

Abstract
Necrotic lesions and necrotic cell death characterize severe autoimmune nephritides, and contribute to local inflammation and to progression of the disease. Poly(ADP-ribose) polymerase-1 (PARP-1), a DNA repair enzyme, is involved in the induction of necrosis and is a key player in the acute and chronic inflammation. Therefore, we hypothesized that PARP-1 controls the severity of nephritis by mediating the induction of necrosis in the kidney. We used lupus and anti-glomerular basement membrane models of nephritis to determine the effects of PARP-1 on the inflammatory response in the kidney. We show in this study that PARP-1 is indeed activated during the course of glomerulonephritis. We also show that the absence of PARP-1 or its pharmacological inhibition results in milder nephritis, with lower blood urea nitrogen levels, reduced necrotic lesions, and higher survival rates. The relevance of PARP-1 showed a strong male sex specificity, and treatment of male mice with 17β-estradiol prolonged their survival during the course of nephritis. PARP-1 also regulated TNF-α expression and up-regulation of adhesion molecules, further supporting a role of PARP-1 in the inflammatory process within the kidney. Our results demonstrate that PARP-1 activation and consequent necrotic cell death play an important role in the pathogenesis of male nephritis, and suggest that PARP-1 can be a novel therapeutic target in glomerulonephritis. The Journal of Immunology, 2009, 182: 7297–7306.
During acute and chronic glomerulonephritis (GN),4 oxidative stress plays a fundamental role in inducing glomerular, tubular, and vascular damage. The acute inflammatory response to the formation of immune complexes in the glomeruli leads to production of mediators such as IL-1 and TNF-α, which are potent stimuli for the generation of reactive oxygen species (ROS) by leukocytes and renal parenchymal cells (1, 2). ROS may lead to tissue damage in autoimmune nephritis by several mechanisms, including irreversible cell injury, generation of proinflammatory cytokines such as TNF-α, and activation of transcription factors such as NF-κB and AP-1 (2, 3).
Irreversible cell injury can lead to necrotic cell death, which is a potent proinflammatory event (4). More recently, much therapeutic effort in autoimmune nephritides such as Lupus, Wegener’s, and anti-glomerular basement membrane (GBM) GN has been concentrated toward modulating the aberrant autoimmune response, with less attention to the downstream effects of local inflammation per se. Necrotic lesions and necrotic cell death contribute to local inflammation leading to renal damage, help define the severity of nephritis, and have prognostic implications (5, 6). Necrotic cell death, traditionally considered to be a passive form of cell death, has been recognized recently as a regulated process, important in certain pathological and physiological processes, including the immune response (7–9). Therefore, the aim of this study was to determine whether necrotic cell death pathway contributes to the pathogenesis of nephritis and whether this pathway can be a target for therapeutic intervention.
ROS induces extensive DNA damage, which activates enzymes involved in DNA damage sensing and repair. Poly(ADP-ribose) (PAR) polymerase-1 (PARP-1) is a nuclear protein activated in response to DNA breaks; it poly-(ADP-ribosyl)-ates proteins using NAD+ as substrate (10–12), thereby facilitating DNA repair. PARP-1 is the best-characterized member of the PARP family of proteins, and accounts for the majority of the poly(ADP-ribose) polymer synthesis in the cell during genotoxic stress (13, 14). PARP-1 overactivation in response to extensive DNA damage results in excessive consumption of NAD+ and ATP depletion, resulting in a sudden reduction of cellular energy and consequent necrosis (7, 8, 15, 16).
In addition to inducing necrosis, PARP-1 also has a profound modulatory effect on the inflammatory response. PARP-1 can form stable complexes with transcription factors such as p53 and AP-2 (17, 18). PARP-1 can also act as a coactivator of NF-κB (17–19) and, therefore, may be involved in the induction of TNF-α, which plays an important role in the development of GN (20–23). Previous studies have also shown that the inhibition of PARP-1 activity reduces the secretion of proinflammatory cytokines as well as neutrophil migration to the inflammatory sites (24, 25), providing further evidence for the role of PARP-1 in regulation of inflammation. Absence of PARP-1 or inhibition of PARP-1 activity protects mice from ischemia/reperfusion injury, septic shock, streptozotocin-induced diabetes, and collagen-induced arthritis (25–30). The role of PARP-1 in autoimmune nephritis, however, has not been studied.
In the present study, we tested the hypothesis that excessive oxidative damage during immune-mediated nephropathy may induce overactivation of PARP-1, leading to necrotic cell death and tissue damage. To this end, we used lupus and anti-GBM models of autoimmune nephritis because recent evidence suggests that, irrespective of the upstream events leading to immune-mediated nephritis, the downstream mechanisms involved in renal tissue damage are similar (31, 32). Specifically, we tested the nephrotoxic serum (NTS)-induced nephritis (NTN) because it allows to bypass the upstream events that lead to the break of tolerance and generation of autoantibodies (31, 32). We hypothesized that due to its role in inducing necrotic cell death, PARP-1 regulates a local inflammatory event leading to tissue damage in immune-mediated nephritis. Our data show that, as predicted, PARP-1 plays an important role in the pathogenesis of immune-mediated nephritis by modulating inflammatory response and inducing necrotic cell death. The protection conferred by the elimination of PARP-1 was significant only in male mice, a result justified by the fact that estrogens physiologically bind and neutralize PARP-1 in females (33). Indeed, treatment of male mice with 17β-estradiol prolonged survival following NTS administration. Our data provide a novel mechanism by which PARP-1-mediated necrotic cell death regulates inflammation and may be a novel therapeutic target in immune-mediated nephritis.
Materials and Methods
Mice
New Zealand Black (NZB)/New Zealand White (NZW)F1, PARP-1−/− mice on 129 background and 129s/sv wild-type mice were obtained from The Jackson Laboratory. PARP-1+/+ and PARP-1−/− mice were bred and maintained in accordance with the guidelines of the University Laboratory Animal Resource Office of the University of Pennsylvania, an American Association for the Accreditation of Laboratory Animal Care-accredited facility. All experimental procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee.
Induction of anti-GBM nephritis
Nephritis was induced in 8- to 10-wk-old PARP-1+/+ or PARP-1−/− mice by injecting 6 ml/kg NTS, raised in sheep (34). A single lot of NTS was used for all experiments. For inhibition of PARP-1 activity, male mice were treated with 5-aminoisoquinolinone (5-AIQ; 3 mg/kg i.p., every 24 h; Alexis) (30) 24 h following NTS injection. Blood urea nitrogen (BUN) was measured using semiquantitative Azostix or by BUN kit (Stanbio Lab). Proteinuria was measured semiquantitatively using Uristix (Bayer). Mice were euthanized at day 6 following NTS administration. H&E staining was performed on paraffin-embedded kidney sections. To induce chronic nephritis, mice were injected with 4 ml/kg NTS. For estrogen treatment, 17β-estradiol pellets (Innovative Research of America) were implanted s.c. and acute nephritis was induced, as previously indicated. Serum levels of 17β-estradiol were determined using 17 β-estradiol RIA (Diagnostic Systems Laboartories), according to manufacturer’s instructions.
Accelerated model of lupus nephritis
Eight-week-old NZB/NZW F1 male mice were injected with 1 × 109 virus particles/mouse AdvCMV-mIFN-α or AdvCMV-Null (Qbiogene) (35). The treatment with 5-AIQ was initiated 13 wk following adenovirus injection. Mice were euthanized 6 wk later.
PARP-1 activation
PARP-1 activation was measured as PAR accumulation in tissues (36–38). Paraffin-embedded tissue sections (6 μm thick) were dewaxed with xylenes and rehydrated with decreasing concentrations of alcohol. The sections were incubated with rabbit anti-PAR Ab (Alexis). PAR accumulation was visualized with rhodamine-conjugated goat anti-rabbit Ab (Molecular Probes). Nuclei were stained using TOTO3 (Molecular Probes). TUNEL assay was performed using In Situ Cell Death detection kit fluorescein (Roche), according to manufacturer’s instructions. For immune complex deposition, sections were stained with FITC-conjugated goat anti-mouse IgG (Fcγ specific; Jackson ImmunoResearch Laboratories). Images were taken with a Nikon TE300 scanning confocal microscope, equipped with Nomarski differential interphase contrast optics, coupled to the Bio-Rad Radiance 2000 laser. A × 100 magnification was used.
Detection of autoantibodies
Autoantibodies were assessed by ELISA, as previously described (39). Briefly, the plates were coated with chicken erythrocyte-derived chromatin at 3 μg/ml, or with calf thymus-derived dsDNA at 2.5 μg/ml in borate-buffered saline (BBS). Following addition of blocking buffer (3% BSA and 1% Tween 80 in 1 × BBS), serum samples diluted 1/250 in BBT (BBS, 0.4% Tween 80, 0.5% BSA) were added in duplicate and incubated overnight at 4°C. For the anti-dsDNA ELISA, plates were coated with poly(L-lysine) (1 μg/ml; Sigma-Aldrich) before coating with Ag. Alkaline phosphatase-conjugated goat anti-mouse IgG (Fcγ specific; Jackson ImmunoResearch Laboratories) was used as secondary Ab. The plates were developed using 1 mg/ml paranitrophenyl phosphate substrate (Sigma-Aldrich) in 0.01 M diethanolamine (pH 9.8). To generate a standard curve for these assays, serum from an older MRL/lpr mouse with high titer of autoantibodies was also assayed at serial 2-fold dilutions from 1/250 to 1/128,000.
Generation of chimeric mice
PARP-1−/− and PARP-1+/+ mice were irradiated with a total dose of 900 rad using Cs-137 and reconstituted with bone marrow (BM) from PARP-1+/+ and PARP-1−/− mice, respectively (i.v. injection of 3 × 107 unsorted BM cells). PARP-1+/+ and PARP-1−/− control chimeras were reconstituted with syngeneic BM. Genomic DNA was extracted from blood, and BM chimerism was determined 6 wk later by PCR for PARP-1 and neomycin genes. Nephritis was induced 7 wk following BM reconstitution by injecting NTS, and the mice were monitored, as described earlier.
Mesangial cell culture and activation
Mouse mesangial cell lines (40) were infected with PARP-1 short hairpin RNA in pBabe-Puro (7), obtained from C. Thompson (University of Pennsylvania, Philadelphia, PA). Single-cell clones were propagated by limited dilution, and PARP-1 protein expression was determined by Western blotting. For Western blots, whole-cell lysates were prepared. Protein concentration was determined by bicinchoninic acid assay (Pierce). Anti-PARP-1 Ab (BD Biosciences) was used to detect PARP-1, and β-actin was used as loading control. Clones with at least 60–70% knockdown were selected, and stimulated with mouse rTNF-α (eBiosciences) at different concentration for 24 h. For immunofluorescence staining, cells were incubated with 2.4G2 to block FcRs and then with anti-VCAM-1 Abs (eBiosciences), fixed in PBS containing 1% paraformaldehyde, and analyzed on a BD Biosciences FACSCalibur or FACSCanto. Relative fluorescence intensity was plotted on a logarithmic scale using Flow-Jo software.
Real-time PCR
Total RNA was isolated using Qiagen RNeasy kit. RNA (1 μg) was reverse transcribed using Applied Biosystems high capacity reverse-transcription kit. TNF-α and IL-10 gene expression, normalized to GAPDH, was detected by real-time PCR using QuantiTect primer assays (Qiagen) and Applied Biosystems 7500 thermal cycler. Relative expression of mRNA transcripts was quantified by relative quantification (2−ΔΔCt) using Applied Biosystems software.
Statistical analysis
ANOVA was performed using GraphPad Prism 4.0c software for Mac (GraphPad). Differences among groups were determined using the Tukey-Kramer post hoc test. Student’s t test and χ2 test were performed where appropriate; statistical significance was defined as p < 0.05.
Results
Absence of PARP-1 protects mice from immune-mediated nephritis
To evaluate the role of PARP-1 in immune-mediated nephritis, we induced anti-GBM nephritis in PARP-1−/− male mice. We used male mice because previous studies have shown a sex specificity in PARP-1 activity (33, 41, 42). After NTS administration, wild-type mice had higher levels of BUN and proteinuria than PARP-1−/− mice (Fig. 1, A and B), and this was associated with lower survival rates (Fig. 1C). Histopathology revealed severe nephritis in wild-type kidneys with thrombotic and necrotic lesions and glomerulosclerosis, whereas the PARP-1−/− kidneys showed minimal disease (Fig. 1D). Both PARP-1+/+ and PARP-1−/− kidneys showed similar sheep IgG and complement (C3) deposition 30 h and 3 days after NTS injection (supplemental Fig. 1,5 and data not shown), and therefore, the benefit was not likely due to differences in the quantity of deposited Abs.
FIGURE 1.
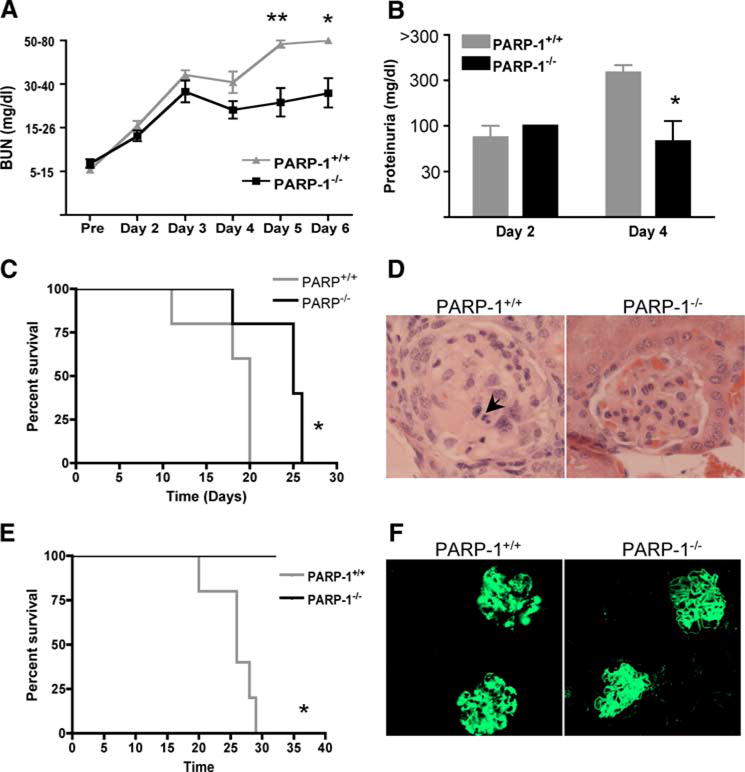
PARP−/− mice are protected from NTS-induced nephritis. PARP-1+/+ and PARP-1−/− male mice were treated with 6 ml/kg NTS and followed for 5 days. A, BUN levels were monitored daily with Axostix. PARP-1−/− mice showed significantly lower level of BUN compared with wild-type mice. Data are represented as mean ± SEM of 10–12 mice of each strain. *, p = 0.0084; **, p = 0.0027. B, Proteinuria was measured semiquantitatively using Uristix. PARP-1−/− mice showed significantly lower proteinuria compared with wild-type mice. *, p = 0.0335. C, Kaplan-Meier survival curves indicate that PARP-1−/− mice survive longer than wild-type mice. Data are represented as mean ± SEM of five to seven mice of each strain. *, p = 0.0317. The results indicate that PARP-1 plays a role in protecting from autoimmune nephritis. D, Renal histology 15 days post-NTS injection is shown. PARP-1+/+ mice showed severe glomerular damage with vacuolization and necrotic (→) and thrombotic lesions. In contrast, PARP-1−/− showed minimal damage. The figure shows representative glomeruli from three mice each strain (5–7 glomeruli per mouse). E, To induce chronic nephritis, mice were injected with 4 ml/kg NTS. PARP-1−/− mice showed higher survival than PARP-1+/+ mice. *, p = 0.0018. F, Both wild-type and PARP-1−/− mice with chronic NTN showed similar immune complex deposition. Frozen kidney sections were stained for deposited mouse IgG using FITC-conjugated goat anti-mouse IgG (Fcγ specific).
NTS-induced nephritis can be distinguished into two phases. The heterologous Ab to GBM (NTS) deposits in the kidney and causes transient injury, leading to acute nephritis. This phase is also known as the heterologous phase. The animal then mounts its own immune response to the foreign Ig, which acts as a planted Ag on the GBM. This phase is known as the autologous phase or chronic nephritis (43).
To determine the role of PARP-1 in the autologous phase of the disease, we induced chronic nephritis by injecting a lower dose of NTS. All wild-type mice with chronic nephritis showed 100% mortality at 1 mo, whereas the PARP-1−/− mice showed 100% survival (Fig. 1E). Both PARP-1+/+ and PARP-1−/− with chronic NTN showed mouse Ig deposition in the kidneys (Fig. 1F). These results confirm the significance of PARP-1 activation in immune complex-mediated disease.
The protection conferred by the absence of PARP-1 was not due to a defective immune system. We have characterized the immune system in the PARP-1−/− mice, and contrary to a recent report (44), have demonstrated that PARP-1−/− mice have normal B and T cell maturation, normal frequency of lymphocytes, macrophages and myeloid and plasmocytoid DC subsets, and normal humoral responses (supplemental Figs. 2 and 3,5 and data not shown).
The protection conferred by the absence of PARP-1 is dependent on the biological sex of mice
To determine whether the protection against nephritis observed in PARP-1−/− male mice shows a similar sex specificity, as previously demonstrated (33, 41, 42), we compared the severity of nephritis in female PARP-1+/+ and PARP-1−/− mice. Indeed, the female PARP-1−/− mice developed nephritis comparable to wild-type mice (Fig. 2). PARP-1 interacts with estrogen receptor (ERα) forming complex with estrogen and ERα, and these interactions inhibit the function/activity of PARP-1 (33). Therefore, we hypothesized that this may account for our observations in female mice. To determine whether the role of PARP-1 in NTS-induced nephritis is estrogen dependent, we treated male wild-type mice with estrogens by implanting 17β-estradiol pellets s.c. We induced nephritis 7 days following pellet implantation, and monitored the mice for 6 days. Estrogen treatment delayed the development of disease (Fig. 3A) and improved survival in male wild-type mice (Fig. 3B). Mice implanted with 17β-estradiol pellets showed higher levels of serum 17β-estradiol (300–900 pg/ml), whereas control mice had levels between 2 and 12 pg/ml (Fig. 3C). The results suggest that the pathways contributing to nephritis in male and female may be different.
FIGURE 2.
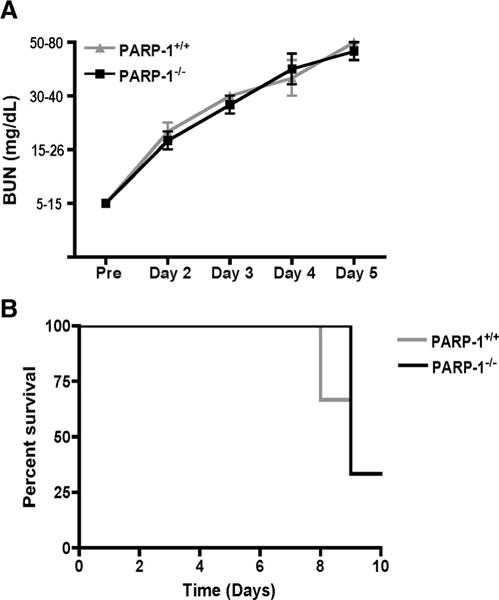
Protection conferred by the absence of PARP-1 is dependent on sex of the mice. PARP-1+/+ and PARP-1−/− female mice were treated with 6 ml/kg NTS and followed for 5 days. A, BUN levels were monitored daily with Axostix. PARP-1−/− mice showed similar levels of BUN as wild-type mice. Data are represented as mean ± SEM of six to seven mice. B, Kaplan-Meier survival curves show no significant difference in survival of PARP-1+/+ and PARP-1−/− female mice. The data show that protection due to absence of PARP-1 is dependent on the biological sex, with only male mice being protected.
FIGURE 3.
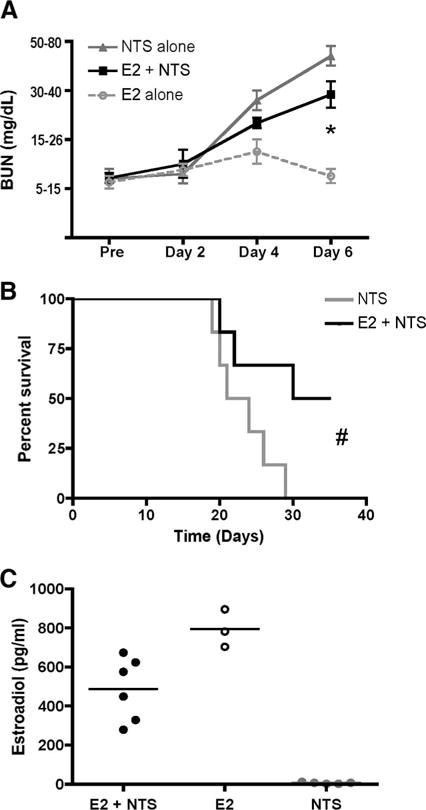
Estrogen delays NTS-induced nephritis and improves survival. To determine whether the role of PARP-1 in NTS-induced nephritis is estrogen dependent, we treated male wild-type mice with estrogens by implanting 17β-estradiol pellets s.c. Nephritis was induced 7 days following pellet implantation, and the mice were monitored for kidney function for 6 days. A, Mice were monitored for kidney function for 6 days. BUN levels were measured by Azostix. *, p = 0.04. Data are represented as mean ± SEM of three to six mice each group. B, Survival curves for mice with or without 17β-estradiol treatment before induction of nephritis. #, 0.0365. C, Mice were bled terminally 6 days after inducing nephritis, and serum 17β-estradiol levels were measured using 17β-estradiol RIA. The data show that presence of estrogens delays the development of nephritis and improves survival in male wild-type mice.
Inhibition of PARP-1 protects mice from immune-mediated nephritis
To determine whether PARP-1 inhibition protects from Ab-mediated nephritis, we induced NTN in PARP-1+/+ male mice (31, 32). We treated a cohort of mice with 5-AIQ to inhibit PARP-1 activity. Six days following NTS administration, mice treated with 5-AIQ had levels of BUN that were significantly lower than untreated mice (Fig. 4A). We obtained similar results with another inhibitor of PARP-1, PJ-34, further supporting the role of PARP-1 during inflammation (data not shown). The treatment with 5-AIQ markedly reduced thrombotic lesions, glomerulosclerosis, and crescent formation (Fig. 4B). PARP-1 was activated during NTS-induced nephritis, and treatment with 5-AIQ inhibited this activation (Fig. 4C).
FIGURE 4.
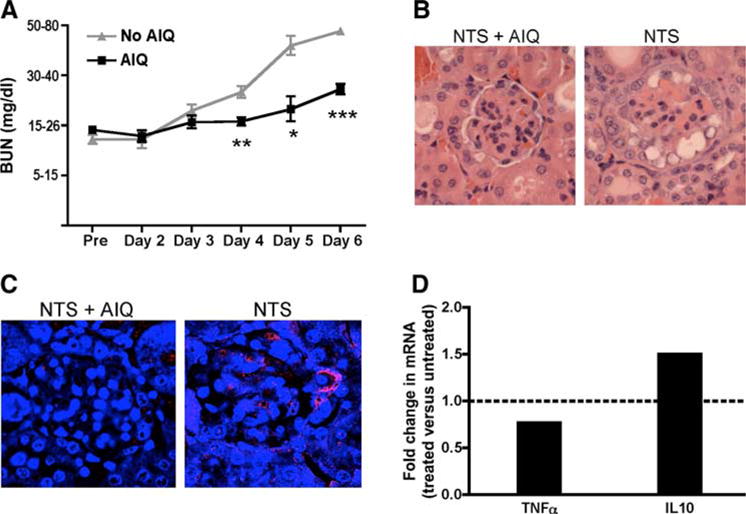
Pharmacological inhibition of PARP-1 protects from NTS-induced nephritis. Nephritis was induced in PARP-1+/+ mice by single i.v. injection of NTS. Mice were treated with 5-AIQ (3 mg/kg i.p., every 24 h) starting 24 h after NTS injection. A, BUN was monitored every 24 h using Azostix. Data are represented as mean ± SEM of 9–11 mice in each group. ***, p < 0.0001; **, p = 0.0005; *, p = 0.0034, when compared with untreated mice. B, H&E stain of paraffin-embedded kidneys 6 days after NTS administration. NTN caused severe glomerular damage that was inhibited by treatment with 5-AIQ. C, PARP-1 is activated in mice treated with NTS. Kidneys were harvested from mice injected with NTS with or without 5-AIQ treatment. PAR staining was performed on paraffin-embedded sections, as described in Materials and Methods. PARP-1+/+ mice showed PAR accumulation indicating PARP-1 activation, which was inhibited by 5-AIQ treatment. D, TNF-α and IL-10 mRNA levels were measured by quantitative real-time PCR in kidneys of PARP-1+/+ and PARP-1−/− mice at day 6 after induction of NTS-induced nephritis. Relative mRNA level was normalized to mice without 5-AIQ treatment. A representative experiment of two experiments with three mice each group per experiment is shown. TNF-α, p = 0.0365; IL-10, p = 0.0246. Treatment with 5-AIQ delays BUN increase and renal failure following NTS-induced nephritis possibly due to the reduced expression of TNF-α.
The protected kidneys from mice treated with 5-AIQ showed reduced TNF-α and increased IL-10 transcripts (Fig. 4D), suggesting that the inhibition of PARP-1 activity may protect mice by reducing the proinflammatory cytokine milieu.
PARP-1 activation in both hematopoietic cells and renal cells influences the severity of nephritis
To elucidate whether PARP-1 activity modulated disease at the level of renal tissue or of BM-derived immune cells, we generated chimeric mice: wild-type mice were reconstituted with either wild-type or PARP-1−/− BM, and PARP-1−/− mice were reconstituted with PARP-1−/− or wild-type BM. As shown in Fig. 5, PARP-1−/− mice reconstituted with syngeneic BM had significantly lower levels of BUN 6 days after NTS administration, as compared with wild-type mice reconstituted with syngeneic BM. The PARP-1−/− or PARP-1+/+ mice reconstituted with PARP-1+/+ or PARP-1−/− BM, respectively, however, had similar levels of BUN as wild-type mice reconstituted with syngeneic BM and were not statistically different. These data suggest that PARP-1 activation in either hematopoietic or renal cells is sufficient to mediate tissue damage during the course of NTN.
FIGURE 5.
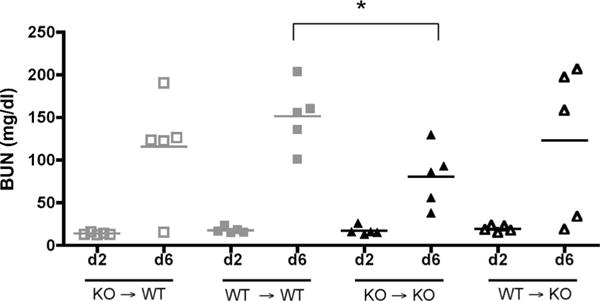
Absence of PARP-1 in both kidney and hematopoietic compartment is important for protection. We generated BM chimera mice in which the immune system was PARP-1−/−, whereas the kidney tissue was PARP-1+/+ and vice versa. PARP-1+/+ and PARP-1−/− control chimeras were reconstituted with syngeneic BM. Nephritis was induced 7 wk later by injecting NTS. BUN levels in sera at day 2 (d2) and day 6 (d6) following induction of nephritis were measured using BUN kit. PARP-1−/− control chimeras showed significantly lower levels of BUN at day 6, whereas the chimeric mice showed similar levels of BUN as the wild-type control chimeras. *, p = 0.0149. The data indicate that activation of PARP-1 is relevant in both renal and hematopoietic compartments.
PARP-1 is activated during the course of lupus nephritis
PARP-1 poly(ADP-ribosyl)-ates target proteins. These ADP-ribose polymers are degraded by PAR glycohydrolase, resulting in the release of free ADP-ribose polymers. The presence of free or bound PAR can be used as a measure of PARP-1 activity (36–38). To determine whether PARP-1 plays a role in spontaneous models of lupus nephritis, we stained formalin-fixed kidney sections from lupus-prone MRL-lpr mice at different ages (between 4 days and 8 mo; donated by R. Eisenberg, University of Pennsylvania, Philadelphia, PA) for accumulation of PAR as a marker of PARP-1 activation. Histological examination of kidney sections from older mice (6 and 8 mo) showed leukocyte infiltration, crescent formation, and glomerular sclerosis (Fig. 6, i and j), whereas the kidneys from young mice were normal (Fig. 6, f–h). Consistent with these observations, kidneys from young mice (4 days to 3 mo) did not show any PAR accumulation, whereas kidneys from older mice showed strong PAR staining (Fig. 6, a–e). We observed similar results in kidneys from two additional spontaneous lupus models characterized by the presence of GN: the NZM2410 (45) (Fig. 6, m and n) and the FcγRIIb−/−/Yaa (46) (Fig. 6, o and p). We used TUNEL assay to determine DNA damage. Fig. 6, k and l, shows that PARP-1 activation corresponds to DNA damage only in kidneys from old MRL/lpr mice, confirming the presence of necrotic cell death during the course of autoimmune nephritis. These data demonstrate that PARP-1 is activated in the course of lupus nephritis, and therefore may play a role in disease progression.
FIGURE 6.
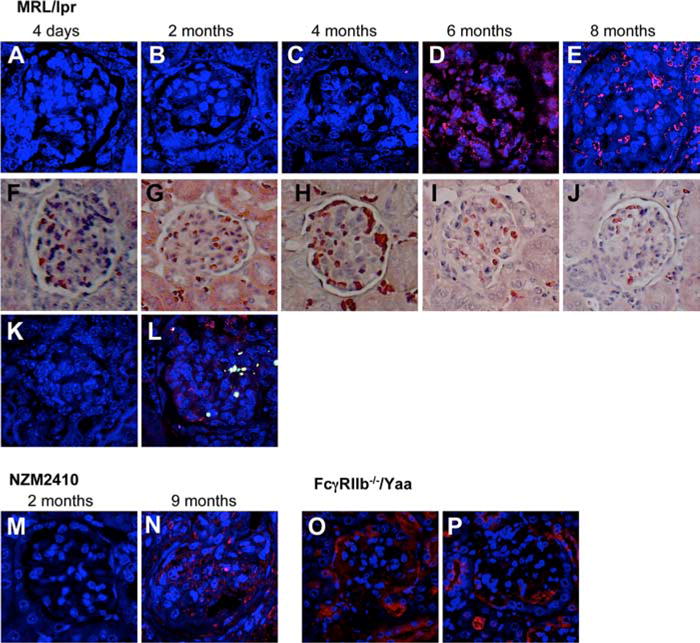
PARP-1 is activated in spontaneous mouse models of lupus. Formalin-fixed kidney sections were stained with anti-PAR as a marker of PARP-1 activation. Tissues were then visualized with goat anti-rabbit rhodamine-conjugated secondary Ab. Nuclei were visualized with TOTO3. A–E, MRL/lpr; M and N, NZM2410; O and P, FcγRIIb−/−/Yaa. A representative glomerulus for each age is shown. F–J, H&E staining for the respective mice. MRL/lpr mice showed increasingly strong PAR staining and, therefore, PARP-1 activation over the course of their renal disease, as demonstrated by the widespread PARP-1 activation in 8-mo-old mouse. These data demonstrate that PARP-1 is activated in the course of lupus GN. Kidneys from MRL/lpr mice, 4 day (K) and 8 mo (L) old, were stained with anti-PAR to detect PARP-1 activation and DNA damage (TUNEL assay). TOTO3 was used to stain nuclei. One example of each is shown. These results suggest that necrosis has occurred.
Inhibition of PARP-1 activity improves survival in accelerated lupus nephritis
Our data with NTS-induced nephritis show that, as expected, PARP-1 regulates local inflammatory events during the pathogenic process that leads to immune-mediated nephritis. To further confirm the role of PARP-1 in lupus nephritis, we used a pharmacological inhibitor of PARP-1 in NZB/NZW F1 male mice. NZB/NZW F1 mice spontaneously develop a lupus-like disease, characterized by high levels of antinuclear Abs, proteinuria, and progressive immune complex GN, by 6–9 mo of age (47). To accelerate disease development, we injected 8-wk-old NZB/NZW F1 mice with adenovirus-expressing mouse IFN-α, as described previously (35). We found anti-dsDNA and anti-chromatin auto-antibodies in mice injected with mouse IFN-α-expressing adenovirus, but not in mice injected with null adenovirus. Autoantibodies were detectable 3 wk following adenovirus injection and reached a plateau by 8 wk (Fig. 7A). We treated mice with the PARP-1 inhibitor 5-AIQ, starting at week 13 after adenovirus injection when the mice were already diseased, and euthanized them 6 wk later. At week 19, mice with accelerated disease without treatment showed 50% survival, whereas the mice treated with inhibitor showed 83% survival (Fig. 7B). Mice treated with 5-AIQ also had lower levels of BUN (Fig. 7C). We choose to treat rather than to prevent lupus GN, and treating is far more difficult that preventing; therefore, these results strongly underscore the relevance of PARP-1 in lupus GN. Fig. 7D shows that PARP-1 activation was effectively blocked by 5-AIQ treatment, and immune complex deposition did not show any difference with or without 5-AIQ treatment. We were unable to detect PAR accumulation or immune complex deposition in mice injected with null adenovirus (data not shown). Fig. 7E shows the histopathology of representative glomeruli from the mice that survived. The renal histopathology showed that PARP-1 inhibition delays lymphocyte infiltration, cellular proliferation, and necrotic cell death. These results strongly suggest that PARP-1 inhibition blocks the development of spontaneous lupus nephritis.
FIGURE 7.
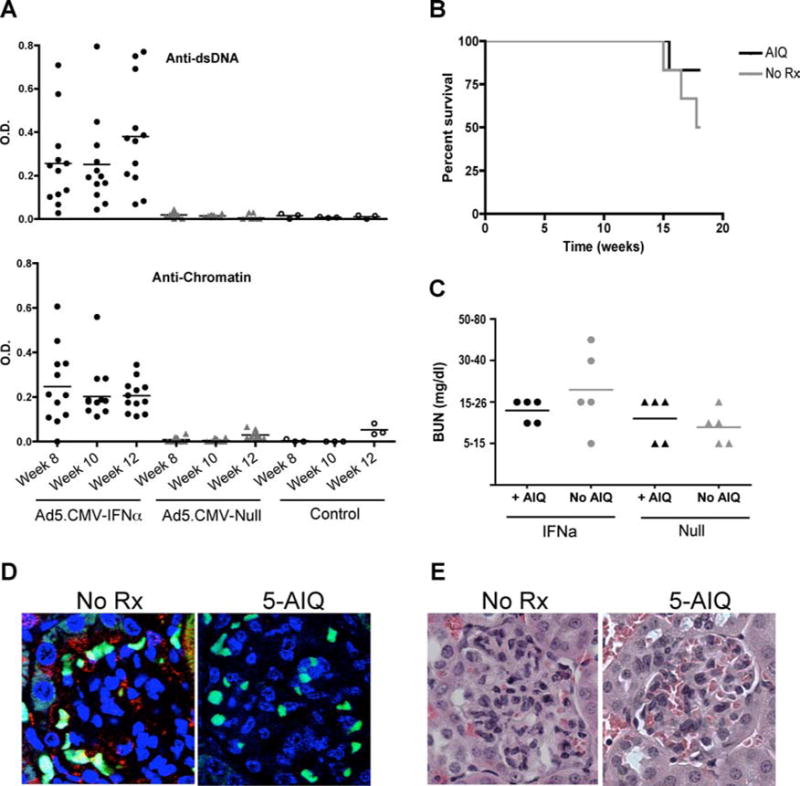
Inhibition of PARP-1 in accelerated model of lupus nephritis improves survival. Eight-week-old NZB/NZW F1 mice were injected with 1 × 109 virus particles/mouse AdvCMV-mIFN-α or AdvCMV-Null (Qbiogene). The autoantibody generation was determined by ELISA. Mice injected with AdvCMV-mIFN-α show significantly higher number of anti-chromatin and anti-dsDNA Abs (A). The treatment with 5-AIQ was initiated 13 wk after adenovirus injection. Mice treated with 5-AIQ showed lower levels of BUN (B). At 20 wk after adenovirus injection, the untreated mice showed 50% survival, whereas the mice treated with 5-AIQ showed 83% survival (C). The 5-AIQ treatment blocked PARP-1 activation, but did not affect immune complex deposition. Blue, nuclei; red, PAR; green, immune complex deposition (D). Representative of three to five mice each group. No AIQ, n = 3; AIQ treatment, n = 5. The renal histopathology showed that PARP-1 inhibition delays lymphocyte infiltration, cellular proliferation, and necrotic cell death (E). Representative of three to five mice each group. No AIQ, n = 3; AIQ treatment, n= 5.
Absence of PARP-1 in mesangial cells inhibits integrin up-regulation upon TNF-α stimulation
The data with chimeric mice suggest that PARP-1 activation in renal cells influences the severity of nephritis. Therefore, we determined the role of PARP-1 in mesangial cells, an important component of the renal tissue. PARP-1 has been reported to regulate the expression of integrins in cochlear lateral wall and endothelial cells (48, 49). Integrin up-regulation by mesangial cells has been suggested as an important regulator of local inflammation in GN (50–52). We determined whether PARP-1 regulates the induction of integrin expression by mesangial cells in response to TNF-α. PARP-1 expression in mesangial cells was inhibited using short hairpin RNA, and cell surface expression of VCAM-1 upon TNF-α stimulation was determined by flow cytometry. As shown in Fig. 8A, clones 3–7 and 4–9 showed 60–90% silencing of PARP-1 expression compared with vector-infected clone V-1. The vector-infected mesangial cells showed increased VCAM-1 expression in response to concentrations of TNF-α as low as 5 ng/ml, whereas in absence of PARP-1 mesangial cells were unable to up-regulate VCAM-1 (Fig. 8B). These data confirm the role of PARP-1 in intrinsic renal cells.
FIGURE 8.
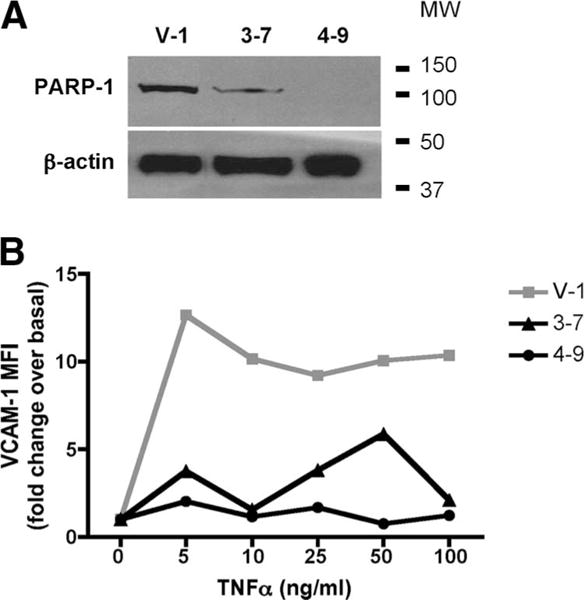
PARP-1−/− mesangial cells show reduced integrin up-regulation upon TNF-α stimulation. A, Mesangial cells were infected with either vector or PARP-1 short hairpin RNA. PARP-1 expression was detected in single-cell clones by immnoblots. A, Clones 3–7 and 4–9 showed silencing of PARP-1 gene compared with the vector-infected cells, V-1. B, Cells were stained with anti-VCAM-1 Abs and analyzed by FACS. A representative of four independent experiments is shown. The data show that inhibition of PARP-1 gene expression inhibits up-regulation of VCAM-1 in response to TNF-α stimulation.
Discussion
Our data demonstrate that PARP-1 plays a fundamental role in the pathogenesis of immune-mediated nephritis in males and suggest that the pathways contributing to nephritis in male and female may be different. We show that PARP-1 is overactivated during autoimmune nephritis. Overactivation of PARP-1 induces necrotic cell death due to energy depletion (7, 8, 15, 16), and PARP-1−/− splenocytes are resistant to necrotic cell death (supplemental Fig. 4).5 We demonstrate that the inhibition or deletion of PARP-1 reduces the damage to the renal tissue by limiting necrotic cell death. Our data indicate that PARP-1 regulates local inflammation in the kidney during nephritis, and therefore, may be target for therapeutic intervention in autoimmune nephritis.
A mechanism by which PARP-1 may be relevant only in males is that PARP-1 interacts with ERα (33). Estrogen facilitates the binding of PARP-1 to ERα and to DNA, forming a tight complex, which inhibits the release of PARP-1 in response to DNA breaks. Our interpretation is supported by our data in which treatment with 17β-estradiol delays the disease progression, and enhances survival of male mice. Our data indirectly demonstrate that PARP-1 is not involved in the pathogenesis of nephritis in females possibly due to the unavailability of PARP-1 that is bound in a complex with estrogens. The involvement of factors other than estrogens in sex specificity of PARP-1 function, however, cannot be excluded. Further studies to identify these regulators of PARP-1 function are being conducted in our laboratory.
GN is one of the most severe features of systemic lupus erythematosus (53). Males with lupus have an increased prevalence of renal disease and a poor prognosis (54, 55). However, due to higher prevalence of lupus in females (female-to-male ratio of 9:1), male lupus nephritis is often underinvestigated. The protection of only male mice in our models demonstrates that different pathways may be responsible for the downstream damage to renal tissue in male and females, and may be the underlying cause for aggravated renal disease in males. Moreover, because anti-GBM nephritis in humans is more prevalent in males, PARP-1 may be a therapeutic target specific for male nephritis.
We have used NTS-induced nephritis and an accelerated model of lupus nephritis as two models for immune-mediated nephritis. Although in these two disease settings the nature or presence of immune complexes and pathological details differs, there is an increasing evidence indicating that the same set of effector molecules determines the degree of tissue damage and renal dysfunction in both disease scenarios, suggesting that many of the pathways leading to disease development are shared (31, 32). Several molecular mediators, including chemokines and cytokines, have been shown to play important roles in the pathogenesis of both NTN and lupus nephritis. The activation of terminal complement components can lead to exacerbation of disease and damage to tissues in both NTN and lupus nephritis. Inhibition of complement activation by Crry-Ig reduced renal injury during experimental nephritis (56, 57) and in a spontaneous mouse model of lupus nephritis (58, 59). FcR-deficient mice showed minimal histopathology and survived longer in response to NTN (60), and FcR-deficient NZB/NZW mice were protected from severe nephritis (61). Therefore, Abdirected complement activation and FcR-mediated injury appear to be necessary in both NTN and lupus nephritis. In addition, several cytokines have also been suggested to be involved in the pathogenesis of both diseases. These include TNF-α, IL-1, and IL-12 (20–23, 62–66). These studies suggest that the local inflammatory response postimmune complex deposition in the renal tissue observed in lupus nephritis may be similar to that during NTN.
Because PARP-1 can act as a coactivator of NF-κB (17–19), it may mediate its effect either by inducing necrotic cell death and/or by inducing NF-κB activation. NF-κB activation may further induce necrosis by inducing proinflammatory cytokine secretion and generation of ROS. PARP-1 may therefore be involved in a feedback loop that exacerbates local inflammation. Absence or inhibition of PARP-1 will, therefore, lead to reduced inflammation and, consequently, reduced tissue damage, by inhibition of necrosis and NF-κB activation. Absence of PARP-1 will, however, inhibit necrosis in both renal cells as well as the infiltrating leukocytes. Indeed, our data with chimeric mice show that PARP-1 activation is relevant in both hematopoietic and renal cells, and the absence of PARP-1 in either one compartment is not sufficient to confer protection. Oliver et al. (67) have demonstrated a defective NF-κB activation in peritoneal macrophages isolated from PARP-1−/− mice injected with LPS, and the mice were protected from endotoxic shock. In our model, the absence of PARP-1 in the hematopoietic compartment may result in defective activation of the infiltrating cells, resulting in reduced proinflammatory cytokine secretion.
We also show that inhibition of PARP-1 expression in mouse mesangial cells, in vitro, reduced the up-regulation of VCAM-1 in response to TNF-α stimulation. VCAM-1 supports adhesion of eosinophils, basophils, monocytes, and lymphocytes through interaction with VLA-4. Induction of VCAM-1 expression is observed on proximal tubule cells in human crescentic nephritis, lupus nephritis, and IgA nephropathy (68), and VCAM-1 is also up-regulated in the kidney of nephritic MRL/lpr mice (51, 54). Evidence suggests that enhanced VCAM-1 expression confers increased adhesiveness to the renal parenchyma in MRL/lpr lupus nephritis, and serves as a pathway by which inflammatory cells adhere to the renal tissue to promote renal injury (51). We propose that PARP-1 contributes to the inflammatory response not only by inducing necrotic cell death and the production of proinflammatory cytokines such as TNF-α, but also by stimulating integrin up-regulation in intrinsic renal cells, which facilitates leukocyte infiltration.
Our data also suggest that PARP-1 may induce proinflammatory cytokine secretion in both infiltrating cells and renal cells, and maintain the inflammatory response. We show in this study that the inhibition of PARP-1 reduces TNF-α mRNA levels in the kidney during NTS-induced nephritis. TNF-α plays a key role in the development of GN (20, 22, 23), which has further been confirmed by the inability to induce anti-GBM GN in TNF-αβ-deficient mice (21). Blockade of TNF-α has also been shown to reduce inflammation and scarring in experimental crescentic GN (22, 23). Our data show that the protection by PARP-1 inhibition is accompanied by reduced TNF-α secretion possibly due to reduced NF-κB activation and inhibition of necrotic cell death. Inhibition of PARP-1 increased the levels of IL-10 transcripts during NTS-induced nephritis. Wilson et al. (69) showed that macrophages genetically modified to secrete IL-10 protected mice from experimental nephritis. Our data further support those findings; however, we cannot exclude the possibility that increased IL-10 is due to decreased TNF-α levels.
In conclusion, our data show that PARP-1 activation and consequent necrotic cell death play an important role in the pathogenesis of lupus and other immune-mediated nephritides in males, and that inhibition of PARP-1 and, consequently, of necrotic cell death and proinflammatory response, may be a novel therapeutic target in male GN.
Supplementary Material
Acknowledgments
We thank Drs. T. L. McGaha, K. R. McLeish, and P. L. Cohen for critically reading the manuscript and for their excellent suggestions. We also thank Dr. R. A. Eisenberg for providing MRL-lpr kidneys, Dr. S. Boland for providing FcγRIIb−/−/Yaa kidneys, and Dr. C. B. Thompson and D. Ditsworth for providing PARP-1 short hairpin RNA constructs. We are grateful to Dr. Mark Birkenbach for evaluating the histopathology of mice treated with 5-AIQ.
Footnotes
This work was supported by the Alliance for Lupus Research (to R.C. and S.G.) and the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (to R.C.).
Abbreviations used in this paper: GN, glomerulonephritis; 5-AIQ, 5-aminoisoquinolinone; BBS, borate-buffered saline; BM, bone marrow; BUN, blood urea nitrogen; Ct, cycle threshold; ER, estrogen receptor; GBM, glomerular basement membrane; NTN, nephrotoxic serum-induced nephritis; NTS, nephrotoxic serum; PAR, poly-(ADP-ribose); PARP-1, PAR polymerase-1; ROS, reactive oxygen species; NZB, New Zealand Black; NZW, New Zealand White.
The online version of this article contains supplemental material
Disclosures
The authors have no financial conflict of interest.
References
- 1.Grande JP. Mechanisms of progression of renal damage in lupus nephritis: pathogenesis of renal scarring. Lupus. 1998;7:604–610. doi: 10.1191/096120398678920721. [DOI] [PubMed] [Google Scholar]
- 2.Gwinner W, Grone HJ. Role of reactive oxygen species in glomerulonephritis. Nephrol Dial Transplant. 2000;15:1127–1132. doi: 10.1093/ndt/15.8.1127. [DOI] [PubMed] [Google Scholar]
- 3.Barchowsky A, Munro SR, Morana SJ, Vincenti MP, Treadwell M. Oxidant-sensitive and phosphorylation-dependent activation of NF-κB and AP-1 in endothelial cells. Am J Physiol. 1995;269:L829–L836. doi: 10.1152/ajplung.1995.269.6.L829. [DOI] [PubMed] [Google Scholar]
- 4.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 5.Weening JJ, D’Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, Balow JE, Bruijn JA, Cook T, Ferrario F, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol. 2004;15:241–250. doi: 10.1097/01.asn.0000108969.21691.5d. [DOI] [PubMed] [Google Scholar]
- 6.Lionaki S, Jennette JC, Falk RJ. Anti-neutrophil cytoplasmic (ANCA) and anti-glomerular basement membrane (GBM) autoantibodies in necrotizing and crescentic glomerulonephritis. Semin Immunopathol. 2007;29:459–474. doi: 10.1007/s00281-007-0093-0. [DOI] [PubMed] [Google Scholar]
- 7.Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. BioEssays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- 11.Nguewa PA, Fuertes MA, Valladares B, Alonso C, Perez JM. Poly(ADP-ribose) polymerases: homology, structural domains and functions: novel therapeutical applications. Prog Biophys Mol Biol. 2005;88:143–172. doi: 10.1016/j.pbiomolbio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 13.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 14.Heeres JT, Hergenrother PJ. Poly(ADP-ribose) makes a date with death. Curr Opin Chem Biol. 2007;11:644–653. doi: 10.1016/j.cbpa.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 15.Berger NA, Berger SJ. Metabolic consequences of DNA damage: the role of poly (ADP-ribose) polymerase as mediator of the suicide response. Basic Life Sci. 1986;38:357–363. doi: 10.1007/978-1-4615-9462-8_39. [DOI] [PubMed] [Google Scholar]
- 16.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 18.Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci. 2002;23:122–129. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- 19.Hassa PO, Hottiger MO. A role of poly(ADP-ribose) polymerase in NF-κB transcriptional activation. Biol Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 20.Le Hir M, Haas C, Marino M, Ryffel B. Prevention of crescentic glomerulonephritis induced by anti-glomerular membrane antibody in tumor necrosis factor-deficient mice. Lab Invest. 1998;78:1625–1631. [PubMed] [Google Scholar]
- 21.Ryffel B, Eugster H, Haas C, Hir MLe. Failure to induce anti-glomerular basement membrane glomerulonephritis in TNF α/β deficient mice. Int J Exp Pathol. 1998;79:453–460. doi: 10.1046/j.1365-2613.1998.00080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karkar AM, Smith J, Pusey CD. Prevention and treatment of experimental crescentic glomerulonephritis by blocking tumor necrosis factor-α. Nephrol Dial Transplant. 2001;16:518–524. doi: 10.1093/ndt/16.3.518. [DOI] [PubMed] [Google Scholar]
- 23.Khan SB, Cook HT, Bhangal G, Smith J, Tam FWK, Pusey CD. Antibody blockade of TNF-α reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. 2005;67:1812–1820. doi: 10.1111/j.1523-1755.2005.00279.x. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan J, O’Connor M, Hake PW, Zingarelli B. Inhibitors of poly(ADP-ribose) polymerase ameliorate myocardial reperfusion injury by modulation of activator protein-1 and neutrophil infiltration. Shock. 2005;23:233–238. [PubMed] [Google Scholar]
- 25.Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly(ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng J, Devalaraja-Narashimha K, Singaravelu K, Padanilam BJ. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am J Physiol. 2005;288:F387–F398. doi: 10.1152/ajprenal.00436.2003. [DOI] [PubMed] [Google Scholar]
- 27.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 28.Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, Verma A, Wang ZQ, Snyder SH. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia S, Bodano A, Gonzalez A, Forteza J, Gomez-Reino JJ, Conde C. Partial protection against collagen antibody-induced arthritis in PARP-1 deficient mice. Arthritis Res Ther. 2006;8:R14. doi: 10.1186/ar1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Rey E, Martinez-Romero R, O’Valle F, Aguilar-Quesada R, Conde C, Delgado M, Oliver FJ. Therapeutic effect of a poly(ADP-ribose) polymerase-1 inhibitor on experimental arthritis by down-regulating inflammation and Th1 response. PLoS ONE. 2007;2:e1071. doi: 10.1371/journal.pone.0001071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu Y, Du Y, Mohan C. Experimental anti-GBM disease as a tool for studying spontaneous lupus nephritis. Clin Immunol. 2007;124:109–118. doi: 10.1016/j.clim.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 32.Du Y, Fu Y, Mohan C. Experimental anti-GBM nephritis as an analytical tool for studying spontaneous lupus nephritis. Archivum Immunologiae et Therapiae Experimentalis. 2008;56:31–40. doi: 10.1007/s00005-008-0007-4. [DOI] [PubMed] [Google Scholar]
- 33.Mabley JG, Horvath EM, Murthy KG, Zsengeller Z, Vaslin A, Benko R, Kollai M, Szabo C. Gender differences in the endotoxin-induced inflammatory and vascular responses: potential role of poly(ADP-ribose) polymerase activation. J Pharmacol Exp Ther. 2005;315:812–820. doi: 10.1124/jpet.105.090480. [DOI] [PubMed] [Google Scholar]
- 34.Christensen M, Su AW, Snyder RW, Greco A, Lipschutz JH, Madaio MP. Simvastatin protection against acute immune-mediated glomerulonephritis in mice. Kidney Int. 2006;69:457–463. doi: 10.1038/sj.ki.5000086. [DOI] [PubMed] [Google Scholar]
- 35.Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-α induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White) F1 but not in BALB/c mice. J Immunol. 2005;174:2499–2506. doi: 10.4049/jimmunol.174.5.2499. [DOI] [PubMed] [Google Scholar]
- 36.Sivarajah A, Chatterjee PK, Patel NS, Todorovic Z, Hattori Y, Brown PA, Stewart KN, Mota-Filipe H, Cuzzocrea S, Thiemermann C. Agonists of peroxisome-proliferator activated receptor-γ reduce renal ischemia/reperfusion injury. Am J Nephrol. 2003;23:267–276. doi: 10.1159/000072088. [DOI] [PubMed] [Google Scholar]
- 37.Chatterjee PK, Patel NS, Sivarajah A, Kvale EO, Dugo L, Cuzzocrea S, Brown PA, Stewart KN, Mota-Filipe H, Britti D, et al. GW274150, a potent and highly selective inhibitor of iNOS, reduces experimental renal ischemia/reperfusion injury. Kidney Int. 2003;63:853–865. doi: 10.1046/j.1523-1755.2003.00802.x. [DOI] [PubMed] [Google Scholar]
- 38.Pieper AA, Blackshaw S, Clements EE, Brat DJ, Krug DK, White AJ, Pinto-Garcia P, Favit A, Conover JR, Snyder SH, Verma A. Poly(ADP-ribosyl)ation basally activated by DNA strand breaks reflects glutamate-nitric oxide neurotransmission. Proc Natl Acad Sci USA. 2000;97:1845–1850. doi: 10.1073/pnas.97.4.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eisenberg R, Choudhury A. The anti-DNA knock-in model of systemic autoimmunity induced by the chronic graft-vs-host reaction. Methods Mol Med. 2004;102:273–284. doi: 10.1385/1-59259-805-6:273. [DOI] [PubMed] [Google Scholar]
- 40.Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int. 2004;65:2223–2227. doi: 10.1111/j.1523-1755.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 41.Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068–1075. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- 42.McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly(ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25:502–512. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- 43.Unanue ER, Dixon FJ. Experimental glomerulonephritis. VI. The autologous phase of nephrotoxic serum nephritis. J Exp Med. 1965;121:715–725. doi: 10.1084/jem.121.5.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambrose HE, Willimott S, Beswick RW, Dantzer F, de Murcia JM, Yelamos J, Wagner SD. Poly(ADP-ribose) polymerase-1 (Parp-1)-deficient mice demonstrate abnormal antibody responses. Immunology. 2008 Sep 8; doi: 10.1111/j.1365-2567.2008.02921.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morel L, Wakeland EK. Lessons from the NZM2410 model and related strains. Int Rev Immunol. 2000;19:423–446. doi: 10.3109/08830180009055506. [DOI] [PubMed] [Google Scholar]
- 46.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcγRIIB−/− mice. J Exp Med. 2002;195:1167–1174. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lambert PH, Dixon FJ. Pathogenesis of the glomerulonephritis of NZB/W mice. J Exp Med. 1968;127:507–522. doi: 10.1084/jem.127.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi X, Nuttall AL. Expression of adhesion molecular proteins in the cochlear lateral wall of normal and PARP-1 mutant mice. Hear Res. 2007;224:1–14. doi: 10.1016/j.heares.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 49.Von Lukowicz T, Hassa PO, Lohmann C, Boren J, Braunersreuther V, Mach F, Odermatt B, Gersbach M, Camici GG, Stahli BE, et al. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc Res. 2008;78:158–166. doi: 10.1093/cvr/cvm110. [DOI] [PubMed] [Google Scholar]
- 50.Wuthrich RP, Jevnikar AM, Takei F, Glimcher LH, Kelley VE. Intercellular adhesion molecule-1 (ICAM-1) expression is up-regulated in autoimmune murine lupus nephritis. Am J Pathol. 1990;136:441–450. [PMC free article] [PubMed] [Google Scholar]
- 51.Wuthrich RP. Vascular cell adhesion molecule-1 (VCAM-1) expression in murine lupus nephritis. Kidney Int. 1992;42:903–914. doi: 10.1038/ki.1992.367. [DOI] [PubMed] [Google Scholar]
- 52.Ikezumi Y, Hurst LA, Masaki T, Atkins RC, Nikolic-Paterson DJ. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003;63:83–95. doi: 10.1046/j.1523-1755.2003.00717.x. [DOI] [PubMed] [Google Scholar]
- 53.Agrawal N, Chiang LK, Rifkin IR. Lupus nephritis. Semin Nephrol. 2006;26:95–104. doi: 10.1016/j.semnephrol.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 54.Adler S, Brady HR. Cell adhesion molecules and the glomerulopathies. Am J Med. 1999;107:371–386. doi: 10.1016/s0002-9343(99)00233-8. [DOI] [PubMed] [Google Scholar]
- 55.Yacoub Wasef SZ. Gender differences in systemic lupus erythematosus. Gend Med. 2004;1:12–17. doi: 10.1016/s1550-8579(04)80006-8. [DOI] [PubMed] [Google Scholar]
- 56.Quigg RJ, He C, Lim A, Berthiaume D, Alexander JJ, Kraus D, Michael Holers V. Transgenic mice overexpressing the complement inhibitor Crry as a soluble protein are protected from antibody-induced glomerular injury. J Exp Med. 1998;188:1321–1331. doi: 10.1084/jem.188.7.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quigg RJ, Kozono Y, Berthiaume D, Lim A, Salant DJ, Weinfeld A, Griffin P, Kremmer E, Holers VM. Blockade of antibody-induced glomerulonephritis with Crry-Ig, a soluble murine complement inhibitor. J Immunol. 1998;160:4553–4560. [PubMed] [Google Scholar]
- 58.Bao L, Haas M, Kraus DM, Hack BK, Rakstang JK, Holers VM, Quigg RJ. Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr mice. J Am Soc Nephrol. 2003;14:670–679. doi: 10.1097/01.asn.0000051597.27127.a1. [DOI] [PubMed] [Google Scholar]
- 59.Bao L, Zhou J, Holers VM, Quigg RJ. Excessive matrix accumulation in the kidneys of MRL/lpr lupus mice is dependent on complement activation. J Am Soc Nephrol. 2003;14:2516–2525. doi: 10.1097/01.asn.0000089831.96794.0b. [DOI] [PubMed] [Google Scholar]
- 60.Park SY, Ueda S, Ohno H, Hamano Y, Tanaka M, Shiratori T, Yamazaki T, Arase H, Arase N, Karasawa A, et al. Resistance of Fc receptor-deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102:1229–1238. doi: 10.1172/JCI3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 62.Brennan DC, Yui MA, Wuthrich RP, Kelley VE. Tumor necrosis factor and IL-1 in New Zealand Black/White mice: enhanced gene expression and acceleration of renal injury. J Immunol. 1989;143:3470–3475. [PubMed] [Google Scholar]
- 63.Tang WW, Feng L, Vannice JL, Wilson CB. Interleukin-1 receptor antagonist ameliorates experimental anti-glomerular basement membrane antibody-associated glomerulonephritis. J Clin Invest. 1994;93:273–279. doi: 10.1172/JCI116956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun H, Liu W, Shao J. Study of immunoregulation by interleukin-1 receptor antagonist in NZB/W F1 mice. Zhonghua Yi Xue Za Zhi. 1996;76:600–602. [PubMed] [Google Scholar]
- 65.Timoshanko JR, Kitching AR, Holdsworth SR, Tipping PG. Interleukin-12 from intrinsic cells is an effector of renal injury in crescentic glomerulonephritis. J Am Soc Nephrol. 2001;12:464–471. doi: 10.1681/ASN.V123464. [DOI] [PubMed] [Google Scholar]
- 66.Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, Kelley VR. IL-12 drives IFN-γ-dependent autoimmune kidney disease in MRL-Faslpr mice. J Immunol. 1999;163:6884–6891. [PubMed] [Google Scholar]
- 67.Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. Am J Hum Genet. 1999;64:1282–1288. doi: 10.1086/302389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seron D, Cameron JS, Haskard DO. Expression of VCAM-1 in the normal and diseased kidney. Nephrol Dial Transplant. 1991;6:917–922. doi: 10.1093/ndt/6.12.917. [DOI] [PubMed] [Google Scholar]
- 69.Wilson HM, Stewart KN, Brown PAJ, Anegon I, Chettibi S, Rees AJ, Kluth DC. Bone-marrow-derived macrophages genetically modified to produce IL-10 reduce injury in experimental glomerulonephritis. Mol Ther. 2002;6:710–717. doi: 10.1006/mthe.2002.0802. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.