

Abstract
Direct conversion of fibroblasts into induced cardiomyocytes (iCMs) offers an alternative strategy for cardiac disease modeling and regeneration. During iCM reprogramming, the starting fibroblasts must overcome existing epigenetic barriers to acquire the CM-like chromatin pattern. However, epigenetic dynamics along this reprogramming process have not been studied. Here, we took advantage of our recently generated polycistronic system and determined the dynamics of two critical histone marks, H3K27me3 and H3K4me3, in parallel with gene expression at a set of carefully selected cardiac and fibroblast loci during iCM reprogramming. We observed reduced H3K27me3 and increased H3K4me3 at cardiac promoters as early as day 3, paralleled by a rapid significant increase in their mRNA expression. In contrast, H3K27me3 at loci encoding fibroblast marker genes did not increase until day 10 and H3K4me3 progressively decreased along the reprogramming process; these changes were accompanied by a gradual decrease in the mRNA expression of fibroblast marker genes. Further analyses of fibroblast-enriched transcription factors revealed a similarly late deposition of H3K27me3 and decreased mRNA expression of Sox9, Twist1 and Twist2, three important players in epithelial-mesenchymal transition. Our data suggest early rapid activation of the cardiac program and later progressive suppression of fibroblast fate at both epigenetic and transcriptional levels. Additionally, we determined the DNA methylation states of representative cardiac promoters and found that not every single CpG was equally demethylated during early stage of iCM reprogramming. Rather, there are specific CpGs, whose demethylation states correlated tightly with transcription activation, that we propose are the major contributing CpGs. Our work thus reveals a differential re-patterning of H3K27me3, H3K4me3 at cardiac and fibroblast loci during iCM reprogramming and could provide future genome-wide epigenetic studies with important guidance such as the appropriate time window and loci to be utilized as positive and negative controls.
Keywords: iCM, epigenetics, reprogramming, H3K4me3, H3K27me3, ChIP, DNA methylation
1. INTRODUCTION
Direct conversion of fibroblasts into a range of cell lineages [1-4] holds great potential for applications in disease modeling and regenerative medicine. In the heart specifically, direct cardiac reprogramming of fibroblasts into induced cardiomyocytes (iCMs) has been achieved in vitro and in vivo through several methods using the transcription factor (TF) cocktail Gata4 (G), Mef2c (M), and Tbx5 (T) [2, 5-7], a microRNA cocktail [8], and other variations [9-16]. Despite many attempts to improve the efficiency of direct cardiac reprogramming [11-16], the conversion rate is still far from the criteria for application. More importantly, across all these different methods, only a fraction of cells expressing reprogramming factors achieved a mature and functional cardiomyocyte (CM) state, while many appeared at various stages of incomplete reprogramming. This asynchronous conversion suggests the existence of a series of molecular barriers, such as the epigenetic barrier that must be overcome during reprogramming in order for cells to acquire complete and stable cell fate changes.
Cell reprogramming is inherently an epigenetic process [17]. Heritable modifications to cytosine bases in the DNA and histones that package the genome constitute the epigenome [18]. The epigenetic status of a cell regulates its chromatin structure and DNA accessibility, and influences expression of the genome across a diverse array of tissue types, developmental stages, and disease states. Consequently, cell fate changes such as those that occur during normal development and cell reprogramming involve dynamic re-patterning of epigenetic landscapes. Existing epigenetic barriers in the starting cell type must be overcome and target cell-like chromatin pattern must be built. Since the discovery of induced pluripotent stem cells (iPSC) [19], epigenetic remodeling along the process of reprogramming somatic cells to pluripotency has been extensively characterized. Early and widespread epigenetic re-patterning was observed and shown to be critical for pluripotency induction [20, 21]. These studies not only improved our understanding of the molecular mechanisms of induced pluripotency and addressed fundamental questions about cell fate decisions, but also enabled rational design of novel reprogramming cocktails. Similarly, elucidating the epigenetic dynamics accompanying direct cardiac reprogramming is critical to improve the efficiency and quality of iCM generation and to better our understanding of the biology of fibroblast plasticity and CM fate determination.
Among the more than 100 different histone modifications, histone3-lysine27-trimethylation (H3K27me3) and histone3-lysine4-trimethylation (H3K4me3) are widely used histone marks associated with inactive and active promoters, respectively [22]. The H3K27me3 methyltransferase polycomb repressive complex 2 is an important regulator of normal heart development [23, 24]. Mutations in MLL2, one of the H3K4 methyltransferases, are a major cause of congenital heart defects in Kabuki Syndrome in human [25]. Recently, two epigenetic studies on embryonic stem cell (ESC) differentiation into CMs revealed distinct temporal patterns of H3K27me3 and H3K4me3 that are coordinated with changes in mRNA expression of functionally related genes [26, 27]. Another important regulator of chromatin state is the methylation status of CpG dinucleotides in DNA, particularly in the promoter regions [28]. Global lack of DNA methylation in ESCs precludes differentiation [29] while DNA methylation-silencing of pluripotency-associated promoters allows differentiation to proceed [30]. Dynamic DNA methylation is also associated with normal cardiac development, such as myocyte maturation and contractile functioning during postnatal transition of CMs to mature adult CMs [31]. Together, epigenetic regulation plays an essential role in a variety of important cellular processes like cardiac development, yet its role in direct cardiac reprograming remains unknown.
Recently, we established a selectable polycistronic system for the delivery of iCM reprogramming factors G, M, and T, where the complete set of constructs resulted in a wide range of reprogramming efficiencies, while infection with each construct led to a relatively homogenous cell population [32]. Since these constructs consistently give rise to differential reprogramming consequences, parallel comparisons among them may reveal important epigenetic changes and identify key regulators and events in iCM reprogramming. In this study, we first characterized H3K27me3 and H3K4me3 patterns in primary neonatal CMs, the HL-1 CM cell line, and three different types of primary fibroblasts to determine their levels at cardiac and fibroblast loci in the starting and target cell types of iCM reprogramming. Interestingly, our data revealed that both H3K27me3 and H3K4me3 marks were present at fibroblast regulatory loci in CMs and cardiac and fibroblast regulatory loci in fibroblasts. We then harnessed our polycistronic system and utilized chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR) to interrogate cardiac and fibroblast promoters for possible H3K27me3 and H3K4me3 re-patterning at two critical time points during iCM reprogramming, and also sought to answer whether and how observed changes in histone marks corresponded to mRNA expression of these genes and reprogramming outcomes. Our data revealed early changes in both histone marks at cardiac promoters and later alterations at fibroblast marker genes. This re-patterning of histone marks coincided with rapid activation of cardiac gene expression and gradual suppression of fibroblast marker genes expression. Next, we generated a list for fibroblast-enriched TFs based on previously published data and examined changes in histone marks at their promoters during iCM reprogramming. We also found a late deposition of H3K27me3 in about one third of the tested fibroblast-enriched TFs, and observed a decrease in mRNA expression in a few of these TFs. Additionally, we determined the DNA methylation states of two select cardiac loci and found that both promoters were demethylated at an early stage of reprogramming. Importantly, we discovered that specific CpGs in these promoters were main contributors to total demethylation and their methylation states closely correlated with transcription activation.
2. MATERIALS AND METHODS
2.1 Mouse lines
Transgenic mice harboring GFP under the control of the αMHC promoter [2, 6, 32] were used for primary neonatal CM, primary cardiac fibroblast (CF), tail-tip fibroblast (TTF), and mouse embryonic fibroblast (MEF) isolation. Animal care was provided in accordance with guidelines established by University of North Carolina, Chapel Hill.
2.2 Plasmids
The six polycistronic constructs expressing M, G, and T (pMXs-MGT/MTG/TMG/TGM/GMT/GTM) and MGT with puro selection marker (pMXs-puro-MGT) were previously constructed in the lab [32]. To generate pMXs-puro-GTM/DsRed, GTM was excised from pGEMT-easy vector and DsRed was excised from pMXs-DsRed (Addgene 22724) and cloned into pMXs-puro vector. To generate pMXs-GFP-Gata4-Tbx5, EGFP was PCR amplified from pLenti-GFP (Cell Biolabs, LTV-400) and inserted into pMXs-MGT to replace Mef2c.
2.3 Retrovirus packaging and transduction
PlatE packaging cells were maintained in growth media: DMEM containing 10% Fetal Bovine Serum (FBS), 50 units/50 μg/ml penicillin/streptomycin, 1 μg/ml puromycin (Sigma), and 100 μg/ml of blasticidin S (Life Technologies). One day before transfection, 4-5×106 cells were seeded onto 10 cm dish in growth media without puromycin and blasticidin. The next day, pMXs-based retroviral vectors were introduced into PlatE cells using lab-prepared Calcium Phosphate (CaP) transfection reagents. Generally, 20 μg of plasmid DNA were combined with 440 μl ddH2O and 40 μl 2.5M CaCl2, and the mixture was added dropwise to 500 μl of 2X HBS (50 mM HEPES, 280 mM NaCl, 10 mM KCl, 1.5 mM Na2HPO4, 12 mM Glucose, pH 7.10) while vortexing. This mixture was kept at room temperature for 3 minutes and then added dropwise to the PlatE cells. Culture media were changed to fresh media right before transfection and all reagents used were warmed up to room temperature before mixing. Transfected cells were then incubated overnight at 37°C with 5% CO2. Medium was changed the next day and virus-containing supernatant was collected 48 hours after transfection, filtered through a 0.45 μm cellulose acetate filter (Thermo Scientific) and incubated with PEG8000 (Sigma, 4 volume of supernatant and 1 volume of 40% PEG8000/PBS) overnight at 4°C. Viruses were then pelleted by centrifuge at 3500 rpm, 4°C for 30 minutes, re-suspended with fibroblast media (IMDM/20% FBS) supplemented with 4 μg/ml polybrene (Life Technologies), and added to target cells immediately. Twenty-four to 48 hours after infection, the virus-containing medium was replaced with iCM medium (10% FBS of DMEM/M199 (4:1)) and changed every 2-3 days after. For positive selection, puromycin was added to target cells (4 μg/ml for MEF and 2 μg/ml for CF and TTF) three days after viral infection and was maintained in iCM medium at a concentration of 1 μg/ml.
2.4 Cell culture and isolation of CM, CF and TTF
HL-1 CM cell line was cultured in Claycomb medium supplemented with 10% Fetal Bovine Serum (FBS), 100 units/100 μg/ml penicillin/streptomycin, 0.1 mM Norepinephrine and 2 mM L-Glutamine (Sigma). Primary neonatal CM was isolated by enzyme digestion followed by negative Magnetic-Activated Cell Sorting (MACS). Briefly, hearts were removed from P0-P2 mice and rinsed and squeezed in chilled PBS to remove blood and other tissues. The hearts were then digested in 10ml enzyme mix (0.5 mg/ml Collagenase II (Worthington) and 20 U/ml DNAseI (USB) in PBS) in 37C water bath with stirring for 5 × 10 minutes. Cells were collected, passed through 40 μm cell strainer and removed of red blood cells (RBC) by lysing in 1 ml of RBC lysis buffer (150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) for 1 minute at room temperature. The resulting RBC-free cells were resuspended in MACS buffer (DPBS with 0.5% BSA and 2 mM EDTA), counted, and resuspend with 10 ul negative selection beads (neonatal CM isolation kit, Miltenyl Biotech) in 90 ul MACS buffer per 10 million cells. After 15 minutes incubation at 4°C, the cell-beads mixture was resuspended with 2 ml MACS buffer and passed through LS column provided in the kit and the column was washed three times with 1 ml MACS buffer each. CM in the flow through and wash through was collected, pelleted, and resuspended in TRIZOL (Invitrogen, for RNA extraction and qRT-PCR), DNAzol (Invitrogen, for genomic DNA extraction and bisulfite sequencing) or 1% paraformaldehyde/PBS (EMS, for ChIP). Isolation of neonatal (day 1.5) explant CF, TTF and fresh CF was performed as previously described [32].
2.5 MEF preparation
E13.5 αMHC-GFP+ MEFs were prepared as previously described [33]. Briefly, E13.5 embryos were isolated from pregnant αMHC-GFP mice. The head and red organs were removed from each embryo and remainder of the embryo was finely minced with a sterile razor blade, digested in 1 ml of 0.05% trypsin/EDTA containing 100 Kunitz units of DNase I for 15 minutes at 37 °C, then resuspended in MEF medium (DMEM containing 10% FBS, and 1x penicillin/streptomycin) and plated onto 0.1% gelatin-coated dish. Unattached cells were removed after 2 hours and the attached fibroblasts (P0, passage 0) were frozen after 24 hours of culturing. Each embryo was also genotyped using the excised red organs and only αMHC-GFP+ MEFs were used for reprogramming. P3-P5 MEFs were predominantly used and the seeding density was 1×104 cells/cm2 for immunofluorescence staining and 3×104 cells/cm2 for ChIP.
2.6 Immunofluorescence staining and flow cytometry
For immunofluorescence staining, at day 10 post-transduction, cells were fixed with 4% paraformaldehyde /PBS at room temperature for 15 minutes, permeabilized with 0.1% Triton/PBS for 15 minutes, blocked with 5% BSA for 1 hour, then incubated with primary antibody (rabbit α-GFP, Invitrogen, 1:500 dilution) at 4°C overnight, secondary antibody (Alexa Fluor 488-conjugated donkey anti-rabbit IgG, Invitrogen, 1:500 dilution) for 1 hour at room temperature, and DAPI nuclei staining in mounting medium Vectashield (Vector labs). Between each step, the cells were washed three times with PBS. Images were acquired using EVOS® FL Auto Cell Imaging System (Life Technologies). For flow cytometry, cells were first dissociated with 0.05% trypsin/EDTA (Life Technologies) for 5 minutes at 37°C and then fixed, permeablized, and stained as described above, except that FACS buffer (PBS supplemented with 2% FBS and 2 mM EDTA) was used for washing in between steps, the blocking step was omitted, and antibody incubation was done at 4°C for 30 minutes. Cells were then run on BD Accuri™ C6 flow cytometer.
2.7 ChIP-qPCR
For primary neonatal CM, ChIP was performed using the Magnify ChIP system (ThermoFisher) according to manufacturers’ instructions. For all other cells, ChIP was performed as previously described [34]. Briefly, 5-10 million cells were cross-linked using 1% paraformaldehyde/PBS at 37°C for 15 minutes and sheared using XL2020 sonicator (MISONIX, intervals 30 seconds on, 60 seconds off for 3-6 minutes). The chromatin-histone complex was then immunoprecipitated using rabbit α-H3K4me3/α-H3K27me3/control IgG (Active Motif # 39915 & 39915) and Pierce™ Protein A/G UltraLink™ Resin. Eluted chromatin-histone complexes were then reverse-crosslinked in 0.2 M NaCl by incubating in 70°C water bath overnight and the DNA was purified by phenol/chloroform extraction followed by ethanol precipitation. DNA quality/size was checked by agarose gel electrophoresis and qPCR was performed with SYBR® Green PCR Master Mix (Applied Biosystems) on ABI ViiA 7 real-time PCR system (Applied Biosystems) as per manufacturer protocols. Primers were designed with Primer3 and their sequences were listed in Table S1. Detailed information of all the tested genes is listed in Table S2.
2.8 Quantitative RT-PCR (qRT-PCR)
RNA extraction, reverse transcription, and qPCR were performed as previously described [32]. Additional primers for SYBR Green qPCR were designed with Primer3 and their sequences are listed in Table S3.
2.9 Bisulfite sequencing
Cells were harvested at day 3 post-transduction by trypsinization, and genomic DNA was extracted using DNAzol and bisulfite converted (~500 ng/reaction) using the EZ DNA Methylation-Gold Kit (Zymo Research). Between 1-4 μl of the 10 μl bisulfite converted DNA was used for PCR amplification of the promoter regions of Myh6 and Nppa genes using previously described primers [2] and the GoTaq polymerase (Promega). The PCR products were then PCR cleaned up and TA-cloned into pGEMT-easy vector (Promega). Ten to 30 clones in each sample were picked, cultured, and sequenced.
2.10 Statistical analyses
Where appropriate, values are expressed as the mean ± standard deviation of triplicate experiments. Statistical analyses were performed with one-way or two-way analyses of variance followed by Bonferroni correction. A P value of < 0.05 was considered statistically significant (*), a P value of < 0.01 was considered highly significant (**), and a P value of < 0.001 was considered strongly significant (***). All data are representative of multiple repeated experiments.
3. RESULTS
3.1 H3K27me3 and H3K4me3 at cardiac and fibroblast loci in fibroblasts and CMs
To understand epigenetic re-patterning along iCM reprogramming, we first determined H3K27me3 and H3K4me3 patterns in fibroblasts and CMs, the initial and target cell types of iCM reprogramming. Three different types of primary fibroblasts including MEF, CF, and TTF, primary neonatal CM and the CM cell line HL-1 were used. We focused on cardiac genes and fibroblast genes because their respective activation and inactivation represent the conversion of fibroblast to CM. The cardiac genes we interrogated included reprogramming factors M, G, T and cardiac structural and functional genes. The β-actin-encoding Actb was used as a negative control for H3K27me3 and as a positive control for H3K4me3; Hoxa10 and Hoxc10 were used as positive controls for both histone marks [35]; an intragenic region in chromatin 8 (Chr_8) was included as the negative control for both histone marks. Up to five pairs of primers were designed for each gene (Table S1) at locations showing H3K27me3 and H3K4me3 peaks in previously published ChIP-seq data [26, 36]; these peaks usually occur around the transcription start site (TSS) of the gene (Fig. 1 & 2, ChIP-seq peaks from heart (the ENCODE database) and MEF [36]). Our ChIP-qPCR results demonstrated that M, G, and T were marked by H3K4me3 only in both primary CM and HL-1, and by both H3K27me3 and H3K4me3 in fibroblasts except for Mef2c, which doesn’t show H3K27me3 peaks in any ChIP-seq data (Fig. 1A, S1A, B, S2A, B). Interestingly, even though all three types of fibroblasts were marked by both H3K27me3 and H3K4me3 at these loci, CFs had the lowest H3K27me3 and highest H3K4me3, while TTF showed the opposite trend (Fig. 1A, S1A, B, S2A, B). Further examination of cardiac structural (Tnnt2 and Myh6) and functional (Ryr2, Nppa and Pln) genes showed that they were marked by H3K4me3 only in CMs and H3K27me3 only (high levels at Tnnt2 and Ryr2 and low levels at Myh6, Nppa and Pln) in fibroblasts (Fig. 1A, S1A, B, S2C, D). H3K27me3 levels at these loci encoding cardiac structural and functional genes were slightly lower in CF than MEF and TTF.
Figure 1. H3K27me3 and H3K4me3 at cardiac promoters and promoters of fibroblast marker genes.
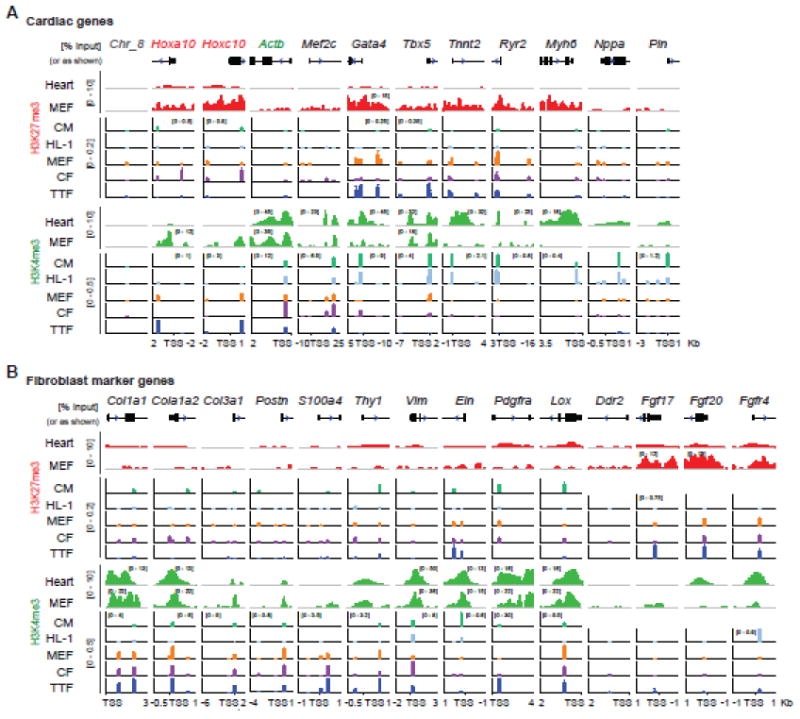
H3K27me3 and H3K4me3 ChIP were performed with primary neonatal CM, HL-1, MEF, CF, and TTF cells. Enrichment of cardiac genes (A) and fibroblast marker genes (B) were determined by quantitative PCR using up to five pairs of primers designed for each gene. For each gene and each cell type, the fold enrichment of each histone mark to ChIP input was plotted against the distance of each primer pair to TSS in kilo bases (Kb). H3K27me3 and H3K4me3 ChIP-seq peaks from mouse heart (the ENCODE project) and MEF [36], based on which the qPCR primers were designed, were shown as reference. Original data can be found in supplementary figures 1, 2, and 4.
Figure 2. H3K27me3 and H3K4me3 at loci of fibroblast-enriched TFs.
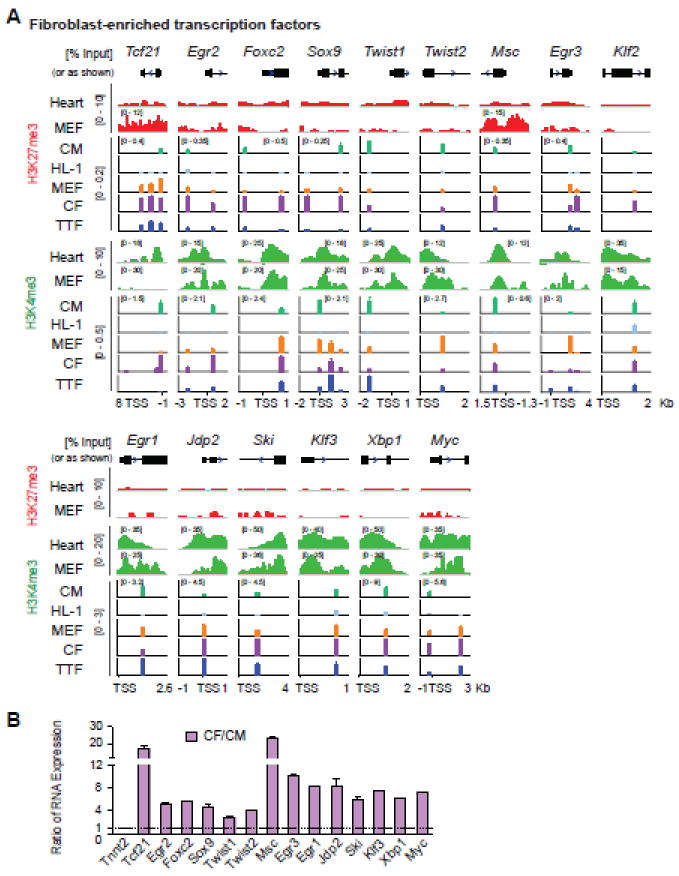
(A) H3K27me3 and H3K4me3 ChIP were performed with CM, HL-1, MEF, CF, and TTF cells, followed by qPCR of fibroblast-enriched TF genes. Plotting is the same as Figure 1. (B) RNA expression ratio of fibroblast-enriched TFs in freshly isolated neonatal CFs to CMs was determined by qRT-PCR. Tnnt2, a CM contractility gene, was included as a negative control. Original data can be found in supplementary figures 4, 5 and 6.
We then determined H3K4me3 and H3K27me3 patterns at fibroblast marker genes. Fibroblasts are heterogeneous in nature, and a set of markers have been reported for use in the identification of fibroblasts [37, 38]. Among them are collagen and related genes (Col1a1, Col1a2, Col3a1 and Lox), mesenchymal markers (Pdgfra, Postn, Vim and Ddr2), fibroblast growth factors and their receptors (Fgf17, Fgf20 and Fgfr4), and other markers (Thy1, Eln and S100a4 that encodes fibroblast-specific protein 1, FSP1) [39-42]. The majority of these tested genes (Col1a1, Col1a2, Col3a1, Postn, S100a4, Thy1, Pdgfra and Lox) were marked by H3K4me3 only in fibroblasts and unmarked in HL-1; some of them (Thy1, Pdgfra and Lox) were marked by H3K27me3 only in primary CM while the others were unmarked (Col1a1, Col1a2, Col3a1, Postn, and S100a4) (Fig. 1B, S1, S3). Interestingly, two well-established fibroblast marker genes Vim and Eln both showed high levels of H3K4me3 in primary CM and HL-1. Some of the tested loci (Ddr2, Fgf17, Fgf20 and Fgfr4) showed high levels of H3K27me3 and/or no H3K4me3 in fibroblasts and were not followed in reprogramming experiments outlined below.
3.2 Identification of fibroblast-enriched TFs and their H3K27me3 and H3K4me3 patterns
TFs are a group of genes residing at the top of the transcriptional hierarchy of the gene regulatory network. TF-mediated cellular reprogramming [1-4, 19] further supports their central roles in cell fate determination. We therefore investigated epigenetic changes at fibroblast TF loci along the cardiac reprogramming process. We started with identifying a more complete list of fibroblast-enriched TFs. Two mouse transcription factor databases (TFDB), Animal TFDB and RIKEN TFDB, were combined to generate a complete list of 3049 mouse TF transcripts. Previously published RNA-seq data [43] was re-analyzed and expression levels of 2163 TF transcripts in neonatal CF and CM were acquired (Table S4, Fig. S4). Twenty-seven TFs were then selected as CF-enriched TFs based on their high expression in CF and high ratio of CF / CM expression (Fig. S4). Among them, Tcf21 is a central regulator of CF lineage specification during development [39, 44]; Twist1 and Twist2 play essential roles in epithelial-mesenchymal transition (EMT, [45]); early growth response gene family (Egr1, 2, 3) is implicated in the pathogenesis of fibrosis [46]. We then determined H3K27me3 and H3K4me3 patterns at the promoters of TFs showing peaks in the above-mentioned ChIP-seq data (Fig. 2A); TFs showing no H3K27me3 peaks from heart tissue (ENCODE, Fig. 2A) were not assessed for H3K27me3 patterns (Egr1, Jdp2, Ski, Klf3, Xbp1, and Myc). Interestingly, ChIP-qPCR demonstrated that H3K27me3 and H3K4me3 patterns at most of these loci were not dramatically different in in primary CMs and fibroblasts (Fig. 2A, S1, S5), which is in great contrast to cardiac loci and fibroblast marker genes (Fig. 1). Most of the loci tested for H3K27me3 and H3K4me3 (Tcf21, Egr2, Foxc2, Sox9, Twist1, and Msc) were shown to be positive for both marks in primary CMs and at least one subtype of fibroblasts, except that Twist2 and Egr3 were marked by H3K27me3 only in primary CMs (Fig. 2A). Similarly, the loci tested for H3K4me3 only (Egr1, Jdp2, Ski, Klf3, Xbp1 and Myc) were marked by H3K4me3 in both primary CMs and fibroblasts (Fig. 2A). Interestingly for the HL-1 CM cell line, all of the tested loci were unmarked or marked by minimal levels of H3K27me3 or H3K4me3 (Fig. 2A), suggesting that despite functional similarities of HL-1 to CM, HL-1 cell line might not have the identical epigenetic states as primary CM and should be used cautiously in future epigenetic studies. In addition, among the three types of fibroblasts, CFs showed consistent higher levels of H3K27me3 at all marked promoters but there seems no specific pattern for the levels of H3K4me3 among the three fibroblast types. Tcf21, a key regulator of the CF lineage, was found to be marked by H3K4me3 in CFs but not in MEFs or TTFs. Consistently, we also found that Tcf21 is expressed only in CFs but not the other two fibroblast cell types (data not shown). Comparison of mRNA expression levels of the selected TFs in freshly isolated neonatal CFs and CMs using qRT-PCR confirmed their enrichment, ranging from 2.4 to 24 fold, in CFs (Fig. 2B, S6).
In summary, CMs are marked by H3K4me3 at cardiac marker and regulatory gene promoters, by H3K27me3 only or unmarked at fibroblast marker promoters, and by both H3K27me3 and H3K4me3 at fibroblast-enriched TFs; fibroblasts are marked by H3K27me3 at cardiac marker loci, by H3K4me3 at fibroblast marker loci, and by both H3K27me3 and H3K4me3 at M, G and T and fibroblast-enriched TFs. This is consistent with the general notion that regulatory genes, such as TFs, are subjected to dynamic controls and more likely to be bivalent than structural/marker genes [27, 47, 48]. Based on the above data (Fig. 1, 2), we selected one pair of primers with the highest enrichment for each gene and each histone modification (Table S1) and continued to investigate how H3K27me3 and H3K4me3 are re-patterned at critical time points during iCM reprogramming.
3.3 Early re-patterning of H3K27me3 and H3K4me3 at cardiac promoters during iCM reprogramming
We recently demonstrated that different order combinations of reprogramming factors in a polycistronic construct give rise to defined ratios of M, G and T protein expression and differential iCM reprogramming outcomes [32]. In the current study, we chose to focus on the optimal reprogramming construct, MGT, and compare it in parallel to the least optimal construct, GTM, in order to determine the epigenetic dynamics during iCM reprogramming and how they correlate with the reprogramming outcome. To facilitate this effort, we modified GTM and control DsRed constructs by adding a puromycin selection marker to further enrich virus-transduced cells, in a way similar to our pMXs-puro-MGT construct [32]. Transduction of fibroblasts using these new constructs yielded expected reprogramming efficiencies: high with MGT and low with GTM (Fig. S7). This system combines defined ratios of reprogramming factor expression, and a single retrovirus transduction and puromycin selection, thus ensuring desired reprogramming outcomes and a relatively homogenous cell population. For all the following experiments, we utilized this system.
M, G, and T-induced conversion of fibroblasts to iCMs is a long process with defined stages. Cardiac markers such as αMHC-GFP start to be activated around day 3 and sarcomere structures begin to form around day 10 [2, 32]. Therefore, we determined the levels of H3K27me3, H3K4me3, and mRNA expression at these two critical time points during iCM reprogramming. We first examined the M, G, and T promoters. ChIP-qPCR results revealed significant decreases in H3K27me3 levels associated with optimal reprogramming (MGT) starting at day 3, and unaltered H3K4me3 under both reprogramming conditions (MGT and GTM) (Fig. 3A, exception: no initial H3K27me3 at Mef2c). Further examination of cardiac structural genes (Tnnt2 and Myh6) and functional genes (Ryr2, Pln and Nppa) revealed significant decreases in H3K27me3 and/or increases in H3K4me3 (except Nppa) starting day 3, and concomitant dramatic increases in RNA expression (Fig. 3B). Among them, Tnnt2 showed the most prominent changes compared to other cardiac structural and functional genes examined, with more than a 3-fold decrease in H3K27me3, an 18-fold increase in H3K4me3, and a180-fold increase in mRNA expression on day 10. While H3K27me3 and H3K4me3 at Myh6 promoter showed similar trends to those at Tnnt2 locus, H3K4me3 at Ryr2 and Pln promoters was more greatly affected than H3K27me3. These changes were observed as early as day 3, continued to be detected on day 10, and were associated with an optimal reprogramming and transcription activation except for Nppa (Fig. 3B), over-activation of which might hinder reprogramming [32]. Increased H3K27me3 at Nppa on day 10 under the optimal reprogramming condition is consistent with its slightly decreased mRNA levels on day 10 (MGT, Fig3B) and also our previous result that Nppa mRNA is upregulated to a greater extent under the least optimal reprogramming condition rather than the optimal one [32]. Together, our data showed an early re-patterning of H3K27me3 and H3K4me3 at cardiac promoters paralleled by a dramatic increase in their mRNA expression during iCM induction.
Figure 3. Re-patterning of H3K27me3 and H3K4me3 at cardiac loci during iCM reprogramming.
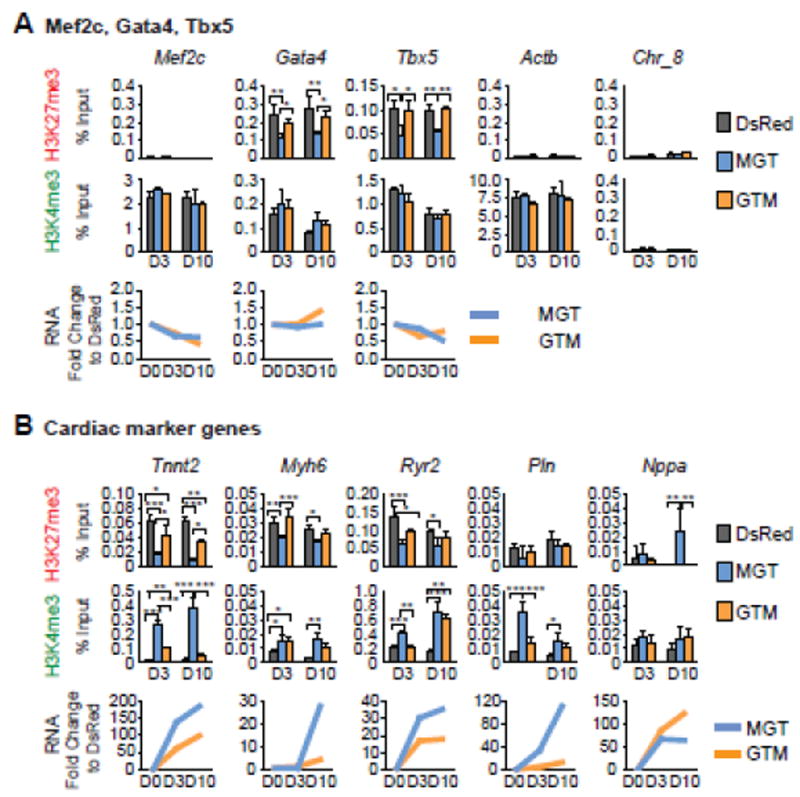
H3K27me3 and H3K4me3 ChIP of iCMs generated under the optimal condition (MGT) and the least optimal one (GTM) were performed on day 3 and day 10, followed by qPCR of M, G, T (A) or cardiac structural and marker genes (B). DsRed: retroviral transduction control. RNA expression levels of these genes were also determined in parallel by qRT-PCR and normalized to Actb first and then to the DsRed control.
3.4 Later H3K27me3 and H3K4me3 re-patterning at fibroblast promoters during iCM reprogramming
Efficient and complete erasure of fibroblast signatures is critical for efficient reprogramming. Early down-regulation of the fibroblast surface marker THY1 at day 1-2 is required for successful iPSC reprogramming [49]. Therefore, we next studied H3K27me3 and H3K4me3 re-patterning at fibroblast loci encoding both fibroblast marker genes and fibroblast-enriched TFs. For fibroblast marker genes, ChIP-qPCR demonstrated decreased H3K4me3 at Col1a2, Col3a1, and Postn paralleled by a decrease in their mRNA expression levels starting from day 3 under both reprogramming conditions (Fig. 4A). On day 10, decreased H3K4me3 was observed at additional loci (Col1a2, Col3a1, Postn, Eln, and Pdgfra); increased H3K27me3 was also detected at partially overlapping gene loci, but only under the optimal reprogramming condition (MGT) (Col3a1, Postn, Eln, Pdgfra, Thy1, and Lox, Fig. 4A). Quantitative RT-PCR revealed a coordinated and gradual decrease in mRNA expression of those genes except for Thy1 (Fig. 4A). Among them, Col1a2, Col3a1, Postn, Eln and Pdgfra transcripts were decreased by 63-93% under the optimal reprogramming condition at day 10 while Lox was decreased by 35%. PDGFRα is a cell surface tyrosine kinase receptor that plays an important role in epicardial EMT and CF fate specification [40]; Lox encodes an enzyme for crosslinking collagen and elastin precursors and is essential for the formation of collagen and elastin fibers [50]. Decreased mRNA levels of Eln, Pdgfa and Lox on day 3 preceded changes in their H3K27me3 and/or H3K4me3 levels on day 10, suggesting the involvement of other regulatory mechanisms for their transcription. It is also possible that following transcription repression, histones were further modified to “lock” the expression pattern of these loci. Similarly, H3K27me3 and H3K4me3 at Col1a1 were unchanged, but Col1a1 mRNA expression was decreased, suggesting the presence of other regulatory mechanisms. Interestingly, increased H3K27me3 on day 10 at Thy1 did not lead to changes in Thy1 mRNA expression under the optimal reprograming condition. Noticeably, Vim and the FSP1-encoding gene S100a4 did not show the expected histone mark re-patterning or a decrease in mRNA expression at either time point. However, this is consistent with Vim’s high level of H3K4me3 in CMs (Fig.1B) and growing evidence indicating that FSP1 is not as specific for fibroblast as its name suggests [51]. These results support the idea that VIM and FSP1 should be used cautiously as fibroblast markers in the future. Taken together, our data showed delayed re-patterning of H3K27me3 and H3K4me3 at fibroblast marker promoters during iCM reprogramming, accompanied by a gradual decrease in corresponding mRNA expression.
Figure 4. Re-patterning of H3K27me3 and H3K4me3 at fibroblast loci during iCM reprogramming.
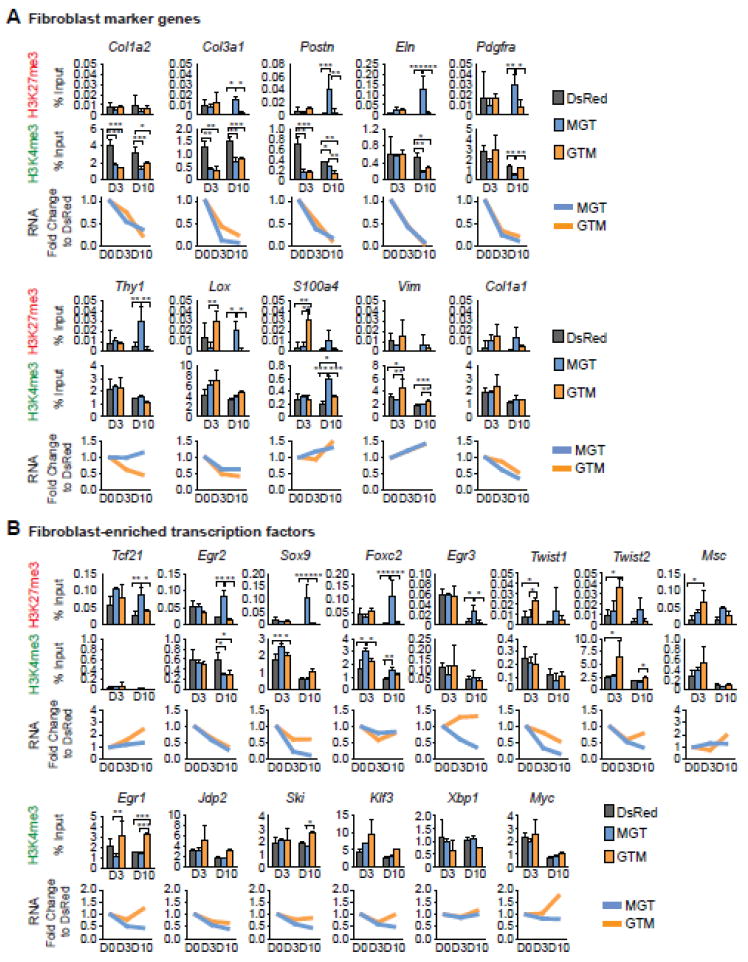
H3K27me3 and H3K4me3 ChIP of iCMs generated under the optimal condition (MGT) and the least optimal one (GTM) were performed on day 3 and day 10, followed by qPCR of fibroblast marker genes (A) or fibroblast-enriched TF genes (B). DsRed: retroviral transduction control. RNA expression levels of these genes were also determined in parallel by qRT-PCR and normalized to Actb first and then to the DsRed control.
Next, we determined the H3K4me3 and H3K27me3 dynamics at loci encoding fibroblast-enriched TF genes. With ChIP-qPCR, we found significantly increased H3K27me3 at about one-third of the tested promoters (Tcf21, Egr2 Sox9, Foxc2, and Egr3) under the optimal reprogramming condition (MGT) on day 10 but not day 3 (Fig. 4B). However, H3K4me3 was not decreased at these genes, except at Egr2, on day 10 (Fig. 4B). Gene expression analysis by qRT-PCR revealed decreased mRNA levels of Egr2, Sox9, Egr3, Twist1, and Twist2 under the optimal reprogramming condition starting from day 3 (Fig. 4B). On day 10, the mRNAlevels of these genes were decreased by 62-86%. Egr2 and Egr3, members of the early growth response family, have been demonstrated to be downstream targets of TGF-β signaling and to mediate pro-fibrotic responses [52, 53]. Sox9, a sex-determining region Y (SRY)-related TF, has been shown previously to induce neural crest EMT and to mediate PDGFR-dependent epicardium EMT [40, 54]. Twist1 and Twist2 are both well-known as central regulators of EMT [45]. Interestingly, H3K27me3 and/or H3K4me3 at some loci were unchanged under the optimal condition (Twist1, Twist2, Egr1, Jdp2, Ski and Klf3), but their mRNA expression was decreased, suggesting the presence of other regulatory mechanisms. H3K27me3 deposition on day 10 at Tcf21 and Foxc2 did not lead to changes in their mRNA expression under the optimal condition, but it is possible that these epigenetic changes have potential long-term impact on complete erasure of fibroblast signatures and eventual successful reprogramming that is not reflected by their gene expression level at the time of detection here. Some of the tested fibroblast-enriched TFs did not show histone mark or gene expression changes (Msc, Xbp1 and Myc), suggesting that they may play minor or no roles in direct cardiac reprogramming; they were selected based on high ratio of CF/CM expression and high CF expression, but a high expression level in CF may not guarantee a key regulatory role in fibroblast/cardiac fate determination. Taken together, our data showed a late increase of H3K27me3 at a subset of fibroblast-enriched TFs during reprogramming, and Egr2, Egr3, Sox9, Twist1 and Twist2 showed decreased mRNA expression.
In summary, the results above revealed a re-patterning of H3K27me3 and H3K4me3 during iCM reprogramming. Overall, cardiac loci lost H3K27me3 and gained H3K4me3 as early as day 3 during iCM generation; fibroblast loci gradually lost H3K4me3 and eventually gained H3K27me3 in this process. Our data are suggestive of early rapid activation of the cardiac program, and later progressive suppression of fibroblast fate.
3.5 Demethylation of cardiac gene promoters during iCM reprogramming
DNA methylation is another important layer of epigenetic regulation in addition to histone modifications. Therefore, we investigated whether and how DNA methylation re-patterns at cardiac promoters during iCM reprogramming. The Myh6 and Nppa promoters have been previously shown to be demethylated in αMHC-GFP+ iCMs at week 4 [2]. Our results above showed dramatic increases in mRNA expression of Myh6 and Nppa as early as day 3, but the levels of H3K4me3 and H3K27me3 at their promoters were relatively low. So we chose Myh6 and Nppa and further characterized their methylation states in the promoters at day 3 of iCM reprogramming using our polycistronic system. Bisulfite sequencing showed that the Myh6 promoter is unmethylated in HL-1 cells, about half-methylated in primary CMs and heavily methylated in CFs (HL-1, CM and CF-DsRed, Fig. 5A). The Nppa promoter is unmethylated in HL-1 and primary CMs and about half-methylated in CFs (HL-1, CM and CF-DsRed, Fig. 5B). Upon introduction of reprogramming factors (MGT or GTM), each promoter was demethylated at day 3 to a similar extent when we calculated the total methylation % of all CpGs (10% decrease in methylation for Myh6 and about 5% decrease for Nppa by MGT/GTM compared to DsRed, Fig. 5A, B). These results were consistent with the activation of Myh6 and Nppa during reprogramming, but failed to reflect the differential gene activation capabilities of the two reprogramming conditions (Fig. 5C, D). So we took a further step to calculate methylation % at each CpG. We found that certain CpG(s) were more demethylated under the optimal reprogramming condition. At the Myh6 promoter, CpG 3 was demethylated by 40% under the optimal condition compared to DsRed control, while demethylation by 10% occurred under the least optimal condition (Fig. 5E, G). Interestingly, CpG3 in the Myh6 promoter is also the least methylated CpG in primary CMs, and its methylation % is the same in primary CM and iCM under the optimal condition (CM and CF-MGT, Fig. 5E, G). At the Nppa promoter, CpG 5 and 6 were more demethylated under the least optimal condition than under the optimal one (30% demethylation v.s. 20%, Fig. 5F, H). Quantitative RT-PCR showed that mRNA expression of Myh6 was more activated under the optimal condition while expression of Nppa was more activated under the least optimal condition (Fig. 5C, D). Combined with these results, our data suggest potentially more important roles for CpG 3 in Myh6 and CpGs 5 and 6 in Nppa than other CpGs, in regulating the expression of Myh6 and Nppa during iCM reprogramming. Together, we show that promoters of cardiac genes Myh6 and Nppa were demethylated as early as day 3 during iCM reprogramming. Moreover, the methylation states of certain defined CpGs at these loci correlated to alterations in their gene expression during reprogramming, suggesting that these CpGs might be the major contributing CpGs to transcription regulation during this process.
Figure 5. Demethylation of cardiac gene promoters during iCM reprogramming.
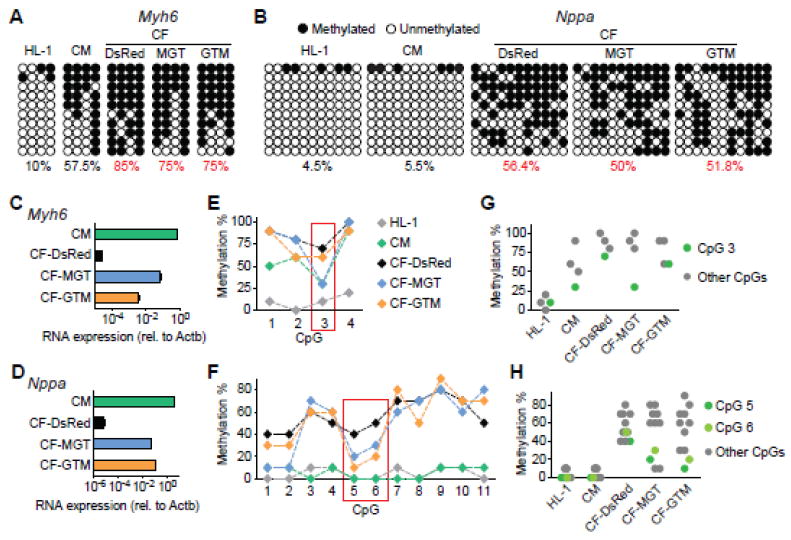
Bisulfite sequencing of iCMs generated under the optimal condition (MGT) and the least optimal one (GTM) were performed on day 3. DNA methylation states at Myh6 (A, E, G) and Nppa (B, F, H) promoters were determined. RNA expression levels of Myh6 (C) and Nppa (D) were also determined by qRT-PCR. (A, B) Methylation states of all 4 (Myh6) or 11 (Nppa) CpGs in the promoters from ten representative clones. Numbers indicate total methylation % combining all clones and all CpGs. (E, F) Methylation % of individual CpGs. (G, H) Methylation % of main contributing CpGs. HL-1 and primary CM: CM controls. DsRed: retroviral transduction control.
4. DISCUSSION
Since the initial report of iCM reprogramming in vitro [2] and in vivo [5, 7, 10], there have been considerable efforts to enhance the efficiency of direct cardiac reprogramming. One of the strategies is to optimize reprogramming efficiency through identifying enhancers of iCM generation or new combinations of reprogramming cocktails. Jayawardena et al. reported iCM induction using a microRNA cocktail consisting of miR-1, -133, -208, and -499 [8]. Others found that different TF /microRNA combinations could efficiently generate iCMs depending on the starting fibroblast type, the viral-delivery system and the cardiac reporter(s) being used. These various combination of reprogramming factors include wild-type G, T, Hand2 in combination with Mef2c fused with the transactivation domain of MyoD [55], the combination of M, T, and Myocd [16], the combination of G, M, T, Hand2, and Nkx2.5 [11], the combination of G, M, T, Myocd, and SRF [12], and the G, M, T cocktail in combination with miR-1 or miR-133 [15]. Additionally, growth factors and small molecules have been used to enhance the efficiency of iCM reprogramming. Pre-conditioning infarcted hearts with VEGF and inhibition of TGFβ signaling has been shown to improve cardiac reprogramming [13, 14]. Furthermore, we recently demonstrated that stoichiometry of G, M, T could greatly affect iCM reprogramming and a relative high level of M and moderate levels of G and T in a MGT polycistronic construct gave rise to enhanced reprogramming [32]. During the review process of our manuscript, two studies were published, reporting enhanced reprogramming efficiency based on the G, M, T and Hand2 cocktail by overexpression of the protein kinase Akt1 [56] or by suppression of pro-fibrotic signaling such as TGFβ or ROCK kinase pathways with small molecule inhibitors [57]. Nevertheless, how the fibroblast epigenome is reset during direct cardiac reprogramming remains largely unknown. Ieda et al examined the epigenetic states of three cardiac promoters (Tnnt2, Ryr2 and Actn2) in 4-week FACS-sorted αMHC-GFP+ iCMs. ChIP –qPCR analysis revealed decreased H3K27me3 and increased H3K4me3 at these three promoters in late-stage reprogrammed cells. However, epigenetic re-patterning has been shown to occur rapidly after the overexpression of reprogramming factors, for example, within a few days for iPSC [21, 36].
In this study, we intended to characterize early epigenetic changes when the cell fate transition was initiated. Therefore, we took advantage of our puro-selectable polycistronic system to enrich cells that have taken all reprogramming factors, and determined the epigenetic resetting at two critical time points at early stages of iCM reprogramming (day 3 and day 10). In addition, in order to gain insights into how fibroblast fate is suppressed during iCM reprogramming, we also expanded the testing loci to include both cardiac promoters and fibroblast promoters; particularly some largely ignored fibroblast regulatory genes in other iCM studies. As critical control experiments, we characterized H3K27me3 and H3K4me3 patterns at cardiac and fibroblast loci in CMs and fibroblasts. Our results revealed that in addition to cardiac regulatory genes, fibroblast regulatory gene promoters were also marked by both H3K27me3 and H3K4me3 in fibroblasts, suggesting the plasticity of fibroblasts. Analysis of H3K27me3 and H3K4me3 dynamics during iCM reprogramming revealed early re-patterning of H3K27me3 and H3K4me3 at cardiac loci and later alterations at fibroblast loci paralleled by a dramatic increase in cardiac markers expression and a gradual decrease in fibroblast markers expression. These results suggest an early activation of the cardiac program and a progressive suppression of fibroblast fate. Additionally, specific CpG(s) in the promoters of Myh6 and Nppa were found to be possible main contributors to total CpG demethylation during iCM reprogramming and their methylation states were correlated with transcriptional activation of Myh6 and Nppa. The methylation states of these CpGs might have important regulatory roles that contribute to the control of DNA accessibility of these two loci during iCM reprogramming. Our study thus reveals a differential re-patterning of H3K27me3, H3K4me3 at cardiac and fibroblast loci, and establishes the platform and conditions that may be used in future genome-wide studies.
To the best of our knowledge, this is the first study to characterize H3K27me3 and H3K4me3 re-patterning accompanying cardiac reprogramming. Recently, it was reported that iPSC reprogramming is characterized by global loss of H3K27me3 in the beginning and later restoration of H3K27me3 at selected sites [58]. Here we observed loss of H3K27me3 at cardiac genes early (day 3) in iCM reprogramming (Fig. 3). However, whether the observed removal of H3K27me3 is specific to the cardiac lineage remains to be answered. In iPSC reprogramming, early down-regulation (at day 1-2) of fibroblast markers such as THY1 is one of the hallmarks and prerequisites for reprogramming to proceed [49]. Here in iCM reprogramming, we showed that H3K27me3 deposition at fibroblast genes was only observed on day 10 and corresponding mRNA expression gradually decreased over time (Fig. 4). More interestingly, Thy1 expression was not completely repressed even at day 10 during iCM reprogramming (Fig. 4A), suggesting that even though both iPSC and iCM reprogramming start from fibroblasts, the routes to pluripotency and to iCM might be different. The difference may reflect differential mechanisms in how the reprogramming factors influence the regulation of histone and DNA modification, or it may suggest that direct cardiac reprogramming allows co-existence of cardiac and certain fibroblast signatures during the early phase of reprogramming. A recent report showed that suppression of pro-fibrotic signaling with small molecule inhibitors led to increased efficiency and accelerated kinetics of iCM reprogramming [57]. Yet, future studies of cell reprogramming including iCM reprogramming are required to address the necessity and mechanisms of erasure of the fibroblast program, which may provide novel insights for acceleration and improved completeness of the reprogramming process. What’s more, to completely understand epigenetic landscape dynamics during cardiac reprogramming and how such changes orchestrate the cell fate conversion, genome-wide mapping of a complete set of histone marks is needed in the future.
From induced pluripotency to the generation of iCM, iNeuron, iHepatocytes and induced multilineage blood progenitors, fibroblasts have often been chosen as the starting cells for reprogramming [1-4]. The rationale behind this choice of starting cell includes easy access to fibroblasts compared with other cell types yet other reasons may apply as well. Studies of ESC revealed that regulatory genes controlling diverse cell fates were largely marked by both H3K27me3 and H3K4me3 in pluripotent cells, which denotes “poising” of these genes for rapid activation or silencing upon differentiation [59]. Depending on the developmental course, these “bivalent” genes resolve to either only H3K4me3-marked or only H3K27me3-marked in terminally differentiated cells [60]. Our data here reveal exclusive marking of cardiac regulatory genes (M, G and T) by H3K4me3 and marking of fibroblast-enriched TFs by both H3K27me3 and H3K4me3 in CMs (Fig. 1, 2). Interestingly, all three types of primary fibroblasts MEF, CF, and TTF were marked by both H3K27me3 and H3K4me3 at cardiac TFs (M, G and T) and at fibroblast-enriched TFs (Fig. 1, 2), similar to findings in ESCs [26]. Our data imply the possibility that fibroblasts are more amenable to be reprogrammed compared to other somatic cell types because of their lower epigenetic barriers and thus higher plasticity. Parallel studies comparing epigenetic re-patterning during cellular reprogramming started from fibroblasts and other cell types are needed to evaluate this possibility.
Even though all categorized as fibroblasts, different type of fibroblasts could have distinct origin, function, gene expression profile and epigenetic pattern [61-63]. Our ChIP-qPCR results showed that H3K27me3/ H3K4me3 states (positive or negative) in the three tested fibroblasts CF, MEF, and TTF were mostly consistent across cardiac and fibroblast loci with a few exceptions but the levels of methylation were different. At certain cardiac genes, CF has higher H3K4me3 (Mef2c, Gata4) and lower H3K27me3 (Gata4, Tnnt2, Ryr2) compared to MEF and TTF while TTF has higher H3K27me3 and lower H3K4me3 at Tbx5 compared to CF and MEF (Fig. 1A). At fibroblast-enriched TF loci, CF generally shows higher H3K27me3 than MEF and TTF (Fig. 2A). In summary, the H3K27me3/ H3K4me3 states in CF resemble most to that in primary CMs while TTF is least similar to CMs among the three. Interestingly, these findings on epigenetic states of CF, MEF and TTF echo the reprogramming efficiencies from the three fibroblasts. Flow cytometry and immunofluorescence staining demonstrated that CF led to the highest percentage of αMHC-GFP+ cells among the three fibroblasts while TTFs showed the lowest (Fig. S7). Together, our data suggest that epigenetic states of the starting fibroblast cells determine the permissiveness of cardiac fate acquisition in these cells.
Most of the observed H3K27me3 and H3K4me3 re-patterning and mRNA expression changes of cardiac genes were associated with the optimal reprogramming condition (MGT, Fig. 3). On the other hand, H3K27me3 deposition at fibroblast genes was observed mostly under the optimal reprogramming condition, while H3K4me3 and expression changes were found under both reprogramming condition (MGT and GTM, Fig. 4). Reprogramming factors have been previously shown to interact with chromatin modifiers and regulate epigenetic modifications during iPSC reprogramming [20, 21, 64]. As central regulators of the cardiac fate during development, MEF2C, GATA4, and TBX5 directly bind to and activate the expression of target genes. In addition, they interact with various chromatin regulators and contribute to epigenetic remodeling in a variety of developmental processes including cardiac development and morphogenesis [65-67]. Yet future studies are needed to clarify the relationships between M, G, T binding, epigenetic re-patterning, and transcription activation of cardiac genes during iCM reprogramming. Similarly, it remains to be answered how M, G, T, and their different protein expression levels in MGT and GTM affect epigenetic remodeling at fibroblast genes during cardiac reprogramming and contribute to the removal of fibroblast signatures.
Supplementary Material
Highlights.
Cardiac transcription factor (TF) promoters are H3K4me+/H3K27me3- in cardiomyocytes
Promoters of fibroblast-enriched TFs are H3K4me3+ AND H3K27me3+ in fibroblasts
↑H3K27me3/↓H3K4me3 at cardiac promoters occur early in iCM reprogramming (day3)
↑H3K27me3 at fibroblast promoters occur later in iCM reprogramming (day10)
Early rapid activation of cardiac fate/later progressive suppression of fibroblast fate
Acknowledgments
We thank Dr. Greg Wang for providing ChIP protocol and helping set up the ChIP experiment. We thank Yi Yao for help on data analysis and heat map generation. We are grateful for expert technical assistance from the UNC Flow Cytometry Core and UNC Microscopy Core. We thank members of the Qian lab and the Liu lab for helpful discussions and critical reviews of the manuscript. This study was supported by NIH/NHLBI R00 HL109079 grant to Dr. Liu and American Heart Association (AHA) Scientist Development Grant 13SDG17060010 and the Ellison Medical Foundation (EMF) New Scholar Grant AG-NS-1064-13 to Dr. Qian.
Footnotes
AUTHOR CONTRIBUTIONS
Ziqing Liu: conception and design, collection of data, data analysis and interpretation and manuscript writing; Olivia Chen: collection of data and manuscript writing; Michael Zheng, Li Wang and Yang Zhou: collection of data; Chaoying Yin: collection of data and administrative support; Jiandong Liu: data analysis and interpretation, supervision, financial support; Li Qian: conception and design, data interpretation, supervision, financial support, administrative support, manuscript writing and final approval of manuscript.
DISCLOSURE OF POTNEITAL CONFLICTS OF INTEREST
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huang P, He Z, Ji S, et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475:386–389. doi: 10.1038/nature10116. [DOI] [PubMed] [Google Scholar]
- 2.Ieda M, Fu JD, Delgado-Olguin P, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szabo E, Rampalli S, Risueno RM, et al. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature. 2010;468:521–526. doi: 10.1038/nature09591. [DOI] [PubMed] [Google Scholar]
- 4.Vierbuchen T, Ostermeier A, Pang ZP, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inagawa K, Miyamoto K, Yamakawa H, et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circulation research. 2012;111:1147–1156. doi: 10.1161/CIRCRESAHA.112.271148. [DOI] [PubMed] [Google Scholar]
- 6.Qian L, Berry EC, Fu JD, et al. Reprogramming of mouse fibroblasts into cardiomyocyte-like cells in vitro. Nature protocols. 2013;8:1204–1215. doi: 10.1038/nprot.2013.067. [DOI] [PubMed] [Google Scholar]
- 7.Qian L, Huang Y, Spencer CI, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayawardena TM, Egemnazarov B, Finch EA, et al. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circulation research. 2012;110:1465–1473. doi: 10.1161/CIRCRESAHA.112.269035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nam YJ, Lubczyk C, Bhakta M, et al. Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development. 2014;141:4267–4278. doi: 10.1242/dev.114025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song K, Nam YJ, Luo X, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Addis RC, Ifkovits JL, Pinto F, et al. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. Journal of molecular and cellular cardiology. 2013;60:97–106. doi: 10.1016/j.yjmcc.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christoforou N, Chellappan M, Adler AF, et al. Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming. PloS one. 2013;8:e63577. doi: 10.1371/journal.pone.0063577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ifkovits JL, Addis RC, Epstein JA, et al. Inhibition of TGFbeta signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PloS one. 2014;9:e89678. doi: 10.1371/journal.pone.0089678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathison M, Gersch RP, Nasser A, et al. In vivo cardiac cellular reprogramming efficacy is enhanced by angiogenic preconditioning of the infarcted myocardium with vascular endothelial growth factor. Journal of the American Heart Association. 2012;1:e005652. doi: 10.1161/JAHA.112.005652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muraoka N, Yamakawa H, Miyamoto K, et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. The EMBO journal. 2014;33:1565–1581. doi: 10.15252/embj.201387605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Protze S, Khattak S, Poulet C, et al. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. Journal of molecular and cellular cardiology. 2012;53:323–332. doi: 10.1016/j.yjmcc.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Theunissen TW, Jaenisch R. Molecular control of induced pluripotency. Cell stem cell. 2014;14:720–734. doi: 10.1016/j.stem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 20.Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nature reviews Genetics. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Papp B, Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324–1343. doi: 10.1016/j.cell.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nature reviews Genetics. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 23.He A, Ma Q, Cao J, et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circulation research. 2012;110:406–415. doi: 10.1161/CIRCRESAHA.111.252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delgado-Olguin P, Huang Y, Li X, et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nature genetics. 2012;44:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature genetics. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wamstad JA, Alexander JM, Truty RM, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paige SL, Thomas S, Stoick-Cooper CL, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature reviews Genetics. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 29.Jackson M, Krassowska A, Gilbert N, et al. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Molecular and cellular biology. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feldman N, Gerson A, Fang J, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nature cell biology. 2006;8:188–194. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]
- 31.Gilsbach R, Preissl S, Gruning BA, et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nature communications. 2014;5:5288. doi: 10.1038/ncomms6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Liu Z, Yin C, et al. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circulation research. 2015;116:237–244. doi: 10.1161/CIRCRESAHA.116.305547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jozefczuk J, Drews K, Adjaye J. Preparation of mouse embryonic fibroblast cells suitable for culturing human embryonic and induced pluripotent stem cells. Journal of visualized experiments : JoVE. 2012 doi: 10.3791/3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang GG, Song J, Wang Z, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459:847–851. doi: 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddington JP, Perricone SM, Nestor CE, et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome biology. 2013;14:R25. doi: 10.1186/gb-2013-14-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koche RP, Smith ZD, Adli M, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell stem cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. Journal of cellular physiology. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lajiness JD, Conway SJ. Origin, development, and differentiation of cardiac fibroblasts. Journal of molecular and cellular cardiology. 2014;70:2–8. doi: 10.1016/j.yjmcc.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Acharya A, Baek ST, Huang G, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith CL, Baek ST, Sung CY, et al. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circulation research. 2011;108:e15–26. doi: 10.1161/CIRCRESAHA.110.235531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore-Morris T, Guimaraes-Camboa N, Banerjee I, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. The Journal of clinical investigation. 2014;124:2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snider P, Standley KN, Wang J, et al. Origin of cardiac fibroblasts and the role of periostin. Circulation research. 2009;105:934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giudice J, Xia Z, Wang ET, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nature communications. 2014;5:3603. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braitsch CM, Combs MD, Quaggin SE, et al. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Developmental biology. 2012;368:345–357. doi: 10.1016/j.ydbio.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiery JP, Acloque H, Huang RY, et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharyya S, Wu M, Fang F, et al. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix biology : journal of the International Society for Matrix Biology. 2011;30:235–242. doi: 10.1016/j.matbio.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyer LA, Plath K, Zeitlinger J, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 48.Lee TI, Jenner RG, Boyer LA, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polo JM, Anderssen E, Walsh RM, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamalainen ER, Jones TA, Sheer D, et al. Molecular cloning of human lysyl oxidase and assignment of the gene to chromosome 5q23.3-31.2. Genomics. 1991;11:508–516. doi: 10.1016/0888-7543(91)90057-l. [DOI] [PubMed] [Google Scholar]
- 51.Kong P, Christia P, Saxena A, et al. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. American journal of physiology Heart and circulatory physiology. 2013;305:H1363–1372. doi: 10.1152/ajpheart.00395.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang F, Ooka K, Bhattacharyya S, et al. The early growth response gene Egr2 (Alias Krox20) is a novel transcriptional target of transforming growth factor-beta that is up-regulated in systemic sclerosis and mediates profibrotic responses. The American journal of pathology. 2011:178, 2077–2090. doi: 10.1016/j.ajpath.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang F, Shangguan AJ, Kelly K, et al. Early growth response 3 (Egr-3) is induced by transforming growth factor-beta and regulates fibrogenic responses. The American journal of pathology. 2013;183:1197–1208. doi: 10.1016/j.ajpath.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakai D, Suzuki T, Osumi N, et al. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development. 2006;133:1323–1333. doi: 10.1242/dev.02297. [DOI] [PubMed] [Google Scholar]
- 55.Hirai H, Katoku-Kikyo N, Keirstead SA, et al. Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain. Cardiovascular research. 2013;100:105–113. doi: 10.1093/cvr/cvt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou H, Dickson ME, Kim MS, et al. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:11864–11869. doi: 10.1073/pnas.1516237112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao Y, Londono P, Cao Y, et al. High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nature communications. 2015;6:8243. doi: 10.1038/ncomms9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hussein SM, Puri MC, Tonge PD, et al. Genome-wide characterization of the routes to pluripotency. Nature. 2014;516:198–206. doi: 10.1038/nature14046. [DOI] [PubMed] [Google Scholar]
- 59.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 60.Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sorrell JM, Baber MA, Caplan AI. Site-matched papillary and reticular human dermal fibroblasts differ in their release of specific growth factors/cytokines and in their interaction with keratinocytes. Journal of cellular physiology. 2004;200:134–145. doi: 10.1002/jcp.10474. [DOI] [PubMed] [Google Scholar]
- 62.Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? Journal of cardiovascular pharmacology. 2011;57:376–379. doi: 10.1097/FJC.0b013e3182116e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodemann HP, R H. Functional Diversity of Fibroblasts. In: Mueller MM, F N, editors. Tumor-Associated Fibroblasts and their Matrix. 2011. pp. 23–36. [Google Scholar]
- 64.Mansour AA, Gafni O, Weinberger L, et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488:409–413. doi: 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- 65.He A, Shen X, Ma Q, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes & development. 2012;26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang CL, McKinsey TA, Chang S, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vallaster M, Vallaster CD, Wu SM. Epigenetic mechanisms in cardiac development and disease. Acta biochimica et biophysica Sinica. 2012;44:92–102. doi: 10.1093/abbs/gmr090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.