

Abstract
Traumatic spinal cord injury (SCI) induces cell cycle activation (CCA) that contributes to secondary injury and related functional impairments such as motor deficits and hyperpathia. E2F1 and E2F2 are members of the activator sub-family of E2F transcription factors that play an important role in proliferating cells and in cell cycle-related neuronal death, but no comprehensive study have been performed in SCI to determine the relative importance of these factors. Here we examined the temporal distribution and cell-type specificity of E2F1 and E2F2 expression following mouse SCI, as well as the effects of genetic deletion of E2F1-2 on neuronal cell death, neuroinflammation and associated neurological dysfunction. SCI significantly increased E2F1 and E2F2 expression in active caspase-3+ neurons/oligodendrocytes as well as in activated microglia/astrocytes. Injury-induced up-regulation of cell cycle-related genes and protein was significantly reduced by intrathecal injection of high specificity E2F decoy oligodeoxynucleotides against the E2F-binding site or in E2F1-2 null mice. Combined E2F1+2 siRNA treatment show greater neuroprotection in vivo than E2F1 or E2F2 single siRNA treatment. Knockout of both E2F1 and E2F2 genes (E2Fdko) significantly reduced neuronal death, neuroinflammation, and tissue damage, as well as limiting motor dysfunction and hyperpathia after SCI. Both CCA reduction and functional improvement in E2Fdko mice were greater than those in E2F2ko model. These studies demonstrate that SCI-induced activation of E2F1-2 mediates CCA, contributing to gliopathy and neuronal/tissue loss associated with motor impairments and post-traumatic hyperesthesia. Thus, E2F1-2 provide a therapeutic target for decreasing secondary tissue damage and promoting recovery of function after SCI.
Keywords: astrocytes, cell cycle pathways, contusive spinal cord injury, E2F1, E2F2, inflammation, motor function, neuropathic pain, neuroprotection
Abbreviations
- BMS
the Basso Mouse Scale
- CCA
cell cycle activation
- CNS
central nervous system
- CDKs
cyclin-activated kinases
- DH
dorsal horn
- ECRC
eriochrome cyanine
- E2F1
E2 promoter binding factor 1
- E2F1-2dko
E2F1-2 double ko
- E2F2ko
E2F2 single ko
- E2Fs ODNs
E2Fs decoy oligodeoxynucleotides
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GFAP
glial fibrillary acid protein
- Iba-1
ionized calcium binding adaptor molecule 1
- ko
knockout
- PCR
polymerase chain reaction
- PCNA
proliferating cell nuclear antigen
- qPCR
quantitative real-time-polymerase chain reaction
- Rb
retinoblastoma
- SCI
spinal cord injury
- VH
ventral horn
- WM
white matter.
Introduction
Traumatic spinal cord injury (SCI) triggers secondary injury processes that contribute to neurological dysfunction - including sensorimotor deficits and chronic neuropathic pain.1,2 Mechanisms of secondary injury include neuronal and oligodendroglial cell death, as well as microglia and astrocyte activation/glial scar formation, among other processes.3-5 Increased expression of cell cycle-related genes/proteins occurs early after experimental SCI, and remains elevated chronically; in post-mitotic neurons, cell cycle activation (CCA) in response to central nervous system (CNS) trauma is associated with neuronal cell death, whereas in microglia or astrocytes it leads to cell proliferation and activation.6 Administration of structurally different cell cycle inhibitors reduces neuronal death and microglial/astrocyte reactivity, attenuates lesion volumes, and improves motor function recovery following SCI. More recent studies have shown that critical cell cycle components, such as E2 promoter binding factor 1 (E2F1), proliferating cell nuclear antigen (PCNA), cyclin-dependent kinases (CDKs) 1,4, and cyclin D1 are up-regulated after SCI not only at the thoracic spinal lesion site, but also in the lumbar spinal dorsal horn and the posterior nucleus of thalamus as well as hippocampus.7-10 Central or systemic administration of a selective CDK inhibitor (CR8) reduced CCA, glial changes, and neuronal activity in the dorsal horn or the posterior nucleus of thalamus, as well as limiting chronic SCI-induced hyperesthesia. However, current drug-based cell cycle inhibitors affect multiple components with diverse actions. More precise dissection of key up-stream regulatory molecules may lead to improved targeted therapy.
E2F transcription factors regulate the progression of the cell cycle by repression or transactivation of genes that encode cyclins, cyclin dependent kinases, checkpoint regulators, and replication proteins.11 Of the 8 known E2F family members, the expression of the repressor subclass E2F4 or E2F5 was not linked to the proliferative state of the cell 12-14 and expression of E2F6-8 in the adult CNS is much lower than for other tissue.14-17 The E2F1, E2F2 and E2F3a are members of the activator sub-family of E2F transcription factors that function in both cell cycle progression and cell death.14,18 For mitotic cells such as microglia and astrocytes in CNS, the normal cell cycle includes the activation of CDKs, the coordinated phosphorylation of retinoblastoma (Rb) and p130 by cyclin/CDK complexes, and the subsequent release and accumulation of E2F activities.14 E2F activators are critical factors important for cell-cycle-dependent gene expression, ultimately regulate cell proliferation 14 and survival.11,19,20 However, the role of E2F activators in glial reactivation following SCI has not been examined.
In the adult CNS, most neurons are post-mitotic, and activation of cell cycle molecules, including E2Fs, may lead to cell death.21 Separation of the Rb-E2F complex not only frees E2F1-3 to activate cell cycle gene expression but also de-represses E2F1-3 regulated transcription of pro-apoptotic molecules such as caspases 3, 9, and 8 and Apaf-1 or pro-apoptotic Bcl−2 family members.22,23 Among E2F activators, E2F1 is believed to potently induce apoptosis directly. Suppression of E2F1 expression and the use of E2F null mice attenuate neuronal death in in vitro 24-26 and in vivo models of Parkinson disease.27 We have provided the first evidence for the upregulation of the E2F1 and its target gene CDK1, early after SCI in rat, which was associated with subsequent neuronal apoptosis in injured spinal cord.28 Molecular or pharmacological targeting of E2F1/CDK1 signaling in vitro was neuroprotective. Although a role for E2F1 in brain ischemia has been shown using the E2F1 constitutive knockout (ko) mice, E2F2 also can independently drive apoptosis.29 A recent study demonstrated distinct E2F1- and E2F2-activated cell death pathways in response to a single tumorigenic insult,29 revealing the distinct properties for these 2 transcription factors. Thus, it is likely that greater protection may occur in the absence of both genes.
The goal of this study was to determine whether genetic targeting E2F1-2 could be an effective strategy for functional repair after SCI. We first examined the temporal distribution and cell-type specificity of E2F1-2 following contusive SCI in a mouse model. E2F1-2 double knockout and wild-type mice were compared to assess the role of activators E2F1-2 and CCA in neuronal cell death, neuroinflammation and associated neurological dysfunction after SCI.
Results
The transcription factors E2F1 and E2F2 are induced after SCI. To examine the induction of E2F1-2 transcription factors after SCI, we assessed levels of E2F1-2 mRNAs and proteins in the injured spinal cord by qPCR and western blot. Levels of E2F1 mRNA significantly increased at as early as 6 hours and further elevated at 1 day after SCI (Fig. 1A). E2F2 mRNA significantly increased at 1 day post-injury (Fig. 1B). Quantitative analysis of western blot showed that expression of both E2F1 and E2F2 protein was significantly increased at 24 hours post SCI (Fig. 1C-E). Immunohistochemistry revealed that extensive E2F1-2 expression was found in neurons (Fig. 2) and glial cells (Fig. 3). Most of E2F1+ cells in the gray matter with neuronal morphology were also co-labeled with E2F2, (Fig. 2C). Caspase-3, the major executioner caspase, is cleaved to an active form during apoptosis. Immunofluorescence double labeling further confirmed that in the ventral gray matter at day 1, cleaved caspase 3-positive cells were also positive for E2F1 or E2F2 (Fig. 2D-E), indicating that E2F1-2 expression correlated with induction of apoptosis. Double-labeling immunohistochemistry revealed that large numbers of E2F1+ or E2F2+ cells in the injured coronal sections at 7 days post-injury were co-labeled with microglia marker Iba-1 at 1 mm rostral to the epicenter (Fig. 3A-D). High magnification images (Fig. 3B and D) showed activated microglial forms displaying cellular hypertrophic or bushy morphologies. GFAP is an indicator of astrocyte reactivity associated with glial scar formation. We observed increased expression of E2F1-2 and GFAP (Fig. 3E and G), and their co-localization (Fig. 3F and H) in the injured tissue at 7 days after SCI. Almost all E2F1+/ED1+ microglia/macrophages also expressed E2F2 (Fig. 3I).
Figure 1.
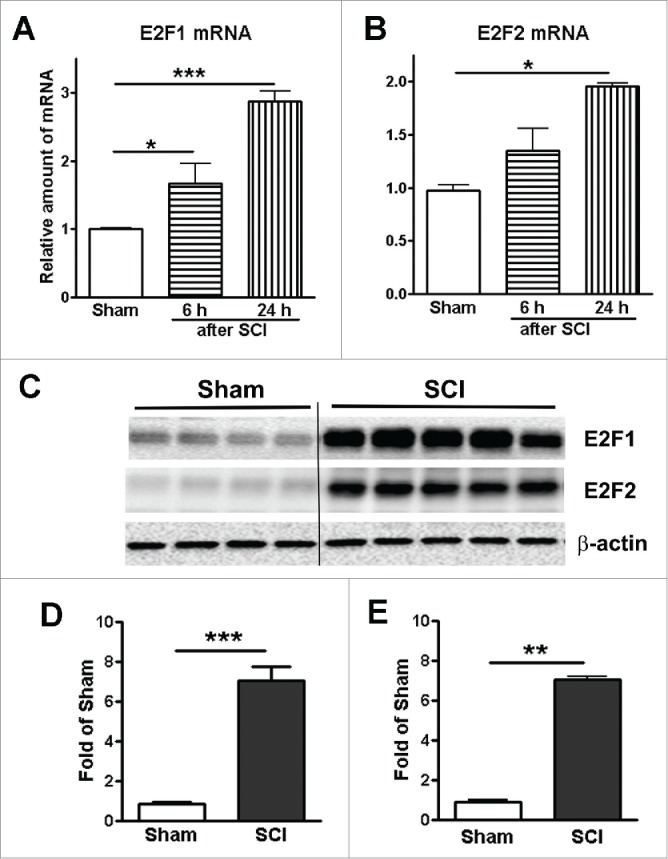
Spinal cord injury induces up-regulation of E2F1 and E2F2 transcription factors. (A-B) Expression of E2F1 and E2F2 mRNA in the mouse injured spinal cord was elevated at 6 and 24 hours after SCI. n=3-4 mice per time point. (C) Representative immunoblots for E2F1, E2F2, and the loading control (β-actin). (D-E) Expression levels of E2F1 and E2F2 protein were normalized by GAPDH, as estimated by optical density measurements, and expressed as a fold of sham spinal cord. Quantification of western blot showed significantly increased E2F1 and E2F2 expression at 24 hours post-injury. n=4-5 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham group.
Figure 2.
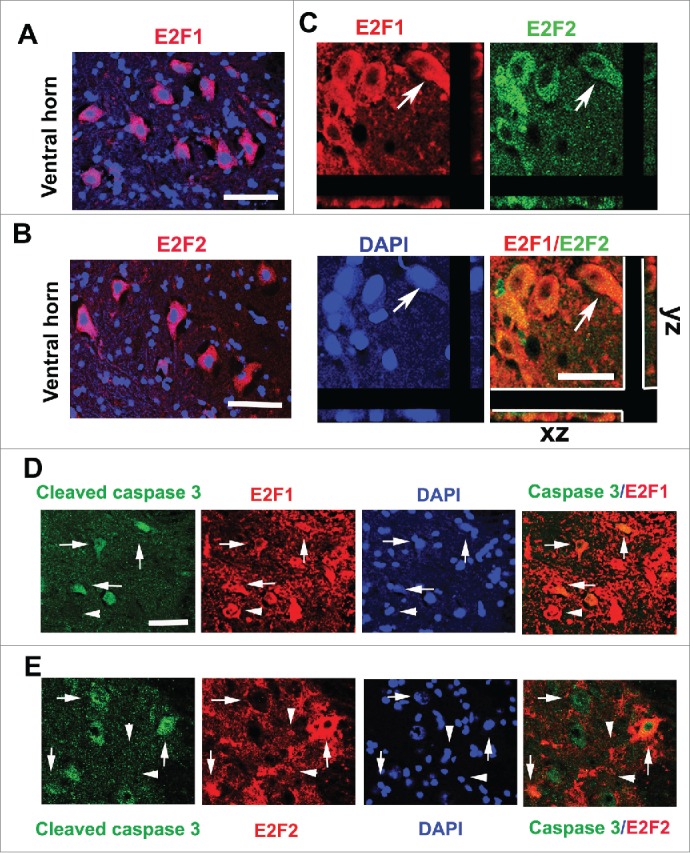
Upregulated E2F1 and E2F2 are associated with cleaved caspase-3+ neurons at 24 hours after SCI. (A-B) E2F1 and E2F2 are induced in neurons of the ventral horn (VH) gray matter at day 1 after SCI. (C) Most of E2F1+ cells (red) in the gray matter were also co-labeled with E2F2 (green), which had neuronal morphology. Arrows indicated E2F1+/E2F2+ cells with z-stacks (x-z axis, y-z axis). (D-E) Co-localization between cleaved caspase-3 (green) and E2F1 (red) or E2F2 (red) is apparent in the VH at day 1 after SCI. Arrows indicated caspase-3+/E2F1-2+; Arrowheads indicated caspase-3−/E2F1-2+. Scale bar is 100 μm.
Figure 3.
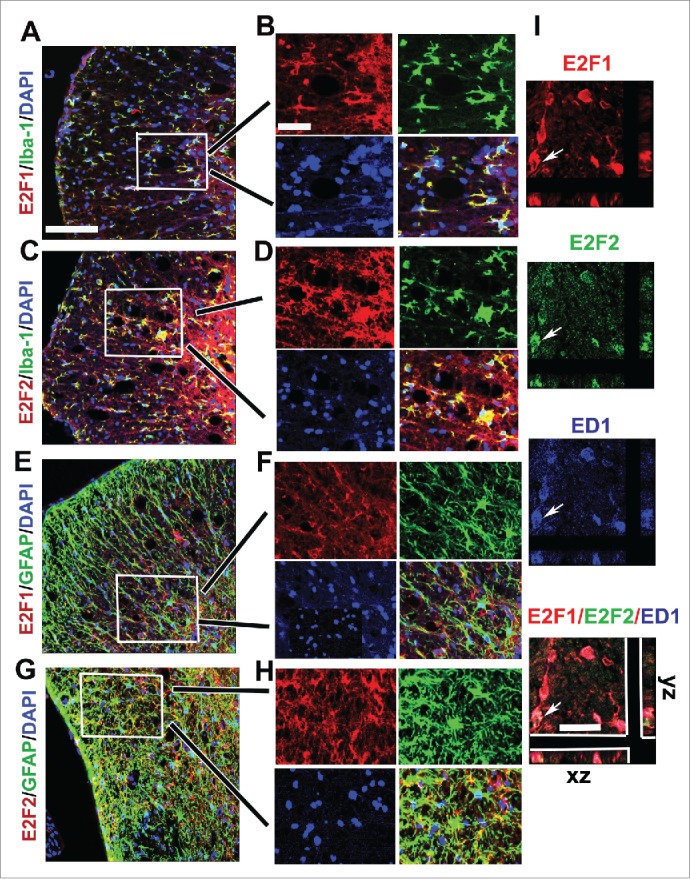
Increased E2F1 and E2F2 are associated with activated microglial and astrocytes at 7 days after SCI. (A-D) E2F1 (red in A) and E2F2 (red in C) are co-labeled with Iba-1+ microglia (green) in the ventral media whiter matter at 1 mm rostral to the epicenter. High magnifications in B,D (from insert in A,C) showed their highly co-localization. (E-H) A subtype of GFAP+ astrocytes (green) expressed E2F1 (red in E) and E2F2 (red in G) in the ventral lateral white matter at 1 mm rostral to the epicenter. F (from insert in E) and H (from insert in G) indicated their strong co-localization. (I) Almost all E2F1+/ED1+ microglia/macrophages also expressed E2F2. Arrows indicated E2F1+(red)/E2F2+(green)/ED1+(blue) cells with z-stacks (x-z axis, y-z axis). Scale bar is 100 μm.
SCI-induced E2F1-2 mediates cell cycle activation. To examine whether induction of E2Fs activity mediates CCA in vivo, we used intrathecal injection of high specificity E2F ODNs against the E2F-binding site. The E2F ODNs are well-described and in the form of Edifoligide have reached clinical testing for certain cardiovascular procedures.30,31 At 24 hours after injection, Western blot analysis demonstrated that E2F ODNs significantly block the up-regulation of E2F1 and E2F2 expression induced in injured tissue (Fig. 4A-B). Moreover, key CCA components, CDK1 and PCNA expression levels were significantly down-regulated by E2F ODNs at 1 day after SCI. In contrast, SCI-induced CCA upregulation was not affected by injection of scrambled ODNs.
Figure 4.
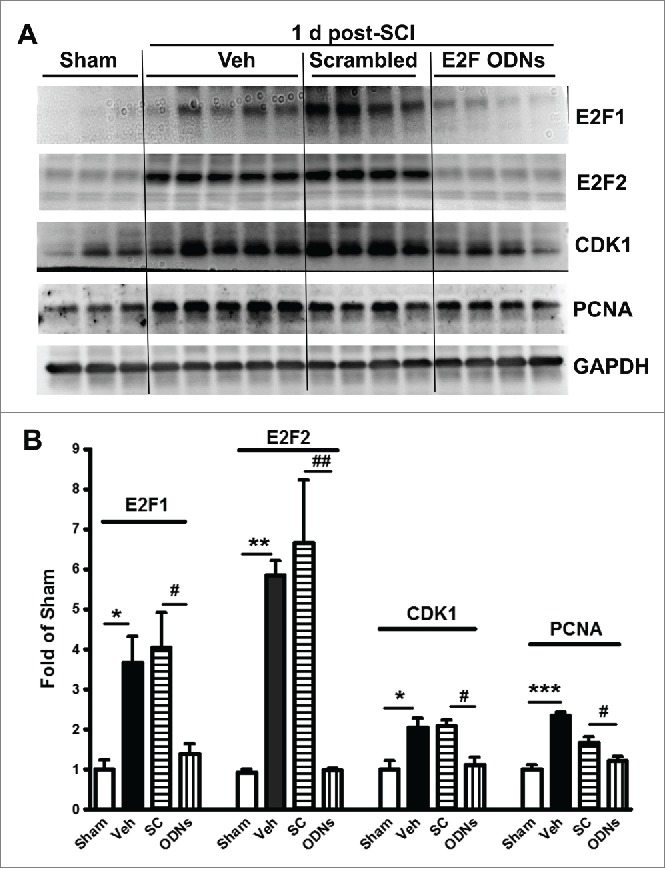
SCI-induced upregulation of cell cycle-relayed proteins at lesion site was significantly attenuated by intrathecal injection of E2Fs decoy oligodeoxynucleotides (ODNs) at day 1 post-injury. (A) Representative western blots of cell cycle-related proteins including E2F1, E2F2, CDK1, and PCNA. (B) Quantification of western blot from A. Veh: Vehicle; SC: scrambled ODN; ODNs: E2Fs ODNs. n=3-5 mice/group. *p <0.05, **p<0.01, ***p< 0.001 vs. sham group; #p<0.05, ##p<0.01 versus SC group.
As E2F ODNs potentially affect all members of E2F family, to further assess whether activation of E2F1-2 is sufficient to mediate CCA, we tested whether genetic deleting E2F2 or E2F1-2 would reduce injury-induced increases in CCA. E2F2ko or E2F1-2dko mice generated from the E2F1+/−E2F2−/−E2F3loxP/loxP breeding pairs develop normally, gain weight at a rate equal to that of WT (C57BL/6) animals.18 Quantitative real-time PCR analysis showed an upregulated expression of multiple cell cycle related genes in the injured spinal cord at 24 h post-injury (Fig. 5). Deletion of E2F2 or E2F1-2 genes significantly reduced the up-regulation of cyclin A1 and A2 expression induced by SCI, whereas elevated expression of other CCA genes including cyclins B2, D1-3, CDK1, and PCNA was significantly downregulated only in the E2F1-2dko mice. Together, these data suggest that E2F1-2 may serve as master regulators of CCA in SCI and greater effects may be achieved in double E2F1-2 KO mice than those in single E2F2 KO mice.
Figure 5.
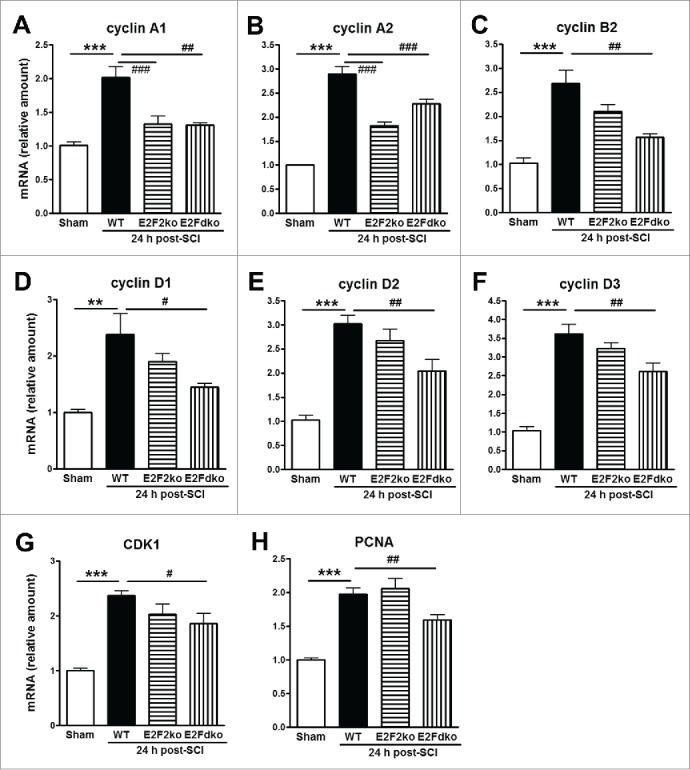
Expression of cell cycle-related genes in the spinal cord from WT, E2F2ko, and E2Fdko mice at 24 hours after SCI. All these CCA genes were up-regulated at 24 h after SCI. Among them, cyclins A1 (A) and A2 (B) were robust down-regulated in injured mice with deletion of E2F2 gene or E2F1+2 genes. Notably, cyclins B2, D1, D2, D3, CDK1, and PCNA (C-H) were significantly reduced in E2F dko mice, but not in the E2F2 ko mice. E2Fdko = E2F1-2 double ko. n=5-8 mice/group. **p <0.01, ***p< 0.001 vs. Sham; #p<0.05, ##p<0.01, ###p<0.001 versus WT group.
E2F1-2 gene silencing reduces SCI-induced neuronal death. To further investigate the neuroprotective role of E2F1-2, we examined whether decreasing E2F1 or E2F2 expression in vivo by intrathecal administration of siRNAs could attenuate neuronal apoptosis. Cleaved and uncleaved fragments of α-fodrin were assessed in the injured spinal cord at 24 h post-injury by Western blotting analysis. 145 or 150 kDa cleavage fragment of the α-fodrin was increased 7- and 13- fold, respectively, whereas uncleaved fragment was decreased 60 %, as compared to sham-injury (Fig. 6). These markers of apoptosis were significantly reversed by E2F1, E2F2, or E2F1+2 siRNA. Notably, E2F1+2 siRNAs show greater effects than E2F1 or E2F2 single siRNA treatment.
Figure 6.
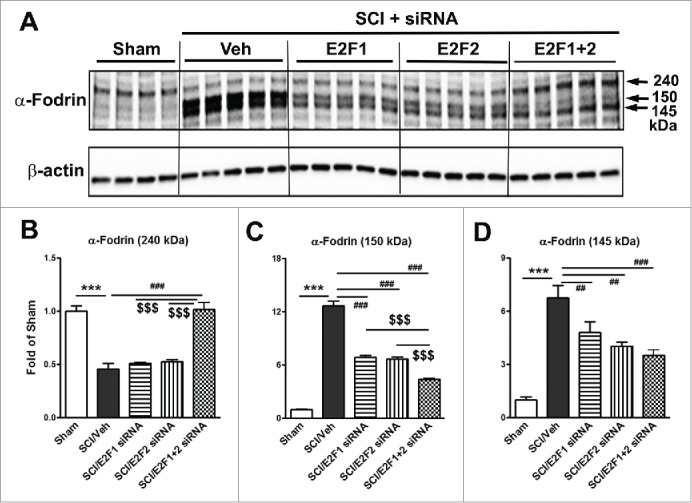
E2F1-2 siRNA reduces neuronal death at 24 h after SCI. E2F1, E2F2, E2F1+2, or scrambled siRNA were intrathecally administrated at 5 min post-injury and cleaved or uncleaved fragments of a-fodrin were assessed in the injured spinal cord at 24 h post-injury by Western blotting analysis. (A) Representative western blots for α-fodrin proteins and the loading control (β-actin). (B-D) Expression levels of α-fodrin (240, 150, 145 kDa) were normalized by β-actin, as estimated by optical density measurements and expressed as a fold of sham spinal cord. Quantitative analysis of western blots showed that 145 or 150 kDa cleavage fragment of the a-fodrin was increased 7- and 13- fold, respectively, whereas uncleaved fragment was decreased 60 %, as compared to sham-injury. These markers of apoptosis were significantly reversed by E2F1, E2F2, or E2F1+2 siRNA. Notably, E2F1+2 siRNA has greater effect than E2F1 or E2F2 single siRNA. n= 4-5 mice/group. ***p <0.001 vs. sham; ##p<0.01, ###p<0.001 versus SCI/Veh; $$$p<0.001 vs. E2F1+2 siRNAs.
E2F1-2 deletion improves functional recovery after SCI. We used an array of behavior tests to assess outcomes in E2F1-2dko or E2F2ko mice and their WT littermates after SCI. At day 1 after SCI, all mice had a BMS locomotor score of 0 or 1, indicating nearly complete loss of motor function (Fig. 7A). By 2 weeks after injury, E2F1-2dko mice had significantly improved BMS scores compared with WT animals which remained through 6 weeks after injury. The errors of step in the grid walk test were significantly reduced in the E2F1-2dko mice at 5 weeks post-SCI as compared to their WT littermates (Fig. 7B). Moreover, the E2F1-2dko mice had enhanced stride length of the hindpaws measured from footprints 5 weeks after injury (Fig. 7C). Notably, the E2F2ko mice did not show significant improvement in all these tests. Together, deletion of E2F1-2 genes significantly improves recovery of locomotor function following SCI.
Figure 7.
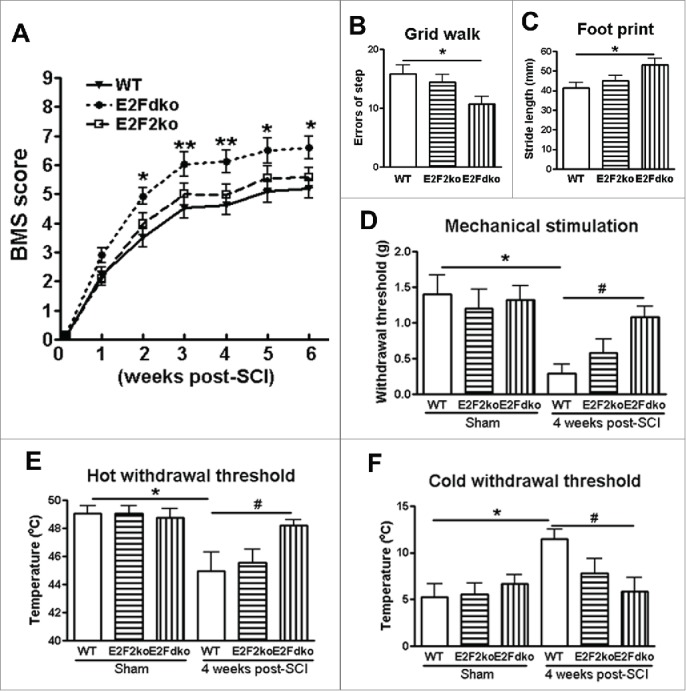
E2F1-2 ablation improves motor functional recovery and limits hyperesthesia after SCI. (A) Basso Mouse Scale (BMS) locomotor scores were significantly improved up to 6 weeks post-SCI in E2Fdko (E2F1-2 double ko) mice as compared to the wild-type (WT) controls. Deletion of E2F2 did not affect BMS scores. n=10 (WT), 8 (E2Fdko), 5 (E2F2ko) mice. *p < 0.05, **p < 0.01, versus WT group. (B) E2Fdko mice significantly decreased grid walk errors at 4 weeks after SCI. n=6 (WT), 6 (E2Fdko), 5 (E2F2ko) mice. *p < 0.05 vs. WT group. (C) E2Fdko mice increased stride length of the hindlimbs at weeks 4 post-injury. n=6 (WT), 6 (E2Fdko), 5 (E2F2ko) mice. *p < 0.05 versus WT mice. (D) Von Frey mechanical test. No significant difference in withdrawal threshold was found in sham groups between the genotypes. On day 28 after SCI, the E2Fdko mice had a higher mechanical threshold than the WT mice. n=5 mice/group. *p < 0.05 vs. sham/WT mice; #p < 0.05 versus SCI/WT mice. (E-F) Thermal test. SCI caused increased thermal sensitivity in WT mice, whereas E2Fdko mice had similar thermal withdrawal threshold compared to sham uninjured mice. n=5 mice/group. *p < 0.05 vs. sham/WT mice; #p < 0.05 versus SCI/WT mice.
No significant difference in withdrawal threshold was found between the genotypes before the SCI (Fig. 7D-F). However, SCI in WT mice caused a significant decrease in mechanical threshold compared to the sham WT animals. On day 28 after SCI, the E2F1-2d ko mice had a higher mechanical threshold than the WT mice (Fig. 7D), indicating less mechanical hyperesthesia. SCI in WT mice caused increased thermal sensitivity, whereas E2F1-2dko mice subjected to SCI had similar thermal withdrawal threshold compared to uninjured mice (Fig. 7E-F), suggesting less thermal hyperesthesia. No significant difference in these tests was observed between the E2F2ko and WT mice following SCI.
Together our data indicate greater functional effects in E2F1-2 double than in E2F2 single KO mice
Genetic ablation of E2F1-2 reduces tissue damage after SCI. To determine if the observed behavioral improvement may relate to increased residual white mater, spinal cord sections from injured mice perfused at 6 weeks were stained with erichromecyanine for myelinated WM area. Histological analysis revealed that there was a significant increase of the area of white matter sparing in the E2F1-2dko mice compared to their WT littermates (Fig. 8A). Representative eriochrome stained sections at the epicenter of each subject illustrate the differences in myelinated WM area between WT and the E2F1-2dko animals (Fig. 8B). SCI-induced lesion volume/cavity formation was measured with GFAP/DAB staining at 6 weeks after SCI. The average lesion volume assessed for WT group was 53.7 ± 4.1 %, and deletion of E2F1 and E2F2 significantly reduced lesion cavity with an average lesion volume of 38.9 ± 3.6 % (Fig. 8C). This reduction occurred in both white and gray matter, with an overall decrease in cavity formation and tissue loss (Fig. 8D). Notably, the E2F2 single ko mice did not show significant improvement in tissue damage (data not shown).
Figure 8.
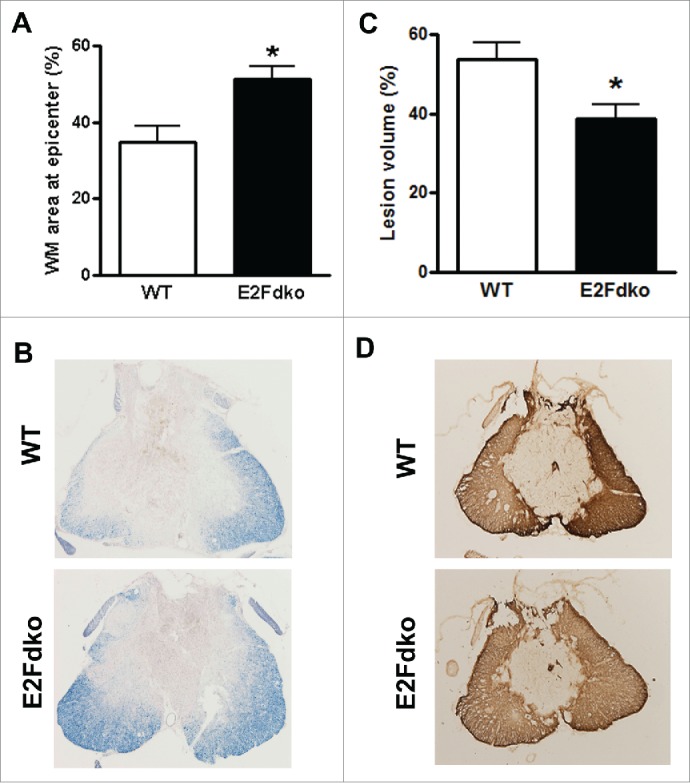
E2F1-2 ablation protects against tissue damage induced by SCI. (A-B) Representative sections show the lesion epicenter stained with eriochrome cyanine (ECRC) for residual white matter (WM) area. Bar graph shows that E2Fdko (E2F1-2 double ko) mice appeared increased spared tissue in whiter matter at epicenter. WM area is expressed as a percentage of ECRC-positive area in total area of each section. (C-D) Assessment of lesion volume at 6 weeks post-injury was performed on GFAP/DAB stained coronal sections. Representative images showed lesion cavity at the injury center. Quantification analysis showed significantly reduced lesion volume in E2Fdko mice. Lesion volume is expressed as a percentage of GFAP absent area in total area of each section. n=6 mice/group. *p < 0.05 vs. WT group.
Deleting E2F1-2 genes confers neuroprotection in the injured spinal cord. To test whether E2F1-2 ablation resulted in neuroprotection against cell death, particularly apoptosis, we examined the level of cleaved (activated) caspase-3, the member of cysteine-aspartic proteases that play essential roles in apoptosis, necrosis, and inflammation.32 Caspase-3 is expressed following SCI and to be critically involved in the execution of the mammalian apoptotic cell death program. At day 1 after SCI, cleaved caspasae-3+ cells were found in both gray matter (GM) and white matter (WM, Fig. 9A). Quantitative image analysis of cleaved caspase-3+ cells was performed in 9 anatomical regions including GM (VH, DH, intermediate gray matter) and WM (ventral medial or lateral white matter). Notably, the total numbers of cleaved caspase-3+ cells in the E2F1-2dko mice were greatly reduced than it in the WT animals at 1 mm rostral to the epicenter (Fig. 9B). Immunohistochemistry showed that cleaved caspasae-3+ cells present in the WM are double immunostained for CC1+ oligodendrocytes (Fig. 9C). In the GM, most of caspasae-3+ cells are also NeuN+ (Fig. 9D). Western blot analysis further revealed that SCI induced a significant increase of cleaved caspase-3 expression in the WT littermates, which was significantly reduced in the E2F1-2dko mice (Fig. 9E-F). Furthermore, we found that at 24 hours after SCI, there was a marked loss of ventral horn neurons at and near the site of injury in the WT littermates (Fig. 9G). In contrast, in the E2F1-2dko mice, the SCI-induced neuronal loss was significantly reduced at 24 hours post-injury.
Figure 9.
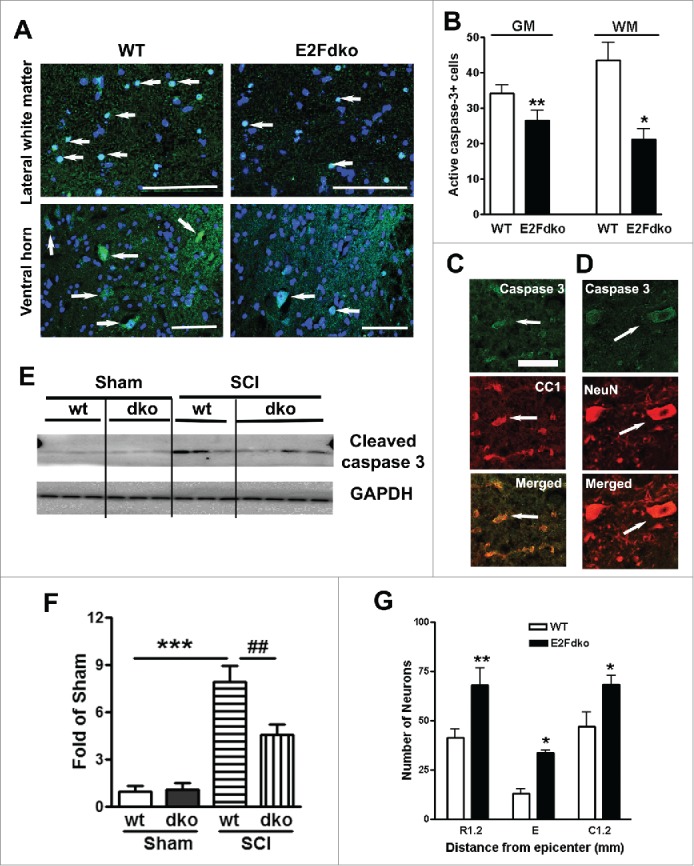
E2F1-2 ablation reduces cell death and increases neuronal survival at 24 hours after SCI. (A) Representative images of IHC staining for cleaved caspase-3 in lateral whiter matter and ventral horn of the gray matter from WT and E2Fdko (E2F1-2 double ko) mice. The images indicated 1 mm rostral to epicenter at 24 hours post-injury. Arrows show caspase-3+ cells (green) in nuclei (blue). (B) Quantification of cleaved caspase-3+ cells in both of gray matter (GM) and whiter matter (WM) at 1 mm rostral to epicenter. Deleting E2F1-2 genes in E2Fdko mice decreases numbers of caspase-3+ cells at day 1 after SCI compared with WT mice (arrows). n=5 mice/group. *p <0.05, **p < 0.01 versus WT group. (C) IHC showed that cleaved caspasae-3+ cells (green) presented in the WM are double immunostained for CC1+ oligodendrocytes (red, arrows). (D) In the GM, most of caspasae-3+ cells (green) are also NeuN+ (red, arrows). (E) Representative immunoblots for cleaved caspase-3 and the loading control (GAPDH). (F) Quantification of western blot showed that E2F1-2 ablation (dko mice) significantly reduced cleaved caspase-3 expression levels induced by SCI. n=3-4 mice/group. ***p < 0.001 vs. sham/WT mice; ##p < 0.01 versus SCI/WT mice. (G) Quantification of NeuN+ cells showed that significantly more neurons survived in the ventral gray matter of the E2Fdko mice at epicenter as well as 1.2 mm rostral or caudal to it compared to their WT littermates; n=5 (WT), 6 (E2Fdko) mice. *p < 0.05, ***p < 0.001 vs. WT. Scale bar is 100 μm.
E2F1-2dko mice have reduced microglial/astrocyte activation in the injured spinal cord. Western blotting was performed for the activated microglia marker Iba-1 after SCI. Very low levels of Iba-1 expression were found in uninjured controls from both the WT mice and the E2F1-2dko mice (Fig. 10A), but 1 day after SCI, Iba-1 protein expression showed a 2.5-fold increase in injured spinal cord extracts from the WT mice compared with sham tissue (Fig. 9B). Spinal cords from the E2F1-2dko mice had significantly less upregulation of Iba-1 protein expression compared with WT mice. Further, Iba-1+ cells in the white matter area were quantified at 1 mm rostral to the epicenter after 7 days post-injury (Fig. 10B). No significant difference was found between the genotypes in sham groups (Fig. 10C). SCI resulted in a significant increase in Iba-1+ microglia in the WT mice (Fig. 10D). Notably, the E2F1-2dko mice had significantly less number of Iba-1+ cells compared with WT mice. Immunohistochemistry at 7 days post-injury showed an increase in immunolabeling of GFAP in contrast to sham tissue (Fig. 11). Quantitative image analysis showed a significant reduction of GFAP+ astrocytes in the white matter from the E2F1-2dko mice compared with WT mice (Fig. 11A). In contrast, no significant difference was found between the genotypes in sham groups. Fig. 11B showed representative images with GFAP staining at 1 mm rostral to the epicenter in WT and E2Fdko mice.
Figure 10.
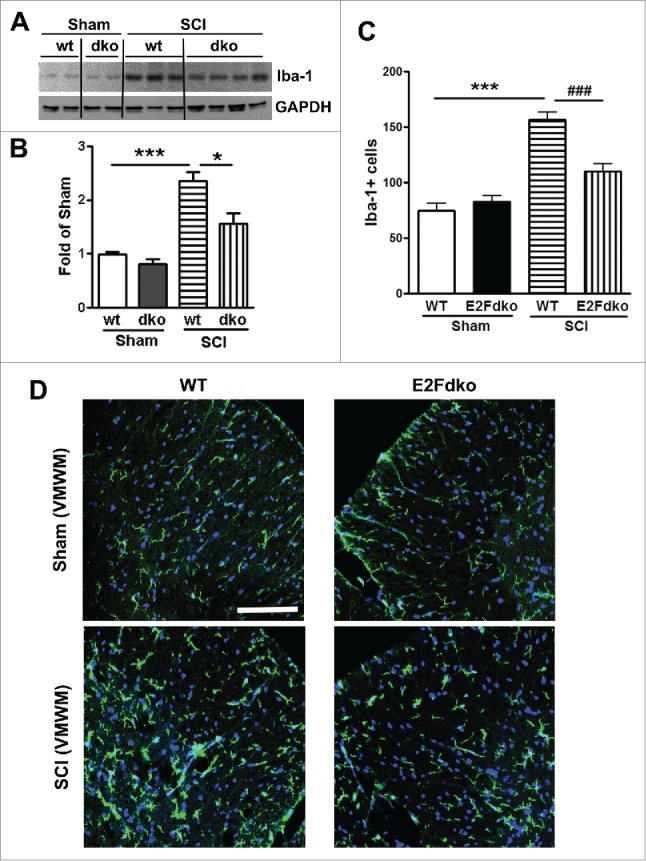
E2F1-2dko mice have reduced microglial activation in the injured spinal cord. (A) Representative immunoblots for Iba-1 and the loading control (GAPDH) at day 1 after SCI. (B) Quantification of western blot from A showed that the E2Fdko mice had significantly less upregulation of Iba-1 protein expression compared with WT mice. (C) Iba-1+ cells in the white matter area were quantified at 1 mm rostral to the epicenter after 7 days post-injury. No significant difference was found between the genotypes in sham groups. The E2Fdko mice had significantly less number of Iba-1+ cells compared with WT mice. n=5 mice/group. ***p < 0.001 versus sham/WT, ###p < 0.001 vs. SCI/WT. (D) Representative images with Iba-1 staining in WT and E2Fdko mice at 7 days post-injury. Scale bar is 100 μm.
Figure 11.
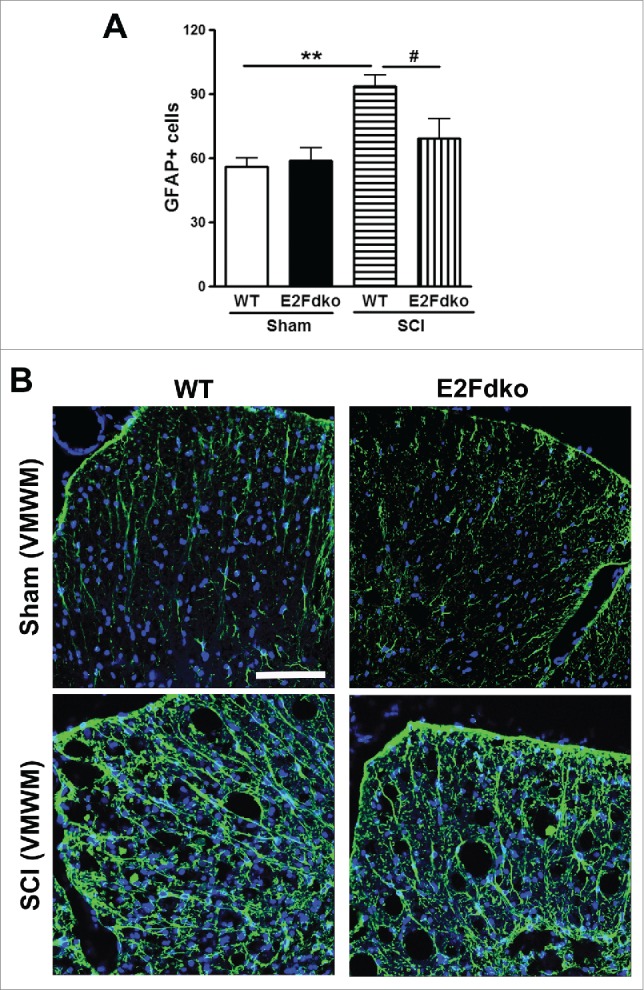
Genetic deletion of E2F1-2 reduces astrogliosis at 7 days after SCI. (A) Quantitative image analysis at 0.5mm rostral to epicenter showed a significant reduction of GFAP+ astrocytes in the white matter from the E2F1-2dko mice compared with WT mice. In contrast, no significant difference was found between the genotypes in sham groups. n=5 mice/group. ***p < 0.001 versus sham/WT, ###p < 0.001 vs. SCI/WT. (B) Representative images with GFAP staining in WT and E2Fdko mice at 7 days post-injury. Scale bar is 100 μm.
Discussion
Our results show that SCI induces elevation of E2F1-2 expression as early as 6 h post-injury, which was localized in active caspase 3+ neurons/oligodendrocytes as well as activated microglia or astrocytes. Blocking E2F1-2 activation by using E2F ODNs or E2Fdko limits SCI-induced CCA. E2F1-2 gene silencing in vivo using double siRNAs demonstrated greater neuroprotection than E2F1 or E2F2 single siRNA treatment. Most importantly, genetic deletion of E2F1-2 significantly reduced neuronal cell death, inflammation, and tissue damage, as well as improved behavioral recovery after SCI. CCA reduction and functional outcome in E2Fdko mice are greater than those in E2F2ko model.
Our prior studies established in CNS trauma that CCA mediates both neuronal injury and neuroinflammatory mechanisms, which may interact in a dynamic and self-propagating manner.28,33-40 It also contributes to chronic functional impairments including motor deficits and neuropathic pain. Previously, we used less specific pharmacological approaches to establish the role of cell cycle pathways in SCI.7,28,33,40 Strong evidence suggests that E2F activators, including E2F1-3, play an important role in proliferating cells as well as in cell cycle related neuronal death of post-mitotic neurons,18,41-43 but no comprehensive study has been performed in SCI to determine the relative importance of these factors. Here we more specifically address the role of E2F1-2 in the pathophysiology following SCI.
E2F1 has long been linked directly to apoptosis in vitro.24-26,44 During development, increased E2F1 causes neuronal apoptosis.45,46 In adult CNS, E2F1 mRNA or protein expression increased in the brain after global or focal ischemia,47,48 traumatic brain injury,38 and in brain from patients with Down syndrome.49 We have reported the up-regulation of the E2F1 protein expression in the injured spinal cord tissue in rat.28 In the present study, we observed elevation of E2F1 mRNA in the contusive mouse spinal cord as early as 6 hours post-injury and further increase at day 1 after SCI. The activated E2F1 was found localized in neurons that are also positive for cleaved caspase-3, resulting in neuronal loss. Genetic deletion of E2F1 limits neuronal damage after mild focal ischemia.50 It is thought that E2F2 only induces cell death indirectly by upregulating E2F1 expression.51 Recent in vivo analysis of cone photoreceptors provides unequivocal evidence that E2F2 can induce neuronal apoptosis independent of E2F1 or E2F3.29 Here we demonstrate increased expression of E2F2 at 24 hours after SCI, co-localizing with elevated cleaved caspase-3+ neurons. Moreover, deleting both of E2F1 and E2F2 genes in the E2Fdko mice caused reduction of cleaved caspase-3+ cells resulting in increased neuronal survival in the ventral horn. Thus, our data suggest that increased E2F1-2 induced, at least in part, caspase-dependent neuronal apoptosis after SCI.
After SCI, microglia and astrocytes proliferate and activate, resulting in astrogliosis and inflammation that contribute to delayed secondary tissue damage. In proliferating cells, E2F activators are required for cellular proliferation. However, the role of E2F1-2 in glial reactivation following SCI has not been reported. We previously demonstrated 28 that at 24 hours after SCI, E2F1 is expressed by a subset of OX42+ microglia/macrophages and the majority of E2F1+ cells in the lesion area likely are infiltrating circulating cells. Here we detected that at 7 days post-injury, both E2F1 and E2F2 are highly expressed by Iba-1+ microglia/macrophages displaying cellular hypertrophic or bushy morphology. In addition, a sub-type of GFAP+ astrocytes also expressed E2F1-2. Furthermore, deleting E2F1-2 reduced total numbers of Iba-1+ or GFAP+ cells. Therefore, our data suggest that SCI-induced upregulation of E2F1-2 transcription factors may contribute to the development of glial scar formation and chronic inflammation. However, glial activation after SCI may have both beneficial and detrimental effects. Controversies regarding the role of glial modulation after SCI reflect failure to consider effects of concurrent astroglial and microglial activation after injury nor to effectively distinguish types of inflammatory response. Recent evidence shows the fibrotic scar formation at the injury site in both mice and rats after contusive SCI.52 These fibroblasts derived from both meningeal cells and the vasculature. We found co-localization of E2F1-2 and fibronectin+ fibroblasts (data not shown), suggesting an involvement of E2F1-2 in other proliferating cells after SCI. Further investigation is required to evaluate the effects of E2F activators on peripheral cellular immune responses as well as fibrotic scar formation to SCI.
E2F activators function at the G1/S transition to regulate entry into S phase and thereby modulate cell-cycle entry or withdrawal 14,41; they also appear critical for cell-cycle-dependent gene expression.14,18 Our data demonstrates that blocking E2F by E2F ODNs in vivo not only inhibits SCI-induced up-regulation of E2F1 and E2F2, but also attenuates CDK1, and PCNA expression at lesion site. Furthermore, genetic deletion of both E2F1 and E2F2 genes reduce the upregulation of cyclins A1-2, B1-2, D1-3, CDK1, and PCNA expression induced by SCI. Thus, our data suggests that E2F activators may serve as master regulators of CCA in SCI. It has been reported 53 that gene expression of E2F1 in the pancreas remains unchanged in the E2F2 ko mice; E2F5 and E2F6 were similarly expressed in the WT and E2F1-2 dko in this model. Cyclin E gene, as well as endogenous inhibitor p21Cip1, was elevated in embryonic fibroblasts cells derived from E2F1-2 dko mice, whereas p53 or p27 remained unchanged.18 However, these changes have yet to be characterized in the central nervous system.
Inhibition of CCA after SCI improves locomotor functional recovery.33,39,40,54,55 More recent data 7,8 suggests that CCA may also play a key role in the pathophysiological mechanisms underlying the development of maladaptive sensory plasticity and central sensitization after SCI. MacManus et al reported that mice lacking the E2F1 transcription factor showed reduced infarct volume and improved motor function after focal ischemia.50,56 A significant finding of present study is that genetic deletion of E2F1-2 resulted in neuroprotection and improved motor recovery following SCI. Moreover, inhibition of CCA by genetic deletion of both E2F1 and E2F2 genes limits SCI-induced hyperesthesia. Therefore, our data indicates that E2F activators are important contributing initiators of apoptotic neuronal cell death and microglial/astrocytes activation, with attenuation of E2F1-2 activity in neurons and microglia/astrocytes showing complementary neuroprotective effects.
Recent study demonstrates distinct E2F1- and E2F2-activated cell death pathways in response to a single tumorigenic insult,29 revealing a differentially biochemical properties between these 2 transcription factors. We found greater reduction of mRNA of PCNA, CDK1 and cyclin D1 in double E2F1-2 ko mice than those in single E2F2 ko mice. Although we did not observe significant differences in locomotor function or hindlimb nocifensive response to mechanical or thermal stimuli between E2F2ko and E2Fdko mice before injury, E2Fdko mice showed improved functional outcome after SCI compared with WT mice, whereas deleting single E2F2 gene did not result in functional improvement. Therefore, our data suggest that greater protection may be achieved following double E2F1-2 ko. E2F3 appears also to be largely responsible for post-mitotic neuronal death following Rb.14 The E2F3 gene expresses 2 alternative proteins, E2F3a and E2F3b. Consistent with shared structure and function, E2F1 or E2F3b can substitute for E2F3a in vivo.57 Indeed, E2F3a-induced apoptosis, either in vitro or in the pituitary gland, requires E2F1 up-regulation. Therefore, E2F3a may induce factors that cooperate with E2F1 to drive p53-independent death. In proliferating cells, mouse embryonic fibroblasts deficient for E2F1 and E2F2 proliferated robustly. However, loss of 3 E2F activators abolished any measurable proliferation, indicating a critical role of E2F3 in the control of cell cycle. These findings indicate that E2F1 and E2F2 cooperate with E2F3 to sustain normal proliferation. Our unpublished data show that E2F3 is co-localized with active caspase-3 in injured spinal cord at 1 day after injury, implicating this protein in post-traumatic cell death. Moreover, E2F3 is predominantly expressed by activated GFAP+ astrocytes and a sub set of CD11b+ microglia/macrophages at day 3 post-injury (data not shown), whereas in sham-mice the E2F3/GFAP signal is weak. Given potential compensatory mechanisms, it is likely that greater protection may be obtained using a triple E2F1,2,3 knockout.
In summary, we provide evidence that SCI induces an elevation of E2F1-2 transcription factors expression that co-localize with apoptotic neurons/oligodendrocytes and activated microglia/astrocytes. Blockade of E2F1-2 by E2F ODNs or genetic deletion of E2F1-2 significantly reduces CCA after SCI, suggesting that E2F1-2 activators may serve as key up-stream regulatory molecules. Administration of E2F1+2 siRNAs shows greater neuroprotective effects than treatment with E2F1 or E2F2 siRNA alone. Deleting E2F1-2 reduces neuronal cell death, limits inflammation and astrogliosis, and subsequent tissue damage, with marked improvement in functional recovery. Thus, E2F1-2 may provide a novel target for neuroprotection following SCI.
Materials and Methods
Animals. Male C57BL/6 mice (12 weeks, 20-25 g) were purchased from Taconic (Rensselaer, NY). Breeding pairs of male and female transgenic E2F1+/−E2F2−/−E2F3loxP/loxP mice 18 were received from Dr. Gustavo Leone (The Ohio State University). The breeding was carried out at the University of Maryland School of Medicine Laboratory Animal Resource Center. E2F1−/−E2F2−/− (E2F1-2 double ko = E2F1-2dko) or E2F2−/− (E2F2 single ko = E2F2ko) mice generated from the E2F1+/−E2F2−/−E2F3loxP/loxP breeding pairs were used in this study. The genotypes of the yielded litters were determined by polymerase chain reaction (PCR). The animals were maintained on a 12/12 hour light/dark cycle with food and water freely available. All procedures were performed under protocols approved by the University of Maryland School of Medicine Animal Care and Use Committee.
Contusive spinal cord injury. Adult male mice were anesthetized with isoflurane and received spinal contusions at the T10 level using the Infinite Horizon Spinal Cord Impactor (Precision Systems and Instrumentation) with a force of 60 kdyn, a moderate injury.8,10 Manual bladder expression was carried out at least 3 times daily until reflex bladder emptying was established. Control animals received laminectomy only. After SCI, mice were assigned to a treatment group according to a randomized block experimental design. The number of mice at various time points in each study is indicated in the figure legends. All SCI animals were assessed at 6 h, 1 d, 7 d, and 6 weeks after injury by the same surgeon and at the same period of time.
Intrathecal injection of E2F ODNs. Highly specificity phosphorothioate double-stranded E2F decoy oligodeoxynucleotides (ODNs) against the E2F-binding site was purchased from Integrated DNA Technologies and reconstituted in saline. Five minutes after SCI, the bilateral microinjections (2 injections/side, 1μl/injection, total 4μl) of the E2F decoy or scrambled ODNs (final concentration 0.5 nmol/mouse) were made intrathecally at 1 mm from the midline. There was a 2mm distance between the 2 injections on each side. The dose of the E2F ODNs was based on the results obtained from pilot studies in vitro and in vivo.
Intrathecal injection of E2F siRNA. Accell mouse E2F1 (13555) siRNA (SMARTpool, cat# E-044993-00-0010), E2F2 (242705) siRNA (SMARTpool, cat# E-057391-00-0010), and scrambled siRNA (non-targeting Pool, cat# D-001910-10-05) were purchased from GE Healthcare Dharmacon Inc.. and diluted in artificial spinal cord fluid (ASCF) to final concentration 0.1 nmol/ul. Five minutes after SCI, the bilateral microinjections (2 injections/side, 1μl/injection, total 4μl) of the E2F siRNA or scrambled siRNA (final concentration 0.5 nmol/mouse) were made intrathecally at 1 mm from the midline. There was a 2mm distance between the 2 injections on each side. The dose of the E2F siRNA was based on the results obtained from pilot studies in vitro and in vivo.
Quantitative Polymerase Chain Reaction (qPCR) analysis. Mouse spinal cord tissue (3-millimeter segments) centered at the injury site was obtained at indicated times after injury. Total RNA was isolated by using a miRNeasy isolation kit (QIAGEN) with on-column DNase treatment (QIAGEN). The Verso cDNA Kit (Thermo Scientific) was used to synthesize cDNA from purified total RNA. Quantitative real-time PCR amplification was performed by using cDNA TaqMan® Universal Master Mix II (Applied Biosystems). In brief, reactions were performed in duplicate containing 2 × TaqMan Universal Master Mix II, 1 μl of cDNA (corresponding to 50 ng RNA/reaction), and TaqMan gene expression assay (Applied Biosystems), 20 × in a final volume of 20 μl. Gene expression assays for the following genes were used: E2F1 (Mm00432936_m1), E2F2 (Mm00624964_m1), cyclin A1 (Mm00432337_m1), cyclin A2 (Mm00438063_m1), cyclin B2 (Mm01171453_m1), cyclin D1 (Mm00432359_m1), cyclin D2 (Mm00438070_m1), cyclin D3 (Mm01612362_m1), PCNA (Mm00448100_g1), CDK1 (Mm00772472_m1). Gene expression was normalized to GAPDH (Mm99999915_g1) and the relative quantity of mRNAs was calculated based on the comparative Ct method.58
Western blots. Mouse spinal cord tissue (3-millimeter segments) centered on the injury site was obtained at indicated times after injury. Western blot analysis was performed as described previously.8 Primary antibodies included: mouse anti-E2F1 (1:500; BD PharMingen); rabbit anti-E2F2 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, UAS); rabbit anti-active caspase-3 (1:200; Cell signaling); rabbit anti-Ionized calcium binding adaptor molecule 1 (Iba-1, 1:1000; Wako Chemicals), and mouse anti-Glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:2000; Millipore). Immune complexes were detected with the appropriate HRP conjugated secondary antibodies (KPL, Inc.., Gaithersburg, MD) and visualized using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL). Chemiluminescence was captured on a Kodak Image Station 4000R station (Carestream Health Inc.., Rochester, NY) and protein bands were quantified by densitometric analysis using Carestream Molecular Imaging Software (Carestream Health Inc.., Rochester, NY). The data presented reflects the intensity of target protein band compared to control and normalized based on the intensity of the endogenous control for each sample (expressed in fold of sham).
Immunohistochemistry and quantification. Following animal perfusion with 4% paraformaldehyde, spinal cord segments containing the lesion area were dissected out, embedded, and cut into 20 μm-thick serial sections. Immunofluorescence staining was followed procedures described previously.8 The following primary antibodies were used: mouse anti-E2F1 (1:500; BD PharMingen); rabbit anti-E2F1 (1:500; Santa Cruz Biotechnology); rabbit anti-E2F2 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, UAS); mouse anti-NeuN (RBFOX3, 1:500, Millipore), rabbit anti-cleaved caspase-3 (1:200; Cell signaling); rabbit anti-Iba-1 (1:1000, Wako Chemicals); rat anti-CD68 (ED1, 1:500; AbDerotec, Raleigh, NC, USA). rabbit anti-glial fibrillary acid protein (GFAP, 1:500, Millipore); and mouse anti-GFAP (1:500, Sigma). Images were acquired using a Leica TCS SP5 II Tunable Spectral Confocal microscope system (Leica Microsystems Inc.). The images were processed using Adobe Photoshop 7.0 software (Adobe Systems). All immunohistological staining experiments were performed with appropriate positive control tissue as well as primary/secondary-only negative controls.
For quantitative image analysis, digital images at 40× magnification were captured from the regions of interest. The number of NeuN labeled cells in the ventral horn (VH) at the epicenter and 1.2 mm rostral or caudal to the center was determined for each animal by a total # of both the left and right sides VH. Active caspase-3+ cells were acquired at 1 mm rostral to the epicenter with total # from GM (both sides of ventral and dorsal horns, intermediate gray matter) and WM (ventral medial white matter and lateral white matter). Iba-1+ or GFAP+ cells were acquired in both sides of ventral medial white matter and lateral white matter as well as dorsal column at 1 mm rostral to the epicenter. All images were captured from n=5–6 mice per group.
Behavioral assessments. All behavioral tests were blindly performed. The Basso Mouse Scale (BMS) locomotor test was performed on day 1 after injury and weekly thereafter for up to 6 weeks according to a method published previously.59 Briefly, mice were placed in an open field chamber (diameter = 40in) and observed for 4 minutes by 2 trained observers. The scores were on a scale of 0 to 9 (9 = normal locomotion; 0 = complete hind limb paralysis), which is based on hind limb movements made in an open field including hind limb joint movement, weight support, plantar stepping, coordination, paw position, and trunk and tail control.
Horizontal grid walk was adapted from the ladder rung walking task to evaluate hindlimbs stepping, placing, and co-ordination.60,61 The apparatus consisted of 2 plexiglass walls with 1m long to hold 30 rungs (round wooden steps, 2 mm in diameter, 10 cm in length) 1 cm apart. Prior to injury, mice will be trained for 3 days on the apparatus. Each mouse will be allowed to cross the ladder walk 3 consecutive times, resting 25 s in a dark box between each trial. The number of footfalls (errors) will be analyzed before injury and on week 4 post-injury. The number of errors of step will be averaged from 3 trials. If an animal is not able to move the hind limbs, a maximum of 20 errors will be given.
Hindpaws of the animals were dipped in non-toxic black ink and the animals were required to cross a 50 cm enclosed runway, the floor of which is covered in white paper, to a darkened box at the end. Each animal crossed the runway twice per testing session. Stride length was measured by using simple scale in mm.
Hindpaws withdrawal from a mechanical stimulus was used to assess the mechanical hyperesthesia according to a method published previously.8 Briefly, each mouse was placed on a wire mesh platform in individual Plexiglas cubicles and allowed to acclimate for one hour. The von Frey filaments (MyNeuroLab, St. Louis, MO), with incremental stiffness ranging from 0.04 g to 2.0 g, were applied serially to the plantar surface of each hind paw in ascending order of stiffness. Each trial was consisted of 5 perpendicular applications (one application every 5 seconds) of a von Frey filament to the hind paw for 2-3 seconds. A positive response is brisk paw withdrawal (at least 3 times out of 5 applications) in response to the filament. Threshold was defined as the filament with the lowest bending force that elicited at least 3 positive responses of 5 trials.62
Hot and cold plate tests were used to assess the thermal hyperesthesia.63 An incremental hot/cold plate (PE34; IITC Life Sciences, Woodland Hills, CA) with a starting temperature of 30°C and the hot and cold ramps, set at the maximum rate of 10 °C/min, was used to induce the nocifensive behaviors of licking a hind paw and jumping to identify the thresholds for noxious heat and cold, respectively. Briefly, each mouse was allowed to acclimate for 30-60 seconds in a Plexiglas cylinder on the 30°C metal plate prior to the onset of the stimulus trial. The temperature of the plate at the time when the licking (hot) or the jumping (cold) occurred was recorded as the outcome measure. Automatic cut-off temperatures of 0°C (cold stimulus) and 50°C (hot stimulus) were used to avoid tissue injury.
Assessment of white matter sparing and lesion volume. Representative slides from each set were stained with eriochrome cyanine (ECRC) for residual white matter (WM) area. Images were taken at 4x magnification and analyzed using NIH ImageJ software.40 WM area is expressed as a percentage of ECRC-positive area in total area of each section which was averaged from 2-3 adjacent sections at the lesion epicenter. The lesion epicenter was defined as the section with the least amount of spared white matter. Sections spaced 1 mm apart from 5 mm caudal to 5 mm rostral the injury epicenter were stained with GFAP and DAB as the chromogen for lesion volume assessment. Images were taken at 4x magnification and analyzed using NIH ImageJ software. Lesion volume at the lesion epicenter is expressed as a percentage of GFAP negative area in total area of each section.
Statistical analysis. Quantitative data are plotted as mean ± standard error of the mean. For the BMS scores repeated measures 2-way analyses of variance (ANOVAs) were conducted, followed by Bonferroni post-hoc test to compare the differences between each group. Statistical significance was evaluated between 2 individual samples using Student unpaired t tests. For multiple comparisons, one-way analysis of variance (ANOVA) followed by Student Newman-Keuls-post-hoc test. Statistical analysis was performed using SigmaPlot Program, Version 12 (Systat Software) or GraphPad Prism software, version 4.00 for windows (GraphPad Software, Inc.). For non-parametric data, the Kruskal–Wallis one-way ANOVA was used followed by multiple pairwise comparisons using Dunn's post hoc test. A p value of <0.05 was considered statistically significant.
Funding Statement
This study was supported by the National Institutes of Health grants R01 NR013601 (AIF), R01 NS052568 (AIF), R21 NR014053 (JW), and a pilot project in P30 NR014129 (JW).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Guanghui Li and Xiaoyi Lin for expert technical support.
Author contributions
JW, BAS, and AIF conceived research; JW designed research; JW, BS, ZZ, SZ, NW, and DY performed research; JW, BS, MML, ZZ, SZ, and NW analyzed data; JW, BAS, and AIF wrote the paper.
References
- 1.Tator CH. Experimental and clinical studies of the pathophysiology and management of acute spinal cord injury. The journal of spinal cord medicine 1996; 19:206-14; PMID:9237787 [DOI] [PubMed] [Google Scholar]
- 2.Hulsebosch CE, Hains BC, Crown ED, Carlton SM. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain research reviews 2009; 60:202-13; PMID:19154757; http://dx.doi.org/ 10.1016/j.brainresrev.2008.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 2001; 24:254-64; PMID:11586110; http://dx.doi.org/ 10.1097/00002826-200109000-00002 [DOI] [PubMed] [Google Scholar]
- 4.Beattie MS, Hermann GE, Rogers RC, Bresnahan JC. Cell death in models of spinal cord injury. Prog Brain Res 2002; 137:37-47; PMID:12440358; http://dx.doi.org/ 10.1016/S0079-6123(02)37006-7 [DOI] [PubMed] [Google Scholar]
- 5.Young W. Secondary injury mechanisms in acute spinal cord injury. J Emerg Med 1993; 11 Suppl 1:13-22; PMID:8445198 [PubMed] [Google Scholar]
- 6.Wu J, Stoica BA, Faden AI. Cell cycle activation and spinal cord injury. Neurotherapeutics 2011; 8:221-8; PMID:21373950; http://dx.doi.org/ 10.1007/s13311-011-0028-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, Raver C, Piao C, Keller A, Faden AI. Cell cycle activation contributes to increased neuronal activity in the posterior thalamic nucleus and associated chronic hyperesthesia after rat spinal cord contusion. Neurotherapeutics 2013; 10:520-38; PMID:23775067; http://dx.doi.org/ 10.1007/s13311-013-0198-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Renn CL, Faden AI, Dorsey SG. TrkB.T1 contributes to neuropathic pain after spinal cord injury through regulation of cell cycle pathways. J Neurosci 2013; 33:12447-63; PMID:23884949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J, Stoica BA, Luo T, Sabirzhanov B, Zhao Z, Guanciale K, Nayar SK, Foss CA, Pomper MG, Faden AI. Isolated spinal cord contusion in rats induces chronic brain neuroinflammation, neurodegeneration, and cognitive impairment: Involvement of cell cycle activation. Cell Cycle 2014; 13:2446-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci 2014; 34:10989-1006; PMID:25122899; http://dx.doi.org/ 10.1523/JNEUROSCI.5110-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wenzel PL, Chong JL, Saenz-Robles MT, Ferrey A, Hagan JP, Gomez YM, Rajmohan R, Sharma N, Chen HZ, Pipas JM, et al.. Cell proliferation in the absence of E2F1-3. Dev Biol 2011; 351:35-45; PMID:21185283; http://dx.doi.org/ 10.1016/j.ydbio.2010.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda MA, Jakoi L, Nevins JR. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci U S A 1996; 93:3215-20; PMID:8622916; http://dx.doi.org/ 10.1073/pnas.93.8.3215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol 1996; 16:1436-49; PMID:8657117; http://dx.doi.org/ 10.1128/MCB.16.4.1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swiss VA, Casaccia P. Cell-context specific role of the E2F/Rb pathway in development and disease. Glia 2010; 58:377-90; PMID:19795505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 2003; 278:42041-9; PMID:12893818; http://dx.doi.org/ 10.1074/jbc.M308105200 [DOI] [PubMed] [Google Scholar]
- 16.Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, Patil K, Tuttle J, Cleghorn W, Leone G. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 2005; 280:18211-20; PMID:15722552; http://dx.doi.org/ 10.1074/jbc.M501410200 [DOI] [PubMed] [Google Scholar]
- 17.Trimarchi JM, Fairchild B, Verona R, Moberg K, Andon N, Lees JA. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc Natl Acad Sci U S A 1998; 95:2850-5; PMID:9501179; http://dx.doi.org/ 10.1073/pnas.95.6.2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et al.. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 2001; 414:457-62; PMID:11719808; http://dx.doi.org/ 10.1038/35106593 [DOI] [PubMed] [Google Scholar]
- 19.Chen D, Pacal M, Wenzel P, Knoepfler PS, Leone G, Bremner R. Division and apoptosis of E2f-deficient retinal progenitors. Nature 2009; 462:925-9; PMID:20016601; http://dx.doi.org/ 10.1038/nature08544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chong JL, Wenzel PL, Saenz-Robles MT, Nair V, Ferrey A, Hagan JP, Gomez YM, Sharma N, Chen HZ, Ouseph M, et al.. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature 2009; 462:930-4; PMID:20016602; http://dx.doi.org/ 10.1038/nature08677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 2007; 8:368-78; PMID:17453017; http://dx.doi.org/ 10.1038/nrn2124 [DOI] [PubMed] [Google Scholar]
- 22.Greene LA, Biswas SC, Liu DX. Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ 2004; 11:49-60; PMID:14647236; http://dx.doi.org/ 10.1038/sj.cdd.4401341 [DOI] [PubMed] [Google Scholar]
- 23.Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, Zhang MQ, Lazebnik Y, Bar-Sagi D, Lowe SW. Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol 2002; 4:859-64; PMID:12389032; http://dx.doi.org/ 10.1038/ncb868 [DOI] [PubMed] [Google Scholar]
- 24.Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci U S A 1994; 91:3602-6; PMID:8170954; http://dx.doi.org/ 10.1073/pnas.91.9.3602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol 1995; 69:2491-500; PMID:7884898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin XQ, Livingston DM, Kaelin WG Jr., Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci U S A 1994; 91:10918-22; PMID:7971984; http://dx.doi.org/ 10.1073/pnas.91.23.10918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoglinger GU, Breunig JJ, Depboylu C, Rouaux C, Michel PP, Alvarez-Fischer D, Boutillier AL, Degregori J, Oertel WH, Rakic P, et al.. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson's disease. Proc Natl Acad Sci U S A 2007; 104:3585-90; PMID:17360686; http://dx.doi.org/ 10.1073/pnas.0611671104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu J, Kharebava G, Piao C, Stoica BA, Dinizo M, Sabirzhanov B, Hanscom M, Guanciale K, Faden AI. Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS One 2012; 7:e42129; PMID:22848730; http://dx.doi.org/ 10.1371/journal.pone.0042129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen D, Chen Y, Forrest D, Bremner R. E2f2 induces cone photoreceptor apoptosis independent of E2f1 and E2f3. Cell Death Differ 2013; 20:931-40; PMID:23558950; http://dx.doi.org/ 10.1038/cdd.2013.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki J, Isobe M, Morishita R, Nagai R. Nucleic acid drugs for prevention of cardiac rejection. J Biomed Biotechnol 2009; 2009:916514; PMID:20069118; http://dx.doi.org/ 10.1155/2009/916514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomita T, Kunugiza Y, Tomita N, Takano H, Morishita R, Kaneda Y, Yoshikawa H. E2F decoy oligodeoxynucleotide ameliorates cartilage invasion by infiltrating synovium derived from rheumatoid arthritis. Int J Mol Med 2006; 18:257-65; PMID:16820932 [PubMed] [Google Scholar]
- 32.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell 1996; 87:171; PMID:8861900; http://dx.doi.org/ 10.1016/S0092-8674(00)81334-3 [DOI] [PubMed] [Google Scholar]
- 33.Byrnes KR, Stoica BA, Fricke S, Di Giovanni S, Faden AI. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain 2007; 130:2977-92; PMID:17690131; http://dx.doi.org/ 10.1093/brain/awm179 [DOI] [PubMed] [Google Scholar]
- 34.Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP, Faden AI. Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol 2003; 53:454-68; PMID:12666113; http://dx.doi.org/ 10.1002/ana.10472 [DOI] [PubMed] [Google Scholar]
- 35.Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A 2005; 102:8333-8; PMID:15923260; http://dx.doi.org/ 10.1073/pnas.0500989102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kabadi SV, Stoica BA, Byrnes KR, Hanscom M, Loane DJ, Faden AI. Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. J Cereb Blood Flow Metab 2012; 32:137-49; PMID:21829212; http://dx.doi.org/ 10.1038/jcbfm.2011.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabadi SV, Stoica BA, Hanscom M, Loane DJ, Kharebava G, Murray Ii MG, Cabatbat RM, Faden AI. CR8, a selective and potent CDK inhibitor, provides neuroprotection in experimental traumatic brain injury. Neurotherapeutics 2012; 9:405-21; PMID:22167461; http://dx.doi.org/ 10.1007/s13311-011-0095-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabadi SV, Stoica BA, Loane DJ, Byrnes KR, Hanscom M, Cabatbat RM, Tan MT, Faden AI. Cyclin D1 gene ablation confers neuroprotection in traumatic brain injury. J Neurotrauma 2012; 29:813-27; PMID:21895533; http://dx.doi.org/ 10.1089/neu.2011.1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu J, Pajoohesh-Ganji A, Stoica BA, Dinizo M, Guanciale K, Faden AI. Delayed expression of cell cycle proteins contributes to astroglial scar formation and chronic inflammation after rat spinal cord contusion. J Neuroinflammation 2012; 9:169; PMID:22784881; http://dx.doi.org/ 10.1186/1742-2094-9-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J, Stoica BA, Dinizo M, Pajoohesh-Ganji A, Piao CS, Faden AI. Delayed cell cycle pathway modulation facilitates recovery after spinal cord injury. Cell Cycle 2012; 11:1782-95; PMID:22510563; http://dx.doi.org/ 10.4161/cc.20153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A 1997; 94:7245-50; PMID:9207076; http://dx.doi.org/ 10.1073/pnas.94.14.7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 1993; 365:349-52; PMID:8377827; http://dx.doi.org/ 10.1038/365349a0 [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 2001; 21:2661-8; PMID:11306619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hou ST, Callaghan D, Fournier MC, Hill I, Kang L, Massie B, Morley P, Murray C, Rasquinha I, Slack R, et al.. The transcription factor E2F1 modulates apoptosis of neurons. J Neurochem 2000; 75:91-100; PMID:10854251; http://dx.doi.org/ 10.1046/j.1471-4159.2000.0750091.x [DOI] [PubMed] [Google Scholar]
- 45.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature 1992; 359:295-300; PMID:1406933; http://dx.doi.org/ 10.1038/359295a0 [DOI] [PubMed] [Google Scholar]
- 46.Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J 1996; 15:6178-88; PMID:8947040 [PMC free article] [PubMed] [Google Scholar]
- 47.Jin K, Mao XO, Eshoo MW, Nagayama T, Minami M, Simon RP, Greenberg DA. Microarray analysis of hippocampal gene expression in global cerebral ischemia. Ann Neurol 2001; 50:93-103; PMID:11456315; http://dx.doi.org/ 10.1002/ana.1073 [DOI] [PubMed] [Google Scholar]
- 48.Osuga H, Osuga S, Wang F, Fetni R, Hogan MJ, Slack RS, Hakim AM, Ikeda JE, Park DS. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci U S A 2000; 97:10254-9; PMID:10944192; http://dx.doi.org/ 10.1073/pnas.170144197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Motonaga K, Itoh M, Hirayama A, Hirano S, Becker LE, Goto Y, Takashima S. Up-regulation of E2F-1 in Down's syndrome brain exhibiting neuropathological features of Alzheimer-type dementia. Brain Res 2001; 905:250-3; PMID:11423103; http://dx.doi.org/ 10.1016/S0006-8993(01)02535-5 [DOI] [PubMed] [Google Scholar]
- 50.MacManus JP, Jian M, Preston E, Rasquinha I, Webster J, Zurakowski B. Absence of the transcription factor E2F1 attenuates brain injury and improves behavior after focal ischemia in mice. J Cereb Blood Flow Metab 2003; 23:1020-8; PMID:12973018; http://dx.doi.org/ 10.1097/01.WCB.0000084249.20114.FA [DOI] [PubMed] [Google Scholar]
- 51.Lazzerini Denchi E, Helin K. E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep 2005; 6:661-8; PMID:15976820; http://dx.doi.org/ 10.1038/sj.embor.7400452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, Krishnan V, Lai-Hsu C, Park KK, Tsoulfas P, et al.. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci 2013; 33:13882-7; PMID:23966707; http://dx.doi.org/ 10.1523/JNEUROSCI.2524-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iglesias A, Murga M, Laresgoiti U, Skoudy A, Bernales I, Fullaondo A, Moreno B, Lloreta J, Field SJ, Real FX, et al.. Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. J Clin Investig 2004; 113:1398-407; PMID:15146237; http://dx.doi.org/ 10.1172/JCI200418879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian DS, Yu ZY, Xie MJ, Bu BT, Witte OW, Wang W. Suppression of astroglial scar formation and enhanced axonal regeneration associated with functional recovery in a spinal cord injury rat model by the cell cycle inhibitor olomoucine. J Neurosci Res 2006; 84:1053-63; PMID:16862564; http://dx.doi.org/ 10.1002/jnr.20999 [DOI] [PubMed] [Google Scholar]
- 55.Ren H, Han M, Zhou J, Zheng ZF, Lu P, Wang JJ, Wang JQ, Mao QJ, Gao JQ, Ouyang HW. Repair of spinal cord injury by inhibition of astrocyte growth and inflammatory factor synthesis through local delivery of flavopiridol in PLGA nanoparticles. Biomaterials 2014; 35:6585-94; PMID:24811262; http://dx.doi.org/ 10.1016/j.biomaterials.2014.04.042 [DOI] [PubMed] [Google Scholar]
- 56.MacManus JP, Koch CJ, Jian M, Walker T, Zurakowski B. Decreased brain infarct following focal ischemia in mice lacking the transcription factor E2F1. Neuroreport 1999; 10:2711-4; PMID:10511428; http://dx.doi.org/ 10.1097/00001756-199909090-00004 [DOI] [PubMed] [Google Scholar]
- 57.Tsai SY, Opavsky R, Sharma N, Wu L, Naidu S, Nolan E, Feria-Arias E, Timmers C, Opavska J, de Bruin A, et al.. Mouse development with a single E2F activator. Nature 2008; 454:1137-41; PMID:18594513; http://dx.doi.org/ 10.1038/nature07066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sabirzhanov B, Stoica BA, Hanscom M, Piao CS, Faden AI. Over-expression of HSP70 attenuates caspase-dependent and caspase-independent pathways and inhibits neuronal apoptosis. J Neurochem 2012; 123:542-54; PMID:22909049; http://dx.doi.org/ 10.1111/j.1471-4159.2012.07927.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 1995; 12:1-21; PMID:7783230; http://dx.doi.org/ 10.1089/neu.1995.12.1 [DOI] [PubMed] [Google Scholar]
- 60.Pajoohesh-Ganji A, Byrnes KR, Fatemi G, Faden AI. A combined scoring method to assess behavioral recovery after mouse spinal cord injury. Neurosci Res 2010; 67:117-25; PMID:20188770; http://dx.doi.org/ 10.1016/j.neures.2010.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metz GA, Merkler D, Dietz V, Schwab ME, Fouad K. Efficient testing of motor function in spinal cord injured rats. Brain Res 2000; 883:165-77; PMID:11074045; http://dx.doi.org/ 10.1016/S0006-8993(00)02778-5 [DOI] [PubMed] [Google Scholar]
- 62.Ren K. An improved method for assessing mechanical allodynia in the rat. Physiology & behavior 1999; 67:711-6; PMID:10604842; http://dx.doi.org/ 10.1016/S0031-9384(99)00136-5 [DOI] [PubMed] [Google Scholar]
- 63.Carozzi VA, Renn CL, Bardini M, Fazio G, Chiorazzi A, Meregalli C, Oggioni N, Shanks K, Quartu M, Serra MP, et al.. Bortezomib-induced painful peripheral neuropathy: an electrophysiological, behavioral, morphological and mechanistic study in the mouse. PLoS One 2013; 8:e72995; PMID:24069168; http://dx.doi.org/ 10.1371/journal.pone.0072995 [DOI] [PMC free article] [PubMed] [Google Scholar]