


Abstract
Pif1 helicases are ubiquitous members of the SF1B family and are essential for maintaining genome stability. It was speculated that Pif1-specific motifs may fold in specific structures, conferring distinct activities upon it. Here, we report the crystal structures of the Pif1 helicase from Bacteroides spp with and without adenosine triphosphate (ATP) analog/ssDNA. BsPif1 shares structural similarities with RecD2 and Dda helicases but has specific features in the 1B and 2B domains. The highly conserved Pif1 family specific sequence motif interacts with and constraints a putative pin-loop in domain 1B in a precise conformation. More importantly, we found that the 2B domain which contains a specific extended hairpin undergoes a significant rotation and/or movement upon ATP and DNA binding, which is absolutely required for DNA unwinding. We therefore propose a mechanism for DNA unwinding in which the 2B domain plays a predominant role. The fact that the conformational change regulates Pif1 activity may provide insight into the puzzling observation that Pif1 becomes highly processive during break-induced replication in association with Polδ, while the isolated Pif1 has low processivity.
INTRODUCTION
DNA helicases are molecular motors that convert the chemical energy of nucleoside triphosphate (NTP) hydrolysis into mechanical force to separate double-stranded DNA into single-stranded DNA (1). ScPif1, as a prototypical member of Pif1 family helicases, was first identified in Saccharomyces cerevisiae due to its important role in maintaining mitochondrial DNA (2). It was then rediscovered as a negative regulator of telomere length, which actively disrupts telomerase from telomere and double-strand break ends, to regulate telomere homeostasis and avoid de novo telomere addition at double-strand breaks (3,4). Its physiological importance was further highlighted by the fact that mutations in human PIF1 are found in families with high risk of breast cancer (5), and Saccharomyces pombe cells with the corresponding mutation are not viable (6,7). Indeed, many intriguing findings in recent Pif1 helicase studies have established that Pif1 helicases plays multiple roles in the maintenance of genomic integrity: it advances fork progression at many nuclear sites, including the regulation of ribosomal DNA replication (8); facilitates replication and suppresses DNA damage at G-quadruplex motifs (9–11); processes Okazaki fragment maturation in cooperation with Dna2 helicase/nuclease (12–14) and promotes break-induced replication (BIR) via bubble migration (15,16).
Pif1 helicases are highly conserved from bacteria to humans and belong to superfamily (SF) 1B as revealed by sequence alignments (17,18). Although BLAST searches for homologs of ScPif1 demonstrated that Pif1 helicases vary in size from 420 to more than 1000 amino acids per polypeptide chain, they can be functionally and structurally divided into three domains: the N-terminal domain of ScPif1 that physically and functionally interacts with yeast SSB protein to coordinate its diverse functions in cell (19), in the human ortholog this domain exhibits a DNA strand annealing activity (20); the helicase core domain is highly conserved in various organisms and shares seven conserved motifs (I, Ia, II, III, IV, V and VI) with helicases of the SF1 family, in which three additional motifs (A, B and C) are found specifically in Pif1 and RecD family helicases (18). However, Pif1 helicases can be distinguished from RecD helicase by the Pif1-family specific sequence (PFSS), which is located between motifs II and III, and appears unique to Pif1 family helicases (17,18). Finally, the C-terminal domain (CTD) is not conserved in sequence and length and is even completely absent from certain species. Truncation of this domain does not affect helicase activities in human and yeast Pif1 proteins (11,21).
Biochemical characterizations of purified Pif1 helicases from several sources show that they are triphosphate (ATP)- and Mg2+- dependent enzymes that unwind DNA with a 5′-3′ polarity (21–23). Both ScPif1 and BsPif1 are monomeric in the absence of DNA and ScPif1 becomes a dimer upon DNA binding (22–24). Although Pif1 helicases display only limited unwinding processivity in vitro (23,25), they are capable of unwinding a variety of DNA structures resembling stalled DNA replication forks and other B-form duplexes such as forked duplexes, DNA/RNA duplexes (23,26), R-loop, D-loop and G-quadruplex DNA (21,23,27). Consistent with Pif1-mediated telomerase disruption activity, we found that BsPif1 dislodges non-nucleosomal protein–DNA complexes in vitro (23).
Lack of structural information on Pif1 helicase has hindered our understanding of the molecular and structural basis for how Pif1 performs its diverse functions. Here, we present the structures of BsPif1 helicase in free and bound states to Adenosine diphosphate (ADP) or ATP analog AMPPNP and that of a ternary complex (BsPif1-ADP·AlF3-ssDNA). From our determined crystal structures, together with a combination of structural, mutational and biochemical analyses, a number of interesting features are revealed, thus shedding light on the mechanism of DNA unwinding and activity regulation of this family of helicases.
MATERIALS AND METHODS
Purification and crystallization of BsPif1 protein
The BsPif1 protein was purified essentially as described previously (23). Crystallization trials of BsPif1 and its complexes with AMPPNP/ADP were performed at 20°C by the sitting-drop vapor diffusion method. The initial crystals were obtained using a precipitant solution containing 0.1 M Bis-Tris propane (pH 6.5), 0.1 M Calcium acetate, 10–15% PEG4000. This condition was optimized by a grid search by using 24-well Linbro plates at 20°C where 1 μl of protein sample and 1 μl of precipitant were mixed together and equilibrated with 0.4 ml of precipitant. Suitable crystals for diffraction experiments were obtained using 0.1 M Bis-Tris propane (pH 6.5), 0.1 M Calcium acetate and 12–15% PEG2000MME or PEG1500. For cocrystals of BsPif1 and AMPPNP/ADP, the protein was mixed with AMPPNP/ADP at a molar ratio of 1–1.5. SeMet-substituted protein and AMPPNP-complex crystals were obtained under the same crystallization conditions. To obtain crystals of BsPif1 in complex with DNA, we incubated the protein with a 8-nt single-stranded DNA (5′- TTTTTTTT-3′), in 10 mM Bis-Tris propane (pH 7.0), 100 mM NaCl, 1 mM Dithiothreitol (DTT), 2 mM MgCl2, 2 mM ADP, 2 mM AlCl3, 10 mM NaF and a protein/DNA ratio of 1:1.2, for 40 min at 16°C and then concentrated to 10 mg/ml. Crystals of the BsPif1-ssDNA complex were obtained using 0.1 M Hepes (pH7.1), 3.5–3.6 M Sodium formate, 5% Glycerol and 0.01 M Spermidine as precipitant.
Data collection, phasing, model building and structure refinement
X-ray diffraction data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) and/or Beijing Synchrotron Radiation Facility (BSRF) and were indexed and scaled using HKL2000 (28) and XDS (29). A BsPif1–AMPPNP SeMet-substituted protein dataset collected at the K edge of Se (0.9792Å) was suitable for SAD phasing using the Autosol module of Phenix (30). The data and refinement statistics are summarized in Supplementary Table S1. With the assumption of one molecule in the asymmetric unit, 10 Se sites were found and after phasing and solvent flattening, the final figure-of-merit was 0.69. The 2.4 Å resolution map was used for automatic model building by Solve/Resolve. Statistics for SAD phasing are given in Supplementary Table S2. The final model was manually adjusted with Coot (31) and refined in Phenix. This model was used to solve all the other structures by molecular replacement with the Phaser module of Phenix. Refinement of all the structures was carried out using Phenix.
Small-angle X-ray scattering
Scattering data were collected at 25°C for BsPif1 (1.0, 3.0, 5.0, 7.0 and 9.0 mg/ml) in 25 mM Hepes (pH 7.5), 500 mM NaCl, 5% Glycerol buffer. Scattering data collection and processing were performed at the National Center for Protein Sciences Shanghai using the BL19U2 beamline. The wavelength (λ) of X-ray radiation was set as 1.033 Å, and a sample-to-detector distance of 2.5 m. The X-ray beam with a size of 0.40 × 0.15 (H × V) mm2 was adjusted to pass through the centers of the capillaries for each experiment. Twenty consecutive frames of 1 s exposure time recorded for each sample were averaged after checking for absence of radiation-induced protein damage. The scattering patterns of the corresponding buffer solution were recorded before and after the measurements of the protein sample, and the averaged buffer patterns were subtracted from the protein patterns. The radius of gyration (Rg) and maximum particle dimension (Dmax) were extracted using the PRIMUS and GNOM programs (32). Ab initio envelopes were determined using DAMMIF with experimental Rg and Dmax values as constraints for protein, DNA and their complex. The quality and uniqueness of the results were assessed further by averaging with DAMAVER and the agreement between replicated models was quantified by the mean normalized spatial discrepancy (NSD). Atomic model of BsPif1 was generated by flexible modeling using DADIMODO (33) where flexibility in domain 1B and between domains 1A and 2A was introduced. Atomic models were fitted in ab initio envelopes with the Supcomb20 algorithm. Comparison of the scattering of the resulting all-atom model with experimental data was computed with CRYSOL. SAXS data collection and analysis are summarized in Supplementary Table S3.
Site-directed mutagenesis
Deletions or site-directed point mutations of BsPif1 on residues which are expected to be critical for the DNA binding and unwinding were performed with site-directed mutagenesis kit (Invitrogen). The primers used for these mutations are available upon request. To facilitate the expected disulfide bond formation in I339C/L95C mutant, both wt and mutant protein (0.5 mg/ml) were stirred with oxygen at pressure 1 bar for 3 h, then the proteins were used for both activity and crystallization assays.
Helicase assay
We used a stopped-flow FRET assay for measuring unwinding amplitude and kinetic rate constants of BsPif1, using doubly labeled DNA substrates, with fluorescein (5′ TTTTTTTTTTTTTTTTTTTTTTTTTTATGTATGTCAAGGAAGG-F) and hexachlorofluorescein (5′HF-CCTTCCTTGACATACAT) as a donor and acceptor, respectively. The stopped-flow FRET assay and data analysis were performed according to Liu et al. (23). In brief, unwinding kinetics were measured in a two-syringe mode, where BsPif1 helicase and DNA substrates were pre-incubated in the unwinding reaction buffer A (25 mM Tris–HCl pH 7.5, 50 mM NaCl, 2 mM MgCl2 and 2 mM DTT) at 25°C for 5 min and the unwinding reaction was initiated by rapid mixing with ATP. The standard reaction was performed with 4 nM DNA substrates, 2 mM ATP and 100 nM BsPif1 in buffer A.
DNA binding assay
A fluorescence anisotropy assay was used to determine the apparent dissociation constants of the proteins under equilibrium DNA-binding condition (34). Protein concentration-dependent changes in fluorescence anisotropy were measured with fluorescein-labeled 16 nt ssDNA (5′CTCTGCTCGACGGATT-F) by fluorescence polarization assay using an Infinite F200 instrument (TECAN). Varying amounts of protein were added to a 150 μl aliquot of binding buffer (25 mM Tris–HCl, pH 7.5, 50 mM NaCl, 2 mM MgCl2 and 2 mM DTT) containing 5 nM fluorescein-labeled DNA. Each sample was allowed to equilibrate in solution for 5 min. After 5 min, the steady-state fluorescence anisotropy (r) was measured. A second reading was taken after 10 min, in order to ensure that the mixture was well equilibrated and stable. Less than 5% change was observed between the 5- and 10-min measurements. The equilibrium dissociation constant was determined by fitting the binding curves using Equation (1):
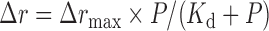 |
(1) |
where Δrmax is the maximal amplitude of the anisotropy ( = rmax,complex – rfree DNA), P is the helicase concentration and Kd is the midpoint of the curve corresponding to the apparent dissociation constant.
ATPase assay
The ATPase activity was determined by measuring the radioactive γ-32Pi liberated during ATP hydrolysis (34). The measurement was carried out at 37°C with ATPase buffer (25 mM Tris–HCl, pH = 7.5; 50 mM NaCl; 2 mM MgCl2; 2 mM DTT; 10% glycerol) in the absence or in the presence of 2 μM of 18 nt ssDNA (dT18) for basal and activated ATPase activities, respectively. The activity of I339C/L95C mutant was also measured in the same buffer, but 2 mM DTT was omitted. The reactions were initiated by the addition of 150 nM protein into 100 μl of reaction mixture and stopped by pipetting 80 μl of aliquots from the reaction mixture every 30 s into a hydrochloric solution of ammonium molybdate. The liberated radioactive 32Pi was extracted with a solution of 2-butanol-benzeneacetoneammonium molybdate (750:750:15:1) saturated with water. An aliquot of the organic phase was counted in 6 ml of Aquasol.
RESULTS
Structure determination and overall structure of BsPif1
Initial efforts to crystallize the full length and different fragments of ScPif1 alone and/or in complex with different DNAs proved to be unsuccessful. We then screened several Pif1 recombinant proteins from different species including human, yeast, thermophilic bacteria and bacteroides for crystallization assays. Finally, the full-length BsPif1 from Bacteroides ssp (431 amino acids) was crystallized alone and in complex with ADP or ATP analogs AMPPNP. The crystal structure of the complex with AMPPNP was determined by single wavelength anomalous dispersion method using a seleno-Met substituted protein. The structure was further refined with data from a native crystal diffracting to 1.38Å to Rwork/Rfree values of 0.167/0.182, respectively. The other crystals of BsPif1, in complex with ADP, and with no nucleotides, belong to the same space group with close unit cell parameters. However, BsPif1 with ADP·AlF3 and ssDNA was crystallized in another space group. These structures were solved by molecular replacement with the BsPif1–AMPPNP structure as a search model. Data collection and refinement statistics are shown in Supplementary Tables S1 and S2.
The domain architecture of BsPif1 resembles that of other SF1B members with known structures, RecD2 from Deinococcus radiodurans (PDB ID: 3GPL) and Dda from bacteriophage T4 (PDB ID: 3UPU), with two domains containing a ‘RecA-like’ α/β domain referred to as 1A and 2A, respectively (Figure 1) (35–37). Domain 1A consists of amino-acids residues (aa) aa 1–71 and aa 89–187 (Supplementary Figure S1) and forms a parallel 5-stranded β-sheet flanked by three and four α-helices on each side. Domain 2A (aa 188–250 and aa 355–401) contains one parallel 3-stranded β-sheet surrounded by 7 α-helices (Figure 1). The seven conserved SFI motifs are located as follows: motifs I, Ia, II, III and IV in domain 1A and motifs V and VI in domain 2A. The two domains are separated by a cleft, where the conformational changes induced by ATP binding and hydrolysis are expected to take place. Similar to RecD2 and Dda, the 1B domain (aa 72–88), which is composed of 18 amino acids, protrudes from 1A and folds as an ordered loop packed against helix α6. Although an SH3-like domain 2B (aa 251–354) comprising four β-strands (β11, β12, β13, β14) arranged in two perpendicular β-sheets is reminiscent of 2B domains of RecD2 and Dda, a significant difference was observed, in which an insertion characterized by a long β-sheet (β15-β16, aa 322–350) forms an unusual long β-hairpin extension of the domain. A small CTD formed by a single α-helix (α14) sometimes terminated by a β-strand depending on the structure, is present at the end of the structure (aa 402–431) and is packed against domain 1A.
Figure 1.

Overall structure of BsPif1. Domains are colored as follows: 1A (blue), 1B (orange), 2A (red), 2B (cyan) CTD (pink), PFSS (green). (A) BsPif1 with AMPPNP (space group P212121). The nucleotide binding site is in the front with the nucleotide shown as ball-and-stick. (B) The view is rotated by 90°. (C) BsPif1 with ADP-AlF3-ssDNA (space group P3121). ssDNA is colored in yellow. BsPif1 is in the same orientation as in (A) with domains 1A superimposed. (D) Rotated view by 90° of BsPif1 with ADP-AlF3-ssDNA.
Nucleotide-binding site
BsPif1 was crystallized with ADP or the ATP analog AMPPNP, respectively. The high quality of the electron density corresponding to AMPPNP (Supplementary Figure S2) allows us to precisely identify the nucleotide binding site. The ATP binding pocket is formed between domains 1A and 2A (Figure 1). The adenine base in the BsPif1–AMPPNP complex is simply stacked against F187 (motif IV) and only its N6 atom forms a hydrogen bond with the carbonyl oxygen of I5 (N-terminus) (Figure 2A). Furthermore, there are no interactions between the hydroxyls of the sugar moiety and the protein, a character resembling PcrA helicase (38). In UvrD, the best known SF1A helicase, the adenine base was sandwiched between hydrophobic and charged residues in addition to a bifurcated hydrogen bond (39). In RecD2, the adenine base is specifically recognized by a Q-motif involving Q343 (3GPL) while the equivalent position is replaced by M10 in BsPif1. The absence of the Q-motif in BsPif1 seems to be an exception in the Pif1 family since a glutamine is conserved at the equivalent Q343 position (Supplementary Figure S1B). Though Dda was crystallized with no nucleotide, a model of a nucleotide bound by a Q-motif was proposed (36). The absence of specificity for adenine has already been described for SF2 helicases in the structures of DHX9 (DEAD-box family) (40), and Prp43 (DEAH/RHA family) (41). Altogether, the lack of Q-motif in the BsPif1 structure solved in this study is consistent with our previous biochemical data showing that BsPif1 can use equally well four deoxy- and ribonucleotide triphosphates without significant preferences (23).
Figure 2.

ATPase site views. Detail of the nucleotide binding site with AMPPNP (A); ADP-AlF3 (B), ADP (C) and no ligand (D). Coloring of 1A and 2A domain is the same as in Figure 1. The amino acids interacting with the nucleotide are explicitly shown in ball-and-stick.
While few residues interact with adenine base, more residues are implicated in the coordination of the phosphate moiety (Figure 2A). α- and β-phosphates are stabilized by residues S32, G33, K34, T35 and T36 from motif I. Five residues, G31 and K34 (Motif I), Q145 (Motif III), R188 (Motif IV) and R390 (Motif VI), make direct contacts with the γ-phosphate. Although both Mg2+ and Ca2+ were included in the crystallization cocktail, Ca2+ instead of Mg2+ is associated with AMPPNP. Mg2+ is essential for hydrolysis and Ca2+ is partially inhibitory (unpublished observations), but they have close structural properties in terms of coordination. Thus, the substitution of Mg2+ by Ca2+ has insignificant effect on the structure. A single Ca2+ is hexacoordinated with an octahedral geometry by three water molecules, oxygens of β- and γ-phosphates and hydroxyl of T35 (motif I). The high resolution enables us to identify one water molecule as the speculative attacking water for ATP hydrolysis. This water, H-bonded to residues Q364, G365 and Q145, is well ordered and close to the γ-phosphorus atom (3.5 Å) and located ideally for a nucleophilic in-line attack (Supplementary Figure S3). Q145, interacting with the γ-phosphate, has similar configuration properties as Q499 in RecD2 and Q194 in RecA (42,43), which have been proposed to detect the presence or absence of the γ-phosphate of ATP and relay the information to other sites within the molecule through conformational changes. Thus, Q145 might function as a ‘γ-phosphate sensor’. Both R188 and R390 from the conserved motifs IV and VI, respectively, coordinate the γ-phosphate in a manner qualitatively similar to R493 and R679 in RecD2, and R287 and R610 in PcrA, which act as an arginine finger to stabilize the transition state, thus facilitating ATP hydrolysis. The importance of R188 and R390 residues in ATP hydrolysis and in DNA unwinding was confirmed by mutagenesis (the location sites of all the mutants presented in this paper are shown in Supplementary Figure S4). As shown in Table 1, both mutants R188A and R390A fail to unwind duplex DNA due to their deficiencies in ATP hydrolysis.
Table 1. DNA unwinding, binding and ATPase activities of wt and BsPif1 variants*.
| Activities | Unwindinga | Bindingb | ATPasec | ||||
|---|---|---|---|---|---|---|---|
| Function and variant | kobs (s−1) | Am | Kd (nM) | kmin (min−1) | kmax (min−1) | kmax/kmin | |
| wt-BsPif1 | 0.6 | 0.94 | 16.0 | 15.5 | 810 | 52 | |
| ATP hydrolysis | R188A | ∼0 | ∼0 | 21.3 | NDd | 4.2 | ND |
| R390A | ∼0 | 0.07 | 20.5 | ND | 3.4 | ND | |
| 1A domain | G56A | 0.23 | 0.74 | 22.6 | 12.7 | 1121 | 88 |
| T66A | 0.05 | 0.72 | 27.1 | 8.1 | 761 | 93 | |
| H68A | 0.17 | 0.88 | 40.4 | 7.9 | 361 | 45 | |
| V149A | 0.09 | 0.93 | 36.9 | 13.7 | 421 | 30 | |
| 2A domain | K237A | ∼0 | 0.25 | 80.3 | 11.0 | 236 | 21 |
| T359A | 0.16 | 0.89 | 26.6 | 14.0 | 659 | 47 | |
| H361A | 0.51 | 0.94 | 12.2 | 9.6 | 523 | 54 | |
| K362A | ∼0 | 0.09 | 52.0 | 11.4 | 75 | 6 | |
| Loop3 | Y332F | ∼0 | 0.01 | 64.2 | 9.0 | 68 | 7 |
| Δaa323–350 | ∼0 | ∼0 | ND | 10.6 | 23 | 2 | |
| I339C | 0.97 | 0.69 | 9.6 | 14.7 | 795 | 54 | |
| L95C | 0.76 | 0.94 | 14.1 | 13.1 | 801 | 61 | |
| I339C/L95Ce | 0.02 | 0.02 | 56.3 | 16.4 | 39 | 2.3 | |
| I339C/L95Cf | 1.65 | 0.71 | 17.2 | 15.8 | 798 | 51 | |
| ABC/PFSS-motifs | Δaa201–204 | 0.02 | 0.49 | 47.4 | 10.7 | 769 | 72 |
| Δaa291–295 | 0.01 | 0.07 | 52.7 | 13.5 | 86 | 6.3 | |
| D289A/K292A | 0.01 | 0.05 | 52.6 | 22.0 | 244 | 11 | |
| I78A/T79A/D82A | 0.12 | 0.57 | 37.9 | 12.6 | 1628 | 129 | |
| PFGG(aa131–134) | ∼0 | 0.02 | ND | 8.6 | 59 | 7 | |
| Pin-loop | F71A | ∼0 | 0.01 | 275 | 12.1 | 50 | 4 |
| F75A | 0.38 | 0.87 | 30.3 | 14.0 | 1002 | 72 | |
| Y81A | 0.41 | 0.74 | 16.9 | 12.8 | 754 | 59 | |
| R83A | 0.24 | 0.92 | 22.9 | 11.4 | 943 | 82 | |
| F88A | 0.25 | 0.91 | 26.6 | 11.9 | 1098 | 92 | |
*Values are the average of two independent experiments.
aUnwinding amplitude (Am) and unwinding rate (kobs) were determined from the unwinding kinetic curves.
bDissociation constant (Kd) was determined form DNA binding curve as described in ‘Materials and Methods’ section.
ckmin and kmax represent the basal and DNA activated ATPase activities, respectively.
dND: values are too low to be determined unambiguously.
eParameters determined without DTT.
fParameters determined with 2 mM DTT.
Structure of the BsPif1–ssDNA complex
Although the crystal structure of the ternary complex (BsPif1-ADP·AlF3-ssDNA) was obtained with 8 base-oligonucleotide, only 6 bases can be identified in contact with the protein thanks to the high quality of electronic density. As shown in Figures 1 and 3, the 6-nt oligo-dT binding site traverses along a channel that runs across the top of domains 2A and 1A in a 5′-to-3′ direction. The conformation of the DNA highly resembles those in RecD2 ternary and Dda binary complexes in which the bases are stacked upon each other, although occasionally separated by amino acids. Therefore, the numbered bases dT3-dT8 have extensive interactions with the protein via the phosphodiester backbone rather than the bases. Within domain 2A, H236 interacts with phosphate groups of dT3, K237 with dT4, T359 with dT5 and K362 with dT5-dT6. The imidazole ring of H361 stacks against the deoxy-ribose group of dT4. Half of the bases (dT6-dT8) mainly interact with domains 1A and 1B. Similarly, within domain 1A, V149 interrupts the dT4 and dT5 base stacking interactions by inserting between them. The side chains of G56, T66 and S69 form hydrogen bonds with phosphate groups of dT6, dT7 and T8 while H68 stacks against the deoxy-ribose of dT6. Three residues (F75, P74 and N86) in the small 1B domain loop are implicated in interactions with bases dT6-dT8. While the side chain amide group of N86 makes a hydrogen bond with O2 group of dT7, F75 and P74 stabilize bases dT6 and dT7 through a π–π stacking and hydrophobic interactions, respectively (Figure 3).
Figure 3.

ssDNA binding site. (A) Details of the interactions between ssDNA and BsPif1. Amino acids are colored according to domain colors defined in Figure 1. (B) Schematic diagram of the protein–DNA interactions.
To determine the roles of interactions identified in the BsPif1–DNA structure, variants of BsPif1 with single alanine substitutions were assayed by fluorescence anisotropy and fluorescence stopped-flow experiments to determine their DNA binding and unwinding activities. Although all variants display certain degrees of reduction in DNA binding and unwinding activities (Table 1), the most significant reduction in the activities is with mutants of residues implicated in the interactions with the phosphate groups, such as T66, K237 and K362, in which the efficiencies of DNA unwinding were reduced by 200–600-fold compared to wild-type (wt). Furthermore, these impairments are accompanied by significant reductions in DNA binding activities (Table 1). However, mutations of these residues that stack against the deoxy-riboses (H68A and H361A) or the bases (F75) only lead to 1–3.5-fold reduction in catalytic efficiency and 1–2 fold in DNA binding. Altogether, these results demonstrate that the residues identified by the crystal structures play essential roles in DNA binding or/and catalysis.
Conformational changes, 2B domain shift and loop3 rotation upon nucleotide and ssDNA binding
In the BsPif1-ADP·AlF3-ssDNA ternary complex structure, BsPif1 undergoes a large conformational change in domains 1A, 2A and 2B compared to the apo-protein. When domains 1A of the binary (BsPif1–AMPPNP) and the ternary structures are carefully superimposed, domains 2A and 2B have a global displacement of about 10.6 Å and a rotation of 36.5° (Supplementary Figure S5). The conformational changes upon nucleotide/ssDNA binding can be described as two sequential steps (Figure 4). First, nucleotide and ssDNA binding induce a 2.33 Å increase in distance between domains 1A and 2A, which is accompanied by an ∼22° clockwise rotation of domain 2A, seen in Figure 4B. The main structural rearrangements in domain 2A occur in the region of aa 219–231, which corresponds to a linker between domains 2A and 2B. Secondly, while domain 2A has rotated, domain 2B is not yet in its definitive position. To bring domain 2B into its final position in the ternary complex, a rotation of nearly 40° and a global displacement of domain 2B of about 12.3 Å are needed (Figure 4D).
Figure 4.

Conformationnal changes upon nucleotide hydrolysis. The Figure shows a two steps sequence to change from the BsPif1–AMPPNP structure (1A, 2A and 2B domains colored in blue, red and cyan, respectively) to BsPif1-ADP-AlF3-ssDNA structure (1A, 2A and 2B domains colored light blue, magenta and green, respectively). Conformational change can be described by a rigid body rotation of domains 2A and 2B. The two structures are superimposed in (A) on 1A domains. (B) The first step in a closure movement of 2A domain by a clockwise rotation of 22° (rotation axis is perpendicular to the plane of the figure). Domain 2A in red is then superimposed on 2A domain in magenta. For clarity, domains 2B are omitted. (C) After 2A domains are superimposed, a second step is necessary to superimpose the 2B domains. An anti-clockwise rotation of nearly 40° is necessary to bring domain 2B in cyan upon domain 2B in green. For clarity, the 1A domains are omitted. The final structure is then perfectly superimposed on BsPif1-ADP-AlF3-ssDNA target structure (D).
The above results revealed the striking feature that the conformational change upon nucleotide and ssDNA binding mainly occurs in domain 2B. Although an SH3-like domain 2B comprising four β-strands (β11, β12, β13, β14) arranged in two perpendicular β-sheets is reminiscent of domains 2B of RecD2 and Dda, a conspicuous difference was observed, in which three additional extension loops termed loop1–3 are inserted into BsPif1 domain 2B (Figure 5A–C). The first loop1 (β8–β9–η5–β10, aa254–277) forms a short β-sheet at the beginning and a loop extension at the surface of the SH3 domain. Loop2 (aa 278–307, β11–β12) contains motifs B and C. Loop3 (β15–β16, aa 322–350) protrudes from the SH3 core domain with an prominent β-hairpin. This region is limited to a short α-helix in RecD2 (aa 632–636) (Figure 5B) and forms the tower and hook structures in Dda (Figure 5C). In BsPif1, β9, β16 and β15 form an antiparallel β-sheet where β15–β16 extends as an unusual long β-hairpin structure in BsPif1. Importantly, in addition to the global conformational change of the whole domain 2B, the loop3 undergoes a more significant movement accompanied by a rotation of the top of the hairpin (aa 331–341) by 36° (Figure 4). The long hairpin β-sheet β15–β16 is then divided into a hinge region, forming a β15′–β16′ β-sheet at the top of the hairpin (Figure 5D). Previous structural and biochemical studies have revealed that the 2B domain in SF1A family helicases including PcrA, Rep and UvrD, can rotate upon nucleotide and/or DNA binding, resulting in ‘open’ or ‘close’ conformations (38,39,44). However, no such significant conformational change in the 2B domain of SF1B has previously been observed.
Figure 5.

The 2B domain of BsPif1. (A) Structure of the BsPif1 2B domain with numbering of the secondary structure elements. (B) Superimposition of domain 2B of BsPif1 (cyan) on domain 2B of RecD2 (magenta). (C) Superimposition of domain 2B of BsPif1 (cyan) on domain 2B of Dda (green). The long hairpin in BsPif1 is equivalent to the tower (α-helix and long two-strand β-sheet) in Dda. There is no equivalent in BsPif1 of the hook structure, the hairpin insertion between β11 and β12. (D) Superimposition of domain 2B of BsPif1–AMPPNP (cyan) on domain 2B of BsPif1-ssDNA-ADP-AlF3 (purple).
We then designed several experiments to probe the roles of loop3 in DNA unwinding/binding as suggested by the structural analysis above (Supplementary Figure S4). Three BsPif1 constructs modified around loop3 were characterized: (i) two mutants near the top of the loop3 (Y332F and I339C) chosen for their proximity to several α-helices in domain 1A of the apo-structure; (ii) complete deletion of loop3 from the SH3 domain and (iii) introduction of a new disulfide bridge between loop3 and domain 1A (using I339C/L95C double mutant) in order to block the potential rotation of loop3 (Supplementary Figure S6). Characterizations of the two mutants showed that while Y332F displays severe impairments in DNA unwinding rate and amplitude with a 4-fold reduction in binding affinity (16 nM versus 64 nM in Kd), the mutant I339C displays a higher unwinding efficiency (kobs) and DNA binding activity than wt-BsPif1, although its unwinding amplitude was moderately reduced (Table 1). This phenotype is reminiscent of the hyperhelicase phenotype UvrD303 which has two point mutations in domain 2B (45). Deletion of loop3 (Δaa323–350) renders the protein unable to bind and unwind DNA as judged from unwinding and binding assays (Table 1), indicating that loop3 plays important roles in both DNA binding and unwinding. Consistent with the above observations, establishing a disulfide bridge between I339C and L95C, therefore blocking the potential rotation of loop3, makes BsPif1 completely inactive in unwinding activity while its DNA binding activity was only reduced 3.5-fold in the absence of DTT. The existence of the disulfide bridge was confirmed on the crystallographic structure of the L95C/I339C mutant in which three-dimensional alignment of protein backbone C-α atoms between wt and mutant BsPif1 are well superimposed except a clear electron density between the two Cys residues (95C-339C) is clearly visible (Supplementary Figure S6). While the unwinding amplitude of wt-BsPif1 was increased 2-fold with low DTT concentration (0.1–0.25 mM) (Figure 6A and C), that of the double mutant (I339C/L95C) could achieve full recovery when DTT concentration was gradually increased to 5 mM (Figure 6B and C). Surprisingly, the unwinding rate of the mutant was even higher than that of wt-BsPif1 (Figure 6D). The observations that mutations increase protein activities or catalytic efficiency have been documented in literatures (45). The molecular mechanism by which the double mutant induces higher rate constant is not understood now; more researches need to be performed to clarify this issue. Taken together, the above results not only stress the importance of loop3 function in DNA binding and unwinding, but also reveal that its rotation/movement during unwinding is absolutely required.
Figure 6.

The unwinding activity of mutant I339C/L95C is increased with increasing concentration of DTT. (A and B) The unwinding kinetic curves of wt-BsPif1 and mutant I339C/L95C, respectively. (C and D) show the fractions of unwinding amplitude and rate determined from the unwinding curves in (A and B). Both wt- and mutant I339C/L95C protein were treated with oxygen for 3 h, then 100 nM BsPif1 proteins and 4 nM fluorescent labeled partial duplex DNA were used for DNA unwinding kinetic assays with increasing DTT concentrations as indicated in the figures.
Pif1-specific motif structures and their functional studies
Pif1 family helicases were distinguished from SF1 family helicases by their characteristic sequences between motif IV and V, referred to as A, B and C, as well as PFSS which appears to be unique to Pif1 helicases (17). The structures and functions of these motifs were totally unknown. Our structural study reveals that these motifs are also structurally conserved between Pif1 and RecD/RecD2, and partially in Dda. Motif A forms an α-helix which links domains 1A and 2A and runs beneath them (Figure 1A and Supplementary Figure S7). The two ends of motif A α-helix are linked by two random coils: one (aa 203–208) further connects the next α-helix, forming a part of motif IV; the other one (aa 183–191) is the linker between 1A and 2A in which an arginine residue (R188) interacts with the γ-phosphate. The structural feature of motif A is conserved with both RecD and RecD2 helicases in domain 2A, in which α-helices of the putative motif A and the 1A-2A domain linker with an arginine residue interacting with the γ-phosphate present in a very similar configuration as motif A in BsPif1. Mutations of the conserved residues L195, N196 and R199 which constitute motif A α-helix resulted in protein degradation or aggregation, probably due to incorrect protein folding. However, deletion of residues downstream of motif A (Δaa201–204) reduces DNA unwinding rate and amplitude 30- and 2-fold, respectively (Table 1).
Motifs B and C identified by sequence alignment are separated by just eight residues and located in a pair of antiparallel β-sheets of the SH3/2B domain (Supplementary Figure S7A). Motifs B and C correspond to β11 and β12 respectively. More interestingly, the eight linker residues further form a short hairpin composed of two antiparallel β-strands linked by a β-turn (Supplementary Figure S7A). Similarly, the putative motifs B and C in RecD2 constitute a hairpin with two antiparallel β-sheets, but not linked with any structured random coil.
The putative PFSS fragment folds as an α-helix with a turn which connects to a β-strand and belongs to one of the seven α-helices that surround the parallel 5-stranded β-sheets in domain 1A (Figure 1). The most striking feature of this α-helix is that it lies vertically in opposite position to the putative strand separation wedge/pin-loop formed by domain 1B. The domain 1B and PFSS/α-helix interact through a set of hydrogen bonds between I78, T79 and D82 on 1B and D113 and R120 on the PFSS α-helix (Supplementary Figure S7B).
We then prepared several mutations to probe functions of these motifs and to identify the potential interactions between PFSS and domain 1B. Deletion of four residues in the B-C motifs/loop1 (Δaa291–295) results in a complete loss of DNA unwinding and a reduction in DNA binding (Table 1). Similarly, the double mutant D289A/K292A displayed impaired DNA unwinding activity (Table 1), indicating that B-C motifs are implicated in DNA unwinding and binding. To identify the potential interactions between PFSS and 1B and to probe which potential aromatic residue wedges into duplex DNA, we first studied the triple-mutant (I78A/T79A/D82A) and found that the unwinding rate, amplitude and DNA binding activities were significantly reduced (Table 1). Furthermore, deletion of the four residues PFGG (Δaa131–134) which are highly conserved in PFSS motif and connect PFSS to its downstream β–strand, completely abolishes helicase activity (Table 1), since the deletion changes PFSS spatial position relative to domain 1B. Further characterization of five variants of aromatic residues within 1B (F71A, F75A, Y81A, R83A and F88A) (supplementary Figures S4 and S7) revealed that only the F71A mutant enzyme was deficient in helicase activity compared to wt and other mutants (F75A, Y81A, R83A and F88A, Table 1). Taken together, all these results indicate that the interactions between residues within domain 1B (I78, T79 and D82) and within PFSS (D113 and R120) are important for maintaining F71 in a precise spatial configuration so as to mechanically split the DNA substrate.
ATPase activity of different mutants
The ATPase activities of the BsPif1 variants were determined in the absence and in the presence of ssDNA (basal and activated ATPase activity, respectively; Table 1). The basal ATPase activity of wt-BsPif1 is about 16 min−1, and is further stimulated >50-fold by the addition of dT30 (kmax ≈ 810 min−1). We observed that the ssDNA stimulation decreases with lower DNA binding affinity (Table 1). However, three of the variants (I78A/T79A/D82A, F75A and F88A) have elevated ssDNA stimulation although their DNA binding affinities are lower than wt-BsPif1 (Table 1). It is noteworthy that the ATPase activity of the mutant I339C/L95C is comparable with that of wt-BsPif1 in the presence of 2 mM DTT (Table 1), in accordance with the prior observation that the mutant unwinding activity was recovered in the presence of DTT. The fact that all variants display comparable basal ATPase activity as wt-BsPif1 and can be further stimulated to different extents, except for R188A and R390A which are implicated in ATP binding or/and hydrolysis, indicates that the mutants fold correctly and that the observed activities reflect the intrinsic properties of the mutants.
Solution structures determinded by SAXS
To rule out the possibility that the ‘apo’ structure is a crystal stacking artefact, SAXS analysis was performed on BsPif1. In the absence of DNA, the molecular weight of BsPif1 estimated from I(0) and Porod volume is 50 kD which is in agreement with a monomeric BsPif1 (Supplementary Figure S8). The fit of the structure in solution with the different crystal structures of BsPif1 gave a χ2 of 5.5 and 7.5 for BsPif1 in P212121 (‘apo’) and P3121 (‘complex’) space group, respectively. DNA and ligands were removed before χ2 calculations since these were not present in solution. The proportion of the two structures in solution has been analyzed with the program MES (46) giving 80% for ‘apo’ and 20% for ‘complex’. The ‘apo’ thus seems to be predominant in solution. The ‘apo’ model can be further improved by introducing flexibility between domains 1A and 2A in the region of aa 185–190 and in domain 1B (aa72–88). The modeling results in a more extended conformation for domain 1B, with a final χ2 of 2.2. Hence BsPif1 has a conformation in solution close to the one observed in the crystal structure of the apo-protein, with the 2B domain packed against domains 1A and 2A.
DISCUSSION
Pif1 family helicases have been identified for many years as important proteins involved in diverse DNA transactions such as maintenance of mitochondrial DNA integrity, disruption of telomerase from the telomeric end, replication at many replication forks and they are especially required at hard-to-replicate regions including ribosomal DNA and G-quadruplex structures. However, the structural basis remained to be elucidated. We present here the first crystal structures, to our knowledge, of the Pif1 DNA helicase from Bacteroides spp with and without ATP analog/ssDNA. A combination of structural, mutational and biochemical analyses reveals a number of interesting features and sheds light on its mechanism of DNA unwinding.
First, BsPif1 structure with AMPPNP enabled us to answer some of the mechanistic questions about BsPif1 ATPase activity. We have previously found that BsPif1 could hydrolyze all types of NTP/dNTP without significant preference. A similar phenomenon has been observed for several DExH helicases including eIF4, HS3 and DHX9 helicases. Our BsPif1 structure revealed that the structural basis for this promiscuity is the missing selectivity filter for the adenine base. While the well studied DExD/H proteins harboring a Q-motif make extensive specific main-chain and side-chain contacts with positions in the adenine rings, these interactions are replaced in BsPif1 by a single π-stacking contact between a phenylalanine 187 and the aromatic ring electron of the base in addition to an isoleucine 5. Therefore, this should provide a structural basis for helicases without a Q-motif acting as general NTPases. The biological advantage of non-selective nucleotide base binding over a selective one should be that in the situation of ATP depletion in a cell, helicases can continue to use other types of nucleotides to ensure their function.
BsPif1 shares its general structural fold with RecD2 and Dda, especially in the core helicase domains, as expected. This is consistent with the general view that the two RecA-like domains act as a molecular engine while the auxiliary domains function in concert with the core to recognize and determine the specificity for DNA/RNA to be unwound (1). Indeed, sequence alignments revealed that Pif1 family helicases are distinct from other helicase proteins by the presence of the specific motifs A, B, C and PFSS. It was not clear whether these structural motifs fold into specific structures and how they confer the Pif1 helicase special functions. Our results show that these sequence-specific motifs are in fact structurally conserved in the SF1B helicase family (i.e. RecD2 and Dda) as well as in other helicase families. Motif A forms an α-helix constituting one of seven α-helices in domain 2A found in most RecA-like domains. It is noteworthy that the motif A localizes to the surface of the cleft between 1A–2A domains and moves as a rigid body with 2A during ATP binding and hydrolysis. Thus, its precise spatial conformation will keep the linker (aa L183-P192) in a correct configuration for ATP binding/hydrolysis, yet remaining flexible enough to allow the cleft to enlarge after ATP hydrolysis. Alteration of the motif A spatial position by truncating aa 201–204 heavily compromised the unwinding activity (Table 1). The putative motifs B-C, forming just two β-strands in domain 2B, which adopts an SH3 domain fold, are equally structurally conserved with RecD2 and Dda. In vitro functional studies show that deletion (Δaa291–295) or mutations (D289A/K292A) of residues in B and C motifs abolish or severely reduce DNA binding and unwinding activities (Table 1). On the basis of structure and mutation results, we speculate that some of the residues in the B-C motifs are implicated in DNA unwinding.
Interestingly, the PFSS motif, which is highly specific to Pif1 sequence, folds in fact as one of the four α-helices which surround the β-strands, found in most RecA-like domains, and does not display any striking structural feature by itself. However its organization with two adjacent loops may contribute to DNA unwinding and binding in an important manner. The interactions between the putative pin-loop in domain 1B and PFSS motif may allow the pin to form a more rigid structure and help the residues on the loop to adopt a precise spatial conformation. Although no conserved sequences in RecD2 corresponding to the Pif1-specific signature sequence can be identified, equivalent α-helices appear to be structurally conserved in RecD2 with similar configuration features (Supplementary Figure S9). Furthermore, the similar structural organization in which both the β-strand of the type PFSS/α-helices and the separation wedge in its opposite position can be found in SF1A helicase family, such as Rep, UvrD and PcrA (Supplementary Figure S9). The interactions between PFSS and pin-loop in domain 1B display three features: (i) the pin-loop/PFSS structure locates at the incoming duplex fork position; (ii) the pin-loop conformation should be stabilized by residues on the PFSS/α-helix through a set of interactions including hydrogen bonds and salt bridges, which appear to be conserved in SF1 family helicases; (iii) by analyzing the available SF1 helicase structures, it appears that there is a correlation between the localization of the comparable wedge/PSFF structure and the SF1 family helicase polarity: while the 5′-3′ SF1B harbor the wedge/PSFF structure in 1A domain, the wedge/PSFF exists in 2A domain for the 3′-5′ SF1A (Supplementary Figure S9), being consistent with the previously proposed molecular mechanism of polarity determination for SF1B helicases (35,38,39,44,47–49). Our mutational studies not only reveal the important interactions between the pin-loop and the PFSS motif, but also allow us to suggest that F71 may be a good candidate as a separating knife to cut duplex- or G4-DNA. Another loop linked PFSS α-helix harbors typical PFSS motif sequences (PFGG, aa 131–134), which is highly conserved in the Pif1 helicase family. Deletion of these residues leads to deficiencies in DNA–binding/unwinding activities. Since our structures show that these residues do not directly contact with DNA, our interpretation is that the shortening of this stretch of residues will affect the PFSS spatial conformation, consequently altering the pin-loop position.
The most striking finding in our present structural study is that the SH3/2B domain, especially loop3, undergoes a significant movement and/or rotation upon DNA binding. The unusually long hairpin constituted by two antiparallel β-strands appears to be unique to Pif1, but conserved from BsPif1 to human Pif1 according to our homology modeling results (not shown). Deletion of loop3 resulted in the complete loss of DNA unwinding/binding activities. Furthermore, while the potential rotation/movement was prevented by introducing a disulfide bridge between the tip of the hairpin and 1A domain, the constrained BsPif1 is completely deficient in unwinding activity, but its DNA binding activity was only modestly reduced. Interestingly, by relieving the disulfide constraint by addition of DTT, DNA unwinding activity could be totally restored, further underlining the importance of the rotation/movement of loop3 for DNA unwinding. More interestingly and importantly, although the B-C motif and the top of loop3 are far away from the DNA binding sites, deletion of 4 residues in the B-C motif or mutation of Y332 in loop3 results in impaired unwinding activities in these variants, while their DNA binding activities are reduced only 2–3 fold. These results suggest that these structural motifs are implicated in DNA unwinding through SH3/loop3 movement and rotation.
According to the above analysis and the previously proposed RecD2 and Dda helicase models, we modeled BsPif1 in complex with a partial duplex and propose a mechanism for DNA unwinding in which 2B domain/loop3 plays a predominant role (Figure 7). In this conformation, the incoming duplex faces the hairpin of the 2B domain. Upon nucleotide binding, the 2B domain can undergo a closing movement on ssDNA. The top of the hairpin (loop3) closes upon domain 1B forming a hole where the ssDNA can pass through. ATP hydrolysis may activate the translocase activity by a relative movement of the two RecA domains 1A and 2A. The ssDNA is moved in the 5′ to 3′ direction. The incoming duplex is blocked by the rigid structure formed by 2B and 1B domains. The translocation force may be sufficient to destabilize the incoming duplex and unwind base pairs. After translocation, the 2B domain may (or may not) move back for another cycle of unwinding (Figure 7).
Figure 7.

Mechanism of BsPif1-catalyzed DNA unwinding. (A) ssDNA is in the binding site, 2B domain is in the upper position. (B) 2B domain rotates upon ssDNA. A tunnel is formed by 2B and 1B domains, where only ssDNA can pass through. (C) Upon nucleotide hydrolysis or exchange, the two RecA domains 1A and 2A act as molecular motor to move ssDNA by one base. The 2B domain acts as a wedge, blocking the incoming duplex. The translocation force may be sufficient to open dsDNA on the 2B wedge. (D) The 2B domain rotates back for a new cycle.
Finally, the discovery that Pif1 helicase activity is highly regulated by the SH3 domain as we observed in this work may provide new insight into the puzzling phenomenon that ScPif1 could unwind more than 10 kb duplex DNA during BIR while the purified Pif1 displays very modest processivity (15,23,25). SH3 domains have been involved in nucleic acid binding as for HIV-1 integrase (50), they are usually involved in protein–protein binding and recognition. Our structures show that the proteins and DNA binding sites do not overlap. The SH3 topology of Pif1 could be available to interact with proteins while it is interacting with DNA. It is logical to suggest that DNA polymerase may interact with Pif1 helicase through the SH3 domain and convert Pif1 helicase from a low activity to a high activity conformation, and consequently enhance its processivity. We are now in an excellent position to simultaneously monitor the loop3 rotation/movement and DNA unwinding activity by combining single-molecule manipulation and smFRET technique.
ACCESSION NUMBERS
5FTB: BsPif1-AMPPNP; 5FTC: BsPif1-ADP; 5FTD: BsPif1 apo; 5FTE: BsPif1-ADP-AlF3-ssDNA; 5FTF: BsPif1 L95C-I339C.
Supplementary Material
Acknowledgments
We thank the staff of BL19U1, BL17U1 and BL19U2 beamlines at the National Center for Protein Sciences Shanghai and Shanghai Synchrotron Radiation Facility (Shanghai, China), and the beam-line staff at 3W1A of BSRF (Beijing, China) for assistance with data collection. We are grateful for access to the SOLEIL (SWING) synchrotron radiation facility for SAXS data collection and acknowledge the support provided by Dr. Aurelien Thureau and his team. The authors wish to acknowledge Dr. Anita Lewit-Bentley for carefully reading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31370798, 11304252, 31301632]; 985 and 211 Projects from the Ministry of Education of China; International associated laboratory (LIA) ‘G-quadruplex-HELI’ (CNRS, ENS-Cachan, Institute of physics of the Chinese Academy of Sciences) (in part). Funding for open access charge: CNRS ( (LIA) ‘G-quadruplex-HELI’).
Conflict of interest statement. None declared.
REFERENCES
- 1.Singleton M.R., Dillingham M.S., Wigley D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 2.Foury F., Kolodynski J. pif mutation blocks recombination between mitochondrial rho+ and rho- genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1983;80:5345–5349. doi: 10.1073/pnas.80.17.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou J., Monson E.K., Teng S.C., Schulz V.P., Zakian V.A. Pif1p helicase, a catalytic inhibitor of telomerase in yeast. Science. 2000;289:771–774. doi: 10.1126/science.289.5480.771. [DOI] [PubMed] [Google Scholar]
- 4.Boule J.B., Vega L.R., Zakian V.A. The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature. 2005;438:57–61. doi: 10.1038/nature04091. [DOI] [PubMed] [Google Scholar]
- 5.Chisholm K.M., Aubert S.D., Freese K.P., Zakian V.A., King M.C., Welcsh P.L. A genomewide screen for suppressors of Alu-mediated rearrangements reveals a role for PIF1. PLoS One. 2012;7:e30748. doi: 10.1371/journal.pone.0030748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabouri N., Capra J.A., Zakian V.A. The essential Schizosaccharomyces pombe Pfh1 DNA helicase promotes fork movement past G-quadruplex motifs to prevent DNA damage. BMC Biol. 2014;12:101–114. doi: 10.1186/s12915-014-0101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J.-Q., Qi H., Schulz V.P., Mateyak M.K., Monson E.K., Zakian V.A. Schizosaccharomyces pombe pfh1(+) encodes an essential 5′ to 3′ DNA helicase that is a member of the PIF1 subfamily of DNA helicases. Mol. Biol. Cell. 2002;13:2180–2191. doi: 10.1091/mbc.02-02-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivessa A.S., Zhou J.Q., Zakian V.A. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell. 2000;100:479–489. doi: 10.1016/s0092-8674(00)80683-2. [DOI] [PubMed] [Google Scholar]
- 9.Paeschke K., Capra J.A., Zakian V.A. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013;497:458–462. doi: 10.1038/nature12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou X.M., Wu W.Q., Duan X.L., Liu N.N., Li H.H., Fu J., Dou S.X., Li M., Xi X.G. Molecular mechanism of G-quadruplex unwinding helicase: sequential and repetitive unfolding of G-quadruplex by Pif1 helicase. Biochem. J. 2015;466:189–199. doi: 10.1042/BJ20140997. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka H., Ryu G.-H., Seo Y.-S., Tanaka K., Okayama H., MacNeill S.A., Yuasa Y. The fission yeast pfh1(+) gene encodes an essential 5′ to 3′ DNA helicase required for the completion of S-phase. Nucleic Acids Res. 2002;30:4728–4739. doi: 10.1093/nar/gkf590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Budd M.E., Reis C.C., Smith S., Myung K., Campbell J.L. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol. Cell. Biol. 2006;26:2490–2500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stith C.M., Sterling J., Resnick M.A., Gordenin D.A., Burgers P.M. Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J. Biol. Chem. 2008;283:34129–34140. doi: 10.1074/jbc.M806668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson M.A., Kwon Y., Xu Y., Chung W.H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P., et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502:393–396. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J.E., Lobachev K.S., Malkova A. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bochman M.L., Sabouri N., Zakian V.A. Unwinding the functions of the Pif1 family helicases. DNA Repair (Amst) 2010;9:237–249. doi: 10.1016/j.dnarep.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bochman M.L., Judge C.P., Zakian V.A. The Pif1 family in prokaryotes: what are our helicases doing in your bacteria. Mol. Biol. Cell. 2011;22:1955–1959. doi: 10.1091/mbc.E11-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramanagoudr-Bhojappa R., Blair L.P., Tackett A.J., Raney K.D. Physical and functional interaction between yeast Pif1 helicase and Rim1 single-stranded DNA binding protein. Nucleic Acids Res. 2013;41:1029–1046. doi: 10.1093/nar/gks1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu Y., Masuda Y., Kamiya K. Biochemical analysis of human PIF1 helicase and functions of its N-terminal domain. Nucleic Acids Res. 2008;36:6295–6308. doi: 10.1093/nar/gkn609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.George T., Wen Q., Griffiths R., Ganesh A., Meuth M., Sanders C.M. Human Pif1 helicase unwinds synthetic DNA structures resembling stalled DNA replication forks. Nucleic Acids Res. 2009;37:6491–6502. doi: 10.1093/nar/gkp671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lahaye A., Leterme S., Foury F. PIF1 DNA helicase from Saccharomyces cerevisiae. Biochemical characterization of the enzyme. J. Biol. Chem. 1993;268:26155–26161. [PubMed] [Google Scholar]
- 23.Liu N.N., Duan X.L., Ai X., Yang Y.T., Li M., Dou S.X., Rety S., Deprez E., Xi X.G. The Bacteroides sp. 3_1_23 Pif1 protein is a multifunctional helicase. Nucleic Acids Res. 2015;43:8942–8954. doi: 10.1093/nar/gkv916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barranco-Medina S., Galletto R. DNA binding induces dimerization of Saccharomyces cerevisiae Pif1. Biochemistry. 2010;49:8445–8454. doi: 10.1021/bi100984j. [DOI] [PubMed] [Google Scholar]
- 25.Ramanagoudr-Bhojappa R., Chib S., Byrd A.K., Aarattuthodiyil S., Pandey M., Patel S.S., Raney K.D. Yeast Pif1 helicase exhibits a one-base-pair stepping mechanism for unwinding duplex DNA. J. Biol. Chem. 2013;288:16185–16195. doi: 10.1074/jbc.M113.470013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boule J.B., Zakian V.A. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007;35:5809–5818. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanders C.M. Human Pif1 helicase is a G-quadruplex DNA-binding protein with G-quadruplex DNA-unwinding activity. Biochem. J. 2010;430:119–128. doi: 10.1042/BJ20100612. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 30.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D.T., Konarev P.V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evrard G., Mareuil F., Bontems F., Sizun C., Perez J. DADIMODO: a program for refining the structure of multidomain proteins and complexes against small-angle scattering data and NMR-derived restraints. J. Appl. Crystallogr. 2011;44:1264–1271. [Google Scholar]
- 34.Dou S.X., Wang P.Y., Xu H.Q., Xi X.G. The DNA binding properties of the Escherichia coli RecQ helicase. J. Biol. Chem. 2004;279:6354–6363. doi: 10.1074/jbc.M311272200. [DOI] [PubMed] [Google Scholar]
- 35.Saikrishnan K., Griffiths S.P., Cook N., Court R., Wigley D.B. DNA binding to RecD: role of the 1B domain in SF1B helicase activity. EMBO J. 2008;27:2222–2229. doi: 10.1038/emboj.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He X.P., Byrd A.K., Yun M.K., Pemble C.W., Harrison D., Yeruva L., Dahl C., Kreuzer K.N., Raney K.D., White S.W. The T4 phage SF1B helicase Dda is structurally optimized to perform DNA strand separation. Structure. 2012;20:1189–1200. doi: 10.1016/j.str.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanya H.S., Bird L.E., Brannigan J.A., Wigley D.B. Crystal structure of a DExx box DNA helicase. Nature. 1996;384:379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- 38.Velankar S.S., Soultanas P., Dillingham M.S., Subramanya H.S., Wigley D.B. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 39.Lee J.Y., Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schutz P., Wahlberg E., Karlberg T., Hammarstrom M., Collins R., Flores A., Schuler H. Crystal structure of human RNA helicase A (DHX9): structural basis for unselective nucleotide base binding in a DEAD-Box variant protein. J. Mol. Biol. 2010;400:768–782. doi: 10.1016/j.jmb.2010.05.046. [DOI] [PubMed] [Google Scholar]
- 41.Martin A., Schneider S., Schwer B. Prp43 is an essential RNA-dependent ATPase required for release of lariat-intron from the spliceosome. J. Biol. Chem. 2002;277:17743–17750. doi: 10.1074/jbc.M200762200. [DOI] [PubMed] [Google Scholar]
- 42.Saikrishnan K., Powell B., Cook N.J., Webb M.R., Wigley D.B. Mechanistic basis of 5′ -3′ translocation in SF1B helicases. Cell. 2009;137:849–859. doi: 10.1016/j.cell.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 43.Story R.M., Weber I.T., Steitz T.A. The structure of the Escherichia-coli reca protein monomer and polymer. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 44.Korolev S., Hsieh J., Gauss G.H., Lohman T.M., Waksman G. Major domain swiveling revealed by the crystal structures of complexes of E-coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 45.Arslan S., Khafizov R., Thomas C.D., Chemla Y.R., Ha T. Engineering of a superhelicase through conformational control. Science. 2015;348:344–347. doi: 10.1126/science.aaa0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelikan M., Hura G.L., Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. General. Physiol. Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernstein D.A., Zittel M.C., Keck J.L. High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 2003;22:4910–4921. doi: 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pike A.C.W., Shrestha B., Popuri V., Burgess-Brown N., Muzzolini L., Costantini S., Vindigni A., Gileadi O. Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1039–1044. doi: 10.1073/pnas.0806908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manthei K.A., Hill M.C., Burke J.E., Butcher S.E., Keck J.L. Structural mechanisms of DNA binding and unwinding in bacterial RecQ helicases. Proc. Natl. Acad. Sci. U.S.A. 2015;112:4292–4297. doi: 10.1073/pnas.1416746112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eijkelenboom A.P.A.M., Lutzke R.A.P., Boelens R., Plasterk R.H.A., Kaptein R., Hard K. The DNA-binding domain of Hiv-1 integrase has an Sh3-like fold. Nat. Struct. Biol. 1995;2:807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.