

Abstract
The incorporation of histone H3 with an acetylated lysine 56 (H3K56ac) into the nucleosome is important for chromatin remodeling and serves as a marker of new nucleosomes during DNA replication and repair in yeast. However, in human cells, the level of H3K56ac is greatly reduced, and its role during the cell cycle is controversial. Our aim was to determine the potential of H3K56ac to regulate cell cycle progression in different human cell lines. A significant increase in the number of H3K56ac foci, but not in H3K56ac protein levels, was observed during the S and G2 phases in cancer cell lines, but was not observed in embryonic stem cell lines. Despite this increase, the H3K56ac signal was not present in late replication chromatin, and H3K56ac protein levels did not decrease after the inhibition of DNA replication. H3K56ac was not tightly associated with the chromatin and was primarily localized to active chromatin regions. Our results support the role of H3K56ac in transcriptionally active chromatin areas but do not confirm H3K56ac as a marker of newly synthetized nucleosomes in DNA replication.
Keywords: Cell cycle, Chromatin, DNA replication, H3K56ac, Mammalian cells, Nucleosome
Abbreviations
- H3K56ac
Histone H3 acetylation at lysine 56
- hESCs
human embryonic stem cells
- FISH
Fluorescent in situ hybridization
- RNAP II
RNA Polymerase II
- HAT
histone acetyltransferase
- SIRT
Sirtuins
- K56
lysine 56
Background
Histones are small basic proteins that stabilize DNA in the chromatin form and help to orchestrate tissue-specific gene expression. The structural conformation and charge of the histones can be modified by different substituents. These substituents allow dynamic communication between histone octamers and DNA. Different histone modifications create harbours for chromatin-modifying complexes. The addition of an acetyl group to the histone structure reduces the electrochemical attraction between positively charged histones and negatively charged DNA. The loosened nucleosomes are more accessible to the DNA recognition motifs of transcription factors. Generally, acetylated histones are associated with chromatin decondensation and transcriptional activation of the nucleosomes. The structure of histone H3 is rich in lysines, which can be modified by an acetyl group. However, nucleosome compactness is not dramatically altered by all histone acetylations. H3 core acetylation at lysine 56 only modestly influences the nucleosome structure compared with the N-tail histone acetylations (on lysines 4, 9, 18, and 27).1
Lysine 56 is positioned at the amino-terminal αN-helix close to the site where the DNA enters and exits the nucleosome.1,2 Lysine 56 acetylation increases the conformation entropy in the αN-helix and destabilizes the entire protein structure, which leads to increases in nucleosome breathing, a dynamic condition in which the DNA is transiently unwrapped from a histone octamer. H3K56ac also increases the affinity of the chromatin-remodelling proteins for the chromatin.3 The pathway of H3K56ac regulation is well defined in yeast, where this modification plays an important role in many nuclear processes. H3K56 acetylation is specifically catalyzed by the histone acetyltransferase (HAT) Rtt109 in complex with the histone chaperone Asf1.4,5 Then, H3K56ac is reintegrated into the new nucleosome during DNA replication or into freshly repaired chromatin after the induction of a double-strand break.6,7 Similarly, histone chaperones reload a histone octamer containing H3K56ac onto the unwrapped DNA during the initiation and elongation steps of transcription. Thus, in yeast, H3K56ac marks newly synthesized H3 histones and chromatin segments with high nucleosome turnover.8-12 Sirtuins are responsible for removing the acetyl group from the histone structure.13,14
The knowledge that is gained from the yeast system is difficult to apply to the mammalian cell system because of the many differences between these species. Mammalian cells do not express HATs with high specificity to K56,4,15 and H3K56ac levels are very low. In mammalian cells, H3K56ac is catalyzed by 3 versatile acetyltransferases: CBP, p300 and Gcn5.16,17 CBP and p300 alone acetylate various proteins in cells. p300/CBP preferentially catalyzes the acetylation of N-terminal lysines on histone H3.16,18 The specificity of p300/CBP for lysine K56 is likely powered by HAT auto-acetylation and the reorganization of their catalytic domains. Proper protein folding enables an interaction between the histone complex and the nucleosome chaperons ASF1A and ASF1B.16,19-21 Similar to yeasts, sirtuins catalyze the removal of the acetyl group from K56.16,22 Despite the low level of this modification in mammalian chromatin, different studies have identified a role for H3K56ac in cancer progression, DNA double-strand break repair, the regulation of gene transcription and pluripotency.16,23-26
H3K56ac levels are elevated in cancer and pluripotent cells16,27,28 compared with normal tissue. Cancer cells are associated with aberrant cell cycle regulation. No changes in H3K56ac levels throughout the cell cycle or its elevations in the S and G2 phases in different cell lines were noted in recent studies that primarily focused on the role of H3K56 in DNA damage and repair.16,29-31 This variability can be explained mainly by the different specificities of H3K56ac antibodies.32 Thus, we focused on H3K56ac regulation and its connection with the S or G2 phase and nuclear processes. Therefore, we aimed to reveal the cell cycle dependency of H3K56ac levels in fast-cycling cell types (embryonic stem cells, hESCs - CCTL1233,34 and in cancer cell lines with different nuclear morphologies, including adherent HeLa and suspension HL-60 lines) using antibody-dependent and -independent methods.
Results
The number of H3K56ac foci is connected to DNA replication activity in cancer cells, but not in hESCs
Cellular H3K56ac levels were measured using a K56ac-specific antibody in 3 different human cell lines (HL-60, HeLa and CCTL12). H3K9ac is a major competitor for antibodies against H3K56ac.32 Therefore, the H3K9ac antigen was masked with a peptide inhibitor of H3K9 before adding the H3K56ac antibodies. In cell nuclei, the H3K56ac signal is distributed in weak, but sharply localized foci (Fig. 1A). A difference in the density of H3K56ac foci was observed in individual nuclei. A weak H3K56ac signal was observed in cytoplasmic areas, possibly due to non-specific antibody binding. Thus, only the nuclear signal and the nuclear histone fraction were used for analysis. In yeasts, H3K56ac regulation is tightly linked to DNA replication; therefore, we determined whether the difference in the density of H3K56ac foci was connected to S phase of the cell cycle. Cells were incubated with EdU to identify DNA replication activity (Fig. 1A). In all of the observed cell lines, H3K56ac levels were elevated in EdU-positive cells (Fig. 1A, 1B and Fig. S1A, S1B). An increase in the H3K56ac foci was significant in cancer cells, but not in embryonic CCTL12 cells (U-test, α=0.05; Fig. 1). These findings indicate a connection between an increase in the number of H3K56ac foci and DNA replication in cancer cell lines.
Figure 1.
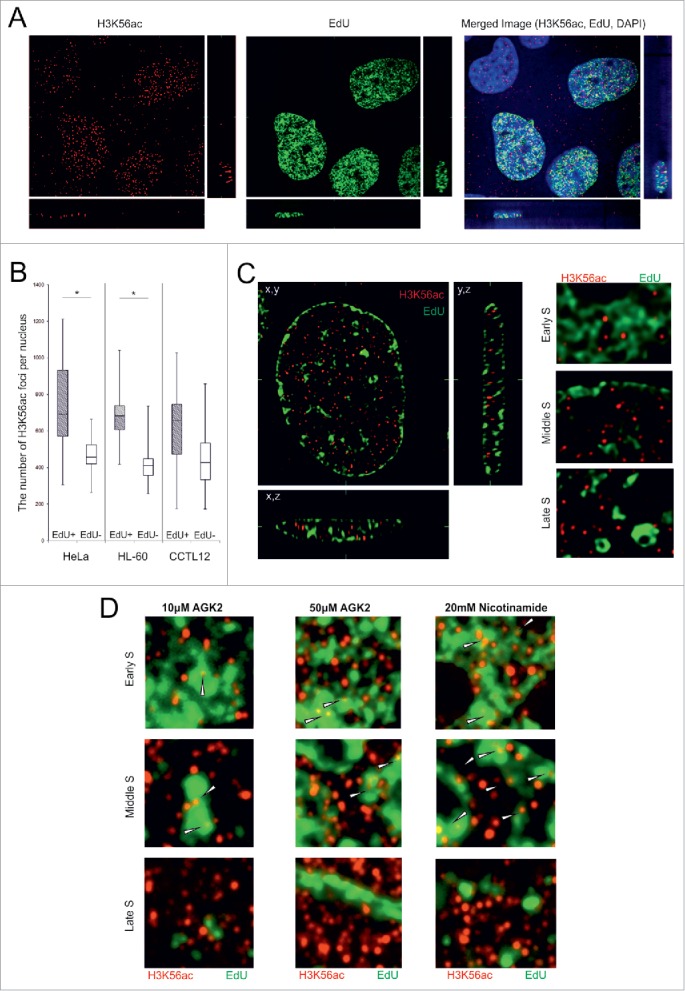
The level of H3K56ac foci are increased in EdU positive cells. (A) HeLa cells were treated with EdU for 2 h before the paraformaldehyde fixation. An increased density of the K56ac (red) foci was observed in the EdU-positive (green) cells. The cell nuclei were counterstained with DAPI (blue). The image represents one slice of the object in the x,y, x,z and y,z planes. (B) The number of H3K56ac foci was increased in EdU-positive cells. The chart represents the number of H3K56ac foci in the EdU-positive (gray box plots) and EdU-negative nuclei (white) in the HeLa, HL-60 and embryonic CCTL12 cell lines. The range of the error bars encompasses the lowest and highest values (n>50 for each measurement; *P value< 0.05; U-test). (C) The H3K56ac foci did not colocalize with the bright EdU-positive areas in the HeLa cells. Representative zoomed images of HeLa nucleus in the early, middle and late S sub-phase are shown (original images in Fig. S2). (D) The colocalization between H3K56ac foci and the EdU bright areas after sirtuin inhibition in the HeLa cells. (zoomed images, original images in Figs. S4-S6).
H3K56ac foci are localized outside of bright EdU-positive areas and late S phase replicating chromatin
Related analysis of the distribution of the signal showed that most of the H3K56ac was positioned outside of bright EdU-positive areas after a pulse (15 min) and long-term (2 h) EdU incubation (Fig. 1C, Fig. S2, S3). Almost no colocalization between H3K56ac and bright EdU-positive areas was observed during the early, middle and late S sub-phases. Prolonged exposure to EdU (6 h) confirmed that EdU incorporation did not interfere with the H3K56ac signal. The H3K56ac foci preferentially colocalized to regions with reduced EdU staining (Fig. S2D).
We next determined whether the absence of H3K56ac in bright EdU-positive areas was caused by the rapid removal of acetyl groups from the newly synthetized chromatin. HeLa cells were pretreated (24 h) with 2 sirtuin inhibitors, AGK-2 and nicotinamide. AGK-2 is specific for SIRT2 at low concentrations (10 μM, IC50=3.5 μM) and inhibits SIRT1, SIRT2 and SIRT3 at higher concentrations (50 μM, IC50=50 μM).35 Nicotinamide inhibits all members of the sirtuin family.36-38 H3K56ac foci were detected inside bright EdU-positive areas during the early and middle S sub-phases after the incubation of HeLa cells with 20 mM nicotinamide and 50 μM AGK-2, not with 10μM AGK-2 (Fig. 1D, Fig. S4 – S6). H3K56ac did not colocalize with late replication chromatin, even after sirtuin inhibition.
The number of H3K56ac foci is greater in S and G2 phase in cancer cells
After the 2-h EdU treatment, some cells showing a late replication pattern were presumably in G2 phase at the time of observation. Hence, we asked whether the number of H3K56ac foci remained stable through G2 phase. A double thymidine block was used to synchronize the cell cycle by blocking cells at the G1/S transition point. The cells were observed to be in S phase at 3 h, G2 at 6 h and G1 at 12 h after the release of the thymidine block (Fig. 2A). The number of H3K56ac foci was significantly greater in the S and G2 phases than in G1 phase in the cancer cell lines (Fig. 2B, C and Fig. S7). The absence of H3K56ac in mitotic chromosomes showed that during late G2 phase, H3K56ac is removed from the chromatin (Fig. 2C). Compared with the cancer cell lines, the number of H3K56ac foci was not significantly regulated by the cell cycle in CCTL12 cells (Fig. S7B).
Figure 2.
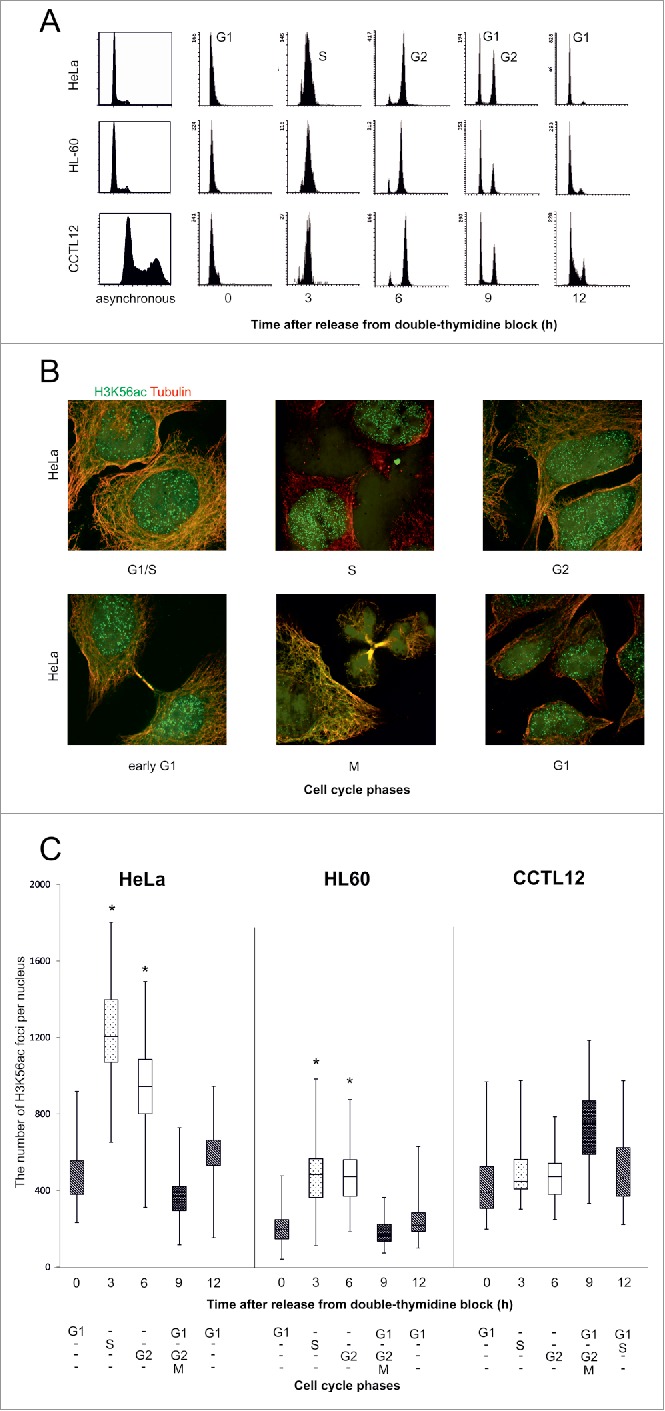
The number of H3K56ac foci during the cell cycle. (A) Flow cytometry analysis of the cell cycle stages. Propidium iodide staining was used to quantify the DNA in the cell nuclei. G1 and M phases are represented by the left peak and G2 phase by the right peak. The cells were synchronized into the G1/S transition point during thymidine blockade. After releasing the thymidine block, the cells were synchronized into purified populations of S (3 h), G2 (6 h) and G1 phase (12 h). The majority of the mitotic cells were observed in G2/M/G1 phase (9 h). (B) HeLa cells were treated with thymidine to induce a G1/S phase block. After thymidine release, the number of H3K56ac (green) foci were analyzed every 3 h in the separate cell cycle phases. Beta-tubulin (red) was used to identify the daughter cells in early G1 phase. H3K56ac did not localize to the chromatin in mitotic cells. (C) The number of H3K56ac foci in distinct cell cycle phases. In the HeLa and HL-60 cancer cell lines, H3K56ac levels were increased in the S (dotted blot) and G2 (white) phases. The error bars represent the maximum and minimum values (n>50 for each measurement, * P value< 0.05, U-test).
Compared with the number of H3K56ac foci, the H3K56ac protein level does not fluctuate during the cell cycle
We speculated that the same cell cycle dynamics regulate H3K56ac protein levels. Histones were isolated from cell nuclei via acid extraction. Amido Black staining was used to normalize protein loading (Fig. 3A). Despite the results of the immunofluorescence experiments, the H3K56ac protein level did not significantly change during interphase stages (Fig. 3B, C). In the cancer cell lines, we observed a second band with increased mobility. The faster band represents N-terminally degraded H3. We next determined the protein level via mass spectrometry (Fig. 3B, C). The ratio of H3K56ac/H3K56 was not significantly changed during the cell cycle (Fig. 3D, E). Average K56ac levels were slightly higher in the cancer cell lines (0.10% ± 0.09 H3K56ac for HeLa cells and 0.14% ± 0.06 for HL-60 cells) than in the CCTL12 cells (0.07% ± 0.02).
Figure 3.

The level of the H3K56ac protein during the cell cycle. (A) Acid-extracted CCTL12 histones were separated on a short Tris-Tricine gel and Amido Black stained. The locations of the major forms of the core histones are noted. (B) Western blot analysis of H3K56ac at different time points after release from the thymidine block. We did not observe a significant change in the H3K56ac level during the cell cycle in HeLa, HL-60 or CCTL12 cells. In the cancer cell lines, a second band with a lower molecular weight appeared. The band represents the H3 histone after N-terminal degradation. (C) The charts show the level of H3K56ac normalized to the total protein (n=3). The total protein was visualized by Amido Black staining. (D) The charts show the relative ratio of H3K56ac and unacetylated H3K56 in different cell cycle phases obtained by mass spectrometry. H3K56ac was not significantly changed during the cell cycle stages in any of the 3 cell lines. The range of the error bars encompasses the standard deviation values (n = 3). (E) An example of the chromatographic profile of the SRM transitions for the native (YQKSTELLIR) and acetylated (YQK[Ac]STELLIR) forms of the peptide.
H3K56ac and ASF1A are loosely bound to chromatin
The rare appearance of the H3K56ac signal in the cell cytoplasm indicates the possibility that 2 H3K56ac states exist. One state is a part of the chaperone complex with ASF1A, and the second is H3K56ac that is incorporated into nucleosomes. We speculated that the elevation detected in S and G2 phase, measured via microscopy, could be influenced by an increase of H3K56ac in the chaperone complex. To evaluate this hypothesis, HeLa cells were treated with cytoskeletal (CSK) buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES, pH 6.8) before fixation. CSK buffer removed most of the soluble proteins in the cells. After the application of CSK buffer, the number of H3K56ac foci in the nuclei was decreased by approximately 10-fold compared with paraformaldehyde-fixed cells (Fig. 4A, B). Similar to H3K56ac, the level of ASF1A was reduced after CSK treatment. Proteins with a distribution similar to that of H3K56ac (p300) or only to the chromatin-bound form (RNAPII-pSer5 CTD) were used as a control for the effect of CSK. H3K56ac, together with ASF1A, binds to chromatin less tightly than active RNA polymerase II and p300. Hence, we examined the number of H3K56ac foci after CSK treatment for each cell cycle stage in HeLa cells. The number of H3K56ac foci was not significantly changed during the cell cycle after CSK treatment (Fig. 4C).
Figure 4. (see previous page).

H3K56ac and ASF1A are not tightly connected to chromatin. (A) HeLa cells were incubated with CSK buffer before formaldehyde fixation. After the CSK buffer treatment, the level of H3K56ac and ASF1A in the nucleus was decreased. We did not observe any change in the distribution of the RNA Polymerase CTD domain (phosphorylated on serine 5) or p300. (B) The H3K56ac foci level (red) at different stages of the HeLa cell cycle. As a control for CSK buffer efficiency, we used the nuclear staining of p300 (green). (C) The chart indicates the number of H3K56ac foci in the HeLa cell nuclei after CSK treatment at different time points after the release from the thymidine block. The error bars represent the maximum and minimum values (n>50 for each measurement). (D) The difference between H3K56ac, H3K9ac and the total protein after acidic and salt extraction of histones. (E) The nuclear histones were concentrated by acid extraction. A small, non-significant increase in the H3k56ac level was observed during G2 phase (6 h). The H3K9ac level decreased at 9 h after release of the thymidine block. The charts indicate the changes in the H3K56ac levels during cell cycle progression in the histones isolated by acid extraction (n=3). The error bars represent the the standard deviation values (n=3). The antigen intensity was normalized to the total protein. The total protein was visualized by Amido Black staining and was used as a loading control. (F) The chromatin fraction of the H3 and H4 histones was isolated by salt extraction. No significant changes in H3K56ac levels were observed. The H3K9ac level decreased at 9 and 12 h after release of the thymidine block. The total protein was used as loading control. The charts represent the changes in the H3K56ac levels during cell cycle progression in the histones isolated by acid extraction (n=3).
Next, we addressed whether it is possible to determine the presence of chaperone-bound H3K56ac in cell nuclei using different protein isolation methods. We compared total protein isolation through RIPA extraction with the extraction of acid-soluble nuclear proteins (acidic extraction) and denaturing salt extraction for separation of H3/H4 nucleosome tetramers. Only a weak H3K56ac signal could be found in the RIPA extract, even under very high protein loading. This weak signal could not be analyzed due to the low signal-to-noise ratio. After densitometric normalization of the H3K56ac signal from the acid and salt extracts (Fig. 4D) according to the total protein control, no significant changes in H3K56ac levels were observed during the cell cycle (Fig. 4E, F). The acid-extracted H3K56ac was more N-terminally degraded than the salt-extracted fraction. Compared with H3K56ac, the level of other histone acetylation regulated by p300/CBP and sirtuins, H3K9ac, was higher in the G1/S, S and G2 cell cycle phases.
The protein level of p300, but not ASF1A, is correlated with H3K56ac dynamics during the cell cycle
H3K56ac biogenesis begins with the transfer of the acetyl group to H3, which is catalyzed by HATs (p300, CBP and GCN5), with the cooperation of ASF1A or ASF1B. The acetyl group is removed by the sirtuins SIRT1, SIRT2 and SIRT6. Hence, we asked whether the expression of the proteins involved in H3K56ac regulation was connected to the cell cycle-dependent H3K56ac changes observed in cancer cell lines (Fig. S8A, B). Similar expression profiles were detected for ASF1A/B and all of the HATs (p300, CBP and GCN5) in HeLa cells. Gene activity was reduced in S phase and then increased to a maximum in G2/M/G1 and the upcoming G1 phase. The maximal expression of ASF1A and the HATs was also observed in G2/M/G1 in HL-60 cells. Among the sirtuin family members, SIRT1 showed S and G2 phase-dependent downregulation, and SIRT6 showed G1/S and S phase-dependent downregulation in HeLa, but not HL-60 cells. The reduction of sirtuin expression could be connected to the increase in H3K56ac foci in the cell nuclei observed in S and G2 phase. In addition, SIRT6, ASF1B and cyclin E showed different expression patterns in HL-60 and HeLa cells. However, western blot analysis revealed no changes in p300 protein levels during the cell cycle (Fig. S8C). The level of the H3K56ac-modulating ASF1A chaperone was slightly elevated during the G1/S transition and G2 phase. Despite the fact that gene expression did not fully correlate with the detected protein levels, the differential expression pattern revealed that the genes involved in the H3K56ac regulation pathway are differentially regulated during the cell cycle in distinct cancer cell lines.
Sirtuins remove H3K56ac from metaphase chromatin
Using different chemical modulators of the H3K56ac regulatory pathway, we investigated whether the change in H3K56ac levels can affect cell cycle progression. The levels of H3K56ac and H3K9ac increased after sirtuin family inhibition (Fig. 5A, 5B and Fig. S9A). We also found that the increase in the truncated form of H3K56ac was correlated with the deacetylation block. Surprisingly, the difference between the number of H3K56ac foci in EdU-positive and EdU-negative nuclei was reduced after SIRT2 inhibition using 10 μM AGK-2. This observation was primarily associated with the preservation of H3K56ac in mitotic chromosomes (Fig. 6D, Fig. S9B). Surprisingly, an activator of SIRT1,39 resveratrol (100 μM), also increased the level of H3K56ac, H3K9ac and ASF1A. In contrast, C646 (10 μM), an inhibitor of p30040 decreased the H3K56ac level (Fig. 5A and Fig. S9C, D). After C646 treatment, nuclear deformation, DNA replication inhibition and the increase in the chromatin condensation was observed in the most of cells. Surprisingly, the H3K56ac foci level reduction was observed in EdU-positive cells after C646 inhibition. As a control for cells in the G1/S transition, the DNA replication inhibitor aphidicolin was used. Similar to the effects observed after thymidine inhibition, we detected no change in H3K56ac levels after aphidicolin inhibition (Fig. 5B).
Figure 5.

Cell cycle changes after inhibition or activation of the H3K56ac regulatory pathway. (A, B) Inhibition of sirtuin activity in HeLa cells (AGK-2, nicotinamide) induced an increase in H3K9ac and truncated form of H3K56ac without influencing ASF1A levels. The SIRT1 activator resveratrol induced an increase in H3K9ac levels together with ASF1A. C646, a p300/CBP inhibitor, reduced H3K56ac levels. No changes in H3K56 levels were observed after DNA replication inhibition by aphidicolin. (C) Increased and decreased H3K56ac levels are both connected to cell cycle perturbations in HeLa cells. The graph represents the proportion of cells in the different cell cycle stages. (D) Increased and decreased H3K56ac levels are both connected to the inhibition of DNA replication in HeLa cells. The cells were treated with EdU for 2 h. The chart represents the ratio of the EdU-positive, EdU-negative and mitotic cells. After 24 h of treatment with resveratrol and C646, the number of EdU-positive cells was reduced. At least 500 cells were analyzed for each inhibitor. (E) The number of H3K56ac foci (red) in the EdU-positive (green) and -negative cells was similar after SIRT2 inhibition. The cells were treated with AGK-2 for 24 h. No noticeable difference between the EdU-positive (green) and -negative cells was observed. H3K56ac localized to mitotic chromosomes after sirtuin inhibition. After 24 h of SIRT2 inhibition, H3K56ac (red) colocalized with the mitotic DNA (blue). (F) A delay in the cell cycle progression in HeLa cells after inhibition or activation of the H3K56ac regulatory pathway. HeLa cells were synchronized using a double thymidine procedure. After release from the thymidine block, the cells were treated with different inhibitors for 12 h. Cell cycle progression was measured by staining with PI and flow cytometry. Twelve hours is sufficient time to reach G1 phase in the control cells treated with DMSO. SIRT1-3 inhibition by AGK-2 does not affect cell cycle progression. A cell cycle delay was observed after sirtuin inhibition by nicotinamide. G1/S and S phase arrest was observed after the HeLa cells were treated with resveratrol and C646. Aphidicolin, a DNA replication inhibitor, was used as the G1/S phase arrest control.
Figure 6.
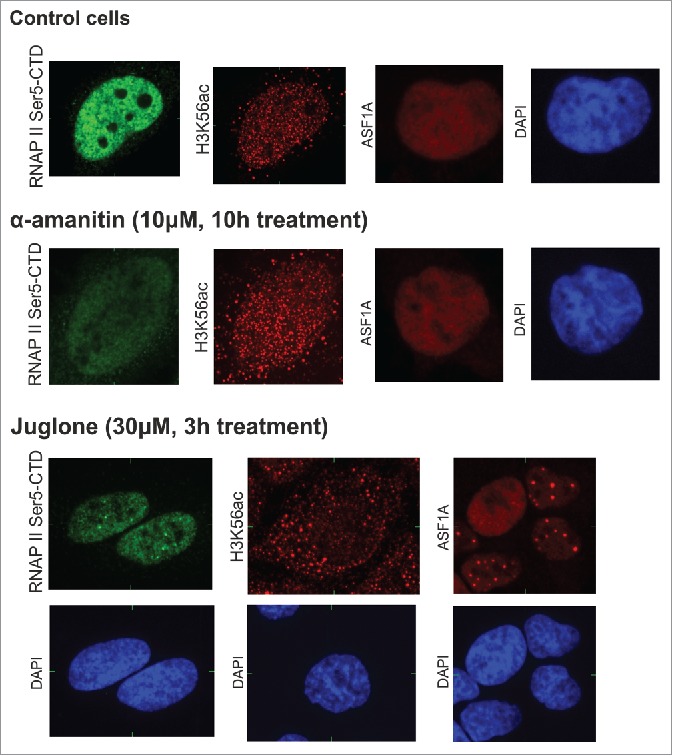
H3K56ac levels after the inhibition of transcriptional activity using α-amanitin and juglone. HeLa cells were treated with the RNA polymerase II inhibitors α-amanitin and juglone. H3K56ac foci, ASF1A and RNAP II Ser5-CTD levels were analyzed by fluorescence microscopy. A decrease in the RNAP II Ser5-CTD, but not ASF1A and H3K56ac, signal was observed after α-amanitin treatment; this result is in contrast to that observed after juglone. Juglone induced the loss of H3K56ac foci in the HeLa cell nuclei. A decrease in the intensity and the presence of large ASF1A and RNAP II Ser5-CTD foci was observed. The image represents one slice of the object in the x,y, x,z and y,z planes.
Inhibition of sirtuin activity is associated with a delay in the cell cycle
The cell cycle distribution of cells was analyzed after 24 h of drug treatment. Significant changes in the cell cycle distribution were noted after resveratrol and C646 administration, but not after the addition of sirtuin inhibitors (Fig. 5C). Similarly, the ratio between EdU-positive and EdU-negative cells was not dramatically affected by inhibition of H3K56ac deacetylation with sirtuin inhibitors (Fig. 5E). The effect of H3K56ac inhibition/activation on cell cycle progression was determined by adding the inhibitors following release from the thymidine block (Fig. 5F). After 12 h, the non-treated cells reached G1 phase. However, nicotinamide delayed cell cycle progression in a concentration-dependent manner. These results suggest that SIRT2 inhibition alone and the presence of H3K56ac in mitotic chromosomes did not influence cell cycle progression. However, the inhibition of other sirtuins and H3K56ac overexpression after nicotinamide inhibition resulted in a cell cycle delay. Resveratrol, C646 and aphidicolin blocked the cell cycle in G1/S or S phase.
H3K56ac does not participate in the repair of dsDNA breaks induced by radiation
In mammalian cells, H3K56ac is associated with dsDNA break repair,16,29-31 a process that, similar to DNA replication, requires a high rate of nucleosome recovery. In our study, double-strand DNA breaks were induced in HeLa cells by radiation (3 Gy). Then, the cells were incubated with CSK buffer after 0.5, 1, 2, and 3 h. H3K56ac did not colocalize with the phosphorylated histone isoform H2A.X (H2AXp), a marker of dsDNA repair (Fig. S10).
H3K56ac foci colocalizes with areas of active transcription
The localized and focal distribution of the H3K56ac signal suggests that this modification is connected to transcriptionally active chromatin regions. H3K56ac is excluded from pericentromeric and highly condensed chromatin regions. To assess the presence of H3K56ac in transcriptionally active areas, we marked the sites of active transcription using RNAPII-pSer5 CTD antibodies. Primary and secondary antibodies against the rare H3K56ac antigen were added prior to RNAPII-pSer5 CTD identification to minimize antigen masking. RNAPII-pSer5 CTD produces a heterogeneous pattern, with mutual exclusivity to pericentromeric heterochromatin regions, primarily in the nucleus and the nucleolar envelope. The overlap of H3K56ac foci with RNAPII-pSer5 CTD-positive areas was measured using the Overlap Map algorithm in Acquiarium software (CBIA, Brno, Czech Republic). Partial overlap with RNAPII-pSer5 CTD-positive areas was observed for the majority of nuclear-localized H3K56ac foci (Fig. S11). An antibody against p300 and CSK buffer were used to determine whether the H3K56ac loci were associated with p300. H3K56ac foci were observed to be highly colocalized with the p300-enhanced territories and were preferentially localized to transcriptionally active chromatin.
To further confirm the role of H3K56ac in transcriptional regulation, we inhibited the activity of RNAPII using α-amanitin and juglone (Fig. 6). The presence of H3K56ac and ASF1A in the nucleus was not influenced by α-amanitin, which inhibits RNAPII-dependent transcriptional elongation. This result contrasts with those obtained using fast-acting juglone, which blocks the assembly of the pre-initiation complex.41 Even after a modest loss of the RNAPII-pSer5 CTD signal, the H3K56ac loci showed no nuclear localization. Similarly, we observed a reduced level of ASF1A associated with the appearance of large foci. Together, these data suggested a possible link between H3K56ac and transcriptional activity. Moreover, disrupting the formation of the functional preinitiation complex by juglone leads to rapid loss of the nuclear localization of ASF1A and H3K56ac.
Discussion
Core histone modifications were unnoticed for many years after the histone code was revealed. The major contributing factor to the inconspicuousness of H3K56ac is the rarity of this modification in mammalian cells. Compared with H3K56ac-overproducing yeast, the evolutionarily advanced eukaryotes upgraded the ancestral H3K56ac-specific HAT regulatory system to the more versatile p300/CBP, with a wide variety of interacting partners. In our work, we aimed to generate a basic description of H3K56ac levels and nuclear distribution during the cell cycle. To determine the level of H3K56ac, we used different methodical approaches with or without specific antibodies. As a model for studying H3K56ac, we chose rapid-cycling cancer and embryonic stem cell lines.
H3K56ac immunofluorescent staining produces a well-separated pattern of sharply bordered foci, enabling their automatic counting by software. The cancer cell lines studied here with on-going DNA replication activity have a higher number of H3K56ac foci than in hESCs. The separation of the cells into distinct cell cycle phases confirmed the persistence of the number of foci from S to G2 phase. A similar distribution was observed in mammary epithelial cells by flow cytometry analysis.42 The difference in the H3K56ac level between the cancer and hESC cells can be explained by the functional and morphological differences between these cells. The chromatin of pluripotent stem cells is a highly dynamic structure with reduced heterochromatin areas.43,44 Moreover, hESCs exhibit a typical, short G1 phase and different regulation of the G1/S transition checkpoint.45 The microscopic images represent the sites of local H3k56ac accumulation but not the basal level of protein modification. The discrepancy between the microscopic and proteomic analyses could be explained by different normalization methods. The number of H3K56ac foci per nucleus was measured by image analysis, whereas the ratio of H3K56ac to total protein (or H3) levels was determined by proteomic analysis. After histone acid extraction, the loading of total protein is not dependent on the levels of histones in the nucleus.
Proteomic analysis showed that the H3K56ac foci level does not correlate with small, non-significant changes in the protein level. In mammalian cells, the level of H3K56ac is significantly lower than in yeasts.27,46 Approximately 0.04% to 0.2%32 of H3 histones are acetylated on H3K56ac. This level of modification is very low in contrast to the approximately 25% rate observed in yeast.16,27,47 In mammalian cells, the relationship between DNA replication and H3K56ac was determined using the S phase increase in the H3K56ac protein level and H3K56 point mutation studies.30,31 Regardless, similar to our observation, the H3K56ac protein levels were not cell cycle dependent.29 Moreover, another study demonstrated that the regulation of the H3K56ac levels during the cell cycle is dependent on cellular aging. IMR90 cells displayed an increase of H3K56ac during S and G2 phase in early passages, but the reduced level of the modification did not change in the later passages.48 Moreover, H3K56me, but not H3K56ac, interacts with the PCNA protein. PCNA binds to H3K56ac during G1 phase, which impairs PCNA recruitment to the chromatin.49 Moreover, thymidine and aphidicolin blocked DNA replication but did not induce changes in H3K56ac levels during cell cycle progression. The inhibition of H3K56ac deacetylation also did not result in a large-scale increase in H3K56ac staining in the nuclear chromatin, but only an increase in the number of well-defined foci. This result indicates that the H3K56ac level is not a marker of the newly associated nucleosomes during DNA replication, as has been shown in yeast.8,9
Nevertheless, our results do not reject the connection between H3K56ac and the DNA replication process. Replication origins are the starting points for DNA replication. Replication origins are primarily located in proximity to tissue-specific transcriptionally active genes. ChIP RNA-Seq showed high levels of H3K56ac at the replication origins.50 The replication origins could be the possible link between H3K56ac and DNA replication. The loss of H3K56ac could induce condensation of these areas and S-phase arrest. Curcumin inhibition of p300 induced cell cycle arrest at the G2/M phase, which is associated with the initiation of senescence-associated heterochromatin foci during S phase.51
To assess the influence of the H3K56ac on cell cycle progression, we inhibited H3K56ac acetylation and deacetylation. After sirtuin inhibition, we observed a concentration-dependent delay in cell cycle progression. More interestingly, we also observed an increased level of the N-terminally degraded H3K56ac product in acid-extracted H3 histones. Histone degradation could be an additional mechanism to restore H3K56ac abundance in the absence of sirtuin activity. Other H3K56ac modulators, resveratrol and C646, induced cell cycle arrest at the G1/S phase. Surprisingly, an activator of SIRT1 increased H3K56ac and H3K9ac levels. The reverse effect is explained by the resveratrol-dependent activation of Tip60 and the increase of the nicotinamide and histone acetylation levels in cells.52 Our results showed that both H3K56ac inhibition/activation may be connected to S phase arrest. p300/CBP and sirtuins exhibit many regulatory functions that hinder the direct connection between the H3K56ac level and cell cycle regulation. Moreover, the cell cycle is regulated by their direct interaction with non-histone proteins.53-55
The appearance of increased H3K56ac degradation in acidic-extracted histones and the discrepancy between the number of foci and the protein level suggest the existence of free and chaperone-bound H3K56ac. The existence of chaperone-bound H3K56ac is supported by the cytosolic release of H3K56ac after CAF-1 chaperone knock-down.16 When HeLa cells were washed in CSK buffer, it revealed that H3K56ac and ASF1A were not as firmly bound to the chromatin as RNAPII and p300. Although the recruitment of p300/CBP and RNAPII to the promoter is a very rapid and dynamic event, the entire complex may remain on the promoter for several hours, even in the case of transient transcription.56 This finding correlates with the high turnover of H3K56ac and indicates the possibility that the chaperone-bound fraction exists in the nucleus. We next determined the changes in the H3K56ac level in the histones in the nuclear and chromatin fractions. We did not observe any significant difference between the acid- and the salt-extracted histones, with the exception of the increase in the degraded H3 product. We expected the H3K56ac level to be reduced in the S and G2 phases in the salt-extracted compared with the acid-extracted histones. The non-significant decrease observed in G2, but not in S phase, does not support our hypothesis that the increase in H3K56ac foci is a result of an increase in the chaperone-bound fraction.
The cleavage of the N-terminal part of H3 histone was catalyzed by glutamate dehydrogenase. The histone clipping is primarily associated with euchromatin.57 We only observed this possible histone clipping in cancer cell lines, but not in hESCs. This fact corresponds with the previously mentioned study that did not observe H3K56ac clipping in chicken brain and liver cells, but only in HeLa cells.57 Our observation is consistent with highly dynamic nature of H3K56ac in the nucleosome. Salt extraction of the chromatin-bound H3/H4 showed a lower ratio of the degraded H3K56ac than in the acid isolation. However, the isolation processes have very different protocols, with longer sample handling and non-denaturing conditions in the acid extraction. Even with the use of protease inhibitors during the nucleus isolation procedure, it was almost impossible to determine whether the shortened H3 form was produced artificially or naturally by histone clipping.
The localization of histone H3K56ac outside of the bright, late S phase EdU-positive areas in the nucleus, as well as the loose binding, corresponds with the well-defined role of H3K56ac in transcriptional regulation. The distribution of H3K56ac negatively correlates with that of H3K56me3.58 H3K56me3 is a marker of heterochromatin and is localized to the bright EdU-positive areas. The bright EdU-positive areas, similar to the intense DAPI-positive areas, were localized to pericentromeric heterochromatin. The intensity of H3K56me3 is higher in EdU-negative cells than in EdU-positive cells.58 In our study, H3K56ac was highly colocalized with RNAPII-pSer5 CTD- and p300-positive transcriptionally active areas. Neither inducing the proteasomal degradation of the largest RNAPII subunit, RPB1,41 nor inhibiting transcriptional elongation with α-amanitin had any effect on H3K56ac levels. The inhibition of preinitiation complex assembly with juglone pointed to a link between H3K56ac foci and transcriptionally active areas. Juglone inhibits the transcription preinitiation complex by modifying sulfhydryl groups. However, juglone is an inhibitor with a broad spectrum of targets, inhibiting the activities of other proteins such as the peptidyl-propyl isomerase Pin1,59 pyruvate decarboxylase and glutathione-S-transferase. More specific inhibitors would help clarify the connection between the loss of H3K56ac and the inhibition of preinitiation complex assembly. Moreover, the loss of H3K56ac foci and the reduced level and reorganization of ASF1A were observed after juglone treatment. Our observations agreed with chromatin immunoprecipitation studies in which increased K56 acetylation was observed on the promoters of genes with very high transcriptional activity, such as histones or tissue-specific genes.27,29 However, the reduction of transcriptional activity could be connected to H3K56ac promoter occupancy.26 Promoter-bound H3K56ac can serve to stabilize the open chromatin conformation of promoters. Oct4, a master regulator of cell pluripotency, directly interacts with H3K56ac in nucleosomes. The H3K56ac-OCT4 complex can stabilize the initiation complex and sustain a high turnover rate for the transcription of pluripotency-related genes.60
In conclusion, although the distribution of H3K56ac was significantly altered during the cell cycle in human cancer cell lines, these changes were not connected to the extensive reconstitution of replicated chromatin as previously demonstrated for yeasts. The connection between this histone modification and transcriptional regulation was supported by the existence of a fraction of H3K56ac that was loosely bound to chromatin and the localization of H3K56ac foci outside of heterochromatin. Our findings illustrate the highly dynamic behavior of this very rare histone modification and provide insights into the regulation of H3K56ac in human cells.
Materials and Method
Cell culture and cell cycle synchronization
The HL-60 cancer cell line (ECACC, Salisbury, UK) was cultured in RPMI-1640, and HeLa (ECACC) cells were cultured in EMEM medium; both media were supplemented with 10% FCS (PAN – Biotech, Aidenbach, Germany ), and the cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. The hESC CCTL12 cell line was kindly provided by the Department of Biology (Faculty of Medicine, Masaryk University, Czech Republic). The CCTL12 cells were cultivated as a monolayer on Matrigel (BD Biosciences, CA, USA) in conditioned media (KD DMEM, KOSR, L-Glut, NEAA, Pen/Strep, 2-mercaptoethanol, and FGF (Life Technologies, CA, USA), after a one-day cultivation with mouse embryonic fibroblasts supplemented with fresh L-glutamine and bFGF.34 For our analysis, we used cells in passages 30–55.
To synchronize the cell cycle, the cells were seeded at 5×104 cells per 1-cm2 dish surface or 1×106 cells per 1 ml of media for the HL-60 suspension. The cells were treated with 2 mM thymidine (20 h for cancer cell lines and 16 h for CCTL12), washed, and cultivated for 8 h in medium w/o thymidine. The cells were inhibited again by the addition of 2 mM thymidine for 19 h (cancer cells) or 16 h (CCTL12 cells). After release from the thymidine block, the cells were analyzed for each 3 h. For the cell cycle analysis, the 1× PBS-washed cells were fixed with ice-cold 95% ethanol and rehydrated by washing in 1× PBS. Interfering RNAs were removed by RNase treatment. The DNA content was visualized by propidium iodide. The cell cycle distribution was measured by a FASC Diva II (BD Biosciences), and the data were evaluated using Flowing Software 2 (Terttu Terho, Turku Center for Biotechnology, http://www.flowingsoftware.com/).
To activate or inhibit the proteins that regulate H3K56ac, HeLa cells were treated with different inhibitors for 24 h before analysis. AGK-2, nicotinamide, resveratrol and C646 (Sigma, MO, USA) were used to activate or inhibit, respectively, the proteins that regulate H3K56ac regulation. Aphidicolin, a DNA polymerase inhibitor, was purchased from Serva (Heidelberg, Germany).
Immunofluorescent staining
To label the DNA replication activity, we used the Click-iT® EdU Alexa Fluor® 488 Imaging Kit (Life Technologies) according to the manufacturer's protocol, with 2 h of EdU incubation. The cells were fixed with 4% paraformaldehyde and permeabilized in 0.5% Triton/1× PBS. Non-specific antigen interactions were blocked by 5% non-fat dry milk/0.1% Tween before incubation with the antibodies. Rabbit anti-H3K56ac (1:400), anti-ASF1A (1:400; Cell Signaling, MA, USA), mouse β-tubulin (1:500), RNA polymerase II CTD p-Ser (1:1000), p300 (1:100; Abcam, UK) and H3K9ac inhibition peptides (1:200; Cell Signaling) were incubated with the cells overnight at 37°C in a moist chamber. The antibodies were visualized by goat anti-rabbit Alexa 555 and goat anti-mouse Alexa 488 (Cell Signaling). The nuclei were counterstained in DAPI solution and sealed in VECTASHIELD.
The chromatin-bound H3K56ac histone fraction was analyzed by a combination of CSK buffer (100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM PIPES, pH 6.8) treatment with immunohistochemistry. After washing with 1× PBS, the cells were incubated with 1× CSK/0.1% Triton for 10 min/RT and then fixed with 4% paraformaldehyde/1× CSK for 15 min before beginning the immunostaining protocol.
Histone isolation and protein gel blotting analysis
For acid isolation of the histones, we used a protocol described by Shechter et al.61. The cell nuclei were isolated in ice-cold hypotonic Extraction buffer: 10 mM Tris-HCl, pH 8.0/1 mM KCl/1.5 mM MgCl2/1 mM DTT with complete proteinase inhibitors (Roche). The histones were extracted from the chromatin with ice-cold 0.2 M H2SO4 overnight, and the extract solutions were subjected to sample preparation prior to mass spectrometric analysis.
For salt extraction of the histones, we modified the protocol described by Rodriguez-Collazo, et al.62. To wash out the nuclear proteins, smaller volumes of the washing buffer were used. During washing, the chromatin pellet was pinned to the tube bottom using a needle.
The 0.2 M H2SO4 histone extracts were processed by the FASP method using the original protocol63 with several modifications. Briefly, the samples were mixed with 8 M urea and 0.1 M DTT in 0.1 M Tris-HCl (pH=8.5) and transferred to an ultrafiltration unit (Microcon 30 kDa, Millipore). After one wash with 8 M Urea in 0.1 M Tris-HCl (pH=8.5), the proteins were alkylated by 55 mM IAA (30 min in the dark, RT). Then, the proteins were washed twice with 8 M Urea in 0.1 M Tris-HCl (pH=8.5) and twice with 50 mM ABC. The proteins on the filter were digested with ArgC protease overnight at 37°C. The peptides were eluted from the filter unit by 50 mM ABC. The peptide mixture was dried under a vacuum. The peptides were subjected to an acid extraction procedure (sonication of the gel in 50 µl of 50% CAN ACN and 2.5% FA) and transferred together with isotopically labeled peptides to autosampler vials containing PEG prior to LC-MS/MS analysis.
For the western blot analysis, the histones in 0.2 H2SO4 were precipitated by trichloroacetic acid, washed with 50 mM HCl/acetone followed by 100% acetone and dried. The dried histone samples were reconstituted in water. The histone concentrations were determined by the A280 and BCA methods and separated on 15% PAGE or Tris-Tricine ELFO according to a previously described protocol.61,64 After transfer, the H3K56ac antigen was detected by incubation with rabbit anti-H3K56ac (1:500, Cell Signaling), H3K9ac (1:1000, Cell Signaling), the C-terminal part of H3 (1:5000, Cell Signaling), ASF1A (1:1000, Cell Signaling), and GAPDH (1:5000, Cell Signaling) in combination with H3K9ac inhibition peptide (1:500, Cell Signaling) overnight/4°C. The total proteins were visualized by Amido Black staining.
RNA isolation and quantitative RT-PCR
The cellular RNA was isolated with QIAShredder™ and RNase Mini Kits (Qiagen) according to the manufacturer's protocol. The RNA quality and quantity were analyzed using a spectrophotometer and RNA electrophoresis. The cDNA was synthesized using M-MuLV RT (Finnzymes) and random nonamers (Sigma). The RT-PCR primers used to determine the genetic expression of the proteins that regulate H3K56ac are summarized in Table S1. The level of gene expression was detected using the FastStart Universal SYBR Green Master (Rox; Roche), and the amplified DNA length was determined by melting curve analysis and DNA electrophoresis.
Mass Spectrometry
The peptides were injected into the nanoLC-MS system, which consists of an Ekspert nanoLC 425 system (Eksigent) coupled with a QTrap 6500 mass spectrometer (AB SCIEX). Prior to LC separation, the peptides were concentrated and desalted using a trapping column (100 μm × 30 mm) filled with 3.5 μm X-Bridge BEH 130 C18 sorbent (Waters). After washing the trapping column with 0.1% FA, the peptides were eluted (flow rate 300 nl/min) from the trapping column onto a Picofrit ProteoPep™ II C18 column (5 µm particles, ID 75 μm × length 10 cm; New Objectives) using the following gradient program (mobile phase A: 0.1% FA in water; mobile phase B: 0.1% FA in 100% acetonitrile): the gradient elution started at 5% of mobile phase B and increased from 5% to 18% during the first 20 min, followed by an increase to 45% in the 50th min, which then increased linearly to 95% of mobile phase B in the next 2 min and remained at this state for the next 8 min. The equilibration of the trapping column and the column was performed prior to the sample injection into the sample loop.
The eluted peptides were introduced into a 6500 QTRAP MS system via electrospray ionization using a NanoSpray® III (AB SCIEX), with temperature of 150°C, spray voltage of 2800 V and ion source gas 1 of 15. The SRM transitions of the specific H3 peptide YQKSTELLIR that contains lysine 56 and its acetylated form were monitored with the Q1 and Q3 setting of unit resolution in the high mass mode. The development of the SRM method and the analysis of the mass spectrometric raw data were performed using the Skyline software tool (http://proteome.gs.washington.edu/software/skyline).65 All statistical analyses were performed using Excel 2010 (version 14.0, Microsoft Office). The details regarding the transitions for the native and synthetized isotopically labeled peptides are summarized in Table S2.
Computational Analysis
Image acquisition was performed using a Zeiss S100 microscope (Carl Zeiss MicroImaging) with a CARV confocal unit (Atto Instruments). The images were captured with a Micromax 1300-YHS camera and a cooled CCD chip (Princeton Instruments, USA). The camera resolution was 1300×1030 pixels. The pixel size of the images was 124×124 nm.
To quantify the number of fluorescent H3K56ac foci inside the nuclei, image analysis and image acquisition were performed using the Acquiarium software (CBIA, Masaryk University). The cell nuclei were segmented based on their overall shape. The rounded HL-60 nuclei were segmented using a method published by Raimondo et al.66 based on a morphological top-hat operation followed by Otsu intensity thresholding. The HeLa and CCTL12 nuclei were less spherical; to segment them, we took the result of the intensity thresholding in the central image plane, followed by the dilation operation from mathematical morphology to segment the entire flattened nucleus.
To detect the signals corresponding to the H3K56ac foci, we first deconvolved the foci images using the Huygens software to better visually separate neighboring foci. After the deconvolution, we used the method proposed by Matula et al.67 A morphological HMax transform was computed after the initial noise was suppressed using a Gaussian blur. This transform identifies the local intensity maxima whose height exceeds a specified threshold. The EMax image is then defined as the regional maxima of the result. After the computation of the EMax transform, the connected components exceeding the size limit for fluorescence spots or those located outside of the nucleus mask obtained by the initial segmentation were discarded. The remaining components then corresponded to the individual H3K56ac foci.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We are grateful to members of CBIA laboratory for generous help thorough this work. We thank Martina Vodinska, Vladimir Rotrekl, Gabriela Lochmanova, Pavel Matula, Pavel Krejčí and Michaela Kunova and reviewers for helpful advices and Dasa Dolezalova for providing cyclins primers.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This study was generously supported by the Ministry of Education, Youth and Sports of the Czech Republic grants (CZ.1.07/2.3.00/30.0030) and by Czech Science Foundation (projects 302/12/G157 and P206-12-G151) and it was carried out with support CEITEC Proteomics Core Facility (CZ.1.05/1.1.00/02.0068) financed from European Regional Development Fund.
References
- 1.Hyland EM, Cosgrove MS, Molina H, Wang D, Pandey A, Cottee RJ, Boeke JD. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol Cell Biol 2005; 25:10060-70; PMID:16260619; http://dx.doi.org/ 10.1128/MCB.25.22.10060-10070.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozdemir A, Spicuglia S, Lasonder E, Vermeulen M, Campsteijn C, Stunnenberg HG, Logie C. Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae. J Biol Chem 2005; 280:25949-52; PMID:15888442; http://dx.doi.org/ 10.1074/jbc.C500181200 [DOI] [PubMed] [Google Scholar]
- 3.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 2005; 121:375-85; PMID:15882620; http://dx.doi.org/ 10.1016/j.cell.2005.03.011 [DOI] [PubMed] [Google Scholar]
- 4.Fillingham J, Recht J, Silva AC, Suter B, Emili A, Stagljar I, Krogan NJ, Allis CD, Keogh MC, Greenblatt JF. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol 2008; 28:4342-53; PMID:18458063; http://dx.doi.org/ 10.1128/MCB.00182-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolonko EM, Albaugh BN, Lindner SE, Chen Y, Satyshur KA, Arnold KM, Kaufman PD, Keck JL, Denu JM. Catalytic activation of histone acetyltransferase Rtt109 by a histone chaperone. Proc Natl Acad Sci U S A 2010; 107:20275-80; PMID:21057107; http://dx.doi.org/ 10.1073/pnas.1009860107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erkmann JA, Kaufman PD. A negatively charged residue in place of histone H3K56 supports chromatin assembly factor association but not genotoxic stress resistance. DNA Repair (Amst) 2009; 8:1371-9; PMID:19796999; http://dx.doi.org/ 10.1016/j.dnarep.2009.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith S, Stillman B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 1989; 58:15-25; PMID:2546672; http://dx.doi.org/ 10.1016/0092-8674(89)90398-X [DOI] [PubMed] [Google Scholar]
- 8.Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, Zhang X, Garcia BA, Shabanowitz J, Burlingame AL, Hunt DF, et al.. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci U S A 2006; 103:6988-93; PMID:16627621; http://dx.doi.org/ 10.1073/pnas.0601676103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 2008; 134:244-55; PMID:18662540; http://dx.doi.org/ 10.1016/j.cell.2008.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008; 134:231-43; PMID:18662539; http://dx.doi.org/ 10.1016/j.cell.2008.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell 2007; 25:703-12; PMID:17320445; http://dx.doi.org/ 10.1016/j.molcel.2007.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 2007; 315:653-5; PMID:17272723; http://dx.doi.org/ 10.1126/science.1133234 [DOI] [PubMed] [Google Scholar]
- 13.Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, Verreault A. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol 2006; 16:1280-9; PMID:16815704; http://dx.doi.org/ 10.1016/j.cub.2006.06.023 [DOI] [PubMed] [Google Scholar]
- 14.Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell 2006; 23:109-19; PMID:16818235; http://dx.doi.org/ 10.1016/j.molcel.2006.06.006 [DOI] [PubMed] [Google Scholar]
- 15.Keck KM, Pemberton LF. Interaction with the histone chaperone Vps75 promotes nuclear localization and HAT activity of Rtt109 in vivo. Traffic 2011; 12:826-39; PMID:21463458; http://dx.doi.org/ 10.1111/j.1600-0854.2011.01202.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009; 459:113-7; PMID:19270680; http://dx.doi.org/ 10.1038/nature07861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Jaramillo-Lambert AN, Yang Y, Williams R, Lee NH, Zhu W. And-1 is required for the stability of histone acetyltransferase Gcn5. Oncogene 2012; 31:643-52; PMID:21725360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al.. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012; 149:214-31; PMID:22464331; http://dx.doi.org/ 10.1016/j.cell.2012.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Black JC, Mosley A, Kitada T, Washburn M, Carey M. The SIRT2 deacetylase regulates autoacetylation of p300. Mol Cell 2008; 32:449-55; PMID:18995842; http://dx.doi.org/ 10.1016/j.molcel.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell 2005; 17:301-11; PMID:15664198; http://dx.doi.org/ 10.1016/j.molcel.2004.12.018 [DOI] [PubMed] [Google Scholar]
- 21.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell 2007; 128:721-33; PMID:17320509; http://dx.doi.org/ 10.1016/j.cell.2007.01.030 [DOI] [PubMed] [Google Scholar]
- 22.Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 2009; 8:2664-6; PMID:19625767; http://dx.doi.org/ 10.4161/cc.8.16.9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu C, Liu M, Zhang W, Xu Q, Ma K, Chen L, Wang Z, He S, Zhu H, Xu N. Upregulation of KLF4 by methylseleninic acid in human esophageal squamous cell carcinoma cells: Modification of histone H3 acetylation through HAT/HDAC interplay. Mol Carcinog 2014; 54(10):1051-9. [DOI] [PubMed] [Google Scholar]
- 24.Im JS, Keaton M, Lee KY, Kumar P, Park J, Dutta A. ATR checkpoint kinase and CRL1βTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev 2014; 28:875-87; PMID:24700029; http://dx.doi.org/ 10.1101/gad.239194.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armas-Pineda C, Arenas-Huertero F, Pérezpeñia-Diazconti M, Chico-Ponce de León F, Sosa-Sáinz G, Lezama P, Recillas-Targa F. Expression of PCAF, p300 and Gcn5 and more highly acetylated histone H4 in pediatric tumors. J Exp Clin Cancer Res 2007; 26:269-76; PMID:17725108 [PubMed] [Google Scholar]
- 26.Liu Y, Wang DL, Chen S, Zhao L, Sun FL. Oncogene Ras/phosphatidylinositol 3-kinase signaling targets histone H3 acetylation at lysine 56. J Biol Chem 2012; 287:41469-80; PMID:22982396; http://dx.doi.org/ 10.1074/jbc.M112.367847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie W, Song C, Young NL, Sperling AS, Xu F, Sridharan R, Conway AE, Garcia BA, Plath K, Clark AT, et al.. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell 2009; 33:417-27; PMID:19250903; http://dx.doi.org/ 10.1016/j.molcel.2009.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stejskal S, Tesařová L, Koutná I. Mysterious Role of H3K56ac in Embryonic Stem Cells. Folia Biol (Praha) 2014; 60 Suppl 1:71-5; PMID:25369345 [PubMed] [Google Scholar]
- 29.Tjeertes JV, Miller KM, Jackson SP. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J 2009; 28:1878-89; PMID:19407812; http://dx.doi.org/ 10.1038/emboj.2009.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vempati RK, Jayani RS, Notani D, Sengupta A, Galande S, Haldar D. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem 2010; 285:28553-64; PMID:20587414; http://dx.doi.org/ 10.1074/jbc.M110.149393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan J, Pu M, Zhang Z, Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 2009; 8:1747-53; PMID:19411844; http://dx.doi.org/ 10.4161/cc.8.11.8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drogaris P, Villeneuve V, Pomiès C, Lee EH, Bourdeau V, Bonneil E, Ferbeyre G, Verreault A, Thibault P. Histone deacetylase inhibitors globally enhance h3/h4 tail acetylation without affecting h3 lysine 56 acetylation. Sci Rep 2012; 2:220; PMID:22355734; http://dx.doi.org/ 10.1038/srep00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krutá M, Bálek L, Hejnová R, Dobšáková Z, Eiselleová L, Matulka K, Bárta T, Fojtík P, Fajkus J, Hampl A, et al.. Decrease in abundance of apurinic/apyrimidinic endonuclease causes failure of base excision repair in culture-adapted human embryonic stem cells. Stem Cells 2013; 31:693-702; http://dx.doi.org/ 10.1002/stem.1312 [DOI] [PubMed] [Google Scholar]
- 34.Kunova M, Matulka K, Eiselleova L, Salykin A, Kubikova I, Kyrylenko S, Hampl A, Dvorak P. Adaptation to robust monolayer expansion produces human pluripotent stem cells with improved viability. Stem Cells Transl Med 2013; 2:246-54; PMID:23486835; http://dx.doi.org/ 10.5966/sctm.2012-0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, et al.. Sirtuin 2 inhibitors rescue α-synuclein-mediated toxicity in models of Parkinson disease. Science 2007; 317:516-9; PMID:17588900; http://dx.doi.org/ 10.1126/science.1143780 [DOI] [PubMed] [Google Scholar]
- 36.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 2002; 277:45099-107; PMID:12297502; http://dx.doi.org/ 10.1074/jbc.M205670200 [DOI] [PubMed] [Google Scholar]
- 37.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 2003; 11:437-44; PMID:12620231; http://dx.doi.org/ 10.1016/S1097-2765(03)00038-8 [DOI] [PubMed] [Google Scholar]
- 38.Yang B, Zwaans BM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 2009; 8:2662-3; PMID:19597350; http://dx.doi.org/ 10.4161/cc.8.16.9329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 2005; 280:17187-95; PMID:15749705; http://dx.doi.org/ 10.1074/jbc.M501250200 [DOI] [PubMed] [Google Scholar]
- 40.Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, et al.. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol 2010; 17:471-82; PMID:20534345; http://dx.doi.org/ 10.1016/j.chembiol.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen VT, Giannoni F, Dubois MF, Seo SJ, Vigneron M, Kédinger C, Bensaude O. In vivo degradation of RNA polymerase II largest subunit triggered by α-amanitin. Nucleic Acids Res 1996; 24:2924-9; PMID:8760875; http://dx.doi.org/ 10.1093/nar/24.15.2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu B, Watanabe K, Dai X. Pygo2 regulates histone gene expression and H3 K56 acetylation in human mammary epithelial cells. Cell Cycle 2012; 11:79-87; PMID:22186018; http://dx.doi.org/ 10.4161/cc.11.1.18402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bártová E, Galiová G, Krejcí J, Harnicarová A, Strasák L, Kozubek S. Epigenome and chromatin structure in human embryonic stem cells undergoing differentiation. Dev Dyn 2008; 237:3690-702; http://dx.doi.org/ 10.1002/dvdy.21773 [DOI] [PubMed] [Google Scholar]
- 44.Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell 2006; 10:105-16; PMID:16399082; http://dx.doi.org/ 10.1016/j.devcel.2005.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barta T, Dolezalova D, Holubcova Z, Hampl A. Cell cycle regulation in human embryonic stem cells: links to adaptation to cell culture. Exp Biol Med (Maywood) 2013; 238:271-5; PMID:23598972; http://dx.doi.org/ 10.1177/1535370213480711 [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Eugeni EE, Parthun MR, Freitas MA. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 2003; 112:77-86; PMID:12937907; http://dx.doi.org/ 10.1007/s00412-003-0244-6 [DOI] [PubMed] [Google Scholar]
- 47.Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. Adenovirus small e1a alters global patterns of histone modification. Science 2008; 321:1084-5; PMID:18719283; http://dx.doi.org/ 10.1126/science.1155544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 2010; 17:1218-25; PMID:20890289; http://dx.doi.org/ 10.1038/nsmb.1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, Song C, Zhang Q, DiMaggio PA, Garcia BA, York A, Carey MF, Grunstein M. Histone H3 lysine 56 methylation regulates DNA replication through its interaction with PCNA. Mol Cell 2012; 46:7-17; PMID:22387026; http://dx.doi.org/ 10.1016/j.molcel.2012.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li B, Su T, Ferrari R, Li JY, Kurdistani SK. A unique epigenetic signature is associated with active DNA replication loci in human embryonic stem cells. Epigenetics 2014; 9:257-67; PMID:24172870; http://dx.doi.org/ 10.4161/epi.26870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prieur A, Besnard E, Babled A, Lemaitre JM. p53 and p16(INK4A) independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat Commun 2011; 2:473; PMID:21915115; http://dx.doi.org/ 10.1038/ncomms1473 [DOI] [PubMed] [Google Scholar]
- 52.Sajish M, Schimmel P. A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol. Nature 2015; 519:370-3; PMID:25533949; http://dx.doi.org/ 10.1038/nature14028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Pan L, Feng Y, Wang Y, Han Q, Han L, Han S, Guo J, Huang B, Lu J. P300 plays a role in p16(INK4a) expression and cell cycle arrest. Oncogene 2008; 27:1894-904; PMID:17906698; http://dx.doi.org/ 10.1038/sj.onc.1210821 [DOI] [PubMed] [Google Scholar]
- 54.Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther 2010; 9:844-55; PMID:20371709; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0971 [DOI] [PubMed] [Google Scholar]
- 55.Fu M, Wang C, Rao M, Wu X, Bouras T, Zhang X, Li Z, Jiao X, Yang J, Li A, et al.. Cyclin D1 represses p300 transactivation through a cyclin-dependent kinase-independent mechanism. J Biol Chem 2005; 280:29728-42; PMID:15951563; http://dx.doi.org/ 10.1074/jbc.M503188200 [DOI] [PubMed] [Google Scholar]
- 56.Byun JS, Wong MM, Cui W, Idelman G, Li Q, De Siervi A, Bilke S, Haggerty CM, Player A, Wang YH, et al.. Dynamic bookmarking of primary response genes by p300 and RNA polymerase II complexes. Proc Natl Acad Sci U S A 2009; 106:19286-91; PMID:19880750; http://dx.doi.org/ 10.1073/pnas.0905469106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mandal P, Verma N, Chauhan S, Tomar RS. Unexpected histone H3 tail-clipping activity of glutamate dehydrogenase. J Biol Chem 2013; 288:18743-57; PMID:23673664; http://dx.doi.org/ 10.1074/jbc.M113.462531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jack AP, Bussemer S, Hahn M, Pünzeler S, Snyder M, Wells M, Csankovszki G, Solovei I, Schotta G, Hake SB. H3K56me3 is a novel, conserved heterochromatic mark that largely but not completely overlaps with H3K9me3 in both regulation and localization. PLoS One 2013; 8:e51765; PMID:23451023; http://dx.doi.org/ 10.1371/journal.pone.0051765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chao SH, Greenleaf AL, Price DH. Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res 2001; 29:767-73; PMID:11160900; http://dx.doi.org/ 10.1093/nar/29.3.767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan Y, Xue Y, Song C, Grunstein M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A 2013; 110:11493-8; PMID:23798425; http://dx.doi.org/ 10.1073/pnas.1309914110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc 2007; 2:1445-57; PMID:17545981; http://dx.doi.org/ 10.1038/nprot.2007.202 [DOI] [PubMed] [Google Scholar]
- 62.Rodriguez-Collazo P, Leuba SH, Zlatanova J. Robust methods for purification of histones from cultured mammalian cells with the preservation of their native modifications. Nucleic Acids Res 2009; 37:e81; PMID:19443446; http://dx.doi.org/ 10.1093/nar/gkp273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wiśniewski JR, Zougman A, Mann M. Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J Proteome Res 2009; 8:5674-8; PMID:19848406; http://dx.doi.org/ 10.1021/pr900748n [DOI] [PubMed] [Google Scholar]
- 64.Činčárová L, Lochmanová G, Nováková K, Šultesová P, Konečná H, Fajkusová L, Fajkus J, Zdráhal Z. A combined approach for the study of histone deacetylase inhibitors. Mol Biosyst 2012; 8:2937-45; http://dx.doi.org/ 10.1039/c2mb25136a [DOI] [PubMed] [Google Scholar]
- 65.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010; 26:966-8; PMID:20147306; http://dx.doi.org/ 10.1093/bioinformatics/btq054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raimondo F, Gavrielides M, Karayannopoulou G, Lyroudia K, Pitas I, Kostopoulos I. utomated evaluation of HER-2/neu status in breast tissue from fluorescent in situ hybridization images. IEEE Trans Image Process 2005; 14:1288-99; PMID:16190465; http://dx.doi.org/ 10.1109/TIP.2005.852806 [DOI] [PubMed] [Google Scholar]
- 67.Matula P, Verissimo F, Wörz S, Eils R, Pepperkok R, Rohr K. Quantification of fluorescent spots in time series of 3-D confocal microscopy images of endoplasmic reticulum exit sites based on the HMAX transform. San Diego: SPIE, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.