

Abstract
Current anticancer chemotherapy relies on a limited set of in vitro or indirect prognostic markers of tumor response to available drugs. A more accurate analysis of drug sensitivity would involve studying tumor response in vivo. To this end, we have developed an implantable device that can perform drug sensitivity testing of several anticancer agents simultaneously inside the living tumor. The device contained reservoirs that released microdoses of single agents or drug combinations into spatially distinct regions of the tumor. The local drug concentrations were chosen to be representative of concentrations achieved during systemic treatment. Local efficacy and drug concentration profiles were evaluated for each drug or drug combination on the device, and the local efficacy was confirmed to be a predictor of systemic efficacy in vivo for multiple drugs and tumor models. Currently, up to 16 individual drugs or combinations can be assessed independently, without systemic drug exposure, through minimally invasive biopsy of a small region of a single tumor. This assay takes into consideration physiologic effects that contribute to drug response by allowing drugs to interact with the living tumor in its native microenvironment. Because these effects are crucial to predicting drug response, we envision that these devices will help identify optimal drug therapy before systemic treatment is initiated and could improve drug response prediction beyond the biomarkers and in vitro and ex vivo studies used today. These devices may also be used in clinical drug development to safely gather efficacy data on new compounds before pharmacological optimization.
INTRODUCTION
The ability to predict the optimal therapy for an individual patient is a major unmet need across many diseases. In most diseases, there are no methods for predicting a patient’s sensitivity to the range of available drugs. A notable exception is bacterial and fungal infections where in vitro testing is routinely performed with high clinical use (1). There have been numerous attempts for complex diseases, such as cancer, to use combinations of in vitro and ex vivo methods to regrow cells or tissue taken from biopsies or tumor resections (2, 3). These methods have, however, failed to gain clinical adoption. Cancer pathogenesis and therapeutic responsiveness are determined not only by genetic mutations but also by epigenetics and other environmental factors that are unique to each patient. For example, mounting evidence suggests that the tumor microenvironment contributes substantially to drug response and resistance (4–6). These and other factors have not been accurately recreated outside of the organism.
Most drugs in clinical cancer treatment, particularly cytotoxics, have no reliable predictor of response, and patients are often treated with multiple lines of standard-of-care therapy without positive results (7). Uninformed therapy selection is highly inefficient and may lead to reduced therapeutic success rates, increased side effects, and excessive economic expenditures (8, 9). Patients do not have the time, and the health care system does not have the resources, to apply several rounds of ineffective therapies.
A related problem exists in the drug discovery process. Testing a drug candidate in humans requires a substantial upfront investment to develop the compound’s pharmacological properties before it can be determined whether it is efficacious. Multiple large studies have shown that the dominant reason for attrition in clinical drug development is a lack of efficacy (10, 11). All too often, vast resources are expended to optimize the delivery, bioavailability, and safety properties of a drug candidate, only to find out in larger clinical trials that the compound is not sufficiently effective in humans (12). Collecting clinical data on the efficacy of anticancer compounds much earlier in the drug development process without risk to the patient is highly desirable.
Bringing the laboratory into the patient may be more promising than removing cells or tissues from their native environment for ex vivo functional analyses. By testing a range of relevant drugs directly inside the living tumor, the native tumor physiology would be preserved, no systemic toxicities would be encountered, and the patient would know his or her individual responsiveness to a drug or combination of drugs. To this end, we have developed an in vivo assay that consists of an implantable microscale device that is placed inside the tumor. This device contains a large number of reservoirs, each with a unique single agent or drug combination in microdose amounts (less than one millionth of a systemic dose). The device allows for rapid, parallel investigation of drug sensitivity in living tumors in only 24 hours.
Implantable drug delivery devices currently in clinical use are most commonly used for therapeutic purposes (13–16). Our fully implanted microscale device is capable of delivering precise doses of different drugs into a tumor for parallel drug efficacy assessments in vivo. Here, we describe the controlled local release of a wide range of anticancer drugs from the device into distinct regions of tumors, the precise measurement of drug release for each reservoir, and the tuning of local intratumor concentrations to systemically relevant drug levels. We also demonstrate the ability to assay drug effect locally and show that this assay has excellent predictive value for systemic efficacy for a range of anticancer drugs and tumor models.
RESULTS
Device concept, design, and implantation
The prototype device was of cylindrical shape measuring 820 μm in diameter and was delivered into tumors through a biopsy needle. Here, we used devices with up to 16 reservoirs, which were located on the cylinder mantle to take maximal advantage of the interface area between the device and tumor. The device was implanted directly into the tumor during a biopsy procedure and remained in situ for ~24 hours (Fig. 1A). Drugs from each reservoir were released passively during this time into distinct regions of 200 to 300 μm of tumor tissue, effectively creating in vivo microreactors for the interaction of tumor with a specific drug.
Fig. 1. In vivo drug sensitivity assay.
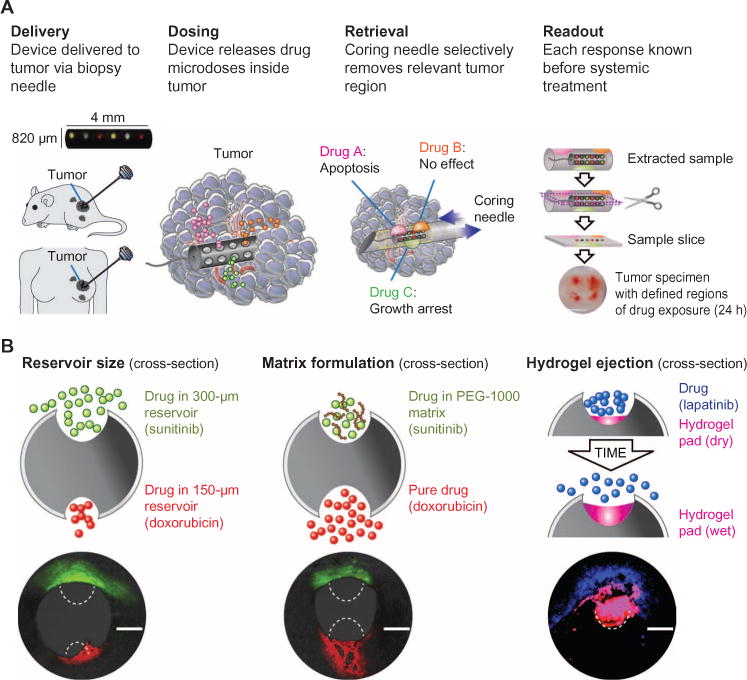
(A) The device is implanted by needle directly into tissue, and drugs diffuse from device reservoirs into confined regions of tumor. Each region is assayed independently to assess the tumor-specific response to a given drug, such as apoptosis or growth arrest. A second biopsy needle selectively retrieves a small column of tissue that immediately surrounds and includes the device. This tissue contains the regions of drug diffusion and is used for determination of drug efficacy. (B) Three methods for precise control over the release profile of a given drug are demonstrated: reservoir opening size affects the rate of transport; the formulation of a drug in a polymer matrix (for example, PEG slows release of sunitinib versus free doxorubicin); and hydrophilic expansive hydrogels (to achieve rapid tissue uptake of highly insoluble drugs, such as lapatinib). Scale bars, 300 μm.
Crosstalk between drugs from different reservoirs was eliminated by appropriate spatial separation of reservoirs and by drug and matrix formulation (Fig. 1B). Drug compounds in pure form have inherently different transport rates, which depend on their chemical properties, including molecular size and solubility. The release properties of compounds often need to be adjusted to compare their relative efficacy at local concentrations that mimic those achieved during systemic dosing. We have developed several methods to obtain adequately sized, yet spatially separated, regions of drug distribution of ~200 to 300 μm in diameter. These techniques are shown in Fig. 1B and included altering the size of the reservoir opening; formulating or embedding compounds in polymer [poly(ethylene glycol) (PEG)] matrices to control their intratumor diffusion properties; or, for difficult-to-dissolve compounds, using hydrophilic expansive polymers to eject drug from reservoirs into the adjacent region of tissue. In the latter, a hydrogel expanded upon contact with fluid in the tumor, which increased the contact area and pressure between drug and tissue.
Implantation of devices into animal tumors was highly reproducible, with greater than 95% of devices (n > 50) successfully placed in their entirety into tumor tissue, including intumors that were 5 mm in diameter. Placement was typically chosen closer to the periphery of tumors to avoid the necrotic core that may be present. The devices used in this study were radiopaque and could be visualized effectively by standard ultrasound and computed tomography (CT) imaging (fig. S2). Visualization of individual reservoirs was achieved in some cases. No device migration was noticed during the implantation period of 1 to 2 days. Retrieval of devices from mouse tumors was performed with success rates >90% (n > 30) and failed only when biopsy gun was improperly aligned with device axis.
Release of drugs from the device into spatially distinct regions of the tumor
Figure 2A shows release of common chemotherapeutic drugs doxorubicin, sunitinib, lapatinib, and the antibody cetuximab from miniature multireservoir devices, identified using immunofluorescence. Dasatinib and gemcitabine were detected in the reservoir region using mass spectrometry (Fig. 2B). Adequate spatial separation of reservoirs ensured that adjacent drugs did not overlap (Fig. 2C).
Fig. 2. Anticancer drugs are delivered into confined regions of tumor.
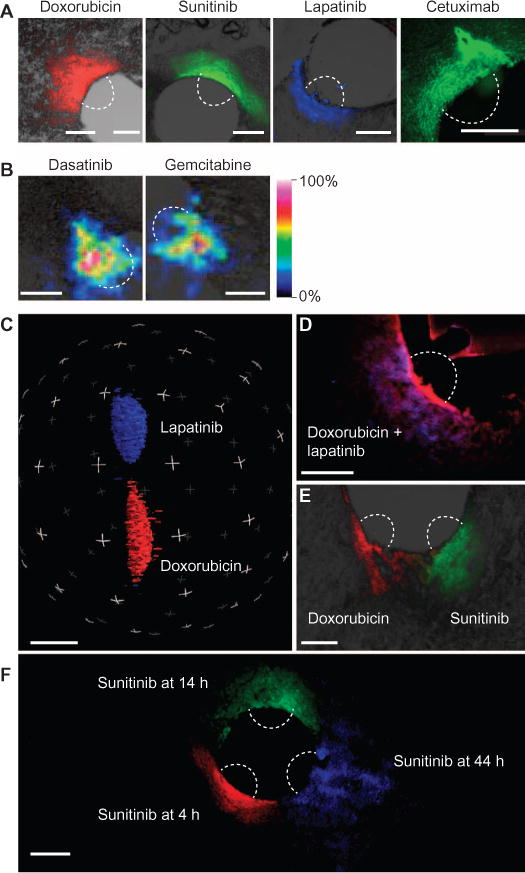
Devices were implanted into BT474 tumors, and drugs (table S1) were passively released. (A) Doxorubicin, sunitinib, and lapatinib autofluorescence were detected by microscopy. Cetuximab conjugated with Alexa 488. (B) Dasatinib and gemcitabine distribution detected by MALDI tissue imaging. (C) Three-dimensional reconstruction of drug release from adjacent reservoirs separated by 750 μm. (D and E) Combinations of drugs are co-delivered to a given region of tumor from one reservoir (red, doxorubicin; blue, lapatinib) (D) or from two adjacent reservoirs 250 μm apart (E). (F) Release of a microdose of sunitinib (50% in PEG-1450) at three time points, demonstrating expanded but confined region of tissue distribution even at longer time points. Scale bars, 300 μm.
Combinations of two or more drugs could be delivered to a given region of tumor to assay their combined in vivo effect on the tumor. Simultaneous exposure to a set of drugs was achieved by loading compounds into the same reservoir, as shown for doxorubicin and lapatinib (Fig. 2D). Alternatively, one can examine the effect of staggering exposure to multiple drugs in a time-dependent manner, which has previously been shown to have a potentially marked effect on efficacy (17). An overlapping region was created in a confined tumor region when first primed with one agent (in this case, doxorubicin) followed by application of a second agent (sunitinib) (Fig. 2E). Drug release occurred upon implantation in a time-dependent manner, with drug distributing over larger regions while still remaining separate from adjacent reservoirs even at longer time points (Figs. 2F and 3A).
Fig. 3. Pharmacokinetics of drug release from device reservoirs.
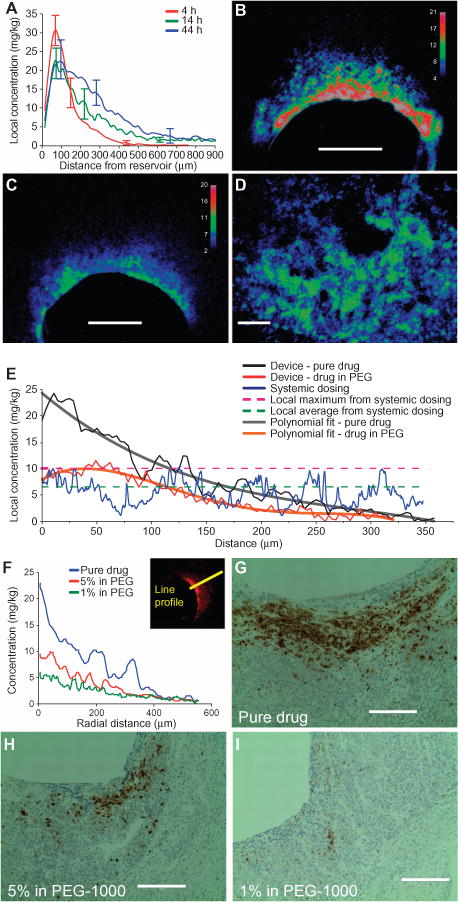
(A) Intratumor transport distances for doxorubicin released from a device in BT474 tumors. Transport was measured as line profiles taken from the center of the reservoir-device interface, moving radially outward, at 4, 14, and 44 hours. Data are averages ± SD (n = 10 distinct reservoirs for each time point). Curves are averaged over 10 samples from five different tumors for each time point. (B and C) Representative tumor cross section showing local doxorubicin distribution at 20 hours after release of the pure drug (B) and drug diluted to 10% (w/w) in PEG-1450 matrix (C) from a device in BT474 tumors. (D) Intratumor doxorubicin distribution after systemic administration of drug (8 mg/kg) in BT474 tumor-bearing mice at 6 hours after injection. Scale bars, 300 μm (B to D). (E) Chart comparing the intratumor concentration of doxorubicin after release as 100% pure drug or 5% drug in PEG-1450 matrix. Device profiles were fit to polynomial curves and compared with systemic dosing. Maximal and average doses after systemic dosing are shown. (F) Drug concentrations for each tumor section in (G) to (I). Concentration profiles are measured as shown in insert. (G to I) Apoptosis after the release of 100% pure drug (F), 5% doxorubicin in PEG-1000 (G), and 1% doxorubicin in PEG-1000 (H). Sections are representative of A375 tumors stained for cleaved caspase-3 (CC3) (brown). Scale bars, 200 μm.
Tuning of device-based doxorubicin release to intratumor concentrations from systemic dosing
The action of chemotherapeutic drugs is often concentration-dependent. When inferring sensitivity of a tumor to a given device-delivered drug, it is critical that the local concentration of drug from a reservoir matches levels that are reached in the tumor after systemic dosing. The widely used anticancer drug doxorubicin was used as a model compound to test drug sensitivity in this manner. Reservoirs loaded with pure doxorubicin were placed in a BT474 (human breast carcinoma) xenografted tumor. Drug was released immediately into tumor tissue upon device implantation, resulting in a steep gradient of drug concentration, shown at three time points in Fig. 3A and as a cross section at 20 hours in Fig. 3B. At distances 0 to 130 μm (Fig. 3B, red region), drug levels were ~15 to 20 mg/kg; in the green region (~130 to 200 μm), drug was present at 8 to 13 mg/kg; whereas in the blue region (~200 to 300 μm), drug levels ranged from 3 to 7 mg/kg. (Tissue drug concentration calibration is described in Materials and Methods.) The concentration of doxorubicin in tissue can be lowered by diluting the drug in a polymer matrix (Fig. 3C).
We administered doxorubicin systemically at 8 mg/kg in a separate cohort of animals. The resulting intratumor drug distribution was measured to provide a calibration for appropriate local drug release from devices. A typical tumor section is shown in Fig. 3D. Drug distribution was heterogeneous, with patches of high-concentration (8 to 13 mg/kg) and low-concentration doxorubicin (3 to 7 mg/kg).
Figure 3E directly compares profiles of device (black, red) and systemically dosed tumor (blue) sections. Reservoirs with pure drug (Fig. 3B) had high drug concentrations in the region 0 to 125 μm from the device-tissue interface, whereas the region 125 to 300 μm represented the range of drug concentrations present when dosing systemically. In the reservoirs with drug diluted in polymer (Fig. 3C), the tissue concentration of the drug was consistently lower and the slope of the drug concentration gradient was reduced compared with reservoirs with pure drug, such that regions within 0 to 200 μm from the device-tissue interface had local drug concentrations similar to those achieved by systemic dosing (Fig. 3, E and F).
Tissue analysis to inform in vivo drug efficacy and pharmacodynamics
Identifying the minimum intratumor drug levels required for apoptosis induction may be useful for understanding potency of compounds during first-in-man efficacy studies in drug development or in the clinic for determining dosages during chemotherapy. Apoptosis-inducing effects (as seen by CC3) of drugs released from reservoirs were observed in tumor regions that were exposed to doxorubicin up to 320 μm from the reservoir in the doxorubicin-sensitive A375 tumor (Fig. 3G). This indicates that apoptosis levels rose substantially above background at intratumor concentrations ofatleast~4mg/kg in this tumor model. The dose dependence of the apoptotic effect of drugs can be investigated by varying the amount of drug that is released from reservoirs into adjacent tumor tissue. We varied formulation and dilution of doxorubicin in reservoirs, which led to a corresponding nonlinear reduction in apoptosis (Fig. 3, G to I). This change in drug effect stems from the different drug levels that are measured for each of the corresponding conditions (Fig. 3F).
Intratumor pharmacodynamic action of doxorubicin over different time points was also tested. Induction of apoptosis was observed in a small subset of cells after 8 hours of drug exposure (Fig. 4A). A larger region of tissue was apoptotic at 14 hours, spreading further away from the reservoir interface at 20 hours. At 20 hours, the tissue 0 to 80 μm from the reservoir no longer expressed CC3 and exhibited the characteristics of post-apoptotic cellular disruption, marked by the absence of cellular morphology and nuclear staining (Fig. 4, A and B). This region contained the cells with the longest exposure to doxorubicin.
Fig. 4. Pharmacodynamics and heterogeneity of drug response based on reservoir.
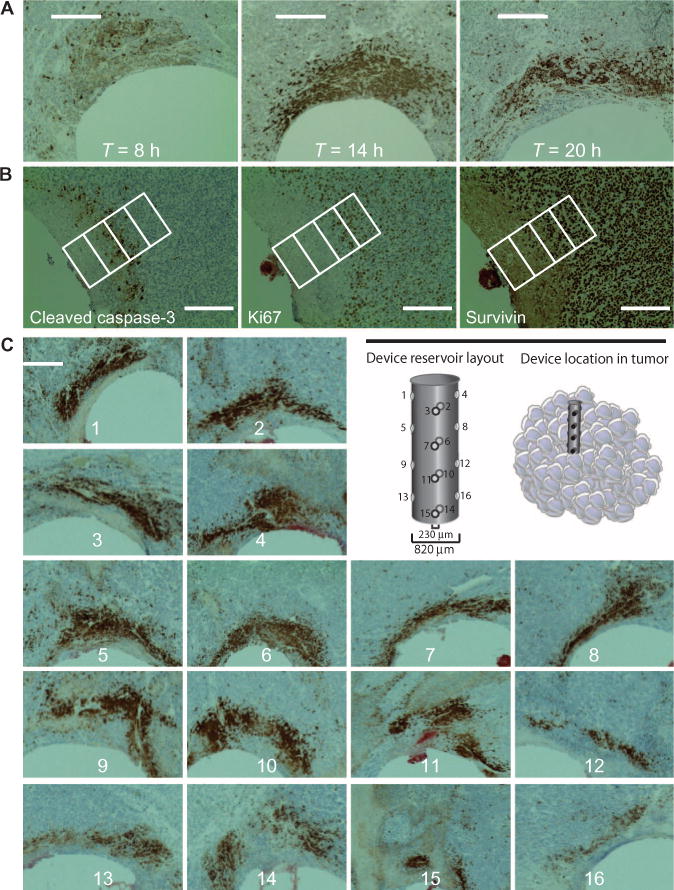
(A) Effect of drug exposure time on CC3 expression in A375 tumors. Tumors were implanted with devices releasing doxorubicin (40% in PEG-1450) and analyzed at 8, 14, and 20 hours. Scale bars, 300 μm. (B) Tissue cross sections stained for multiple biomarkers of drug efficacy: CC3 (apoptosis), Ki67 (cell proliferation), and survivin (apoptosis inhibitor). Images were taken 20 hours after doxorubicin (40% in PEG-1450) exposure and are representative of n = 12 A375 tumors. Scale bars, 200 μm. Individual rectangles represent 100 μm. (C) Panel of 16 distinct reservoirs from a single device, each eluting doxorubicin (40% in PEG-1450) in A375 tumors after 20 hours of implantation. Scale bar, 300 μm.
In the A375 tumor (Fig. 4A), application of doxorubicin for 14 hours led to apoptosis of nearly all cells within 250 μm, which corresponds to local concentrations above 4 mg/kg (Fig. 3E). Histological artifacts were limited to mechanical damage to the tissue caused by the needle insertion and were present within the nearest 20 to 30 μm of the reservoir-tissue interface in under 20% of samples, but did not affect the ability to analyze a given section. No observable adverse effects, such as a reduction in animal body score (18), owing to device implantation or incubation were observed.
Insight into the pharmacodynamic action of drugs can be gained by simultaneously analyzing multiple biomarkers. The apoptotic effect of doxorubicin in BT474 tumors, seen in Fig. 4A, was corroborated by multiparameter immunohistochemistry (IHC) (Ki67, pH2AX), demonstrating concordant growth arrest and DNA damage (fig. S3). Figure 4B shows adjacent tissue sections from a doxorubicin reservoir implanted for 20 hours. At distances 100 to 200 μm, corresponding to local drug concentrations equivalent to 8 to 12 mg/kg, a subset of cells underwent apoptosis (CC3+), no cells were proliferating (Ki67), and some cells still expressed survivin, a protein inhibitor of apoptosis. At greater distances of 200 to 300 μm, corresponding to local concentration of doxorubicin (4 to 8 mg/kg), only small numbers of cells were undergoing apoptosis and growth arrest, whereas most expressed survivin (Fig. 4B). On the basis of these data, in this BT474 tumor, higher concentrations of doxorubicin or, alternatively, the addition of another drug to treatment may be required to achieve more durable antitumor effects.
Assay reproducibility and heterogeneity of drug response across multiple regions in a single tumor
Tumor heterogeneity at the genetic and phenotypic level is an increasingly important consideration in understanding tumor evolution and resistance to drug treatment (19–21). Six devices with identically loaded drug reservoirs (n = 16) were implanted into multiple tumor locations to assess how drug response varied within a single tumor. Local drug exposure during systemic dosing cannot be precisely known over time owing to a heterogeneous drug distribution across the tumor (22, 23) (Fig. 3D). The release of drug from the device, however, is highly controlled and consistent in exposing tumor to drug (Fig. 3A).
Clinically, the confounding effect of tumor heterogeneity, which affects virtually all diagnostics, can be reduced in such a way by using replicates of identically loaded reservoirs on different locations of the device or by implanting multiple identical devices into the same tumor. Sixteen identically loaded reservoirs, each separated from the nearest neighbor by at least 700 μm (Fig. 4C), released doxorubicin microdoses into a human melanoma (A375) xenografted tumor, in both core and peripheral regions. Local efficacy, measured by CC3 expression, was similar across reservoirs except at reservoirs 15 and 16, which are closest to the core of the tumor (Fig. 4C). Regions of tumor that were closer to the core showed a higher incidence of necrotic areas compared to regions near the periphery (panels 13 to 16 in Fig. 4C).
Local microdose response as a predictor of systemic response to drug
Doxorubicin (Adriamycin) is an anthracycline drug that attacks tumor cells by intercalating DNA, leading to apoptosis. It is used in many types of cancer as first- or second-line chemotherapy, but its use is accompanied by serious side effects, including heart damage (24, 25). No clinical test currently exists to predict patients’ antitumor response to doxorubicin. Such a test may help avoid the serious side effects and shortened treatment window associated with a potentially ineffective doxorubicin treatment. We chose three widely used, established mouse models of human cancer—A375 melanoma, BT474 breast, and PC3 prostate—to test whether the device-based intratumor (local) response to doxorubicin can serve as a predictor of systemic efficacy.
Cross sections of tumors containing reservoirs were evaluated for CC3 expression. The percentage of cells in a given region that express CC3 is termed the apoptotic index (AI). A375 cells show the highest apoptotic response to doxorubicin (AI = 55%), BT474 intermediate (AI = 18%), and PC3 the lowest (AI = 8%) (Fig. 5A), corroborating reports that A375 is highly sensitive to doxorubicin [median inhibitory concentration (IC50) = 9.2 nM], BT474 is moderately sensitive (IC50 = 779 nM), and PC3 is insensitive (IC50 = 957 nM) (26, 27).
Fig. 5. Local and systemic sensitivity to doxorubicin in three human tumor models.
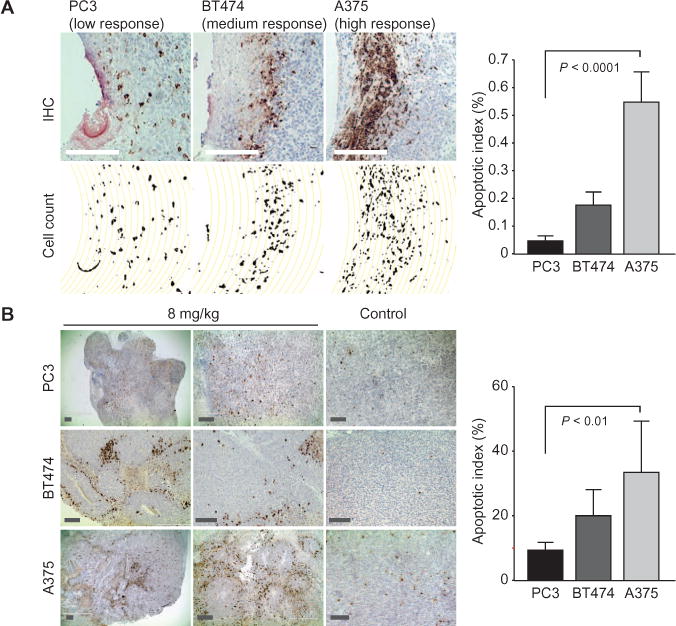
(A) Differential response of three human cell line tumor models to pure doxorubicin as measured by CC3+ cells. Data are averages ± SD (n = 18 to 22 unique reservoirs from 12 tumors for each model). Scale bars, 250 μm. (B) Apoptosis induction (CC3+ cells) after systemic administration of doxorubicin in A375, BT474, and PC3 tumors. Representative sections of tumors are shown 24 hours after treatment with doxorubicin (8 mg/kg) or control (saline injection). Data are averages ± SD CC3 expression from 12 sections scored per tumor model (4 sections each from three tumors for each model), and were scored in a blinded manner. Scale bars, 250 μm.
We treated a separate cohort of animals systemically with doxorubicin to confirm these findings. The intratumor apoptotic response was measured from multiple random slices of excised tumors 24 hours after dosing. In PC3 and A375 tumors, local apoptosis levels followed systemic dosing, with significantly higher tumor response in animals harboring human A375 (AI = 34.9%) versus PC3 (8.7%) (Fig. 5B). Variability in tumor response, as measured by coefficient of variation (CV), was greater in systemic (CV = 43%) than in device sections (CV = 18%).
Device validation with multiple drugs and tumor models
We tested the ability of our device assay to predict response of several widely used tumor models to other cytotoxic and targeted anticancer agents. Vemurafenib is an enzyme inhibitor that specifically targets the BRAF V600E mutation, which is present in A375 tumors, but not in PC3 tumors (26). Response to device-delivered vemurafenib, as measured by intratumor apoptosis, was significantly higher in animals harboring A375 tumors versus PC3 tumors (Fig. 6A).
Fig. 6. Differential drug sensitivity in human tumor models.
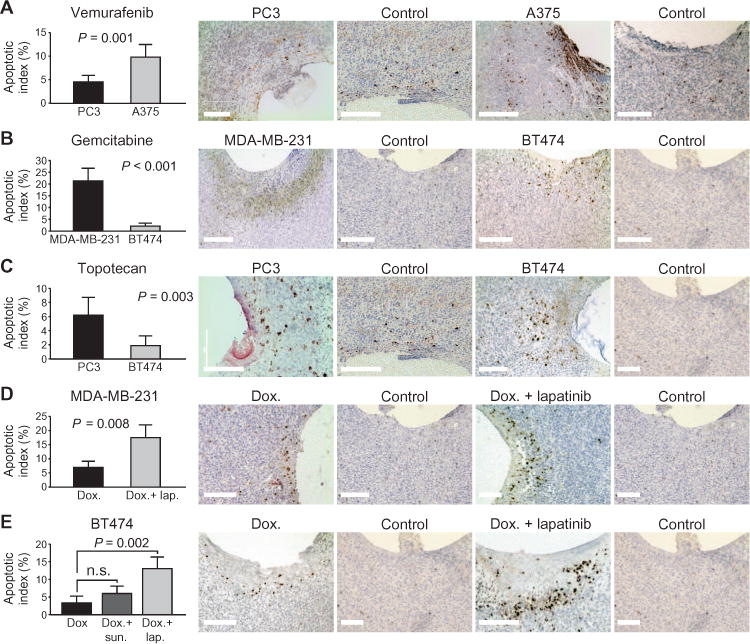
(A to C) Differential apoptotic response of human tumors to vemurafenib (50%), gemcitabine (60%), and topotecan (30%), all in PEG-1450, released from the device and assessed by CC3 expression after 20 to 24 hours. Data are averages ± SD [n = 12 spatially distinct reservoirs (biological replicates) from at least four tumors for each drug/tumor combination]. Scale bars, 200 μm. (D and E) Enhancement of apoptotic response by addition of targeted agents lapatinib and sunitinib to doxorubicin in the same device reservoir at 24 hours. Data are averages ± SD [n = 10 spatially distinct reservoirs (biological replicates) from at least four tumors for each drug/tumor combination]. Scale bars, 200 μm. P values were determined using Student’s t test for all graphs. n.s., not significant.
Gemcitabine, an inhibitor of DNA synthesis, demonstrated significantly greater apoptotic response from MDA-MB-231 (AI = 21.8%) versus BT474 (AI = 3.7%) tumors (Fig. 6B), in accordance with one published study (26). Topotecan, a topoisomerase inhibitor and a derivative of camptothecin that is frequently used in lung, cervical, and ovarian cancers, demonstrated the greatest effect on PC3 tumors (AI = 9.3%) and a significantly lower response in BT474 (2.6%) models (Fig. 6C), in accordance with (26).
Investigation of synergistic effects of combination therapy
Combining cytotoxic with targeted agents as a method for overcoming drug resistance is a promising clinical strategy even in tumors that show only slightly elevated levels of inhibitory targets (17, 28, 29). Our device could be used to predict which combinations are effective. We tested whether addition of the multikinase inhibitor sunitinib or the dual epidermal growth factor receptor (EGFR)/human EGFR-2 (HER2) inhibitor lapatinib to reservoirs already loaded with doxorubicin would exhibit an increase in apoptosis, as has been reported versus treatment with doxorubicin alone (28–30). The apoptotic response in MDA-MB-231 (human triple-negative breast) tumors, as tested by the device, was significantly elevated to 17.5% from 7.4% by the addition of lapatinib to doxorubicin in reservoirs (Fig. 6D). In BT474, addition of sunitinib to doxorubicin reservoirs leads to a moderate increase in apoptotic response, whereas lapatinib addition to doxorubicin led to a significant increase in local apoptosis (Fig. 6E), as expected, given the BT474 model’s HER2-positive status.
Device analysis of drug sensitivities in patient-derived model of triple-negative breast cancer
Triple-negative breast cancer (TNBC) is an indication with very poor prognosis in patients, which is usually treated with a range of drugs that include several cytotoxic agents for which no predictive biomarker exists (31, 32). We used a primary patient-derived xenograft of TNBC to demonstrate the potential value of our implantable device in this indication. This mouse model of human cancer was characterized by slight EGFR amplification and PTEN loss, and high Ki67 expression (33), and was obtained from a patient that had been treated with an EGFR inhibitor and then relapsed. Such a model, transferred only as tissue, is considered to be one of the best available approximations of human tumors in animals with regard to tumor structure and heterogeneity.
Device microdose testing revealed a wide range of intratumor efficacies, with highest sensitivities to paclitaxel in this tumor (AI = 54%), followed by doxorubicin (36%), cisplatin (25%), gemcitabine (12%), and lapatinib (4%) (Fig. 7). We performed systemic dosing studies with the same set of drugs delivered at the same proportions to corroborate these sensitivity findings. The relative orders of sensitivities of these drugs were identical to those observed in local device studies (Fig. 7), indicating a fundamental difference in drug sensitivity that is not dependent on route of administration; however, the extent of response to drug differed between device and systemic administration (for example, AI of 54% for the device versus 25% systemically for paclitaxel). These overall lower levels in systemic studies might be due to heterogeneity of drug distribution throughout the tumor, with regions of tissue not receiving drug at levels sufficient for inducing apoptosis. The very weak response to lapatinib may be explained by the patient’s relapse after treatment with an EGFR inhibitor. These measurements provide a proof of principle of how local microdose testing reflects the overall tumor responsiveness to intravenously administered drugs, and could therefore be used to discover inherent drug sensitivities in the tumor and as a predictive biomarker in TNBC.
Fig. 7. Efficacy of five drugs in a patient-derived TNBC tumor model.
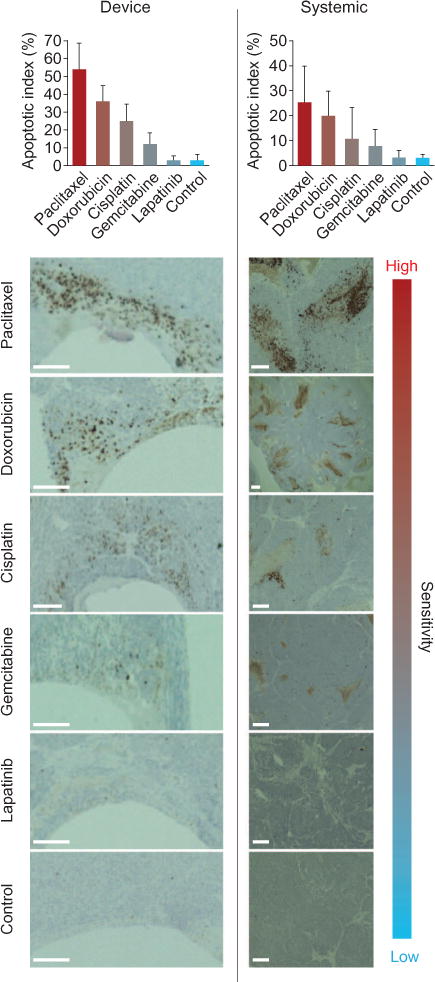
(Top) Differential response of TNBC PDX tumor model to five commonly used drugs. AI was calculated from the number of CC3+ cells divided by total cells. Data are averages ± SD (n = 16 unique reservoirs from 8 tumors for device studies, 8 animals for systemic studies). (Bottom) Representative images of TNBC tumor sections removed 24 hours after exposure to microdose of each drug from the device or after systemic injections. Device formulations (w/w) in PEG-1450 are in table S1. Systemic doses: paclitaxel, 16 mg/kg; doxorubicin,8 mg/kg; cisplatin,20 mg/kg; gemcitabine,30 mg/kg; lapatinib,50 mg/kg. Scale bars, 200 μm.
DISCUSSION
Implantable devices have been demonstrated to deliver exact quantities of drugs to different regions of the body for therapeutic purposes (13–16, 34). This study presents one such device that delivers precise quantities of a microdose of about one millionth of the systemic dose of a drug into confined regions of tumor in the living organism and allows for diagnostic assessment of each drug effect. Because of the miniscule doses that are used, one can obtain in vivo efficacy data on a range of compounds without the side effects that are associated with most systemic therapies. Our method enabled selective removal of device, drug, and affected tissue in a minimally invasive manner, using a standard biopsy needle, which fits into the clinical treatment paradigm. The major advantage of our device over existing ones is the ability to deliver many different drugs or drug combinations (up to 16) into distinct regions of tumor, without overlap between adjacent reservoirs, so that each drug effect can be observed in isolation, but all within the native tumor microenvironment.
Our data suggest that the local drug activity readout obtained from releasing drugs into confined tumor regions at clinically relevant doses may be used as a prognostic marker of drug sensitivity of tumors. In a patient-derived xenograft model of TNBC, tissue apoptotic response to five different drugs mirrored that observed when delivered systemically to the same model, possibly even reflecting the patient’s resistance to EGFR inhibitor. Larger-scale in vivo screens for candidate compounds could therefore be performed more efficiently on such tumor models that are limited in scalability.
In addition, the precise and tunable pharmacokinetic parameters of drug release from the device may offer new insight into exact intratumor pharmacodynamics, especially when coupled with analysis of the local tumor microenvironment and biomarkers. For instance, knowledge of the minimum dose and length of treatment to elicit a certain level of tumor response, such as shown in Fig. 4 (A and B), may be used to enable optimization of experimental time points or effective staggering of treatments. Clinically, it may be used to optimally reduce drug doses given to patients without compromising treatment efficacy. Obtaining such data for multiple drugs on a previously untreated tumor—for example, in the neoadjuvant setting—is not possible with traditional systemic dosing assays and is difficult even for single agents when dosing systemically owing to the heterogeneity of drug distribution, which can cause nearby tumor regions within 100 μm to have vastly different local drug concentrations, as shown in Fig. 3D. Such an assay can also be effectively combined with existing tumor genomic testing (35–37). The most likely risk factor associated with use of the device in humans is adverse events from biopsy procedures. Device implantation may be coordinated with an already scheduled small-needle biopsy. The retrieval of the device after the incubation period currently requires an additional biopsy procedure. Such biopsies are performed routinely in many indications, for example, breast cancer, with excellent safety record. Applicability of the device assay may be limited in other organs, most notably in intrathoracic cancers (38).
We observed that measurements of AI from device reservoirs were generally higher than from systemic dosing, and variability was reduced in the device, presumably because drug distribution is more heterogeneous in systemically dosed tumors (39). Although the sensitivity of the tumor to certain drugs may be inferred from device measurements, it is unlikely that a precise value of AI for systemically dosed tumors can always be predicted from device AI measurements.
Tumor heterogeneity is a potential problem associated with virtually all cancer diagnostics and is thought to be greater in human tumors than xenografts (19, 40–42). Our device assay seeks to address tumor heterogeneity at the cellular and regional levels. At the cellular level, a given tumor/device section contains hundreds of cells from a given reservoir that have been exposed to a specific drug (thousands of cells if using serial sections). Analysis of AI for one drug reservoir integrates over all the genetically distinct cell types that are present in this tumor region. At the regional level, one strategy to both study and mitigate the potentially varying efficacy of drugs across different regions of tumor is to have identically loaded reservoirs on multiple locations on the device or, additionally, to have multiple devices (with replicates of reservoirs) implanted into the tumor. Fiducial imaging markers, which are widely used in breast, prostate, and various other cancers, may provide a precedent for such procedures (43, 44). We demonstrated that a consistent readout can be obtained from replicates of 16 identically loaded reservoirs in the same tumor. The impact of heterogeneity on device readout may be reduced by choosing the neoadjuvant setting as an initial area of clinical application. The neoadjuvant setting is often considered by physicians to be the time in treatment when the tumor may be most homogeneous (45).
Future improvements to the device may include a greater number of reservoirs or further miniaturization and integrated tissue retrieval methods to reduce the needle size that is required, thus increasing the assay’s applicability to more organs and tumor types. Clinical care arguably holds the greatest transformative potential for this assay. TNBC, for example, requires various combinations of several cytotoxic drugs that are usually chosen empirically by trial and error. No robust biomarkers exist for these highly toxic therapies, but the described device could provide a phenotypic readout of in vivo drug efficacy that could be used to prioritize given drugs for treatment. The broader paradigm of testing tiny amounts of therapies for in vivo response in an individual patient rapidly and nonsystemically, and using the local efficacy for selecting the optimal therapy, may also be applicable to diseases beyond cancer.
It remains to be seen how the predictive ability of the described device approach translates to use in humans. A pervasive problem in clinical drug development is that vast resources are expended to develop a drug candidate’s bioavailability, safety, and stability before its efficacy can be tested, or therapeutic target validated, in humans (46, 47). This requirement for human testing contributes to the high failure rate and costs in cancer drug development (48, 49). Obtaining early efficacy data in humans in a safe manner, without systemic effects, could allow drug developers to focus the considerable resources required for pharmacological optimization and full clinical development only on the most promising drug candidates and in the optimal patient population. Recent regulatory developments may be favorable for such studies. Several U.S. Food and Drug Administration (FDA) initiatives seek to streamline the incorporation of novel biomarkers into clinical trials (47, 50), for example, in adaptive trials (51). The use of microdosing is also viewed favorably by the FDA (52, 53). Furthermore, the FDA has proposed to revise standards for investigational devices in early feasibility studies (54). Together, these developments may help pave a feasible path for incorporating microdose phenotypic efficacy data, as measured by our device assay, into clinical drug development and decision-making.
MATERIALS AND METHODS
Study design
The objectives of the studies were to show biological response to release of drugs from an implanted microdosing device and to test whether this response was different between different tumor types, drugs, and delivery methods in animals. Sample sizes were chosen to demonstrate statistical significance by Student’s t test between biologically distinct conditions or outcomes. Tissue sections were scored by an ImageJ image analysis algorithm in a blinded manner. Only biological replicates were used in data analysis. Average values and SDs are from 10 to 24 samples, as indicated in each figure legend. At most, two biological replicates were from the same device at distant reservoir sites (excluding Fig. 4C). Data from tissue sections were only excluded in the rare event that the tissue section was damaged during retrieval or was found to be entirely necrotic by IHC.
Device manufacturing and loading
Cylindrical, microscale devices with 820 μm (diameter) × 3 mm (length) were manufactured from medical-grade Delrin acetal resin blocks (DuPont) by micromachining (CNC Micro Machining Center, Cameron). This material was selected for its combination of structural rigidity, machinability, and biocompatibility (compliance to ISO 10993-1 and USP Class IV). Circular reservoirs (3 to 30 per device) were shaped on the outer surface of devices in dimensions ranging from 150 to 350 μm (diameter) × 150 to 250 μm (depth). Adjacent reservoirs were positioned 400 to 750 μm apart to prevent the intersection of compounds in tissue. All drugs were purchased in powder form (Selleckchem) and stored according to the manufacturer’s instructions. Cetuximab labeled with Alexa 488 was given by K. D. Wittrup (Massachusetts Institute of Technology). For doses with specific concentrations, compounds in the appropriate amounts were added to PEG-1000 or PEG-1450 (Alfa Aesar) and vortexed for 5 min above its melting point (37°C). For insoluble drugs, a mixture of drug, PEG, and an organic solvent (ethanol or acetone) was heated to ~45°C until completely dissolved (table S1). The solution was placed on a rotary evaporator (Yamato Scientific) for ~30 to 40 min at below respective vapor pressures to completely evaporate the solvent, leaving a homogeneous mixture of drug and PEG. Pure powders and concentrations in PEG were packed in solid form into device reservoirs using a tapered, metal needle (Electron Microscopy Sciences) until the reservoirs were completely filled. Before implantation, the devices were rotated on adhesive tape to remove excess compounds on the exterior of the device.
Device implantation
Mice were briefly anesthetized during implantation with 1 to 3% isoflurane. A 19-gauge spinal biopsy needle (Angiotech) containing an inner stylet and outer cannula was inserted into the tumor at a precise depth beyond the length of the device to be introduced. As the cannula remained secured in the tissue, the stylet was retracted and the device was placed into the orifice at the back of the needle. The stylet was then reinserted into the cannula to move the device down the length of the needle and into the tissue. The site of insertion was marked with surgical dye to maintain the correct orientation for tissue processing after tumor dissection.
Device removal
To determine antineoplastic effect for each treatment being evaluated, a small region of tissue was removed at 24 hours from the tumor and analyzed by IHC. Device removal was accomplished by using a larger coring biopsy needle (Cassi Beacon, Scion Medical Technologies), which was inserted into the tumor and positioned concentric to the device using ultrasound imaging. The coring needle protruded over and beyond the device, capturing the device itself and a cylindrical column of tissue 1.6 mm thick and 4 to 5 mm long, including ~400-μm thickness radially outward from the device along its entire length. This represented nearly the entire region of drug distribution over the incubation period of 1 to 2 days (fig. S1) and was thus sufficient for examining drug effect without residual drug remaining in the tumor.
Imaging of drug compounds in tissue
The autofluorescent compounds—doxorubicin, lapatinib, and sunitinib—were detectable and quantifiable in tissue sections by fluorescence microscopy. Devices with reservoirs containing these drugs were inserted into tumors for 12 to 48 hours to analyze drug release into tissue. Frozen sections (15 to 25 μm) were cryosectioned (CM1950 cryostat, Leica) from optimal cutting temperature (OCT) blocks at the cross section of drug-containing reservoirs at an angle perpendicular to the device, and imaged using an EVOS FL Cell Imaging System (Advanced Microscopy Group). Doxorubicin was visualized using a 531-nm excitation and 593-nm emission red fluorescent protein filter; lapatinib was visualized using a 357-nm excitation and 447-nm emission 4′,6-diamidino-2-phenylindole filter; sunitinib and cetuximab-Alexa 488 were visualized using a 470-nm excitation and 525-nm emission green fluorescent protein filter. For nonfluorescent drugs gemcitabine and paclitaxel, drug distribution was imaged in tumor sections by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry on a Bruker solariX 12T FTMS with Helix cell instrument. Images were obtained at 50-μm resolution. Before imaging, tumors were frozen and cryosectioned at 20 μm and thaw-mounted on glass slides. Slides were coated with a-cyano-4-hydroxycinnamic acid matrix in a Bruker ImagePrep coating instrument before MALDI imaging.
Diffusion profiles of the fluorescent compounds in tissue were generated using ImageJ Plot Profile with a line starting from the center of the device, crossing through the middle of a reservoir, and passing into the region of tissue containing drug. Profiles were averaged across multiple sections.
Drug dosing calibrations
Small pieces of excised fresh tumors were weighed and then placed into a well with 600 μl of doxorubicin (10 or 30 mg/kg) in phosphate-buffered saline for 72 hours [protocol extracted from (55)]. The tumors were removed from solution and prepared for frozen sectioning as described in the previous section. Doxorubicin concentration remaining in the wells was measured with a plate reader, and the value was subtracted from the control wells to calculate the concentration of doxorubicin in the tumor tissue.
Systemic dosing studies
For studies testing intratumor effect or drug concentration of systemically treated mice, animals were dosed with doxorubicin (8 mg/kg) by tail vein injection. At 6 hours, a subset of tumors were harvested, fixed, and processed exactly as for device studies. Concentration of doxorubicin in tumor tissue was measured exactly as for device-based studies. At 24 hours, a subset of tumors was harvested for IHC studies to assess drug effect. These tumors were processed, sliced, and stained exactly as for device-based studies. For Fig. 5B, CC3 expression by IHC was scored using the same ImageJ algorithm described below.
Statistical analysis
AI was calculated as % CC3+ cells/total cells within 300 μm from reservoir-tissue interface for device sections. For systemic studies, AI = % CC3+ cells/total cells for the entire tumor section. All error bars represent 1 SD. CV is defined as SD divided by mean. Significance tests and P values were calculated using Student’s t test, using normal-based 95% confidence intervals. Two-sided testing was used. The data sets were verified to be normally distributed.
Supplementary Material
Fig. S1. Removal of drug-exposed tumor region by a coring needle.
Fig. S2. Visibility of device for in vivo imaging with ultrasound and CT.
Fig. S3. Device/tissue cross sections stained for multiple drug sensitivity markers.
Table S1. Solvents and concentrations of drug-polymer mixtures for device reservoirs.
Acknowledgments
We thank J. Ross, M. Scaltriti, E. Lander, and M. Levin for helpful discussions; N. Morse for help with tumor models; and K. Cormier and W. Zhang for help and advice with histology. We thank K. Kellersberger from Bruker Daltonics Inc. for help with MALDI tissue imaging.
Funding: National Cancer Institute Innovative Molecular Analysis Technologies program (R21-CA177391) and Kibur Medical Inc.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/7/284/284ra57/DC1
Materials and Methods
Author contributions: O.J. designed and performed experiments, analyzed data, and wrote the manuscript. H.M.L. performed experiments and analyzed data. R.L. and M.J.C. supervised the research and preparation of the manuscript. R.I.T., J.E.F., J.T.S., and J.B. helped with experimental design, analysis, and preparation of the manuscript.
Competing interests: Kibur Medical Inc. holds intellectual property related to this technology.
Data and materials availability: Requests from commercial entities for devices may require review from Kibur Medical Inc.
REFERENCES AND NOTES
- 1.Mouton Y, Dubreuil L. In vitro antibiotics testing and its relationship to clinical activity. J Chemother. 1997;9(Suppl 1):93–99. [PubMed] [Google Scholar]
- 2.Merz F, Gaunitz F, Dehghani F, Renner C, Meixensberger J, Gutenberg A, Giese A, Schopow K, Hellwig C, Schäfer M, Bauer M, Stöcker H, Taucher-Scholz G, Durante M, Bechmann I. Organotypic slice cultures of human glioblastoma reveal different susceptibilities to treatments. Neuro Oncol. 2013;15:670–681. doi: 10.1093/neuonc/not003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fang C, Avis I, Salomon D, Cuttitta F. Novel phenotypic fluorescent three-dimensional platforms for high-throughput drug screening and personalized chemotherapy. J Cancer. 2013;4:402–415. doi: 10.7150/jca.6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA, Golub TR. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the micro-environment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meric-Bernstam F, Mills GB. Overcoming implementation challenges of personalized cancer therapy. Nat Rev Clin Oncol. 2012;9:542–548. doi: 10.1038/nrclinonc.2012.127. [DOI] [PubMed] [Google Scholar]
- 8.Sun X, Vilar S, Tatonetti NP. High-throughput methods for combinatorial drug discovery. Sci Transl Med. 2013;5:205rv1. doi: 10.1126/scitranslmed.3006667. [DOI] [PubMed] [Google Scholar]
- 9.Schilsky RL. Personalized medicine in oncology: The future is now. Nat Rev Drug Discov. 2010;9:363–366. doi: 10.1038/nrd3181. [DOI] [PubMed] [Google Scholar]
- 10.Pammolli F, Magazzini L, Riccaboni M. The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov. 2011;10:428–438. doi: 10.1038/nrd3405. [DOI] [PubMed] [Google Scholar]
- 11.Vaidyanathan G. Redefining clinical trials: The age of personalized medicine. Cell. 2012;148:1079–1080. doi: 10.1016/j.cell.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 12.Kummar S, Kinders R, Rubinstein L, Parchment RE, Murgo AJ, Collins J, Pickeral O, Low J, Steinberg SM, Gutierrez M, Yang S, Helman L, Wiltrout R, Tomaszewski JE, Doroshow JH. Compressing drug development timelines in oncology using phase “0” trials. Nat Rev Cancer. 2007;7:131–139. doi: 10.1038/nrc2066. [DOI] [PubMed] [Google Scholar]
- 13.LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat Biotechnol. 2003;21:1184–1191. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 14.Farra R, Sheppard NF, Jr, McCabe L, Neer RM, Anderson JM, Santini JT, Jr, Cima MJ, Langer R. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci Transl Med. 2012;4:122ra21. doi: 10.1126/scitranslmed.3003276. [DOI] [PubMed] [Google Scholar]
- 15.Grayson ACR, Shawgo RS, Johnson AM, Flynn NT, Yawen L, Cima MJ, Langer R. A BioMEMS review: MEMS technology for physiologically integrated devices. Proc IEEE. 2004;92:6–21. [Google Scholar]
- 16.Chirra HD, Desai TA. Multi-reservoir bioadhesive microdevices for independent rate-controlled delivery of multiple drugs. Small. 2012;8:3839–3846. doi: 10.1002/smll.201201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, Yaffe MB. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foltz CJ, Ullman-Cullere M. Guidelines for assessing the health and condition of mice. Lab Anim. 1999;28:28–32. [PubMed] [Google Scholar]
- 19.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almendro V, Kim HJ, Cheng YK, Gönen M, Itzkovitz S, Argani P, van Oudenaarden A, Sukumar S, Michor F, Polyak K. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014;74:1338–1348. doi: 10.1158/0008-5472.CAN-13-2357-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez-Farre X, Muñoz M, Russnes HG, Helland A, Rye IH, Borresen-Dale AL, Maruyama R, van Oudenaarden A, Dowsett M, Jones RL, Reis-Filho J, Gascon P, Gönen M, Michor F, Polyak K. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014;6:514–527. doi: 10.1016/j.celrep.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker JH, Lindquist KE, Huxham LA, Kyle AH, Sy JT, Minchinton AI. Direct visualization of heterogeneous extravascular distribution of trastuzumab in human epidermal growth factor receptor type 2 overexpressing xenografts. Clin Cancer Res. 2008;14:2171–2179. doi: 10.1158/1078-0432.CCR-07-4465. [DOI] [PubMed] [Google Scholar]
- 23.Huxham LA, Kyle AH, Baker JHE, Nykilchuk LK, Minchinton AI. Microregional effects of gemcitabine in HCT-116 xenografts. Cancer Res. 2004;64:6537–6541. doi: 10.1158/0008-5472.CAN-04-0986. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 25.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Colon Cancer. National Comprehensive Cancer Network; Fort Washington, PA: 2011. [DOI] [PubMed] [Google Scholar]
- 26.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holbeck SL, Collins JM, Doroshow JH. Analysis of Food and Drug Administration-approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol Cancer Ther. 2010;9:1451–1460. doi: 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res. 2000;6:4885–4892. [PubMed] [Google Scholar]
- 29.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 30.Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CR, Jr, Huang Y, Robey RW, Liang YJ, Chen LM, Shi CJ, Ambudkar SV, Chen ZS, Fu LW. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: Disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7:683–692. doi: 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- 32.Hudis CA, Gianni L. Triple-negative breast cancer: An unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 33.Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, Weitsman G, Barber P, Vojnovic B, Ellis H, Morse N, Viola-Villegas NT, Bosch A, Juric D, Hazra S, Singh S, Kim P, Bergamaschi A, Maheswaran S, Ng T, Penault-Llorca F, Lewis JS, Carey LA, Perou CM, Baselga J, Scaltriti M. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. 2014;7:ra29. doi: 10.1126/scisignal.2005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fung LK, Ewend MG, Sills A, Sipos EP, Thompson R, Watts M, Colvin OM, Brem H, Saltzman WM. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998;58:672–684. [PubMed] [Google Scholar]
- 35.Ross JS, Wang K, Al-Rohil RN, Nazeer T, Sheehan CE, Otto GA, He J, Palmer G, Yelensky R, Lipson D, Ali S, Balasubramanian S, Curran JA, Garcia L, Mahoney K, Downing SR, Hawryluk M, Miller VA, Stephens PJ. Advanced urothelial carcinoma: Next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod Pathol. 2014;27:271–280. doi: 10.1038/modpathol.2013.135. [DOI] [PubMed] [Google Scholar]
- 36.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, Barrette T, Everett J, Siddiqui J, Kunju LP, Navone N, Araujo JC, Troncoso P, Logothetis CJ, Innis JW, Smith DC, Lao CD, Kim SY, Roberts JS, Gruber SB, Pienta KJ, Talpaz M, Chinnaiyan AM. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arteaga CL, Baselga J. Impact of genomics on personalized cancer medicine. Clin Cancer Res. 2012;18:612–618. doi: 10.1158/1078-0432.CCR-11-2019. [DOI] [PubMed] [Google Scholar]
- 38.Overman MJ, Modak J, Kopetz S, Murthy R, Yao JC, Hicks ME, Abbruzzese JL, Tam AL. Use of research biopsies in clinical trials: Are risks and benefits adequately discussed? J Clin Oncol. 2013;31:17–22. doi: 10.1200/JCO.2012.43.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 40.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 41.Di Cosimo S, Baselga J. Management of breast cancer with targeted agents: Importance of heterogeneity. Nat Rev Clin Oncol. 2010;7:139–147. doi: 10.1038/nrclinonc.2009.234. [DOI] [PubMed] [Google Scholar]
- 42.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Piña V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sterzing F, Engenhart-Cabillic R, Flentje M, Debus J. Image-guided radiotherapy: A new dimension in radiation oncology. Dtsch Arztebl Int. 2011;108:274–280. doi: 10.3238/arztebl.2011.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habermehl D, Henkner K, Ecker S, Jäkel O, Debus J, Combs SE. Evaluation of different fiducial markers for image-guided radiotherapy and particle therapy. J Radiat Res. 2013;54(Suppl 1):i61–i68. doi: 10.1093/jrr/rrt071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denisov EV, Litviakov NV, Zavyalova MV, Perelmuter VM, Vtorushin SV, Tsyganov MM, Gerashchenko TS, Garbukov EY, Slonimskaya EM, Cherdyntseva NV. Intratumoral morphological heterogeneity of breast cancer: Neoadjuvant chemotherapy efficiency and multidrug resistance gene expression. Sci Rep. 2014;4:4709. doi: 10.1038/srep04709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol Oncol. 2012;6:155–176. doi: 10.1016/j.molonc.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orloff J, Douglas F, Pinheiro J, Levinson S, Branson M, Chaturvedi P, Ette E, Gallo P, Hirsch G, Mehta C, Patel N, Sabir S, Springs S, Stanski D, Evers MR, Fleming E, Singh N, Tramontin T, Golub H. The future of drug development: Advancing clinical trial design. Nat Rev Drug Discov. 2009;8:949–957. doi: 10.1038/nrd3025. [DOI] [PubMed] [Google Scholar]
- 48.Adams CP, Brantner VV. Spending on new drug development. Health Econ. 2010;19:130–141. doi: 10.1002/hec.1454. [DOI] [PubMed] [Google Scholar]
- 49.Rubin EH, Gilliland DG. Drug development and clinical trials—The path to an approved cancer drug. Nat Rev Clin Oncol. 2012;9:215–222. doi: 10.1038/nrclinonc.2012.22. [DOI] [PubMed] [Google Scholar]
- 50.U.S. Food and Drug Administration. Paving the Way for Personalized Medicine: FDA’s Role in a New Era of Medical Product Development. U.S. Food and Drug Administration; Silver Spring, MD: 2013. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/PersonalizedMedicine/UCM372421.pdf. [Google Scholar]
- 51.Berry DA. Adaptive clinical trials in oncology. Nat Rev Clin Oncol. 2011;9:199–207. doi: 10.1038/nrclinonc.2011.165. [DOI] [PubMed] [Google Scholar]
- 52.Garner RC, Lappin G. The phase 0 microdosing concept. Br J Clin Pharmacol. 2006;61:367–370. doi: 10.1111/j.1365-2125.2006.02575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahajan R, Gupta K. Food and Drug Administration’s critical path initiative and innovations in drug development paradigm: Challenges, progress, and controversies. J Pharm Bioallied Sci. 2010;2:307–313. doi: 10.4103/0975-7406.72130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.U.S. Food and Drug Administration. Investigational Device Exemptions (IDEs) for Early Feasibility Medical Device Clinical Studies, Including Certain First in Human (FIH) Studies—Guidance for Industry and Food. U.S. Food and Drug Administration; Silver Spring, MD: 2011. pp. 1–40. [Google Scholar]
- 55.Zheng JH, Chen CT, Au JL, Wientjes MG. Time- and concentration-dependent penetration of doxorubicin in prostate tumors. AAPS PharmSci. 2001;3:E15. doi: 10.1208/ps030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Removal of drug-exposed tumor region by a coring needle.
Fig. S2. Visibility of device for in vivo imaging with ultrasound and CT.
Fig. S3. Device/tissue cross sections stained for multiple drug sensitivity markers.
Table S1. Solvents and concentrations of drug-polymer mixtures for device reservoirs.