: 10.1111/1755-0998.12406. [DOI] [PubMed] [Google Scholar]
- 74.Chen Q, et al. Genotyping by Genome Reducing and Sequencing for Outbred Animals. Plos One. 2013;8 doi: 10.1371/journal.pone.0067500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Evans BJ, Zeng K, Esselstyn JA, Charlesworth B, Melnick DJ. Reduced Representation Genome Sequencing Suggests Low Diversity on the Sex Chromosomes of Tonkean Macaque Monkeys. Molecular Biology and Evolution. 2014;31:2425–2440. doi: 10.1093/molbev/msu197. [DOI] [PubMed] [Google Scholar]
- 76.Larson WA, Seeb JE, Pascal CE, Templin WD, Seeb LW. Single-nucleotide polymorphisms (SNPs) identified through genotyping-by-sequencing improve genetic stock identification of Chinook salmon (Oncorhynchus tshawytscha) from western Alaska. Canadian Journal of Fisheries and Aquatic Sciences. 2014;71:698–708. doi: 10.1139/cjfas-2013-0502. [DOI] [Google Scholar]
- 77.Candy JR, et al. Population differentiation determined from putative neutral and divergent adaptive genetic markers in Eulachon (Thaleichthys pacificus, Osmeridae), an anadromous Pacific smelt. Molecular Ecology Resources. 2015 doi: 10.1111/1755-0998.12400. [DOI] [PubMed] [Google Scholar]
- 78.Dann TH, Habicht C, Baker TT, Seeb JE. Exploiting genetic diversity to balance conservation and harvest of migratory salmon. Canadian Journal of Fisheries and Aquatic Sciences. 2013;70:785–793. doi: 10.1139/cjfas-2012-0449. [DOI] [Google Scholar]
- 79.Emerson KJ, et al. Resolving postglacial phylogeography using high-throughput sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16196–16200. doi: 10.1073/pnas.1006538107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Combosch DJ, Vollmer SV, Trans-Pacific RAD-Seq. population genomics confirms introgressive hybridization in Eastern Pacific Pocillopora corals. Molecular Phylogenetics and Evolution. 2015;88:154–162. doi: 10.1016/j.ympev.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 81.Gaither MR, et al. Genomic signatures of geographic isolation and natural selection in coral reef fishes. Molecular Ecology. 2015;24:1543–1557. doi: 10.1111/mec.13129. [DOI] [PubMed] [Google Scholar]
- 82.Eaton DAR, Ree RH, Inferring Phylogeny. Introgression using RADseq Data: An Example from Flowering Plants (Pedicularis: Orobanchaceae) Systematic Biology. 2013;62:689–706. doi: 10.1093/sysbio/syt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ford AGP, et al. High levels of interspecific gene flow in an endemic cichlid fish adaptive radiation from an extreme lake environment. Molecular Ecology. 2015;24:3421–3440. doi: 10.1111/mec.13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chutimanitsakun Y, et al. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. Bmc Genomics. 2011;12 doi: 10.1186/1471-2164-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wagner CE, et al. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Molecular Ecology. 2013;22:787–798. doi: 10.1111/mec.12023. [DOI] [PubMed] [Google Scholar]
- 86.Schlöetterer C, Tobler R, Kofler R, Nolte V. Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nature Reviews Genetics. 2014;15:749–763. doi: 10.1038/nrg3803. [DOI] [PubMed] [Google Scholar]
- 87.Futschik A, Schlöetterer C. The Next Generation of Molecular Markers From Massively Parallel Sequencing of Pooled DNA Samples. Genetics. 2010;186:207–218. doi: 10.1534/genetics.110.114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gautier M, et al. Estimation of population allele frequencies from next-generation sequencing data: pool-versus individual-based genotyping. Molecular Ecology. 2013;22:3766–3779. doi: 10.1111/mec.12360. [DOI] [PubMed] [Google Scholar]
- 89.Anderson EC, Skaug HJ, Barshis DJ. Next-generation sequencing for molecular ecology: a caveat regarding pooled samples. Molecular Ecology. 2014;23:502–512. doi: 10.1111/mec.12609. [DOI] [PubMed] [Google Scholar]
- 90.Zhu Y, Bergland AO, Gonzalez J, Petrov DA. Empirical validation of pooled whole genome population re-sequencing in. Drosophila melanogaster Plos One. 2012;7 doi: 10.1371/journal.pone.0041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lynch M, Bost D, Wilson S, Maruki T, Harrison S. Population-genetic inference from pooled-sequencing data. Genome biology and evolution. 2014;6:1210–1218. doi: 10.1093/gbe/evu085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ferretti L, Ramos-Onsins SE, Perez-Enciso M. Population genomics from pool sequencing. Molecular Ecology. 2013;22:5561–5576. doi: 10.1111/mec.12522. [DOI] [PubMed] [Google Scholar]
- 93.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kayser M, Brauer S, Stoneking M. A genome scan to detect candidate regions influenced by local natural selection in human populations. Molecular Biology and Evolution. 2003;20:893–900. doi: 10.1093/molbev/msg092. [DOI] [PubMed] [Google Scholar]
- 95.Nielsen R, et al. Genomic scans for selective sweeps using SNP data. Genome Research. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Montgomery SB, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. American Journal of Human Genetics. 2013;93:641–651. doi: 10.1016/j.ajhg.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Briggs AW, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 101.Hodges E, et al. Genome-wide in situ exon capture for selective resequencing. Nature Genetics. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 102.Gnirke A, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mamanova L, et al. Target-enrichment strategies for next-generation sequencing. Nature Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 104.Henning F, Lee HJ, Franchini P, Meyer A. Genetic mapping of horizontal stripes in Lake Victoria cichlid fishes: benefits and pitfalls of using RAD markers for dense linkage mapping. Molecular Ecology. 2014;23:5224–5240. doi: 10.1111/mec.12860. [DOI] [PubMed] [Google Scholar]
- 105.Good JM, et al. Comparative population genomics of the ejaculate in humans and the Great Apes. Molecular Biology and Evolution. 2013;30:964–976. doi: 10.1093/molbev/mst005. [DOI] [PubMed] [Google Scholar]
- 106.Hedtke SM, Morgan MJ, Cannatella DC, Hillis DM. Targeted Enrichment: Maximizing Orthologous Gene Comparisons across Deep Evolutionary Time. Plos One. 2013;8 doi: 10.1371/journal.pone.0067908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bi K, et al. Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics. 2012;13:403. doi: 10.1186/1471-2164-13-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Faircloth BC, et al. Ultraconserved Elements Anchor Thousands of Genetic Markers Spanning Multiple Evolutionary Timescales. Systematic Biology. 2012;61:717–726. doi: 10.1093/sysbio/sys004. [DOI] [PubMed] [Google Scholar]
- 109.McCormack JE, et al. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Research. 2012;22:746–754. doi: 10.1101/gr.125864.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Burbano HA, et al. Targeted investigation of the Neandertal genome by array-based sequence capture. Science. 2010;328:723–725. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bos KI, et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. 2011;478:506–510. doi: 10.1038/nature10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Avila-Arcos MC, et al. Application and comparison of large-scale solution-based DNA capture-enrichment methods on ancient DNA. Scientific Reports. 2011;1 doi: 10.1038/srep00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bos KI, et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature. 2014;514:494–+. doi: 10.1038/nature13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Carpenter ML, et al. Pulling out the 1%: Whole-Genome Capture for the Targeted Enrichment of Ancient DNA Sequencing Libraries. American Journal of Human Genetics. 2013;93:852–864. doi: 10.1016/j.ajhg.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Castellano S, et al. Patterns of coding variation in the complete exomes of three Neandertals. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6666–6671. doi: 10.1073/pnas.1405138111. [DOI] [PMC free article] [PubMed] [Google Scholar]