

The fibroblast growth factor (Fgf) family of ligands and receptor tyrosine kinases is required throughout embryonic and postnatal development and also regulates multiple homeostatic functions in the adult. Here, Brewer et al. review the mechanisms of Fgf signaling by focusing on genetic strategies that enable in vivo analysis.
Keywords: fibroblast growth factor, Fgfr, receptor tyrosine kinase, Erk1/2, signaling
Abstract
The fibroblast growth factor (Fgf) family of ligands and receptor tyrosine kinases is required throughout embryonic and postnatal development and also regulates multiple homeostatic functions in the adult. Aberrant Fgf signaling causes many congenital disorders and underlies multiple forms of cancer. Understanding the mechanisms that govern Fgf signaling is therefore important to appreciate many aspects of Fgf biology and disease. Here we review the mechanisms of Fgf signaling by focusing on genetic strategies that enable in vivo analysis. These studies support an important role for Erk1/2 as a mediator of Fgf signaling in many biological processes but have also provided strong evidence for additional signaling pathways in transmitting Fgf signaling in vivo.
The fibroblast growth factor (Fgf) family of signaling proteins includes 22 members that have been identified based on sequence homology. Eighteen of these Fgfs function as ligands, which bind four receptor tyrosine kinases (RTKs) in mice and humans. The four remaining Fgfs (Fgf11–14) are intracellular proteins that do not interact with Fgf receptors (Fgfrs) (Smallwood et al. 1996; Olsen et al. 2003). A fifth Fgfr-like protein (FgfrL1) has also been identified that lacks an intracellular tyrosine kinase domain and likely negatively regulates Fgfrs by sequestering ligand (for review, see Trueb et al. 2013). Fgf signaling is required throughout metazoans and is commonly studied in organisms ranging from cnidarians to humans (Tulin and Stathopoulos 2010). These studies indicate that Fgf signaling is required pleiotropically during development and also regulates multiple homeostatic and reparative functions in adults (Ornitz and Itoh 2015). Additionally, pathological activation of Fgfrs underlies many congenital disorders and cancer types. Several therapeutic strategies are currently being developed to modulate Fgfr signaling in various pathologies (Carter et al. 2015; Degirolamo et al. 2016). Understanding the mechanisms that govern Fgf signaling is therefore important to appreciate many aspects of Fgf biology and disease.
Many of the developmental functions of Fgf signaling seem to be conserved between mice and humans. This is evident by the striking phenotypic similarities between human congenital disorders caused by alterations in Fgf signaling and their corresponding mouse models. Conserved developmental requirements have been demonstrated in skeletal growth, palate closure, limb patterning, ear development, cranial suture ossification, neural development, and the hair cycle (Hebert et al. 1994; Rousseau et al. 1994; Shiang et al. 1994; Wilkie et al. 1995; Partanen et al. 1998; Chen et al. 1999; Li et al. 1999; Wang et al. 1999, 2005; Dode et al. 2003; Tsai et al. 2005; Gill and Tsai 2006; Mason 2007; Riley et al. 2007; Falardeau et al. 2008; Mansour et al. 2009; Stanier and Pauws 2012; Simonis et al. 2013; Higgins et al. 2014; Ornitz and Marie 2015). These conserved developmental functions and accessible genetics make the mouse an excellent model for studying the mechanisms that Fgf signaling uses in vivo, which we discuss in this review. Valuable information pertaining to Fgf signaling has also been gained from studies of invertebrate organisms, Xenopus, and zebrafish, which have been reviewed elsewhere (Huang and Stern 2005; Itoh 2007; Dorey and Amaya 2010).
Ligand binding specificity
Ligand binding represents the first step in initiating the Fgfr signaling cascade. Fgfrs contain three extracellular immunoglobulin-like domains (IgI–IgIII) with an eight-residue acid box in the linker region between IgI and IgII (Lee et al. 1989). IgI and the acid box play an inhibitory role in ligand–receptor complex formation (Kalinina et al. 2012), while IgII and IgIII cooperate in ligand binding. In Fgfr1–3, ligand binding specificity is largely determined by alternative splicing of the C terminus of the IgIII domain, which is encoded by either exon 8 or 9 to generate the Fgfrb or Fgfrc isoform (Fig. 1A; Johnson et al. 1991; Chellaiah et al. 1994; Ornitz et al. 1996; Zhang et al. 2006). These b and c isoforms are generally restricted to epithelial and mesenchymal tissues, respectively. In this way, alternative splicing of the receptors allows ligands to activate receptors in the adjacent mesenchymal or epithelial tissue without activating autocrine signaling (Fig. 1B,C; Miki et al. 1992; MacArthur et al. 1995; Min et al. 1998; Xu et al. 1998b). However, there are several exceptions to this general principle of paracrine signaling, as some biological processes depend on ligand–receptor interactions within the same tissue. For example, mesenchymal Fgf9 influences development of both the epithelium and mesenchyme during lung development (del Moral et al. 2006; White et al. 2006). Additionally, Fgf20 is required in an autocrine fashion during development of the kidney and organ of Corti (Barak et al. 2012; Huh et al. 2012). Finally, a recent study has demonstrated that Fgf10, expressed in the lung mesenchyme, engages Fgfr1b and Fgfr2b in the same tissue during the formation of lipofibroblasts (Al Alam et al. 2015).
Figure 1.
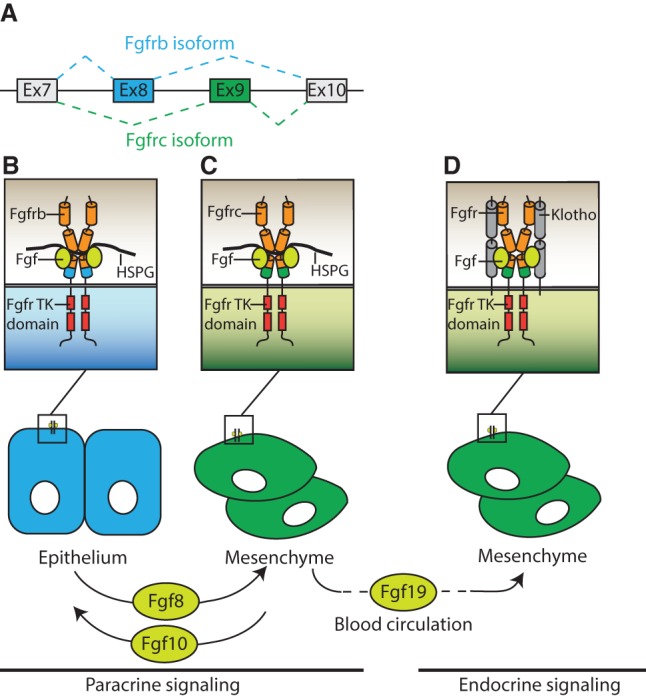
Fgfr alternative splicing facilitates interactions between epithelial and mesenchymal tissues. (A) Alternative splicing of exons 8 and 9 generates b and c isoforms of Fgfr1–3, while exon 10 encodes an invariant transmembrane domain. (B,C) Fgf ligands expressed in epithelium engage Fgfrc isoforms in the adjacent mesenchyme (B), while ligands expressed in the mesenchyme activate Fgfrb isoforms in the epithelium (C). Paracrine signaling also depends on the heparan sulfate proteoglycan (HSPG) coreceptor. (D) Endocrine Fgf ligands use Klotho coreceptors rather than HSPGs. (Ex) exon; (TK) tyrosine kinase. The exon, Fgfr isoform, and cell type specificity are color coded, with blue and green representing epithelium and mesenchyme, respectively.
Proper splicing of the Fgfrb isoforms is achieved by a splicing complex that includes the epithelial-specific Esrp1 and Esrp2 RNA-binding proteins (Warzecha et al. 2009). Accordingly, combined genetic ablation of Esrp1 and Esrp2 leads to aberrant splicing of Fgfr1–3b in vivo and causes defects in multiple epithelial contexts that require Fgf signaling (Bebee et al. 2015). Genetic disruption of specific b and c isoforms individually has demonstrated that Fgfr1 and Fgfr3 primarily function in the mesenchyme, while Fgfr2 is more important in epithelial contexts (Partanen et al. 1998; De Moerlooze et al. 2000; Hajihosseini et al. 2001; Eswarakumar et al. 2002; Zhang et al. 2004; Eswarakumar and Schlessinger 2007). However, each receptor also possesses functions in the reciprocal cell type.
Heparan sulfate proteoglycans (HSPGs) also regulate multiple properties of Fgf ligands and receptors (Ornitz 2000). These cell surface and extracellular matrix macromolecules are composed of a protein core to which heparan sulfate (HS) glycosaminoglycan (GAG) disaccharide polymers are added (Nelson and Cox 2005). HS molecules are differentially O- or N-sulfated in a tissue-specific manner, and these sulfation patterns facilitate distinct ligand–receptor associations (Guimond et al. 1993; Pye et al. 1998; Allen and Rapraeger 2003; Qu et al. 2011). HSPG affinity influences ligand dispersal to shape morphogen gradients (Harada et al. 2009; Makarenkova et al. 2009; Qu et al. 2012). These HS chains can also be cleaved to spread ligand between cells or release ligand sequestered by the extracellular matrix (Patel et al. 2007; Shimokawa et al. 2011).
The FGF19 subfamily lacks the ability to bind HSPGs, enabling them to escape the HS-rich cell surface and function as endocrine hormones (Fig. 1D; Itoh et al. 2015). The endocrine subfamily of Fgf ligands regulates multiple processes in the adult, including phosphate homeostasis, adipocyte metabolism, and bile acid synthesis (Shimada et al. 2004; Inagaki et al. 2005; Kharitonenkov et al. 2005; Schoenberg et al. 2011). Two homologous proteins, Klotho and βklotho, serve as coreceptors in place of HSPGs to facilitate ligand–receptor interactions (Kurosu et al. 2006; Urakawa et al. 2006; Ogawa et al. 2007). Recent studies suggest that modulating the homeostatic functions of Fgf signaling may be of therapeutic value in multiple pathologies (Degirolamo et al. 2016).
Ligand–receptor binding affinities are therefore determined by multiple properties, including alternative splicing of the receptor, the presence of specific HSPG modifications, and the expression of Klotho coreceptors. Several fundamental studies have determined each ligand's receptor specificity in vitro using mitogenic assays or by directly measuring complex affinities (MacArthur et al. 1995; Ornitz et al. 1996; Kurosu et al. 2006; Olsen et al. 2006; Zhang et al. 2006; Ogawa et al. 2007). A comprehensive review of ligand–receptor binding specificities has been discussed recently elsewhere (Ornitz and Itoh 2015).
Fgfrs function individually and in combination
All of the Fgf ligands and receptors have been genetically knocked out in mice, producing phenotypes at virtually every stage of life, from the preimplantation blastocyst to the adult organism. The phenotypes caused by genetic disruption of the Fgf ligands have been extensively reviewed elsewhere (Ornitz and Itoh 2015). Fgfr knockout phenotypes have demonstrated that these receptors have both essential and redundant roles throughout development. Fgfr1−/− mutant mice fail to undergo the epithelial-to-mesenchymal transition required for mesoderm formation (Deng et al. 1994; Yamaguchi et al. 1994; Ciruna et al. 1997; Ciruna and Rossant 2001; Hoch and Soriano 2006). However, this phenotype is dependent on genetic background, since Fgfr1 was shown to regulate primitive endoderm formation on a 129S4 genetic background, while the same null allele caused mesoderm defects on a mixed genetic background (Hoch and Soriano 2006; Brewer et al. 2015). Other studies have also demonstrated that ear defects caused by an ENU-induced mutation in Fgfr1 are also modified by genetic background (Pau et al. 2005; Calvert et al. 2011). For Fgfr2, different targeting strategies have produced distinct phenotypes. Deletion of exons 9–12 (Fgfr2Δ9–12 allele) or exon 5 (Fgfr2Δ5 allele) caused perimplantation lethality, likely due to defects in extraembryonic lineages (Arman et al. 1998; Blak et al. 2007). Fgfr2 mutants that lack exons 7–9 (Fgfr2Δ7–9 allele) or exons 8–10 (Fgfr2Δ8–10 allele) die around embryonic day 10 (E10) and exhibit defects in limb induction, chorioallantoic fusion, and the labyrinth component of the placenta (Xu et al. 1998b; Yu et al. 2003). The Fgfr2Δ7–9/Δ7–9 phenotype was consistent across different genetic backgrounds, suggesting that second site modifiers do not underlie this phenotypic discrepancy (Xu et al. 1998b). Further work is therefore necessary to resolve the issue of the Fgfr2-null mutant phenotype. Genetic loss of Fgfr3 causes long bone overgrowth and deafness (Colvin et al. 1996; Deng et al. 1996). The discrete developmental requirements of Fgfr1–3 likely reflect differences in Fgfr expression, ligand binding affinities, and signaling potentials, which have been documented (Orr-Urtreger et al. 1991; Ornitz and Leder 1992; Vainikka et al. 1994; Shaoul et al. 1995; Ornitz et al. 1996; Yaylaoglu et al. 2005). Fgfr4−/− mutant mice are viable and develop normally. However, analysis of Fgfr3−/−; Fgfr4−/− double mutants indicates that these receptors cooperate during alveolar development in the lung (Weinstein et al. 1998).
Several additional contexts have been shown to require signaling through multiple Fgfrs. Here, Fgfrs are largely thought to function as homodimers in vivo. However, two studies have provided biochemical evidence that suggests that Fgfrs are capable of forming heterodimers. First, Fgfr2 is capable of phosphorylating Fgfr1 intracellular tyrosines (Bellot et al. 1991). Second, an Fgfr1 dominant-negative (Fgfr1DN) allele that lacks the cytoplasmic tail is capable of suppressing activation of Fgfr1–3 (Ueno et al. 1992). The absence of the cytoplasmic tail prevents receptor transphosphorylation following ligand binding and therefore results in a nonproductive dimerization event. The ability of the Fgfr1DN protein to suppress activation of Fgfr2 and Fgfr3 therefore suggests that Fgfr1 is capable of forming a heterodimer with other Fgfrs (Ueno et al. 1992). However, the Fgfr1DN construct could inhibit activation of wild-type Fgfrs by sequestering ligand without forming a heterodimer. These studies have been conducted using overexpression assays in cultured cells or Xenopus oocytes. The existence of Fgfr heterodimers in vivo at endogenous expression levels therefore remains to be demonstrated.
Intracellular signaling
Fgfrs engage multiple signaling pathways, including Erk1/2, PI3K/Akt, Plcγ, Pkc, and Stats. This is achieved mostly through an adaptor-mediated mechanism in which the receptor recruits nonenzymatic proteins that function as a scaffold to engage additional signaling proteins (Fig. 2). Fgfr-recruited proteins, their known signaling capabilities, and their in vivo significance are discussed below.
Figure 2.
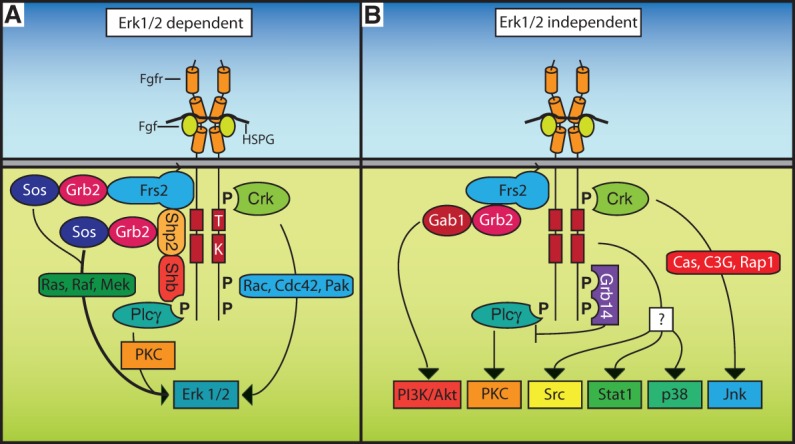
Schematic representation of Fgfr signaling functions. (A) Fgfrs are capable of engaging Erk1/2 through multiple mechanisms, including the Frs2, Shb, and Crk adaptor proteins as well as Plcγ. For simplicity, CrkI, CrkII, and CrkL adaptor proteins are referred to as Crk. Please see the text for further discussion of the role of these signaling proteins. (B) Fgfrs are also capable of engaging several additional signaling pathways, including PI3K/Akt, Pkc, Src, Stat1, p38, and Jnk.
The Frs (Fgf-regulated substrate) family of adaptor proteins engages Erk1/2 downstream from Fgfrs
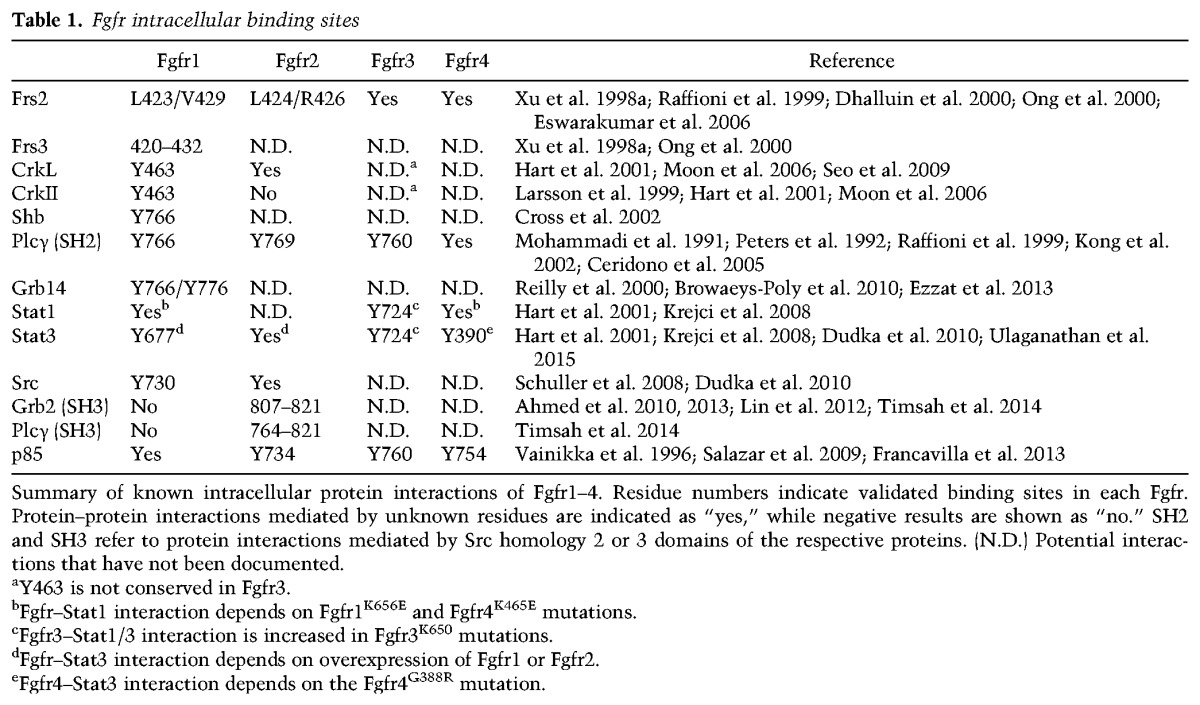
Engagement of Fgfrs leads to the phosphorylation of several Frs. Frs2 and Frs3 are myristyl-anchored membrane adaptor proteins that bind the juxtamembrane domain of Fgfrs (Table 1; Kouhara et al. 1997; Xu et al. 1998a; Dhalluin et al. 2000; Ong et al. 2000). This interaction is mediated by the phosphotyrosine-binding domain of Frs2 and Frs3; however, this complex is formed constitutively independently of receptor phosphorylation (Dhalluin et al. 2000; Ong et al. 2000). Upon receptor activation, Frs2 and Frs3 are phosphorylated on multiple tyrosine residues, enabling these adaptor proteins to bind Shp2 and Grb2–Sos to activate the Ras–Erk1/2 signaling pathway (Fig. 2A; Ong et al. 1996; Kouhara et al. 1997; Gotoh et al. 2004b). Frs3 overexpression is capable of rescuing Fgf-mediated Erk1/2 activation in Frs2−/− fibroblasts, indicating that the two adaptor proteins share similar functions in activating this pathway (Gotoh et al. 2004b). During embryonic development, Frs2 is broadly expressed in many tissues, while Frs3 expression is more restricted and not detectable by in situ hybridization until E11.5 (Gotoh et al. 2004b).
Table 1.
Fgfr intracellular binding sites
Several phenotypes caused by loss of Frs2 function have also indicated that Frs2 is required for Fgf-mediated Erk1/2 activation in vivo. Frs2−/− mutants are not recovered at the expected Mendelian frequency at E6.5 and have defects in anterior/posterior patterning (Gotoh et al. 2005). Erk1/2 activation is decreased in the extraembryonic ectoderm of Frs2−/− mutants at E6.5 (Gotoh et al. 2005). Fgfr2 is expressed in the extraembryonic ectoderm (Ciruna and Rossant 1999) and is believed to engage Erk1/2 through Frs2 in this tissue. However, similar defects have not been documented in Fgfr2−/− mutants (Arman et al. 1998; Xu et al. 1998b), suggesting that Frs2 may function downstream from multiple Fgfrs or other RTKs in the E6.5 extraembryonic ectoderm. Chimeric analysis demonstrated that Frs2−/− cells accumulate at the primitive streak (Gotoh et al. 2005). This phenotype was also observed in Fgfr1−/− mutants, since this receptor is required to initiate the epithelial-to-mesenchymal transition required to form mesoderm (Yamaguchi et al. 1994; Ciruna et al. 1997; Ciruna and Rossant 2001).
While Frs2 is an important mediator of Fgfr signaling, several studies have indicated that Frs2 regulates only a subset of Fgfr functions. Mice containing amino acid substitutions in Fgfr1 that prevent Fgfr1–Frs2 binding die at birth with multiple developmental defects, including cleft palate, post-axial polydactyly, hypoplasia of multiple middle ear bones, and anterior/posterior patterning defects of the thoracic vertebrae (Brewer et al. 2015). This phenotype is much less severe than the Fgfr1−/− phenotype on the same genetic background, characterized by fewer primitive endoderm cells at the blastocyst stage and perimplantation lethality (Brewer et al. 2015). Another strategy to uncouple Fgfr1 and Frs2 signaling deleted the juxtamembrane domain responsible for both Frs2 and Frs3 binding (Fgfr1ΔFrs allele) (Hoch and Soriano 2006). Again, Fgfr1ΔFrs/ΔFrs mice failed to recapitulate the phenotype of Fgfr1−/− mutants, indicating that Frs2 and/or Frs3 are required for only a subset of Fgfr1 signaling functions in vivo (Hoch and Soriano 2006). Fgfr1ΔFrs/ΔFrs embryos exhibited neural tube closure defects, posterior truncations, and defects in multiple pharyngeal arch derivatives (Hoch and Soriano 2006). However, some Fgfr1ΔFrs/ΔFrs ear phenotypes affecting the number of cochlear and vestibular hair cells as well as inner ear morphology were as severe as complete loss of Fgfr1 function (Ono et al. 2014). This suggests that the importance of Frs adaptor proteins downstream from Fgfr1 may differ depending on the context. Phenotypes associated with the Fgfr1ΔFrs allele were considerably more severe than those observed in mice containing amino acid substitutions that prevented only Frs2 from binding Fgfr1 (Hoch and Soriano 2006; Brewer et al. 2015). This phenotypic disparity is likely due in part to allele design, since the Fgfr1ΔFrs allele relied on a partial cDNA knock-in strategy that failed to completely recapitulate normal expression or function of Fgfr1 (Hoch and Soriano 2006).
Surprisingly, mice containing L424A and R426A mutations in Fgfr2 (Fgfr2LR allele) that disrupt Frs2 binding are viable (Eswarakumar et al. 2006; Sims-Lucas et al. 2009). Fgfr2–Frs2 signaling is therefore dispensable for embryonic development. However, Fgfr2 signaling through Frs2 is required for multiple phenotypes in a mouse model of Crouzon syndrome (Eswarakumar et al. 2006). Mice heterozygous for a constitutively active allele of Fgfr2 (Fgfr2C342Y allele containing the C342Y mutation) approximate Crouzon syndrome, which is characterized by premature fusion of cranial sutures (craniosynostosis). Mice containing three amino acid substitutions, C342Y, L424A, and R426A (Fgfr2CLR allele), designed to disrupt Fgfr2–Frs2 binding in the constitutively active receptor were phenotypically indistinguishable from wild-type littermates. This result indicates that Fgfr2 signaling requires Frs2 during pathologic cranial suture ossification. Mice homozygous for the activated allele of Fgfr2 exhibited several additional phenotypes, including cleft palate, formation of a tracheal cartilaginous sleeve, and fused knees and elbows. Fgfr2CLR/CLR mice displayed no defects at the knee or elbow joints but retained the cleft palate and tracheal phenotypes of the Fgfr2C342Y/C342Y mutant (Eswarakumar et al. 2006). This context-specific attenuation of Fgfr2C342Y/C342Y phenotypes may be due to differential requirements for Frs2 signaling in each developmental process or distinct threshold effects between the palate, trachea, and elbow/knee joints.
Engineering Fgfrs to disrupt their ability to activate Frs2 provides a major advantage in understanding the relative importance of this signaling protein downstream from specific Fgfrs. This is because Frs2 binds a number of RTKs in addition to Fgfrs that include Trks, Ret, Alk, and Vegfrs (Rabin et al. 1993; Ong et al. 1996, 2000; Dhalluin et al. 2000; Kurokawa et al. 2001; Melillo et al. 2001; Degoutin et al. 2007; Chen et al. 2014b). Loss of Frs2 therefore alters signaling downstream from multiple RTKs, making the phenotypes of Frs2−/− mutants difficult to attribute to an individual RTK. This point is emphasized by a series of studies focused on kidney development. Fgfr2, Frs2, and Ret are each required for kidney development (Schuchardt et al. 1994; Zhao et al. 2004; Sims-Lucas et al. 2009). However, Fgfr2 mutants that lack the ability to bind Frs2 develop normal kidneys, while Ret mutants that are unable to signal through Frs2 recapitulate the kidney defects observed when Frs2 is conditionally disrupted in the ureteric bud (Jijiwa et al. 2004; Zhao et al. 2004; Sims-Lucas et al. 2009). Therefore, specifically uncoupling Frs2 signaling from Fgfr2 or Ret helped to clarify the contribution of each receptor's signaling function in kidney development. More information on how Fgf signaling regulates kidney development can be found in several recent review articles (Bates 2011; Trueb et al. 2013).
Shp2 is a critical mediator of Frs2-dependent Erk1/2 activation
Shp2 is a tyrosine phosphatase that also functions as an adaptor protein downstream from multiple RTKs. Frs2-mediated Fgfr signal transduction is reinforced by the constitutive Shb–Shp2 complex (Fig. 2A; Cross et al. 2002). Following receptor activation, Shb binds the Fgfr tyrosine kinase domain, enabling Shp2 to bind Frs2 (Cross et al. 2002). Shp2 is also tyrosine-phosphorylated following Fgf treatment, which allows Shp2 to bind Grb2–Sos and engage the Ras–Erk1/2 signaling pathway in PC12 cells (Hadari et al. 1998). Of note, prolonged Erk1/2 activation depends on Grb2 that is recruited by Shp2 rather than Grb2 that binds Frs2 directly (Hadari et al. 1998). It is not known whether the phosphatase function of Shp2 is involved in this or another process in Fgfr-mediated signal transduction, but this phosphatase activity is required for sustained Fgf-mediated Erk1/2 activation (Hadari et al. 1998).
Genetic studies also indicate that Shp2 is an important mediator of Frs2-dependent functions downstream from Fgfrs. To better understand the requirement of specific signaling functions of Frs2, mutations were engineered into Frs2 that disrupt the ability of this adaptor protein to interact with Shp2 (Frs22F allele) or Grb2 (Frs24F allele) (Gotoh et al. 2004a). Analysis of mice engineered with these mutations demonstrated that the Frs2–Shp2 protein complex is required for Fgf-dependent lens placode induction but that Frs2–Grb2 binding was dispensable for eye development (Faber et al. 2001; Gotoh et al. 2004a). Frs22F/2F mutant mice also exhibited decreased Erk1/2 activation during lens induction, indicating that Frs2-mediated Erk1/2 activation depends on Shp2 binding (Gotoh et al. 2004a). Similar experiments have demonstrated the importance of Shp2 in Fgf-mediated closure of the optic fissure (Cai et al. 2013). Conditional loss of both Fgfr1 and Fgfr2 in the optic vesicle caused ocular coloboma. This phenotype was also observed when both Frs2 and Ptpn11 (the gene encoding murine Shp2) were conditionally disrupted in the optic vesicle or when the Frs2–Shp2 protein complex was disrupted in a Ptpn11-deficient background (in Frs22F/cKO; Ptpn11cKO/cKO mice). This result indicates that Shp2-independent functions of Frs2 are not sufficient for closure of the optic fissure (Cai et al. 2013). Additionally, loss of Fgfr1 and Fgfr2 or Frs2 and Ptpn11 was rescued by introducing a constitutively active KrasG12D allele, indicating that Fgf signaling primarily depends on Erk1/2 in this context. Collectively, these studies support the model that Frs2-mediated Erk1/2 activation depends on Shp2 in Fgf-mediated developmental processes.
Loss of Ptpn11 causes phenotypes reminiscent of decreased Fgf signaling in other developmental processes as well. Genetic disruption of Ptpn11 causes gastrulation defects that are reminiscent of Fgfr1−/− mutant phenotypes (Deng et al. 1994; Yamaguchi et al. 1994; Saxton et al. 1997). Conditional deletion of Ptpn11 in neural crest cells or Fgf8 in the facial epithelium also leads to agenesis of multiple craniofacial structures (Trumpp et al. 1999; Nakamura et al. 2009). These phenotypes are consistent with the concept that Shp2 is required in multiple developmental contexts regulated by Fgf signaling.
The Crk family of adaptor proteins engages Erk1/2 and Jnk downstream from Fgfrs
The Crk gene encodes two distinct proteins (named CrkI and CrkII) by alternative splicing (Feller 2001). These proteins function as adaptors and form multiprotein complexes via their SH2 and SH3 domains (Feller 2001). CrkII binds the juxtamembrane domain of activated Fgfr1 at phosphotyrosine 463 to mediate Erk1/2 and Jnk activation (Table 1; Larsson et al. 1999). Disruption of the Fgfr1–CrkII complex also decreases Fgfr1-mediated Frs2 tyrosine phosphorylation in vitro, suggesting that CrkII enhances Frs2 activation by Fgfrs (Larsson et al. 1999). Therefore, Crk-mediated activation of Erk1/2 downstream from Fgfrs may be Frs2-dependent to some extent. CrkII activates Jnk by recruiting Cas and activating the C3G, Rap1 axis (Fig. 2B; Larsson et al. 1999). A related protein, Crk-like (CrkL), contains amino acid composition, domain structure, and functional similarities to CrkI and CrkII (Feller 2001). CrkL also interacts with phosphotyrosine 463 of Fgfr1 and phosphotyrosine 466 of Fgfr2 with a greater affinity than Crk proteins (Seo et al. 2009). CrkL has been shown to engage the Erk1/2 pathway independently of Ras through Rac1, Cdc42, and Pak (Fig. 2A; Seo et al. 2009).
Genetic analysis suggests that CrkL signaling is required to mediate Fgf8-dependent development of the pharyngeal arches (Moon et al. 2006). While Fgf8+/− or CrkL+/− heterozygous mutants exhibit normal development of the pharyngeal arches, Fgf8+/−; CrkL+/− compound heterozygous mice have defects in multiple pharyngeal arch derivatives, including the vasculature, cardiac outflow tracts, thymus, and parathyroid glands. The penetrance and severity of these defects are enhanced in Fgf8+/−; CrkL−/− mutants. Decreased Erk1/2 activation was observed in the pharyngeal arches and correlated with the severity of gene dosage and phenotypic outcome, suggesting that CrkL is required for Fgf8-mediated Erk1/2 activation. Alterations in Fgf8 and CrkL gene dosage also affected development of the femur, palate, and mandible, suggesting that CrkL is required for Fgf8-mediated signaling in multiple developmental contexts. These phenotypes are likely dependent on multiple Fgfrs, since CrkL interacts with both Fgfr1 and Fgfr2 in mouse embryonic fibroblasts (Moon et al. 2006). Mice engineered to disrupt the ability of Fgfr1 to signal through Crk adaptor proteins were viable and fertile without developmental or homeostatic defects (Brewer et al. 2015). This result may be consistent with the hypothesis that multiple Fgfrs signal through Crk adaptor proteins in the pharyngeal arches and other Fgf-dependent developmental contexts.
Erk1/2 is required in many Fgf-mediated developmental processes
Surprisingly, despite being activated by many cell surface receptors, Erk1/2 is phosphorylated at high levels in discrete tissues during development rather than being uniformly activated (Corson et al. 2003). Erk1/2 activation is decreased in many of these contexts when embryos are cultured in Fgfr inhibitors, suggesting that Fgf signaling is a major driver of Erk1/2 activation in multiple developmental processes (Corson et al. 2003). Additionally, Fgf, Fgfr, and Erk1/2 loss-of-function phenotypes are often similar, suggesting that Fgfrs primarily signal through Erk1/2 in vivo. Fgfrs have been shown to function through Erk1/2 in many biological processes, and several of these are discussed below.
Fgf4–Erk1/2 signaling regulates primitive endoderm specification
The preimplantation blastocyst is composed of an inner cell mass surrounded by trophectoderm (Fig. 3). The inner cell mass is made up of primitive endoderm and epiblast cells, which give rise to the yolk sac and embryo, respectively. Initially, cells of the inner cell mass express both epiblast and primitive endoderm markers (Plusa et al. 2008). These lineages subsequently become restricted to an epiblast or primitive endoderm cell fate as the expression of lineage-specific genes becomes mutually exclusive (Plusa et al. 2008). This cell fate decision can be modulated to generate an inner cell mass completely composed of primitive endoderm or epiblast cells by culturing embryos in exogenous FGF4 or a Fgfr inhibitor, respectively (Fig. 3; Yamanaka et al. 2010). Mek inhibition also results in an inner cell mass devoid of primitive endoderm (Nichols et al. 2009), indicating that Erk1/2 signaling is also required for primitive endoderm formation and thus is likely responsible for mediating Fgfr function.
Figure 3.
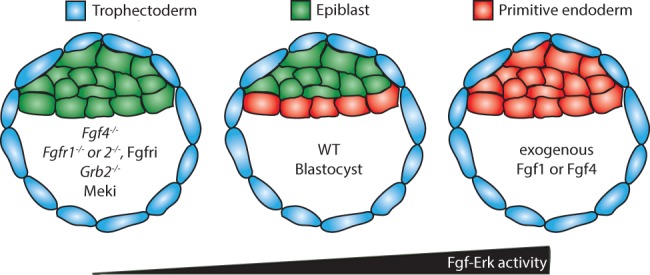
Fgf–Erk1/2 signaling regulates the composition of the inner cell mass. The inner cell mass of the blastocyst is composed of epiblast (green) and primitive endoderm (red) cells. Decreasing Fgf or Erk1/2 signaling through pharmacological inhibition or genetically disrupting components of the pathway produces blastocysts with fewer primitive endoderm cells. Conversely, the composition of the inner cell mass can be shifted toward the primitive endoderm cell fate by culturing embryos in an excess of exogenous Fgf ligands.
Genetic studies have also supported the model that Fgf uses Erk1/2 in the formation of primitive endoderm. Fgf4−/− mutants fail to express primitive endoderm markers at implantation and die around this time (Feldman et al. 1995; Goldin and Papaioannou 2003; Kang et al. 2013). Genetic disruption of Fgfr1 shifts the composition of the inner cell mass in favor of the epiblast lineage and produces fewer primitive endoderm cells (Brewer et al. 2015). Fgfr2 is also thought to mediate this process, since this receptor is expressed in the primitive endoderm, and some Fgfr2−/− mutants die at implantation (Arman et al. 1998; Blak et al. 2007). This raises the possibility that Fgfr1 and Fgfr2 function together during primitive endoderm formation, although this possibility needs to be tested. Genetic loss of Mapk1 or Mapk3 (the genes encoding Erk2 and Erk1, respectively) has not been associated with primitive endoderm defects to date (Pages et al. 1999; Saba-El-Leil et al. 2003). However, it is possible that Erk1 and Erk2 function redundantly in this context. The inner cell mass of Grb2−/− mutants is also composed entirely of epiblast cells and is devoid of primitive endoderm (Chazaud et al. 2006). Grb2 associates with Frs2 and Shp2, allowing Fgfrs to engage the Ras–Erk1/2 pathway through Sos (Fig. 2A; Ong et al. 1996; Kouhara et al. 1997; Hadari et al. 1998). Grb2 is also capable of engaging PI3K downstream from Fgfrs (Fig. 2B; Ong et al. 2001), suggesting that loss of PI3K activation may also contribute to the failure of Grb2−/− mutants to form primitive endoderm. However, no defects in primitive endoderm have been associated with decreased PI3K activity in the blastocyst (Brachmann et al. 2005; Riley et al. 2005, 2006).
Fgfr1 functions through Erk1/2 in the segmentation clock
Fgf signaling also functions through Erk1/2 during axial elongation and periodic somite formation. Axial elongation depends on cell movements in the presomitic mesoderm that facilitate posterior outgrowth (Hubaud and Pourquié 2014). Modulating the activity of Fgf8, Fgfr1, or Erk1/2 influences cell movements and therefore axis elongation in zebrafish and chicks (Dubrulle et al. 2001; Sawada et al. 2001; Delfini et al. 2005). In mice, an Fgf8 gradient observed in the presomitic mesoderm was shown to correlate with a gradient of Akt activity, raising the possibility that Fgfr1 also functions through PI3K in this context (Dubrulle and Pourquié 2004). However, pharmacological inhibition of PI3K did not affect cell movements or axial elongation in chicks, suggesting that PI3K activity is dispensable for Fgf-mediated axial elongation in this species (Delfini et al. 2005). No functional interrogation of PI3K activity in murine axial elongation has been described to date.
Periodic somite formation involves oscillating activity of multiple signaling pathways (Dubrulle and Pourquié 2004). During this process, the most anterior presomitic mesoderm condenses and forms somites. Erk1/2 activity oscillates, but a similar oscillating expression has not been described for Fgf ligands or receptors (Niwa et al. 2011). Instead, Fgf8 is present in a gradient throughout the presomitic mesoderm, while Sprouty2/4 and Dusp4/6 feedback inhibitors oscillate and may regulate Erk1/2 activity (Dequeant et al. 2006; Niwa et al. 2007; Hayashi et al. 2009). In addition, Shp2 oscillations have been described, suggesting that constant receptor activation and differential expression of this signaling protein may also contribute to oscillating Erk1/2 activity (Dequeant et al. 2006). Conditional disruption of Fgf4 and Fgf8 or Fgfr1 produces characteristic segmentation defects in which expression of cyclic genes is lost and presomitic mesoderm prematurely differentiates into disorganized somite structures, resulting in truncation of the embryo's posterior end (Niwa et al. 2007; Wahl et al. 2007; Naiche et al. 2011). Similarly, inhibition of Fgfrs or Erk1/2 results in reduced random cell motility and abolished expression of cyclic genes belonging to multiple pathways (Delfini et al. 2005; Niwa et al. 2007; Benazeraf et al. 2010). Collectively, these results indicate that Fgf functions through Erk1/2 in axial elongation and periodic somite formation.
Fgf8–Erk1/2 is required for development of the facial prominences
A functional requirement for Erk1/2 activity downstream from Fgf signaling has also been demonstrated in the developing pharyngeal arches. Erk1/2 is highly activated in an Fgfr-dependent fashion in the pharyngeal arches (Corson et al. 2003). Additionally, conditional inactivation of Fgf8 in the ectoderm of the first pharyngeal arch or Mapk1 and Mapk3 in the neural crest-derived mesenchyme produces similar phenotypes, characterized by agenesis of the maxillary and mandibular prominences and clefting of the nasal prominences (Trumpp et al. 1999; Newbern et al. 2008; Griffin et al. 2013). Conditional inactivation of Fgfr1 in the neural crest-derived mesenchyme produced a milder phenotype of midline facial clefting and normal development of the mandible (Trokovic et al. 2003; Wang et al. 2013; Brewer et al. 2015). Combined deletion of Fgfr1 and Fgfr2 in neural crest cells did produce a more severe facial cleft, although these mutants still fail to recapitulate the facial agenesis caused by conditional loss of Fgf8 (Park et al. 2008). Therefore, Fgf8 likely functions through multiple Fgfrs to engage Erk1/2 during development of the pharyngeal mesenchyme.
Fgf–Erk1/2 signaling regulates epithelial–mesenchymal interactions in the limb
Reciprocal Fgf signaling between the epithelium and mesenchyme during limb development is also mediated by Erk1/2. Fgf10 expressed in the limb bud mesenchyme activates Fgfr2b in the presumptive apical ectodermal ridge (AER), which in turn induces Fgf8 expression in the AER (Fig. 4; Min et al. 1998; Xu et al. 1998b; De Moerlooze et al. 2000). Fgf4, Fgf9, and Fgf17 are subsequently expressed in the AER together with Fgf8, and these ligands engage Fgfr1c and Fgfr2c in the mesenchyme to reinforce Fgf10 expression (Fig. 4; Mariani et al. 2008; Yu and Ornitz 2008). In this way, reciprocal Fgf signaling regulates induction and proximal/distal patterning of the limb. Consequently, genetic disruption of Fgf10 or Fgfr2b results in complete agenesis of the limbs (Min et al. 1998; Xu et al. 1998b; De Moerlooze et al. 2000). Conditional ablation of Fgf8 and Fgf4 or of Fgfr2 in the epithelium also causes a near complete agenesis of the hindlimb (Sun et al. 2002; Yu and Ornitz 2008). A less dramatic phenotype was observed in the forelimbs, characterized by missing distal elements (Sun et al. 2002; Yu and Ornitz 2008). The difference in severity between the forelimb and hindlimb defects may be attributed to the activity of the Msx2-Cre driver, which is observable in the hindlimb at an earlier stage than the forelimb (Sun et al. 2000). Activated Erk1/2 is observable in the limb bud mesenchyme, and AER and has been shown to depend on Fgfr signaling (Corson et al. 2003). Hindlimb agenesis accompanied by distal forelimb defects is also observed in conditional mutants that lack Mapk1 in the embryo proper (Fremin et al. 2015). It is not known why loss of Mapk1 produces more severe defects in hindlimb development, although this may suggest that forelimbs and hindlimbs require different levels of Erk1/2 signaling. It is also unknown whether disruption of both Mapk1 and Mapk3 would recapitulate the limb agenesis phenotypes reported in Fgf10−/− and Fgfr2b−/− mutants (Min et al. 1998; Xu et al. 1998b; De Moerlooze et al. 2000).
Figure 4.

Fgf mediates reciprocal tissue interactions during induction and proximal/distal patterning of the limb. Limb induction depends on mesenchyme-derived Fgf10 engaging Fgfr2b in the adjacent AER (blue). Fgfr2b then functions through Erk1/2 to induce expression of Fgf4, Fgf8, Fgf9, and Fgf17 in the AER, which activate Fgfr1c and Fgfr2c in the adjacent mesenchyme (green). Fgfr1c and Fgfr2c then function to reinforce expression of Fgf10 and instruct limb outgrowth through Erk1/2. (P/D) Proximal/distal.
Fgfr3 functions through Erk1/2 to inhibit chondrocyte hypertrophic differentiation
In growth plate chondrocytes, Fgfr3 functions through Erk1/2 to regulate postnatal hypertrophic differentiation. Fgfr3 limits long bone growth by inhibiting chondrocyte proliferation and hypertrophic differentiation (Colvin et al. 1996; Deng et al. 1996). Loss of Fgfr3 function therefore causes long bone overgrowth, while activating mutations in Fgfr3 cause skeletal dwarfism (Colvin et al. 1996; Deng et al. 1996; Naski et al. 1998; Chen et al. 1999; Li et al. 1999; Wang et al. 1999). Similarly, genetic inactivation of Mapk1 and Mapk3 in chondrocytes causes long bone overgrowth (Sebastian et al. 2011). Transgenic expression of a constitutively active Map2k1 allele (the gene encoding Mek1) in chondrocytes also causes skeletal dwarfism associated with fewer hypertrophic chondrocytes but normal chondrocyte proliferation (Murakami et al. 2004). Activation of Mek1 is also capable of rescuing the long bone overgrowth caused by loss of Fgfr3, indicating that Erk1/2 functions downstream from Fgfr3 to regulate long bone growth through hypertrophic differentiation (Murakami et al. 2004). Some studies have also proposed that Erk1/2 regulates Fgfr3-mediated inhibition of chondrocyte proliferation (Raucci et al. 2004; Krejci et al. 2008). However, other studies have suggested that this process is mediated by Stat1 (Sahni et al. 1999, 2001; Murakami et al. 2004).
Fgfrs function through PI3K in GnRH-producing neurons and during lens cell survival
PI3Ks contain p85 regulatory and p110 catalytic subunits that function as heterodimers (Thorpe et al. 2015). Fgfrs activate the PI3K/Akt pathway through Frs2. This occurs through Grb2-mediated recruitment of Gab1 independently of Ras (Fig. 2B; Ong et al. 2001). However, disruption of the Fgfr1–Frs2 protein complex fails to reduce Fgfr1-mediated phosphorylation of Akt, suggesting that Fgfrs may also possess an Frs2-independent mechanism to engage this pathway (Hoch and Soriano 2006; Brewer et al. 2015). Fgfr1–4 have been shown to recruit p85 directly (Table 1), although here p85 is thought to function independently of the PI3K/Akt pathway (Fig. 6A, below; Salazar et al. 2009; Francavilla et al. 2013).
Figure 6.
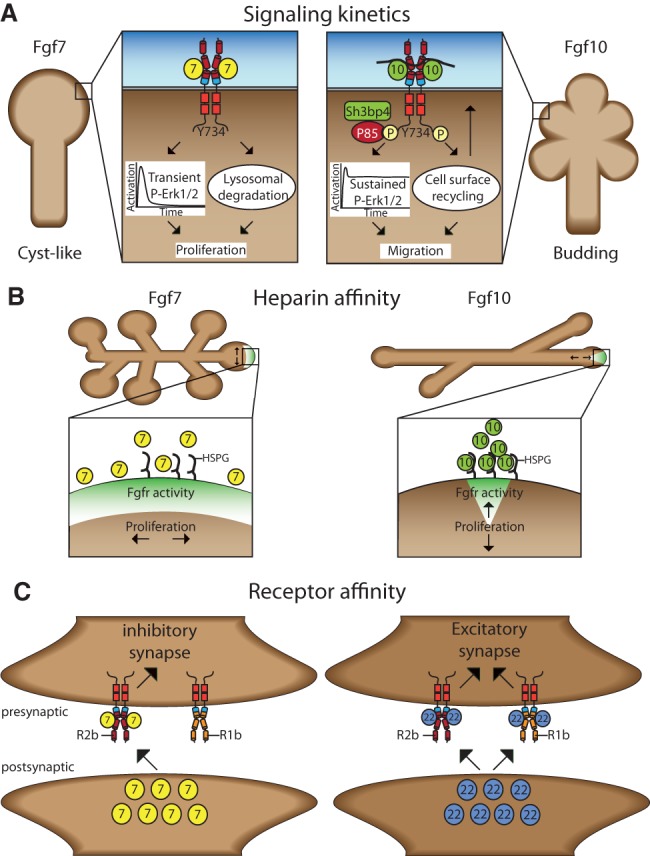
Fgf ligands encode distinct biological responses through diverse mechanisms. (A) FGF7 and FGF10 induce distinct lung explant morphologies and cellular responses through differential signal durations. (B) Differential HSPG affinity influences the shape of FGF7 and FGF10 gradients to influence the pattern of proliferating cells and tissue morphology in submandibular gland explants. (C) Fgf7 and Fgf22 instruct differentiation of inhibitory or excitatory presynaptic terminals by engaging distinct Fgfrs in hippocampal CA3 pyramidal neurons.
PI3K is required for Fgf-mediated development of GnRH-secreting neurons, which regulate the production of gonadotropin to control puberty and gametogenesis. In humans, loss-of-function mutations in FGF8 and FGFR1 cause hypogonadism that is characterized by stunted puberty and infertility (Dode et al. 2003; Pitteloud et al. 2006; Falardeau et al. 2008). Transgenic mice that express a dominant-negative allele of Fgfr1 exhibit delayed puberty and compromised fertility and have fewer GnRH-expressing neurons with less projections (Tsai et al. 2005; Gill and Tsai 2006). Decreases in fertility have also been documented in mice that conditionally lack Pik3r1 (the murine gene encoding p85α) in GnRH-expressing neurons (Acosta-Martinez et al. 2009). Conditional loss of Mapk3 and Mapk1 in these neurons had no affect on fertility (Wierman et al. 2012). Studies in chicks have also demonstrated that pharmacological inhibition of Fgfrs or PI3K signaling affected GnRH migration in ovo, but this process was not altered by Mek inhibition (Hu et al. 2013). Collectively, these studies suggest that Fgfr1 mediates GnRH neural migration through PI3K signaling.
In the eye, Fgfr2 is required for cell survival and differentiation of the lens. Conditional disruption of Fgfr2 in the lens results in increased cell death, which can be rescued by concurrent loss of Pten, a negative regulator of the PI3K/Akt pathway. However, disruption of Pten failed to rescue subsequent differentiation defects observed in Fgfr2 conditional mutants. These results suggest that Fgfr2 functions through the PI3K/Akt pathway to regulate cell survival and that additional pathways are involved in differentiation (Chaffee et al. 2016).
Plcγ functions in Fgfr1-mediated vertebral patterning and Fgfr4-induced cardiac hypertrophy
Plcγ binds the Fgfr1 C-terminal tail at phosphotyrosine 766 via the Plcγ SH2 domain (Table 1; Mohammadi et al. 1991; Peters et al. 1992). Subsequent tyrosine phosphorylation of Plcγ results in activation of the enzyme and hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol (1,4,5) trisphosphate (IP3) (Mohammadi et al. 1992; Peters et al. 1992). IP3 is soluble and diffuses to the endoplasmic reticulum, where it binds IP3 receptors to release Ca2+ from the endoplasmic reticulum. The resulting elevated cytosolic Ca2+ concentration, in cooperation with the membrane-bound DAG, activates Pkc (Fig. 2B; Huang et al. 1995).
It has been proposed that Plcγ also contributes to Erk1/2 activation by acting at the level of Raf1, based on the analysis of Y766F mutations engineered in Fgfr1 to abrogate Fgfr1–Plcγ binding (Huang et al. 1995). Another study has demonstrated that Shb also interacts with Fgfr1 at phosphorylated Y766 to recruit Shp2 (Cross et al. 2002). The Shb–Shp2 protein complex is required for maximal activation of Frs2 and recruitment of additional Grb2 molecules (Hadari et al. 1998; Cross et al. 2002). This may provide an alternative, Plcγ-independent mechanism by which Fgfr1 phosphotyrosine 766 is required for maximal Erk1/2 activation.
Fgfr1–Plcγ signaling negatively regulates the duration of Fgfr signaling by initiating internalization of the receptor in vitro (Sorokin et al. 1994). This model was also supported in vivo by generating mice harboring a Y766F amino acid substitution (Fgfr1Y766F) that prevents Plcγ from binding the receptor (Partanen et al. 1998). Fgfr1Y766F/Y766F mice exhibit posteriorization of the vertebral column, while Fgfr1Y766F/+ mice present a similar phenotype with lower penetrance. The opposite homeotic transformation (vertebral anteriorization) is present in mice homozygous for an Fgfr1 hypomorphic allele (Fgfr1hypo/hypo) and transheterozygous mice containing hypomorphic and null alleles (Fgfr1hypo/−). This indicates that Fgfr1Y766F is a semidominant, gain-of-function mutation and that Plcγ or downstream pathway members such as Pkc may act as negative regulators of Fgfr1 (Partanen et al. 1998).
Fgfr4 functions through Plcγ in cardiomyocytes during disease progression of left ventricular hypertrophy (LVH) (Faul et al. 2011; Grabner et al. 2015). Fgf23 functions as an endocrine hormone to regulate phosphate homeostasis and is found at high levels in individuals with chronic kidney disease (Faul et al. 2011). Elevated levels of Fgf23 cause LVH by activating Fgfr4–Plcγ signaling independently of Frs2–Erk1/2 (Faul et al. 2011; Grabner et al. 2015). Additionally, pharmacological inhibition of Plcγ prevented Fgf23-induced hypertrophy of neonatal rat ventricular cardiomyocytes to a greater extent than Mek inhibition in vitro (Faul et al. 2011). Genetic disruption of Fgfr4 also prevented LVH and Plcγ activation in an Fgf23-dependent model of chronic kidney disease (Grabner et al. 2015). LVH and Plcγ activation were observed in mice homozygous for a G385R-activating mutation in Fgfr4, collectively indicating that Fgfr4 signaling is necessary and sufficient for LVH pathogenesis (Grabner et al. 2015). Plcγ functions in LVH pathogenesis by regulating Ca2+ and the calcineurin/NFAT pathway, a potent inducer of cardiac hypertrophy (Molkentin et al. 1998; Faul et al. 2011; Grabner et al. 2015).
Pkcδ is required during ossification
Pkcs are a family of serine threonine kinases organized into three categories based on mechanisms of activation (Hage-Sleiman et al. 2015). Conventional (c) Pkcs (α, β, and γ) are activated by DAG, Ca2+, and phorbol esters, while novel (n) Pkcs (δ, ε, η, and θ) are activated by DAG and phorbol esters but not Ca2+. Atypical (a) Pkcs (ζ, ι, and μ) are activated by protein–protein interactions rather than secondary messengers (Hage-Sleiman et al. 2015). Fgfrs engage Pkc through Plcγ (Fig. 2), although little is known about this signaling function in vivo. In osteoblast cell lines, FGF2 enhances Runx2 expression and DNA-binding activity in a Pkc-dependent fashion (Kim et al. 2003). Inhibition of individual Pkc isoforms indicates that this process primarily relies on Pkcδ (Niger et al. 2013). Accordingly, Pkcδ−/− mutant mice exhibit delayed ossification of many skeletal structures, although Pkcδ activity is thought to be downstream from the noncanonical Wnt pathway in this context (Tu et al. 2007). Similar delays in ossification have also been reported in Fgf18−/− mutant mice, suggesting that Fgf18 could initiate a Pkcδ-dependent pathway in osteogenesis (Liu et al. 2002; Ohbayashi et al. 2002). This hypothesis is speculative, however, as Pkcδ activity has not been evaluated in Fgf18−/− mutants to determine whether this signaling pathway is regulated by Fgf signaling.
Adaptor protein Grb14
Grb14 was identified as an Fgfr1-binding protein in a yeast two-hybrid screen (Reilly et al. 2000). This interaction is mediated by the Grb14 SH2 domain and C-terminal Fgfr1 phosphotyrosines 766 and 776 (Table 1; Fig. 2B). Overexpression of Grb14 inhibited FGF2-mediated proliferation, suggesting that Grb14 functions as a negative regulator of Fgfr signaling (Reilly et al. 2000). It has been proposed that Grb14 inhibits Fgfr signaling by preventing recruitment and activation of Plcγ to phosphotyrosine 766 (Browaeys-Poly et al. 2010). Grb14−/− mice are viable and fertile with metabolic phenotypes that are generally attributed to alterations in signaling through the insulin receptor (Cooney et al. 2004). It is therefore not known whether Grb14 contributes to Fgf-mediated biological processes in vivo.
Stat1 functions downstream from Fgfr3 to inhibit chondrocyte proliferation
Stats are a family of proteins that bind transmembrane receptors and function in the nucleus as transcription factors. Stats are tyrosine-phosphorylated, often by Jak nonreceptor tyrosine kinases, which allows them to dimerize and translocate to the nucleus. In vitro studies have demonstrated that Stat1, Stat3, and Stat5 can be activated by Fgfrs (Hart et al. 2000; Deo et al. 2002; Yang et al. 2009; Dudka et al. 2010).
Fgfr3 functions through Stat1 to regulate chondrocyte proliferation during postnatal endochondral ossification. During this process, chondrocytes proliferate, exit the cell cycle, and undergo hypertrophic differentiation (Ornitz and Marie 2015). Fgfr3 signaling regulates bone growth by inhibiting both chondrocyte proliferation and hypertrophic differentiation (Colvin et al. 1996; Deng et al. 1996). Therefore, loss of Fgfr3 causes skeletal overgrowth, while activating mutations in Fgfr3 cause skeletal dwarfism in mice and humans (Colvin et al. 1996; Deng et al. 1996; Naski et al. 1998; Chen et al. 1999; Li et al. 1999; Wang et al. 1999). It has been proposed that Fgfr3 uses Stat1 to inhibit chondrocyte proliferation and Erk1/2 to restrict hypertrophic differentiation (Murakami et al. 2004). Stat1 is activated by FGF1 treatment in primary chondrocytes and is required for FGF1-mediated growth arrest in these cells (Sahni et al. 1999). Genetic loss of Stat1 also rescues the shortening of long bones induced through transgenic overexpression of FGF2 in mice by restoring normal proliferation rates (Sahni et al. 2001). Intriguingly, genetic loss of Stat1 restores normal chondrocyte proliferation in mice expressing an activating Fgfr3G374R mutant allele but does not restore normal long bone length (Murakami et al. 2004).
STAT3 binds phosphotyrosine 677 of FGFR1 in cell lines containing genomic amplification of the receptor but not in cells that express FGFR1 at endogenous levels (Table 1; Dudka et al. 2010). Thus, FGFR1-mediated activation of STAT3 may represent a cancer-specific signaling function of FGFR1. Consistent with this hypothesis, Stat3 is dispensable for Fgfr1-mediated murine facial morphogenesis (Brewer et al. 2015). Similarly, STAT3 preferentially binds a germline G388R variant of FGFR4 that has been associated with multiple cancer types (Ulaganathan et al. 2015). Here, the FGFR4G388R allele alters the transmembrane domain of FGFR4, creates a membrane-proximal STAT3-binding site, and facilitates increased STAT3 activation (Ulaganathan et al. 2015). Therefore, both FGFR1 and FGFR4 possess cancer-specific signaling functions through Stat3.
p38 functions in Fgfr2-mediated pathological skin and bone development
The p38 serine threonine kinases represent a family of MAPKs activated by cellular stress and several growth factors. FGF1 or FGF18 treatment is capable of activating p38 in chondrocyte cell lines (Shimoaka et al. 2002; Raucci et al. 2004). Little is known about the mechanism by which Fgfrs engage the p38 pathway, although it has been shown to depend on Ras (Tan et al. 1996). p38 contributes to some aspects of congenital disorders caused by activating alleles of Fgfr2. Beare-Stevenson cutis gyrata syndrome is caused by constitutively active mutations in FGFR2 and is associated with craniosynostosis, epidermal hyperplasia, and other skin and skeletal abnormalities in humans (Beare et al. 1969; Stevenson et al. 1978; Hall et al. 1992). Many of these phenotypes were also observed in mice engineered with the analogous mutation (Fgfr2Y394C allele) (Wang et al. 2012). p38 activity was higher in both the skin and skull of Fgfr2Y394C/+ mutants, while Erk1/2 activity was elevated only in the skull. In utero inhibition of p38 allowed for normal skin development by restoring epidermal proliferation to wild-type levels but did not attenuate the skull defects caused by constitutive Fgfr2 signaling. Treatment with a Mek inhibitor did not modify the skin or skull phenotypes in mutant mice (Wang et al. 2012), suggesting that signaling through p38 but not Erk1/2 is required downstream from Fgfr2Y394C during epidermal hyperplasia.
Other studies have evaluated the requirement for specific signaling pathways during pathologic endochondral ossification in a mouse model of Apert syndrome (Yin et al. 2008; Chen et al. 2014a). Apert syndrome is caused by S252W- or P253R-activating mutations in FGFR2 and is characterized by craniosynostosis and syndactyly (Wilkie et al. 1995). Fgfr2S252W/+ and Fgfr2P253R/+ mice are smaller than their wild-type counterparts, with decreased bone length and mass (Yin et al. 2008; Chen et al. 2014a). Both p38 and Erk1/2 activity was higher in bone mesenchyme stem cells derived from mutant mice, and inhibition of either pathway was capable of restoring normal length in cultured long bones (Yin et al. 2008; Chen et al. 2014a). The craniosynostosis associated with Apert syndrome is Erk1/2-dependent and can be prevented or treated using a Mek inhibitor (Shukla et al. 2007; Yin et al. 2008).
Fgfr4 functions through Jnk to regulate bile acid synthesis
The Jnk serine threonine kinases represent a family of MAPKs activated by cellular stress and growth factors. Treatment of FGF19 on primary human hepatocytes represses the rate-determining enzyme of bile acid synthesis, CYPZA1, in a JNK-dependent fashion (Holt et al. 2003). Additionally, Fgfr4−/− mice exhibited higher levels of bile acid production and Cypza1 expression, while transgenic mice expressing a constitutively active allele of Fgfr4 in the liver had decreased bile acid synthesis and lower Cypza1 expression and exhibited greater Jnk activity (Yu et al. 2000, 2005). Fgfr1 has been shown to engage Jnk through the Crk adaptor proteins C3G and Rap1 (Fig. 2B; Larsson et al. 1999). It is not known whether Fgfr4 uses a similar mechanism to activate Jnk.
Utilization of signaling functions
Fgfrs possess many signaling functions, raising the question of whether these effectors work individually or in combination. For example, Frs2 appears to be important in Fgfr1-mediated mesoderm formation (Gotoh et al. 2005), while CrkL has been implicated in pharyngeal arch development downstream from Fgfr1 and Fgfr2 (Moon et al. 2006). Does this suggest that Fgfrs use distinct effectors to activate Erk1/2 during different biological processes? Several studies addressing these questions have demonstrated that Fgfrs use diverse signaling mechanisms throughout development.
Fgfr1 requires the cumulative effect of multiple signaling effectors that converge on downstream pathways (Fig. 5, left panel). This model was developed by analyzing the phenotype of mice with knock-in mutations designed to disrupt the ability of Fgfr1 to bind and therefore activate a subset of signaling functions. Loss of the ability to engage Frs2, Crk proteins, or Plcγ individually produced only subtle defects relative to the Fgfr1-null phenotype (Partanen et al. 1998; Hoch and Soriano 2006; Brewer et al. 2015). Importantly, loss of individual signaling functions influenced similar Fgfr1-mediated developmental processes, most notably anterior/posterior patterning of the thoracic vertebrae (Partanen et al. 1998; Brewer et al. 2015). Disruption of multiple signaling functions simultaneously produced more severe developmental defects, including developmental retardation, posterior truncations, and agenesis of the second pharyngeal arch, indicating that Fgfr1 uses these signaling functions additively (Brewer et al. 2015). Similarly, Erk1/2 activation was only modestly decreased in primary cells when Fgfr1 was unable to engage CrkL, Plcγ, and Grb14 collectively or Frs2 individually (Hoch and Soriano 2006; Brewer et al. 2015). Combined disruption of these signaling functions led to decreases in Erk1/2 activation that were similar to complete loss of Fgfr1 function (Brewer et al. 2015). This supports the idea that Erk1/2 is engaged downstream from Fgfr1 through the combination of multiple effectors (Fig. 5, left panel). Plcγ is also likely to be engaged through multiple mechanisms by Fgfr1, since this signaling molecule is activated both directly by the receptor and in an Frs2-dependent fashion (Brewer et al. 2015). Additionally, Akt phosphorylation was not decreased by mutations engineered to disrupt the Fgfr1–Frs2 protein complex, suggesting that Fgfr1 also possesses additional mechanisms to activate the PI3K/Akt pathway (Hoch and Soriano 2006; Brewer et al. 2015).
Figure 5.
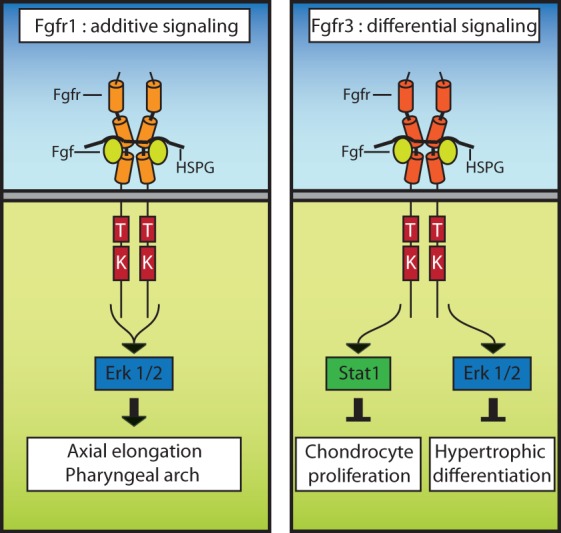
Fgfrs use diverse mechanisms of signaling. Fgfr1 functions through multiple effectors that converge on downstream Erk1/2 signaling in multiple contexts, including axial elongation and development of the pharyngeal arches. In contrast, Fgfr3 uses differential signaling during distinct cellular responses in growth plate chondrocytes. Here, Stat1 is engaged to limit chondrocyte proliferation, and Erk1/2 is subsequently used to inhibit hypertrophic differentiation.
Similar allelic series of signaling mutations have not yet been described for other Fgfrs. However, Fgfr3 may use a distinct mechanism in growth plate chondrocytes that relies on differential signaling to inhibit proliferation and differentiation (Fig. 5, right panel). Stat1 is used by Fgfr3 in order to increase expression of p21 and inhibit chondrocyte proliferation (Sahni et al. 1999, 2001; Murakami et al. 2004). Fgfr3 then functions through Erk1/2 to restrict hypertrophic differentiation (Murakami et al. 2004). In contrast to Fgfr1, Fgfr3 may therefore use differential signaling functions during distinct developmental processes. Similar studies of Pdgfrs have also demonstrated that Pdgfrα has distinct requirements for individual signaling functions, while Pdgfrβ requires the additive effect of multiple pathways (Tallquist et al. 2000, 2003; Klinghoffer et al. 2002). This supports the notion that evolutionarily related RTKs can function through distinct mechanisms.
Ligand-specific cellular responses
An emerging theme in Fgf signaling is that cellular responses are often encoded in the identity of the ligand. Different Fgf ligands can therefore initiate distinct developmental responses in the same tissue. This is achieved through multiple mechanisms, some of which function by initiating distinct properties of intracellular signaling.
Culture of lung explants with FGF7 or FGF10 generates cyst-like or branched structures by inducing proliferation or migration, respectively (Fig. 6A; Bellusci et al. 1997; Francavilla et al. 2013). These ligands induce distinct tissue morphologies by initiating different kinetics of FGFR2b signaling (Francavilla et al. 2013). FGF10 but not FGF7 stimulation induces phosphorylation of intracellular Tyr734 on FGFR2b. Phosphotyrosine 734 functions as a docking site for p85 bound to SH3BP4, which enables receptor recycling back to the cell surface and sustained receptor activation. Mutation of Y734F switches the kinetics of FGF10-activated FGFR2b to a transient signal and the structure of lung explants to resemble an FGF7-induced morphology (Francavilla et al. 2013).
An alternative mechanism has been proposed in the submandibular and lacrimal glands based on each ligand's HSPG affinity (Fig. 6B). Here, FGF7 or FGF10 produces branched or elongated structures in explants (Steinberg et al. 2005; Makarenkova et al. 2009). FGF7 binds HSPGs with a lower affinity than FGF10, allowing FGF7 to diffuse more extensively through the tissue, while FGF10 forms sharp gradients restricted to the tips (Igarashi et al. 1998; Makarenkova et al. 2009). These gradients influence the spatial pattern of proliferation within the tissue to regulate morphogenesis. Mutation of the FGF10 HSPG-binding domain functionally mimics FGF7 HSPG-binding properties to generate diffuse gradients and branched tissue structures (Makarenkova et al. 2009).
Distinct Fgf8 isoforms have also been shown to initiate different developmental programs that correlate with each isoform's receptor affinity. Eight Fgf8 isoforms (Fgf8a through Fgf8h) are generated by alternative splicing in mice (Crossley and Martin 1995; MacArthur et al. 1995). Fgf8a and Fgf8b induce distinct cellular responses when ectopically expressed in the chick neural plate. Fgf8a expression causes an expansion of the midbrain into the presumptive forebrain, while Fgf8b switches the fate of the midbrain to the cerebellum (Sato et al. 2001). Similar phenotypes have also been observed in transgenic mice that ectopically express Fgf8a or Fgf8b in the midbrain (Lee et al. 1997; Liu et al. 1999a). It is not known whether the Fgf8 isoforms induce distinct cellular programs by initiating different intracellular signaling profiles. However, Fgfr2c forms a larger hydrophobic interface with Fgf8b than Fgf8a, providing a greater affinity of the ligand to the receptor (Olsen et al. 2006). It therefore seems possible that Fgf8b may initiate a stronger signal than Fgf8a.
In the hippocampus, Fgf7 and Fgf22 have been shown to differentially promote the formation of inhibitory or excitatory presynaptic terminals, respectively (Fig. 6C; Terauchi et al. 2010). Mice genetically lacking Fgf7 or Fgf22 therefore have fewer inhibitory or excitatory synapses on hippocampal CA3 pyramidal neurons. Altering the balance of inhibitory/excitatory synapses influences the predisposition to experimentally induced epileptic seizures. Loss of Fgf7 therefore causes increased seizure susceptibility, while loss of Fgf22 protects against seizures. The ability to induce different synapse identities is dependent on differences in each ligand's receptor affinity (Terauchi et al. 2010; Dabrowski et al. 2015). Fgf7 primarily engages Fgfr2b, while Fgf22 can activate both Fgfr2b and Fgfr1b (Zhang et al. 2006). Genetic disruption of Fgfr2b caused decreases in inhibitory and excitatory synapses, while loss of Fgfr1b prevented only excitatory synapse development (Dabrowski et al. 2015). This supports the model that the differential presynaptic responses to each ligand are dependent on activation of distinct receptor profiles.
Of course, some Fgf ligands are likely to initiate similar cellular programs. For example, multiple ligands are expressed in the AER to regulate induction and proximal/distal patterning of the limb (Lewandoski et al. 2000; Sun et al. 2000, 2002; Mariani et al. 2008). Nevertheless, these studies collectively demonstrate that Fgf ligands can also induce distinct biological responses. This can be achieved by influencing intracellular signaling kinetics, altering the pattern of cellular responses within a tissue, or engaging distinct Fgfrs.
Considerations and future directions
Many genetic and pharmacological studies have demonstrated that Erk1/2 mediates many Fgfr functions in diverse biological contexts. However, these in vivo studies do have limitations to consider. One difficulty of these studies is the inability to definitively connect an individual RTK to specific signaling pathways in vivo. A commonly used approach is to determine whether reducing pathway activity phenocopies loss of a given RTK. However, results from this approach may be difficult to interpret, as many biological contexts require signaling through multiple RTKs. For example, the labyrinth compartment of the placenta requires Fgfr2, Met, Egfr, Igf1r, and many signaling pathways, including p38α, Erk1/2, and Akt (Liu et al. 1993; Bladt et al. 1995; Sibilia and Wagner 1995; Threadgill et al. 1995; Xu et al. 1998b; Adams et al. 2000; Mudgett et al. 2000; Hatano et al. 2003; Yang et al. 2003, 2005; Fremin et al. 2015). Since Fgfr2 is capable of engaging all of these pathways, it is difficult to know whether Fgfr2-associated placental defects are simply due to decreases in Erk1/2 signaling or whether these phenotypes are the result of lowering the activity of multiple signaling pathways. Similarly, it seems likely that phenotypes caused by decreased Erk1/2 activity alter signaling downstream from multiple RTKs. This problem is challenging given the technical difficulties associated with modulating multiple signaling pathways simultaneously in vivo.
Combining in vitro and in vivo strategies may be helpful in resolving this problem. One recent study has analyzed the transcriptional response to FGF1 treatment in primary cells derived from the mouse palatal mesenchyme (Vasudevan et al. 2015). Of note, only half of the genes that are transcriptionally regulated following FGF1 treatment depend on Erk1/2 activity (Vasudevan et al. 2015). It would therefore be interesting to determine whether any of the Fgf-regulated, Erk1/2-independent genes are required for Fgf-mediated palate morphogenesis.
Redundancy also complicates interpretation of genetic studies. Erk1 and Erk2 proteins are functionally equivalent kinases (Fremin et al. 2015), making it difficult but still genetically tractable to disrupt both the Mapk3 and Mapk1 genes. This is considerably more challenging for the PI3K/Akt pathway, since there are five isoforms of the p85 regulatory subunit, three isoforms of the p110 catalytic subunit, and three isoforms of Akt (Thorpe et al. 2015). Similarly, Fgfrs have been reported to signal through Src in vitro (Klint et al. 1999; Liu et al. 1999b; Li et al. 2004; Cunningham et al. 2010); however, genetic interrogation of this axis is not practical, since there are eight Src family kinases in mammals, four of which are broadly expressed. Additional strategies that rely on pharmacological inhibition or dominant-negative constructs may therefore be helpful in overcoming this issue.
Despite the prominent role for Erk1/2 in mediating Fgf-regulated biological processes, this review has also discussed several studies that have identified Erk1/2-independent signaling pathways used by Fgfrs. For example, Fgfr4 functions through Plcγ during cardiac hypertrophy and through Jnk when regulating bile acid synthesis (Holt et al. 2003; Inagaki et al. 2005; Yu et al. 2005; Faul et al. 2011; Grabner et al. 2015). Although a comprehensive analysis of all Fgfrs has not been performed to date, biochemical studies have suggested that Fgfrs possess different signaling potentials in vitro. Most notably, Fgfr1 possesses a greater ability to activate Frs2, Erk1/2, and Plcγ than Fgfr4 in vitro (Vainikka et al. 1994; Wang et al. 1994; Shaoul et al. 1995). This idea has also been supported in vivo, since Fgfr1 seems to primarily function through Erk1/2, while Fgfr4 uses Plcγ or Jnk. Therefore, there is substantial evidence to support the notion that Fgfr1 and Fgfr4 have qualitatively different signaling requirements. Less is known about qualitative or quantitative differences between other Fgfrs in vivo. Biochemical studies have also demonstrated that Fgfr1 is capable of initiating a greater magnitude of Erk1/2 activation than Fgfr2 (Shaoul et al. 1995), suggesting that these receptors exhibit quantitative differences in their signaling potentials. It may therefore be interesting to determine how the differential signaling potentials of Fgfrs are used in vivo.
Signaling kinetics represent another quantitative aspect of signaling that should be further investigated in vivo. The importance of signaling kinetics was initially demonstrated in studies of PC12 cells, which proliferate or differentiate in response to transient or sustained Erk1/2 activity, respectively (for review, see Marshall 1995). Recent phosphoproteomic studies have demonstrated that differential FGFR2b signaling kinetics instruct a proliferation or migration response during branching morphogenesis of the lung (Francavilla et al. 2013). Here, the duration of FGFR2b signaling is determined by ligand identity and differential phosphorylation of Y734 (Francavilla et al. 2013). It may therefore be possible to engineer mice that lack this phosphorylation site or contain a phosphomimetic allele to experimentally force a transient or prolonged FGFR2b signal in vivo. This approach would be useful in identifying how signaling kinetics influence development and adult homeostasis. Another study has used a FRET-based system to monitor the spatial and temporal dynamics of Erk1/2 activation in the skin (Hiratsuka et al. 2015). This strategy may be particularly useful to monitor the kinetics of Erk1/2 signaling in contexts amenable to live imaging, such as preimplantation development or explant culture systems.
Conclusion
In the many years that have followed the identification of Fgfrs, multiple studies have shed light on the diversity of Fgf signaling mechanisms in numerous developmental and homeostatic processes. In many biological contexts, Fgf signaling functions through the Erk1/2 pathway, although there is also strong evidence implicating Erk1/2-independent signaling functions in vivo. Additionally, quantitative differences in the magnitude or duration of Erk1/2 activation may be used to instruct diverse cellular responses. Collectively, these studies have provided new insights into signal transduction, informed the developmental etiologies of many congenital disorders, and may form the basis to develop novel therapeutic strategies.
Acknowledgments
We thank Stuart Aaronson, Robert Krauss, Ramon Parsons, and our laboratory colleagues for discussions and critical comments on the manuscript. We apologize to authors whose work we did not cite due to space limitations. Work from the author's laboratory was supported by National Institutes of Health (NIH)/National Institute of Dental and Craniofacial Research (NIDCR) grant RO1 DE022778 and New York State Stem Cell Science (NYSTEM) grant IIRP N11G-131 to P.S. J.R.B. was supported by NIH/NIDCR fellowship F31 DE023686.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.277137.115.
References
- Acosta-Martinez M, Luo J, Elias C, Wolfe A, Levine JE. 2009. Male-biased effects of gonadotropin-releasing hormone neuron-specific deletion of the phosphoinositide 3-kinase regulatory subunit p85α on the reproductive axis. Endocrinology 150: 4203–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda AR. 2000. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol Cell 6: 109–116. [PubMed] [Google Scholar]
- Ahmed Z, George R, Lin CC, Suen KM, Levitt JA, Suhling K, Ladbury JE. 2010. Direct binding of Grb2 SH3 domain to FGFR2 regulates SHP2 function. Cell Signal 22: 23–33. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Lin CC, Suen KM, Melo FA, Levitt JA, Suhling K, Ladbury JE. 2013. Grb2 controls phosphorylation of FGFR2 by inhibiting receptor kinase and Shp2 phosphatase activity. J Cell Biol 200: 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Alam D, El Agha E, Sakurai R, Kheirollahi V, Moiseenko A, Danopoulos S, Shrestha A, Schmoldt C, Quantius J, Herold S, et al. 2015. Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 142: 4139–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Rapraeger AC. 2003. Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J Cell Biol 163: 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. 1998. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci 95: 5082–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak H, Huh SH, Chen S, Jeanpierre C, Martinovic J, Parisot M, Bole-Feysot C, Nitschke P, Salomon R, Antignac C, et al. 2012. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev Cell 22: 1191–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates CM. 2011. Role of fibroblast growth factor receptor signaling in kidney development. Pediatr Nephrol 26: 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare JM, Dodge JA, Nevin NC. 1969. Cutis gyratum, acanthosis nigricans and other congenital anomalies. A new syndrome. Br J Dermatol 81: 241–247. [PubMed] [Google Scholar]
- Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, Rohacek AM, Xing Y, Carstens RP. 2015. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 4: e08954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot F, Crumley G, Kaplow JM, Schlessinger J, Jaye M, Dionne CA. 1991. Ligand-induced transphosphorylation between different FGF receptors. EMBO J 10: 2849–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. 1997. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124: 4867–4878. [DOI] [PubMed] [Google Scholar]
- Benazeraf B, Francois P, Baker RE, Denans N, Little CD, Pourquie O. 2010. A random cell motility gradient downstream of FGF controls elongation of an amniote embryo. Nature 466: 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. 1995. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376: 768–771. [DOI] [PubMed] [Google Scholar]
- Blak AA, Naserke T, Saarimaki-Vire J, Peltopuro P, Giraldo-Velasquez M, Vogt Weisenhorn DM, Prakash N, Sendtner M, Partanen J, Wurst W. 2007. Fgfr2 and Fgfr3 are not required for patterning and maintenance of the midbrain and anterior hindbrain. Dev Biol 303: 231–243. [DOI] [PubMed] [Google Scholar]
- Brachmann SM, Yballe CM, Innocenti M, Deane JA, Fruman DA, Thomas SM, Cantley LC. 2005. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol Cell Biol 25: 2593–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer JR, Molotkov A, Mazot P, Hoch RV, Soriano P. 2015. Fgfr1 regulates development through the combinatorial use of signaling proteins. Genes Dev 29: 1863–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browaeys-Poly E, Blanquart C, Perdereau D, Antoine AF, Goenaga D, Luzy JP, Chen H, Garbay C, Issad T, Cailliau K, et al. 2010. Grb14 inhibits FGF receptor signaling through the regulation of PLCγ recruitment and activation. FEBS Lett 584: 4383–4388. [DOI] [PubMed] [Google Scholar]
- Cai Z, Tao C, Li H, Ladher R, Gotoh N, Feng GS, Wang F, Zhang X. 2013. Deficient FGF signaling causes optic nerve dysgenesis and ocular coloboma. Development 140: 2711–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert JA, Dedos SG, Hawker K, Fleming M, Lewis MA, Steel KP. 2011. A missense mutation in Fgfr1 causes ear and skull defects in hush puppy mice. Mamm Genome 22: 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter EP, Fearon AE, Grose RP. 2015. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol 25: 221–233. [DOI] [PubMed] [Google Scholar]
- Ceridono M, Belleudi F, Ceccarelli S, Torrisi MR. 2005. Tyrosine 769 of the keratinocyte growth factor receptor is required for receptor signaling but not endocytosis. Biochem Biophys Res Commun 327: 523–532. [DOI] [PubMed] [Google Scholar]
- Chaffee BR, Hoang TV, Leonard MR, Bruney DG, Wagner BD, Dowd JR, Leone G, Ostrowski MC, Robinson ML. 2016. FGFR and PTEN signaling interact during lens development to regulate cell survival. Dev Biol 410: 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazaud C, Yamanaka Y, Pawson T, Rossant J. 2006. Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2–MAPK pathway. Dev Cell 10: 615–624. [DOI] [PubMed] [Google Scholar]
- Chellaiah AT, McEwen DG, Werner S, Xu J, Ornitz DM. 1994. Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J Biol Chem 269: 11620–11627. [PubMed] [Google Scholar]
- Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV, Yayon A, Deng CX. 1999. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest 104: 1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zhang L, Weng T, Zhang S, Sun S, Chang M, Li Y, Zhang B, Zhang L. 2014a. A Ser252Trp mutation in fibroblast growth factor receptor 2 (FGFR2) mimicking human Apert syndrome reveals an essential role for FGF signaling in the regulation of endochondral bone formation. PLoS One 9: e87311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Qin L, Zhuang ZW, Tellides G, Lax I, Schlessinger J, Simons M. 2014b. The docking protein FRS2α is a critical regulator of VEGF receptors signaling. Proc Natl Acad Sci 111: 5514–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruna BG, Rossant J. 1999. Expression of the T-box gene Eomesodermin during early mouse development. Mech Dev 81: 199–203. [DOI] [PubMed] [Google Scholar]
- Ciruna B, Rossant J. 2001. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell 1: 37–49. [DOI] [PubMed] [Google Scholar]
- Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP, Rossant J. 1997. Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenetic movement through the primitive streak. Development 124: 2829–2841. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. 1996. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 12: 390–397. [DOI] [PubMed] [Google Scholar]
- Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, Biden TJ, Ormandy CJ, James DE, Daly RJ. 2004. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J 23: 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson LB, Yamanaka Y, Lai KM, Rossant J. 2003. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development 130: 4527–4537. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Lu L, Magnusson P, Nyqvist D, Holmqvist K, Welsh M, Claesson-Welsh L. 2002. The Shb adaptor protein binds to tyrosine 766 in the FGFR-1 and regulates the Ras/MEK/MAPK pathway via FRS2 phosphorylation in endothelial cells. Mol Biol Cell 13: 2881–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley PH, Martin GR. 1995. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121: 439–451. [DOI] [PubMed] [Google Scholar]
- Cunningham DL, Sweet SM, Cooper HJ, Heath JK. 2010. Differential phosphoproteomics of fibroblast growth factor signaling: identification of Src family kinase-mediated phosphorylation events. J Proteome Res 9: 2317–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski A, Terauchi A, Strong C, Umemori H. 2015. Distinct sets of FGF receptors sculpt excitatory and inhibitory synaptogenesis. Development 142: 1818–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degirolamo C, Sabba C, Moschetta A. 2016. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov 15: 51–69. [DOI] [PubMed] [Google Scholar]
- Degoutin J, Vigny M, Gouzi JY. 2007. ALK activation induces Shc and FRS2 recruitment: signaling and phenotypic outcomes in PC12 cells differentiation. FEBS Lett 581: 727–734. [DOI] [PubMed] [Google Scholar]
- Delfini MC, Dubrulle J, Malapert P, Chal J, Pourquié O. 2005. Control of the segmentation process by graded MAPK/ERK activation in the chick embryo. Proc Natl Acad Sci 102: 11343–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Moral PM, De Langhe SP, Sala FG, Veltmaat JM, Tefft D, Wang K, Warburton D, Bellusci S. 2006. Differential role of FGF9 on epithelium and mesenchyme in mouse embryonic lung. Dev Biol 293: 77–89. [DOI] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. 2000. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 127: 483–492. [DOI] [PubMed] [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. 1994. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev 8: 3045–3057. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. 1996. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84: 911–921. [DOI] [PubMed] [Google Scholar]
- Deo DD, Axelrad TW, Robert EG, Marcheselli V, Bazan NG, Hunt JD. 2002. Phosphorylation of STAT-3 in response to basic fibroblast growth factor occurs through a mechanism involving platelet-activating factor, JAK-2, and Src in human umbilical vein endothelial cells. Evidence for a dual kinase mechanism. J Biol Chem 277: 21237–21245. [DOI] [PubMed] [Google Scholar]
- Dequeant ML, Glynn E, Gaudenz K, Wahl M, Chen J, Mushegian A, Pourquié O. 2006. A complex oscillating network of signaling genes underlies the mouse segmentation clock. Science 314: 1595–1598. [DOI] [PubMed] [Google Scholar]
- Dhalluin C, Yan KS, Plotnikova O, Lee KW, Zeng L, Kuti M, Mujtaba S, Goldfarb MP, Zhou MM. 2000. Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Mol Cell 6: 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, et al. 2003. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33: 463–465. [DOI] [PubMed] [Google Scholar]
- Dorey K, Amaya E. 2010. FGF signalling: diverse roles during early vertebrate embryogenesis. Development 137: 3731–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrulle J, Pourquié O. 2004. fgf8 mRNA decay establishes a gradient that couples axial elongation to patterning in the vertebrate embryo. Nature 427: 419–422. [DOI] [PubMed] [Google Scholar]
- Dubrulle J, McGrew MJ, Pourquié O. 2001. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 106: 219–232. [DOI] [PubMed] [Google Scholar]
- Dudka AA, Sweet SM, Heath JK. 2010. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res 70: 3391–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Schlessinger J. 2007. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proc Natl Acad Sci 104: 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. 2002. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development 129: 3783–3793. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Ozcan F, Lew ED, Bae JH, Tome F, Booth CJ, Adams DJ, Lax I, Schlessinger J. 2006. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci 103: 18603–18608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzat S, Zheng L, Florez JC, Stefan N, Mayr T, Hliang MM, Jablonski K, Harden M, Stancakova A, Laakso M, et al. 2013. The cancer-associated FGFR4-G388R polymorphism enhances pancreatic insulin secretion and modifies the risk of diabetes. Cell Metab 17: 929–940. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Faber SC, Dimanlig P, Makarenkova HP, Shirke S, Ko K, Lang RA. 2001. Fgf receptor signaling plays a role in lens induction. Development 128: 4425–4438. [DOI] [PubMed] [Google Scholar]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, et al. 2008. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 118: 2822–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Aguillon-Prada R, Lincoln J, Hare JM, et al. 2011. FGF23 induces left ventricular hypertrophy. J Clin Invest 121: 4393–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. 1995. Requirement of FGF-4 for postimplantation mouse development. Science 267: 246–249. [DOI] [PubMed] [Google Scholar]
- Feller SM. 2001. Crk family adaptors-signalling complex formation and biological roles. Oncogene 20: 6348–6371. [DOI] [PubMed] [Google Scholar]
- Francavilla C, Rigbolt KT, Emdal KB, Carraro G, Vernet E, Bekker-Jensen DB, Streicher W, Wikstrom M, Sundstrom M, Bellusci S, et al. 2013. Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol Cell 51: 707–722. [DOI] [PubMed] [Google Scholar]
- Fremin C, Saba-El-Leil MK, Levesque K, Ang SL, Meloche S. 2015. Functional redundancy of ERK1 and ERK2 MAP kinases during development. Cell Rep 12: 913–921. [DOI] [PubMed] [Google Scholar]
- Gill JC, Tsai PS. 2006. Expression of a dominant negative FGF receptor in developing GNRH1 neurons disrupts axon outgrowth and targeting to the median eminence. Biol Reprod 74: 463–472. [DOI] [PubMed] [Google Scholar]
- Goldin SN, Papaioannou VE. 2003. Paracrine action of FGF4 during periimplantation development maintains trophectoderm and primitive endoderm. Genesis 36: 40–47. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Ito M, Yamamoto S, Yoshino I, Song N, Wang Y, Lax I, Schlessinger J, Shibuya M, Lang RA. 2004a. Tyrosine phosphorylation sites on FRS2α responsible for Shp2 recruitment are critical for induction of lens and retina. Proc Natl Acad Sci 101: 17144–17149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh N, Laks S, Nakashima M, Lax I, Schlessinger J. 2004b. FRS2 family docking proteins with overlapping roles in activation of MAP kinase have distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett 564: 14–18. [DOI] [PubMed] [Google Scholar]
- Gotoh N, Manova K, Tanaka S, Murohashi M, Hadari Y, Lee A, Hamada Y, Hiroe T, Ito M, Kurihara T, et al. 2005. The docking protein FRS2α is an essential component of multiple fibroblast growth factor responses during early mouse development. Mol Cell Biol 25: 4105–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, et al. 2015. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab 22: 1020–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JN, Compagnucci C, Hu D, Fish J, Klein O, Marcucio R, Depew MJ. 2013. Fgf8 dosage determines midfacial integration and polarity within the nasal and optic capsules. Dev Biol 374: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. 1993. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J Biol Chem 268: 23906–23914. [PubMed] [Google Scholar]
- Hadari YR, Kouhara H, Lax I, Schlessinger J. 1998. Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol 18: 3966–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage-Sleiman R, Hamze AB, Reslan L, Kobeissy H, Dbaibo G. 2015. The novel PKCθ from benchtop to clinic. J Immunol Res 2015: 348798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajihosseini MK, Wilson S, De Moerlooze L, Dickson C. 2001. A splicing switch and gain-of-function mutation in FgfR2-IIIc hemizygotes causes Apert/Pfeiffer-syndrome-like phenotypes. Proc Natl Acad Sci 98: 3855–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BD, Cadle RG, Golabi M, Morris CA, Cohen MM Jr. 1992. Beare-Stevenson cutis gyrata syndrome. Am J Med Genet 44: 82–89. [DOI] [PubMed] [Google Scholar]
- Harada M, Murakami H, Okawa A, Okimoto N, Hiraoka S, Nakahara T, Akasaka R, Shiraishi Y, Futatsugi N, Mizutani-Koseki Y, et al. 2009. FGF9 monomer–dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet 41: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ. 2000. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 19: 3309–3320. [DOI] [PubMed] [Google Scholar]
- Hart KC, Robertson SC, Donoghue DJ. 2001. Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol Biol Cell 12: 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, Niwa H, Miyazaki J, Hamaoka T, Ogata M. 2003. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 8: 847–856. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Shimoda T, Nakajima M, Tsukada Y, Sakumura Y, Dale JK, Maroto M, Kohno K, Matsui T, Bessho Y. 2009. Sprouty4, an FGF inhibitor, displays cyclic gene expression under the control of the notch segmentation clock in the mouse PSM. PLoS One 4: e5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert JM, Rosenquist T, Gotz J, Martin GR. 1994. FGF5 as a regulator of the hair growth cycle: evidence from targeted and spontaneous mutations. Cell 78: 1017–1025. [DOI] [PubMed] [Google Scholar]
- Higgins CA, Petukhova L, Harel S, Ho YY, Drill E, Shapiro L, Wajid M, Christiano AM. 2014. FGF5 is a crucial regulator of hair length in humans. Proc Natl Acad Sci 111: 10648–10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka T, Fujita Y, Naoki H, Aoki K, Kamioka Y, Matsuda M. 2015. Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. Elife 4: e05178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch RV, Soriano P. 2006. Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development 133: 663–673. [DOI] [PubMed] [Google Scholar]
- Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, et al. 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 17: 1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Poopalasundaram S, Graham A, Bouloux PM. 2013. GnRH neuronal migration and olfactory bulb neurite outgrowth are dependent on FGF receptor 1 signaling, specifically via the PI3K p110α isoform in chick embryo. Endocrinology 154: 388–399. [DOI] [PubMed] [Google Scholar]
- Huang P, Stern MJ. 2005. FGF signaling in flies and worms: more and more relevant to vertebrate biology. Cytokine Growth Factor Rev 16: 151–158. [DOI] [PubMed] [Google Scholar]
- Huang J, Mohammadi M, Rodrigues GA, Schlessinger J. 1995. Reduced activation of RAF-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem 270: 5065–5072. [DOI] [PubMed] [Google Scholar]
- Hubaud A, Pourquié O. 2014. Signalling dynamics in vertebrate segmentation. Nat Rev Mol Cell Biol 15: 709–721. [DOI] [PubMed] [Google Scholar]
- Huh SH, Jones J, Warchol ME, Ornitz DM. 2012. Differentiation of the lateral compartment of the cochlea requires a temporally restricted FGF20 signal. PLoS Biol 10: e1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi M, Finch PW, Aaronson SA. 1998. Characterization of recombinant human fibroblast growth factor (FGF)-10 reveals functional similarities with keratinocyte growth factor (FGF-7). J Biol Chem 273: 13230–13235. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2: 217–225. [DOI] [PubMed] [Google Scholar]
- Itoh N. 2007. The Fgf families in humans, mice, and zebrafish: their evolutional processes and roles in development, metabolism, and disease. Biol Pharm Bulletin 30: 1819–1825. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ohta H, Konishi M. 2015. Endocrine FGFs: evolution, physiology, pathophysiology, and pharmacotherapy. Front Endocrinol 6: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jijiwa M, Fukuda T, Kawai K, Nakamura A, Kurokawa K, Murakumo Y, Ichihara M, Takahashi M. 2004. A targeting mutation of tyrosine 1062 in Ret causes a marked decrease of enteric neurons and renal hypoplasia. Mol Cell Biol 24: 8026–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DE, Lu J, Chen H, Werner S, Williams LT. 1991. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol 11: 4627–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinina J, Dutta K, Ilghari D, Beenken A, Goetz R, Eliseenkova AV, Cowburn D, Mohammadi M. 2012. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure (London, England: 1993) 20: 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Piliszek A, Artus J, Hadjantonakis AK. 2013. FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development) 140: 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, et al. 2005. FGF-21 as a novel metabolic regulator. J Clin Invest 115: 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim JH, Bae SC, Choi JY, Kim HJ, Ryoo HM. 2003. The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J Biol Chem 278: 319–326. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, Soriano P. 2002. An allelic series at the PDGFαR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell 2: 103–113. [DOI] [PubMed] [Google Scholar]
- Klint P, Kanda S, Kloog Y, Claesson-Welsh L. 1999. Contribution of Src and Ras pathways in FGF-2 induced endothelial cell differentiation. Oncogene 18: 3354–3364. [DOI] [PubMed] [Google Scholar]
- Kong M, Wang CS, Donoghue DJ. 2002. Interaction of fibroblast growth factor receptor 3 and the adapter protein SH2-B. A role in STAT5 activation. J Biol Chem 277: 15962–15970. [DOI] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J. 1997. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89: 693–702. [DOI] [PubMed] [Google Scholar]
- Krejci P, Salazar L, Goodridge HS, Kashiwada TA, Schibler MJ, Jelinkova P, Thompson LM, Wilcox WR. 2008. STAT1 and STAT3 do not participate in FGF-mediated growth arrest in chondrocytes. J Cell Sci 121: 272–281. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Iwashita T, Murakami H, Hayashi H, Kawai K, Takahashi M. 2001. Identification of SNT/FRS2 docking site on RET receptor tyrosine kinase and its role for signal transduction. Oncogene 20: 1929–1938. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, et al. 2006. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281: 6120–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson H, Klint P, Landgren E, Claesson-Welsh L. 1999. Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the Src homology (SH) 2/SH3 domain-containing adaptor protein Crk. J Biol Chem 274: 25726–25734. [DOI] [PubMed] [Google Scholar]
- Lee PL, Johnson DE, Cousens LS, Fried VA, Williams LT. 1989. Purification and complementary DNA cloning of a receptor for basic fibroblast growth factor. Science 245: 57–60. [DOI] [PubMed] [Google Scholar]
- Lee SM, Danielian PS, Fritzsch B, McMahon AP. 1997. Evidence that FGF8 signalling from the midbrain-hindbrain junction regulates growth and polarity in the developing midbrain. Development 124: 959–969. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Sun X, Martin GR. 2000. Fgf8 signalling from the AER is essential for normal limb development. Nat Genet 26: 460–463. [DOI] [PubMed] [Google Scholar]
- Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. 1999. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet 8: 35–44. [DOI] [PubMed] [Google Scholar]
- Li X, Brunton VG, Burgar HR, Wheldon LM, Heath JK. 2004. FRS2-dependent SRC activation is required for fibroblast growth factor receptor-induced phosphorylation of Sprouty and suppression of ERK activity. J Cell Sci 117: 6007–6017. [DOI] [PubMed] [Google Scholar]
- Lin CC, Melo FA, Ghosh R, Suen KM, Stagg LJ, Kirkpatrick J, Arold ST, Ahmed Z, Ladbury JE. 2012. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell 149: 1514–1524. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. 1993. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75: 59–72. [PubMed] [Google Scholar]
- Liu A, Losos K, Joyner AL. 1999a. FGF8 can activate Gbx2 and transform regions of the rostral mouse brain into a hindbrain fate. Development 126: 4827–4838. [DOI] [PubMed] [Google Scholar]
- Liu J, Huang C, Zhan X. 1999b. Src is required for cell migration and shape changes induced by fibroblast growth factor 1. Oncogene 18: 6700–6706. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. 2002. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev 16: 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur CA, Lawshe A, Xu J, Santos-Ocampo S, Heikinheimo M, Chellaiah AT, Ornitz DM. 1995. FGF-8 isoforms activate receptor splice forms that are expressed in mesenchymal regions of mouse development. Development 121: 3603–3613. [DOI] [PubMed] [Google Scholar]
- Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA, Mohammadi M. 2009. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci Signal 2: ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SL, Twigg SR, Freeland RM, Wall SA, Li C, Wilkie AO. 2009. Hearing loss in a mouse model of Muenke syndrome. Hum Mol Genet 18: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani FV, Ahn CP, Martin GR. 2008. Genetic evidence that FGFs have an instructive role in limb proximal-distal patterning. Nature 453: 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179–185. [DOI] [PubMed] [Google Scholar]
- Mason I. 2007. Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci 8: 583–596. [DOI] [PubMed] [Google Scholar]
- Melillo RM, Santoro M, Ong SH, Billaud M, Fusco A, Hadari YR, Schlessinger J, Lax I. 2001. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol Cell Biol 21: 4177–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Bottaro DP, Fleming TP, Smith CL, Burgess WH, Chan AM, Aaronson SA. 1992. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc Natl Acad Sci 89: 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. 1998. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev 12: 3156–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J. 1991. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-γ1. Mol Cell Biol 11: 5068–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Dionne CA, Li W, Li N, Spivak T, Honegger AM, Jaye M, Schlessinger J. 1992. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature 358: 681–684. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. 1998. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93: 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. 2006. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell 10: 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudgett JS, Ding J, Guh-Siesel L, Chartrain NA, Yang L, Gopal S, Shen MM. 2000. Essential role for p38α mitogen-activated protein kinase in placental angiogenesis. Proc Natl Acad Sci 97: 10454–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. 2004. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev 18: 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiche LA, Holder N, Lewandoski M. 2011. FGF4 and FGF8 comprise the wavefront activity that controls somitogenesis. Proc Natl Acad Sci 108: 4018–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Gulick J, Colbert MC, Robbins J. 2009. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci 106: 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naski MC, Colvin JS, Coffin JD, Ornitz DM. 1998. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 125: 4977–4988. [DOI] [PubMed] [Google Scholar]
- Nelson DL, Cox MM. 2005. Lehninger principles of biochemistry. W.H. Freeman and Company, New York. [Google Scholar]
- Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, Cherosky N, Karlo JC, O'Loughlin B, Wikenheiser J, et al. 2008. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc Natl Acad Sci 105: 17115–17120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Silva J, Roode M, Smith A. 2009. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development 136: 3215–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niger C, Luciotti MA, Buo AM, Hebert C, Ma V, Stains JP. 2013. The regulation of runt-related transcription factor 2 by fibroblast growth factor-2 and connexin43 requires the inositol polyphosphate/protein kinase Cδ cascade. J Bone Miner Res 28: 1468–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa Y, Masamizu Y, Liu T, Nakayama R, Deng CX, Kageyama R. 2007. The initiation and propagation of Hes7 oscillation are cooperatively regulated by Fgf and notch signaling in the somite segmentation clock. Dev Cell 13: 298–304. [DOI] [PubMed] [Google Scholar]
- Niwa Y, Shimojo H, Isomura A, Gonzalez A, Miyachi H, Kageyama R. 2011. Different types of oscillations in Notch and Fgf signaling regulate the spatiotemporal periodicity of somitogenesis. Genes Dev 25: 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Kurosu H, Yamamoto M, Nandi A, Rosenblatt KP, Goetz R, Eliseenkova AV, Mohammadi M, Kuro-o M. 2007. βKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci 104: 7432–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. 2002. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev 16: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, Mohammadi M. 2003. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem 278: 34226–34236. [DOI] [PubMed] [Google Scholar]
- Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL, Mohammadi M. 2006. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev 20: 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SH, Goh KC, Lim YP, Low BC, Klint P, Claesson-Welsh L, Cao X, Tan YH, Guy GR. 1996. Suc1-associated neurotrophic factor target (SNT) protein is a major FGF-stimulated tyrosine phosphorylated 90-kDa protein which binds to the SH2 domain of GRB2. Biochem Biophys Res Commun 225: 1021–1026. [DOI] [PubMed] [Google Scholar]
- Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, Lax I. 2000. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol 20: 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. 2001. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci 98: 6074–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono K, Kita T, Sato S, O'Neill P, Mak SS, Paschaki M, Ito M, Gotoh N, Kawakami K, Sasai Y, et al. 2014. FGFR1–Frs2/3 signalling maintains sensory progenitors during inner ear hair cell formation. PLoS Genet 10: e1004118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM. 2000. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays 22: 108–112. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. 2015. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4: 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Leder P. 1992. Ligand specificity and heparin dependence of fibroblast growth factor receptors 1 and 3. J Biol Chem 267: 16305–16311. [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. 2015. Fibroblast growth factor signaling in skeletal development and disease. Genes Development 29: 1463–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. 1996. Receptor specificity of the fibroblast growth factor family. J Biol Chem 271: 15292–15297. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. 1991. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development 113: 1419–1434. [DOI] [PubMed] [Google Scholar]
- Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. 1999. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 286: 1374–1377. [DOI] [PubMed] [Google Scholar]
- Park EJ, Watanabe Y, Smyth G, Miyagawa-Tomita S, Meyers E, Klingensmith J, Camenisch T, Buckingham M, Moon AM. 2008. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development 135: 3599–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J. 1998. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev 12: 2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VN, Knox SM, Likar KM, Lathrop CA, Hossain R, Eftekhari S, Whitelock JM, Elkin M, Vlodavsky I, Hoffman MP. 2007. Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development 134: 4177–4186. [DOI] [PubMed] [Google Scholar]
- Pau H, Fuchs H, de Angelis MH, Steel KP. 2005. Hush puppy: a new mouse mutant with pinna, ossicle, and inner ear defects. Laryngoscope 115: 116–124. [DOI] [PubMed] [Google Scholar]
- Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, Mirda D, Williams LT. 1992. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature 358: 678–681. [DOI] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, et al. 2006. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci 103: 6281–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plusa B, Piliszek A, Frankenberg S, Artus J, Hadjantonakis AK. 2008. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development 135: 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye DA, Vives RR, Turnbull JE, Hyde P, Gallagher JT. 1998. Heparan sulfate oligosaccharides require 6-O-sulfation for promotion of basic fibroblast growth factor mitogenic activity. J Biol Chem 273: 22936–22942. [DOI] [PubMed] [Google Scholar]
- Qu X, Carbe C, Tao C, Powers A, Lawrence R, van Kuppevelt TH, Cardoso WV, Grobe K, Esko JD, Zhang X. 2011. Lacrimal gland development and Fgf10–Fgfr2b signaling are controlled by 2-O- and 6-O-sulfated heparan sulfate. J Biol Chem 286: 14435–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Pan Y, Carbe C, Powers A, Grobe K, Zhang X. 2012. Glycosaminoglycan-dependent restriction of FGF diffusion is necessary for lacrimal gland development. Development 139: 2730–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin SJ, Cleghon V, Kaplan DR. 1993. SNT, a differentiation-specific target of neurotrophic factor-induced tyrosine kinase activity in neurons and PC12 cells. Mol Cell Biol 13: 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffioni S, Thomas D, Foehr ED, Thompson LM, Bradshaw RA. 1999. Comparison of the intracellular signaling responses by three chimeric fibroblast growth factor receptors in PC12 cells. Proc Natl Acad Sci 96: 7178–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucci A, Laplantine E, Mansukhani A, Basilico C. 2004. Activation of the ERK1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J Biol Chem 279: 1747–1756. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Mickey G, Maher PA. 2000. Association of fibroblast growth factor receptor 1 with the adaptor protein Grb14. Characterization of a new receptor binding partner. J Biol Chem 275: 7771–7778. [DOI] [PubMed] [Google Scholar]
- Riley JK, Carayannopoulos MO, Wyman AH, Chi M, Ratajczak CK, Moley KH. 2005. The PI3K/Akt pathway is present and functional in the preimplantation mouse embryo. Dev Biol 284: 377–386. [DOI] [PubMed] [Google Scholar]
- Riley JK, Carayannopoulos MO, Wyman AH, Chi M, Moley KH. 2006. Phosphatidylinositol 3-kinase activity is critical for glucose metabolism and embryo survival in murine blastocysts. J Biol Chem 281: 6010–6019. [DOI] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, et al. 2007. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci 104: 4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A. 1994. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 371: 252–254. [DOI] [PubMed] [Google Scholar]
- Saba-El-Leil MK, Vella FD, Vernay B, Voisin L, Chen L, Labrecque N, Ang SL, Meloche S. 2003. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep 4: 964–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D, Basilico C. 1999. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev 13: 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni M, Raz R, Coffin JD, Levy D, Basilico C. 2001. STAT1 mediates the increased apoptosis and reduced chondrocyte proliferation in mice overexpressing FGF2. Development 128: 2119–2129. [DOI] [PubMed] [Google Scholar]
- Salazar L, Kashiwada T, Krejci P, Muchowski P, Donoghue D, Wilcox WR, Thompson LM. 2009. A novel interaction between fibroblast growth factor receptor 3 and the p85 subunit of phosphoinositide 3-kinase: activation-dependent regulation of ERK by p85 in multiple myeloma cells. Hum Mol Genet 18: 1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Araki I, Nakamura H. 2001. Inductive signal and tissue responsiveness defining the tectum and the cerebellum. Development 128: 2461–2469. [DOI] [PubMed] [Google Scholar]
- Sawada A, Shinya M, Jiang YJ, Kawakami A, Kuroiwa A, Takeda H. 2001. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development 128: 4873–4880. [DOI] [PubMed] [Google Scholar]
- Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, Feng GS, Pawson T. 1997. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 16: 2352–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg KM, Giesy SL, Harvatine KJ, Waldron MR, Cheng C, Kharitonenkov A, Boisclair YR. 2011. Plasma FGF21 is elevated by the intense lipid mobilization of lactation. Endocrinology 152: 4652–4661. [DOI] [PubMed] [Google Scholar]
- Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. 1994. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367: 380–383. [DOI] [PubMed] [Google Scholar]
- Schuller AC, Ahmed Z, Levitt JA, Suen KM, Suhling K, Ladbury JE. 2008. Indirect recruitment of the signalling adaptor Shc to the fibroblast growth factor receptor 2 (FGFR2). Biochem J 416: 189–199. [DOI] [PubMed] [Google Scholar]
- Sebastian A, Matsushita T, Kawanami A, Mackem S, Landreth GE, Murakami S. 2011. Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. J Orthop Res 29: 375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JH, Suenaga A, Hatakeyama M, Taiji M, Imamoto A. 2009. Structural and functional basis of a role for CRKL in a fibroblast growth factor 8-induced feed-forward loop. Mol Cell Biol 29: 3076–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaoul E, Reich-Slotky R, Berman B, Ron D. 1995. Fibroblast growth factor receptors display both common and distinct signaling pathways. Oncogene 10: 1553–1561. [PubMed] [Google Scholar]
- Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ. 1994. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78: 335–342. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. 2004. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoaka T, Ogasawara T, Yonamine A, Chikazu D, Kawano H, Nakamura K, Itoh N, Kawaguchi H. 2002. Regulation of osteoblast, chondrocyte, and osteoclast functions by fibroblast growth factor (FGF)-18 in comparison with FGF-2 and FGF-10. J Biol Chem 277: 7493–7500. [DOI] [PubMed] [Google Scholar]
- Shimokawa K, Kimura-Yoshida C, Nagai N, Mukai K, Matsubara K, Watanabe H, Matsuda Y, Mochida K, Matsuo I. 2011. Cell surface heparan sulfate chains regulate local reception of FGF signaling in the mouse embryo. Dev Cell 21: 257–272. [DOI] [PubMed] [Google Scholar]
- Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. 2007. RNA interference and inhibition of MEK–ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet 39: 1145–1150. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Wagner EF. 1995. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 269: 234–238. [DOI] [PubMed] [Google Scholar]
- Simonis N, Migeotte I, Lambert N, Perazzolo C, de Silva DC, Dimitrov B, Heinrichs C, Janssens S, Kerr B, Mortier G, et al. 2013. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J Med Genet 50: 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims-Lucas S, Cullen-McEwen L, Eswarakumar VP, Hains D, Kish K, Becknell B, Zhang J, Bertram JF, Wang F, Bates CM. 2009. Deletion of Frs2α from the ureteric epithelium causes renal hypoplasia. Am J Physiol Renal Physiol 297: F1208–F1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood PM, Munoz-Sanjuan I, Tong P, Macke JP, Hendry SH, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. 1996. Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc Natl Acad Sci 93: 9850–9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin A, Mohammadi M, Huang J, Schlessinger J. 1994. Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J Biol Chem 269: 17056–17061. [PubMed] [Google Scholar]
- Stanier P, Pauws E. 2012. Development of the lip and palate: FGF signalling. Front Oral Biol 16: 71–80. [DOI] [PubMed] [Google Scholar]
- Steinberg Z, Myers C, Heim VM, Lathrop CA, Rebustini IT, Stewart JS, Larsen M, Hoffman MP. 2005. FGFR2b signaling regulates ex vivo submandibular gland epithelial cell proliferation and branching morphogenesis. Development 132: 1223–1234. [DOI] [PubMed] [Google Scholar]
- Stevenson RE, Ferlauto GJ, Taylor HA. 1978. Cutis gyratum and acanthosis nigricans associated with other anomalies: a distinctive syndrome. J Pediatr 92: 950–952. [DOI] [PubMed] [Google Scholar]
- Sun X, Lewandoski M, Meyers EN, Liu YH, Maxson RE Jr, Martin GR. 2000. Conditional inactivation of Fgf4 reveals complexity of signalling during limb bud development. Nat Genet 25: 83–86. [DOI] [PubMed] [Google Scholar]
- Sun X, Mariani FV, Martin GR. 2002. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature 418: 501–508. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Klinghoffer RA, Heuchel R, Mueting-Nelsen PF, Corrin PD, Heldin CH, Johnson RJ, Soriano P. 2000. Retention of PDGFR-β function in mice in the absence of phosphatidylinositol 3′-kinase and phospholipase Cγ signaling pathways. Genes Dev 14: 3179–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist MD, French WJ, Soriano P. 2003. Additive effects of PDGF receptor β signaling pathways in vascular smooth muscle cell development. PLoS Biol 1: E52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. 1996. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J 15: 4629–4642. [PMC free article] [PubMed] [Google Scholar]
- Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H. 2010. Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465: 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ. 2015. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15: 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. 1995. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269: 230–234. [DOI] [PubMed] [Google Scholar]
- Timsah Z, Ahmed Z, Lin CC, Melo FA, Stagg LJ, Leonard PG, Jeyabal P, Berrout J, O'Neil RG, Bogdanov M, et al. 2014. Competition between Grb2 and Plcγ1 for FGFR2 regulates basal phospholipase activity and invasion. Nat Struct Mol Biol 21: 180–188. [DOI] [PubMed] [Google Scholar]
- Trokovic N, Trokovic R, Mai P, Partanen J. 2003. Fgfr1 regulates patterning of the pharyngeal region. Genes Dev 17: 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueb B, Amann R, Gerber SD. 2013. Role of FGFRL1 and other FGF signaling proteins in early kidney development. Cell Mol Life Sci 70: 2505–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. 1999. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev 13: 3136–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PS, Moenter SM, Postigo HR, El Majdoubi M, Pak TR, Gill JC, Paruthiyil S, Werner S, Weiner RI. 2005. Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population. Mol Endocrinol (Baltimore, Md) 19: 225–236. [DOI] [PubMed] [Google Scholar]
- Tu X, Joeng KS, Nakayama KI, Nakayama K, Rajagopal J, Carroll TJ, McMahon AP, Long F. 2007. Noncanonical Wnt signaling through G protein-linked PKCδ activation promotes bone formation. Dev Cell 12: 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulin S, Stathopoulos A. 2010. Extending the family table: insights from beyond vertebrates into the regulation of embryonic development by FGFs. Birth Defects Res C Embryo Today 90: 214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, Gunn M, Dell K, Tseng A Jr, Williams L. 1992. A truncated form of fibroblast growth factor receptor 1 inhibits signal transduction by multiple types of fibroblast growth factor receptor. J Biol Chem 267: 1470–1476. [PubMed] [Google Scholar]
- Ulaganathan VK, Sperl B, Rapp UR, Ullrich A. 2015. Germline variant FGFR4 p.G388R exposes a membrane-proximal STAT3 binding site. Nature 528: 570–574. [DOI] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. 2006. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444: 770–774.17086194 [Google Scholar]
- Vainikka S, Joukov V, Wennstrom S, Bergman M, Pelicci PG, Alitalo K. 1994. Signal transduction by fibroblast growth factor receptor-4 (FGFR-4). Comparison with FGFR-1. J Biol Chem 269: 18320–18326. [PubMed] [Google Scholar]
- Vainikka S, Joukov V, Klint P, Alitalo K. 1996. Association of a 85-kDa serine kinase with activated fibroblast growth factor receptor-4. J Biol Chem 271: 1270–1273. [DOI] [PubMed] [Google Scholar]
- Vasudevan HN, Mazot P, He F, Soriano P. 2015. Receptor tyrosine kinases modulate distinct transcriptional programs by differential usage of intracellular pathways. Elife 4: e07186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MB, Deng C, Lewandoski M, Pourquie O. 2007. FGF signaling acts upstream of the NOTCH and WNT signaling pathways to control segmentation clock oscillations in mouse somitogenesis. Development 134: 4033–4041. [DOI] [PubMed] [Google Scholar]
- Wang JK, Gao G, Goldfarb M. 1994. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol 14: 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Spatz MK, Kannan K, Hayk H, Avivi A, Gorivodsky M, Pines M, Yayon A, Lonai P, Givol D. 1999. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc Natl Acad Sci 96: 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, et al. 2005. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development 132: 3537–3548. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhou X, Oberoi K, Phelps R, Couwenhoven R, Sun M, Rezza A, Holmes G, Percival CJ, Friedenthal J, et al. 2012. p38 Inhibition ameliorates skin and skull abnormalities in Fgfr2 Beare-Stevenson mice. J Clin Invest 122: 2153–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chang JY, Yang C, Huang Y, Liu J, You P, McKeehan WL, Wang F, Li X. 2013. Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J Biol Chem 288: 22174–22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. 2009. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein M, Xu X, Ohyama K, Deng CX. 1998. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125: 3615–3623. [DOI] [PubMed] [Google Scholar]
- White AC, Xu J, Yin Y, Smith C, Schmid G, Ornitz DM. 2006. FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development 133: 1507–1517. [DOI] [PubMed] [Google Scholar]
- Wierman ME, Xu M, Pierce A, Bliesner B, Bliss SP, Roberson MS. 2012. Extracellular signal-regulated kinase 1 and 2 are not required for GnRH neuron development and normal female reproductive axis function in mice. Neuroendocrinology 95: 289–296. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. 1995. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 9: 165–172. [DOI] [PubMed] [Google Scholar]
- Xu H, Lee KW, Goldfarb M. 1998a. Novel recognition motif on fibroblast growth factor receptor mediates direct association and activation of SNT adapter proteins. J Biol Chem 273: 17987–17990. [DOI] [PubMed] [Google Scholar]
- Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C. 1998b. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 125: 753–765. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. 1994. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8: 3032–3044. [DOI] [PubMed] [Google Scholar]
- Yamanaka Y, Lanner F, Rossant J. 2010. FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development 137: 715–724. [DOI] [PubMed] [Google Scholar]
- Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, Hemmings BA. 2003. Protein kinase B α/Akt1 regulates placental development and fetal growth. J Biol Chem 278: 32124–32131. [DOI] [PubMed] [Google Scholar]
- Yang ZZ, Tschopp O, Di-Poi N, Bruder E, Baudry A, Dummler B, Wahli W, Hemmings BA. 2005. Dosage-dependent effects of Akt1/protein kinase Bα (PKBα) and Akt3/PKBγ on thymus, skin, and cardiovascular and nervous system development in mice. Mol Cell Biol 25: 10407–10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Qiao D, Meyer K, Friedl A. 2009. Signal transducers and activators of transcription mediate fibroblast growth factor-induced vascular endothelial morphogenesis. Cancer Res 69: 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. 2005. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn 234: 371–386. [DOI] [PubMed] [Google Scholar]
- Yin L, Du X, Li C, Xu X, Chen Z, Su N, Zhao L, Qi H, Li F, Xue J, et al. 2008. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone 42: 631–643. [DOI] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. 2008. FGF signaling regulates mesenchymal differentiation and skeletal patterning along the limb bud proximodistal axis. Development 135: 483–491. [DOI] [PubMed] [Google Scholar]
- Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, McKeehan WL. 2000. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem 275: 15482–15489. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. 2003. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130: 3063–3074. [DOI] [PubMed] [Google Scholar]
- Yu C, Wang F, Jin C, Huang X, McKeehan WL. 2005. Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J Biol Chem 280: 17707–17714. [DOI] [PubMed] [Google Scholar]
- Zhang H, Dessimoz J, Beyer TA, Krampert M, Williams LT, Werner S, Grose R. 2004. Fibroblast growth factor receptor 1-IIIb is dispensable for skin morphogenesis and wound healing. Eur J Cell Biol 83: 3–11. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. 2006. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem 281: 15694–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, Baum M, Bates CM. 2004. Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276: 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]