

It is increasingly recognized that metabolic remodeling is integral to heart failure development and progression.1,2 In particular, impairments in the ability of cardiac mitochondria to oxidize fatty acids have been noted along with an increase in glycolysis that is uncoupled from glucose oxidation.3,4 This overall reduction in the myocardial oxidative capacity is purported to be the root cause of energy deficiency in the failing heart. Although past research has primarily focused on myocardial use of glucose and fatty acids, the heart is an omnivore and capable of oxidizing other substrates such as lactate, ketone bodies, and amino acids. The current understanding of the contribution of lactate, ketone bodies, and amino acids to cardiac metabolism is limited, particularly in the setting of heart failure. In this issue of Circulation, two independent studies shed new insights on the reliance of the failing heart on ketone bodies for energy supply. Proteomics analysis in mouse models of heart failure by Aubert et al 5 and metabolomics analysis of end-stage human failing hearts by Bedi et al 6 demonstrate strong and concordant evidence of increased ketone oxidation in the failing heart.
Ketone bodies, i.e. acetoacetate, beta-hydroxybutyrate (βOHB) and acetone (in low abundance) are produced in the liver and used in peripheral tissues as an energy source when glucose is not readily available, either due to limited exogenous supply or impaired insulin signaling, or when fatty acids are in surplus, such that occurs in marked activation of lipolysis. Upon entering the cell, they rapidly form acetyl CoA via a series of reactions catalyzed by βOHB dehydrogenase (BDH1), succinyl-CoA:3-oxoacid-CoA transferase (SCOT) and mitochondrial acetyl-CoA acetyltransferase 1 (ACAT1; also known as thiolase) as shown in Figure 1. All enzymes are present in the heart and the reactions are driven primarily by substrate availability, save the SCOT reaction which is a succinyl CoA dependent process.
Figure 1.
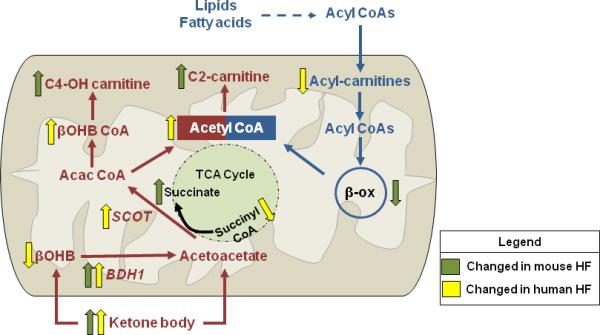
Ketone metabolism is increased in failing hearts of mice and humans. The major findings of altered ketone body (red) and fatty acid (blue) metabolism from current studies in failing mice and humans are summarized. Decreased fatty acid oxidation was evidenced by a reduction in long-chain acylcarnitines and reduced protein abundance of several enzymes in beta-oxidation (β-ox). Elevations in serum ketone body concentration were matched with alterations in metabolites and enzymes that are consistent with the upregulation of the ketone oxidation pathway. βOHB, beta-hydroxybutyrate; BDH1, mitochondrial beta-hydroxybutyrate dehydrogenase; SCOT, succinyl-CoA:3-oxoacid-CoA transferase; Acac CoA, acetoacetyl CoA; βOHB CoA, beta-hydroxybutyryl CoA; C4-OH carnitine, hydroxybutyrylcarnitine; C2-carnitine, acetyl-carnitine. Green arrow, findings made in mouse heart failure (HF); Yellow arrow, findings made in human HF.
In their work, Aubert, et al.,5 examined longitudinal changes of cardiac energy metabolism during the development of heart failure in murine models of compensated hypertrophy (CH), induced by transverse aortic constriction (TAC), and heart failure (HF), generated by superimposing an apical infarction on the TAC model. Using a proteomic approach, the group identified significant downregulation of proteins involved in fatty acid utilization in both CH and HF hearts with a concurrent 2-3-fold increase in BDH1, the enzyme that catalyzes the initial step in the ketone oxidation pathway. The notable finding of increased BDH1 protein prodded an additional search to identify metabolic signatures of cardiac ketone body utilization. In particular, the authors found increases in hydroxybutyrylcarnitine (C4OH-carnitine) and acetylcarnitine (C2-carnitine) in the HF group (Figure 1). In addition, a significant increase in succinate was noted, consistent with the succinyl CoA-dependent SCOT reaction (Figure 1). Increased levels of C4-OH-carnitine and C2-carnitine were also observed in the heart of young, healthy mice fed a 4-week ketogenic diet, supporting their utility as the metabolic signature of ketone metabolism.
Also in this issue of Circulation, the observations in mice are complimented by an independent study of end-stage human heart failure by Bedi et al.,6 in which metabolomics analysis showed increased ketogenic βOHB-CoA and evidence for enhanced myocardial utilization of βOHB, e.g. decreases of βOHB and succinyl-CoA levels in the cardiac tissue (Figure 1). These changes were accompanied by an upregulation of BDH1, BHD2 (cytosolic isoform) and SCOT (also called OXCT1), key enzymes in ketone oxidation pathway. Furthermore, a negative correlation of OXCT1 expression and succinyl-CoA level lent further support for the increased SCOT reaction in the failing heart. These studies, collectively, provide direct evidence of increased reliance on ketone body metabolism in the failing hearts of both mice and men, and successfully introduce a novel player to the game of metabolic remodeling in heart failure.
The studies also sought to determine whether the upregulation of the ketone oxidation pathway was a driver of metabolic remodeling by comparing the stages of compensated hypertrophy and heart failure. Bedi et al.6 found that the metabolite profile characteristic of ketone oxidation was only present in the failing hearts and not observed in tissues with left ventricular hypertrophy and apparent normal function. Moreover, gene expression of enzymes for ketone oxidation was unaltered in patients with cardiac hypertrophy suggesting that increased ketone utilization is a late event in heart failure. Elevations in serum levels of both fatty acids and ketone bodies inferred that the upregulation of ketone oxidation is a consequence of enhanced hepatic ketogenesis and, hence, increased substrate delivery to the failing heart. Since ketone availability is a primary determinant of ketone utilization, these observations suggest that impaired systemic metabolism is an important contributor to remodeling of cardiac metabolism in advanced heart failure.
Data obtained from mouse hearts provided a different perspective to this question, however. Aubert, et al.5 observed a consistent upregulation of BDH1 in mouse hearts with pathological hypertrophy and failure despite similar circulating ketone levels, suggesting that increased BDH1 level could accelerate ketone metabolism during the development of heart failure. Using stable isotope labeling techniques in isolated perfused hearts, the group showed a significant increase in the relative oxidation of βOHB in hypertrophied mouse hearts which coincided with the upregulation of BDH1. As the concentration of βOHB (1mM) used in perfusion experiments was several folds higher than serum levels in mice or men with heart failure, future studies are warranted to determine whether the upregulation of BDH1 increases the use of ketone supplied at an in-vivo concentration. Nevertheless, the efforts of Aubert, et al. 5 and Bedi et al. 6 have identified a novel aspect of cardiac metabolism in the failing heart that has high clinical relevance and opens up new opportunities for translational research in metabolic therapy for heart failure.
Despite unambiguous observations, the mechanism for increased myocardial ketone oxidation remains unclear. Both studies showed evidence of impaired fatty acid oxidation (FAO) in the failing heart suggesting that increased ketone use could be a part of the metabolic remodeling that shifts the substrate utilization towards non-fatty acids sources. The mouse study found marked downregulation of proteins involved in FAO, implying a decrease in the capacity of FAO in the failing heart. This is consistent with prior studies demonstrating accumulation of intermediate metabolites of fatty acid metabolism in the heart of animal models and patients of heart failure.7,8 The human study, however, found a depletion rather than accumulation of myocardial lipid intermediates, particularly long-chain acylcarnitines. In contrast to the proteomics data from mice, gene expression for proteins involved in fatty acid β-oxidation or PPARα was unchanged, whereas transporters for carnitine were downregulated in cardiac tissue of advanced heart failure patients. Thus, FAO is decreased in the failing mouse heart because the system is “jammed” while in the human failing heart the system is “deprived”. Although increased ketone utilization is coupled with decreased FAO, the underlying mechanisms for impaired FAO in the failing hearts of mice and human could be distinct. It does not, however, rule out the possibility that observations made in the cardiac tissue obtained from patients of advanced heart failure are unique to the end-stage disease, which is not comparable to the stable heart failure stage represented by the mouse model. Future studies are necessary to clarify this point, and more importantly, to determine whether impaired FAO is the cause of increased reliance on ketone as an energy source in heart failure. Furthermore, the current studies focused on non-diabetic, non-obese heart failure models/patients, which allow for the demonstration of a diabetes-independent increase of cardiac ketone utilization in heart failure. As diabetes is present in approximately 40% of patients hospitalized with HF,9 it will be important to consider the influence of comorbidities on ketone metabolism in future experimental models.
An important question raised by the studies is whether the increased reliance on ketone body metabolism is adaptive or maladaptive for the failing heart. Deletion of SCOT in mouse hearts promotes pathological remodeling and cardiac dysfunction after TAC,10 suggesting that the ability to oxidize ketone bodies is important for the failing heart. An elevation in serum ketone levels has now been observed by several studies in patients with heart failure.6,11,12 Therefore, the current data would suggest that the increase in myocardial ketone body utilization is a consequence of increased systemic availability. In end-stage heart failure, insulin resistance and cardiac cachexia are common which increases the likelihood of ketone production by the liver and renders cardiac ketone utilization unavoidable. As the metabolite profile associated with increased ketone oxidation is indicative of impaired TCA cycle fluxes in the failing hearts,6 measures to reduce the reliance on ketone bodies is likely desirable. In this regard, preventing the decrease in myocardial FAO during the development of pathological hypertrophy is worth consideration.13 In turn, measures to tailor nutritional interventions or optimize systemic metabolism in order to attenuate hepatic ketone body production may also be a potential therapeutic approach.
Acknowledgments
Funding Sources: This work was supported in part by grants from the National Institute of Health (HL088634, HL110349, HL118989, and HL129510 to R.T.) and the American Heart Association (14SDG18590020 to S.C.K. and 15POST22560002 to S.A.).
Footnotes
Disclosures: None.
References
- 1.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 3.Fukushima A, Milner K, Gupta A, Lopaschuk GD. Myocardial energy substrate metabolism in heart failure : From pathways to therapeutic targets. Curr Pharm Des. 2015;21:3654–3664. doi: 10.2174/1381612821666150710150445. [DOI] [PubMed] [Google Scholar]
- 4.Kolwicz SC, Jr., Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–616. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aubert G, Martin O, Horton J, Lai L, Vega R, Leone T, Koves T, Gardell S, Kruger M, Hoppel C, Lewandowski E, Crawford P, Muoio D, Kelly D. The failing heart relies on ketone bodies as fuel. Circulation. 2016;133 doi: 10.1161/CIRCULATIONAHA.115.017355. XX-XXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedi K, Snyder N, Brandimarto J, Aziz M, Mesaros C, Worth A, Wang L, Javheri A, Blair I, Margulies K, Rame J. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133 doi: 10.1161/CIRCULATIONAHA.115.017545. XX-XXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012;125:2844–2853. doi: 10.1161/CIRCULATIONAHA.111.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: A multisystems approach. Circ Heart Fail. 2014;7:1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg BH, Abraham WT, Albert NM, Chiswell K, Clare R, Stough WG, Gheorghiade M, O'Connor CM, Sun JL, Yancy CW, Young JB, Fonarow GC. Influence of diabetes on characteristics and outcomes in patients hospitalized with heart failure: A report from the organized program to initiate lifesaving treatment in hospitalized patients with heart failure (optimize-hf). Am Heart J. 2007;154:277, e271–278. doi: 10.1016/j.ahj.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Schugar RC, Moll AR, Andre d'Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. 2014;3:754–769. doi: 10.1016/j.molmet.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du Z, Shen A, Huang Y, Su L, Lai W, Wang P, Xie Z, Xie Z, Zeng Q, Ren H, Xu D. 1h-nmr-based metabolic analysis of human serum reveals novel markers of myocardial energy expenditure in heart failure patients. PLoS One. 2014;9:e88102. doi: 10.1371/journal.pone.0088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. doi: 10.1016/0735-1097(96)00214-8. [DOI] [PubMed] [Google Scholar]
- 13.Kolwicz SC, Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl coa carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111:728–738. doi: 10.1161/CIRCRESAHA.112.268128. [DOI] [PMC free article] [PubMed] [Google Scholar]