

Abstract
Analysis of patients with severe congenital neutropenia (SCN) may shed light on the delicate balance of factors controlling differentiation, maintenance, and decay of neutrophils. We identify 9 distinct homozygous mutations in the gene encoding Jagunal homolog 1 (JAGN1) in 14 SCN patients. JAGN1-mutant granulocytes are characterized by ultrastructural defects, paucity of granules, aberrant N-glycosylation of multiple proteins, and increased apoptosis. JAGN1 participates in the secretory pathway and is required for granulocyte-colony stimulating factor receptor-mediated signaling. JAGN1 emerges as a factor necessary in differentiation and survival of neutrophils.
Severe congenital neutropenia (SCN), first described by Kostmann1, is characterized by life-threatening bacterial infections due to paucity of mature neutrophils. Studies of patients with SCN have highlighted principles governing differentiation, homeostasis, and functions of neutrophils2, illustrated by roles for ELANE4,5 and G6PC36 in endoplasmic reticulum (ER) stress or HAX1 for mitochondrial function7. Genetic defects affecting the endosomal/lysosomal system have been associated with congenital neutropenia (AP3B18, LAMTOR29, VPS13B10, VPS4511,12); other genes mutated in monogenic SCN include GFI113 and WASP14.
We here report that JAGN1 is an ER-resident protein with a function in the early secretory pathway and is critical for differentiation and maintenance of human neutrophils.
We studied two sibships of Algerian origin (Family A, Figure 1a) that originate from the same sephardic community and share the family name. Consanguinity cannot be proven, but several familial links were found. Five children in family A had SCN associated with recurrent, severe bacterial infections (Table 1). Histological analysis of bone marrow smears revealed maturation arrest at the promyelocyte/myelocyte stage (Supplementary Fig. 1). Sequencing of ELANE, HAX1, and G6PC3 yielded no mutations.
Figure 1.
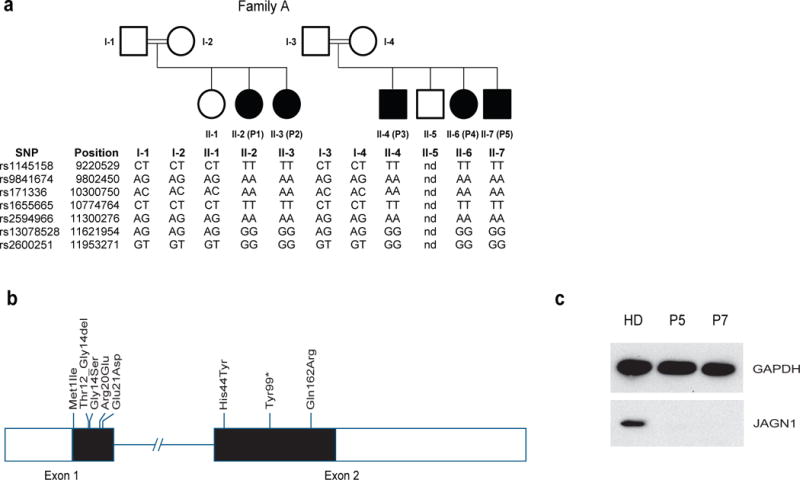
Identification of mutations in JAGN1. (a) Genotypes of affected (filled symbols) and unaffected individuals (open symbols) from Family A on chromosome 3p. (b) Exon/intron scheme of the JAGN1 gene locus with all mutations identified in this study. (c) JAGN1 protein expression was determined by Western blot analysis using an antibody directed against the N-terminal end of JAGN1 as outlined in Material and Methods. Px denotes patient number x in this study, PxF the respective father.
Table 1. Clinical and molecular characteristics of patients.
For the two families used in the linkage analysis (see also Figure 1), consanguinity was suggested as they belong to the same ethnic subgroup and originate from the same village.
| Individual | Sex | Country of origin (consanguinity) | ANCs (/μl) prior to G-CSF therapy | Bone marrow smear (prior to G-CSF therapy) | ANCs (/μl) at recent follow up | JAGN1 genotype (homozygous) | Infections | Extrahematopoietic symptoms | Age and status in mid-2013 |
|---|---|---|---|---|---|---|---|---|---|
| P1 (AII-2) | F | Algeria (consanguinity suggested) | 830 | Maturation arrest | 370 | c. 3G>A p. Met1Ile |
Ear nose and throat (ENT) infections, aphtosis, perianal cellulitis, skin abscesses | None | 23, Alive and well |
| P2 (AII-3) | F | Algeria (consanguinity suggested) | 800 | No maturation arrest | 800 | c. 3G>A p. Met1Ile |
ENT infections | Short stature (height 1.46m) | 17, Alive and well |
| P3 (AII-4) | M | Algeria (consanguinity suggested) | 570 | Maturation arrest (intermittent) | 430 | c. 3G>A p. Met1Ile |
Aphtosis, skin abscesses, balanitis, pneumonitis, lung abcess, osteitis perianal cellulitis | Pyloric stenosis | 19, Alive and well |
| P4 (AII-6) | F | Algeria (consanguinity suggested) | 501 | Maturation arrest (intermittent) | 270 | c. 3G>A p. Met1Ile |
Otitis, Paraodontopathy | Scoliosis, dental malformations | 17, Alive and well |
| P5 (AII-7) | M | Algeria (consanguinity suggested) | 165 | Maturation arrest | 230 | c. 3G>A p. Met1Ile |
ENT infections, aphtosis, skin abscesses, pneumonitis, lung abcess, perianal cellulitis | None | 5, Alive and well |
| P6 (BII-1) | F | Iran (consanguineous) | 892 | Maturation arrest | 501 | c. 59G>A p. Arg20Glu |
Upper respiratory tract infections, pneumonia, skin abscesses | Febrile convulsion, focal epilepsy | 12, Alive and well |
| P7 (CII-1) | M | Turkey (consanguineous) | 191 | Maturation arrest | 819 | c. 130C>T p. His44Tyr |
Upper respiratory tract infections, pneumonia, skin and perianal abscesses, sepsis (Haemophilus Influenza) | Extramedullary hematopoiesis with thickening of skull bones | 10, Alive and well |
| P8 (CII-2) | F | Turkey (consanguineous) | missing | Maturation arrest | 3587 | c. 130C>T p. His44Tyr |
Upper respiratory tract infections, skin abscess | Bilateral hip dysplasia, extra-medullary hematopoiesis withthickening of skull bones | 7, Alive and well |
| P9 (DII-1) | F | Iran (consanguineous) | 920 | Maturation arrest | 1479 | c. 40G>A p. Gly14Ser |
Skin abscesses, onycholysis | None | 28, Alive and well |
| P10 (EII-1) | M | Israel (consanguineous) | 130 | Maturation arrest | n/a | c. 297C>G p. Tyr99* |
Aspergillosis (none after HSCT) | Severe osteoporosis and repeated bone fractures (continuing after HSCT) | 13, HSCT at the age of 9 months due to therapy-refractory neutropenia, alive and well |
| P11 (FII-1) | F | Morocco (consanguineous) | 70 | Maturation arrest (intermittent) | 563 | c. 485A>G p. Gln162Arg |
Skin abscesses, omphalitis, pancolitis | Lipomatosis, pancreatic insufficiency, bone abnormalities, dental malformations | Died at age 5 due to pancolitis and septicemia |
| P12 (GII-1) | F | Albania (consanguineous) | 408 | Maturation arrest | 555 | c. 63G>T p. Glu21Asp |
Upper respiratory tract infections, pneumonia, skin abscess | Short stature (5 cm below 3rd percentile), amelogenesis imperfecta, neurodevelopmental delay | 16, Alive and well |
| P13 (HII-3) | F | Pakistan (no known consanguinity) | 290 | Maturation arrest, slight dyserythropoiesis | 1491 | c. 485A>G p.Gln162Arg |
ENT infections, upper respiratory tract infections, pneumonia, sepsis (E. coli) | Failure to thrive (height 5cm below 3rd percentile, weight 3.8 kg below 3rd percentile), coarctation of the aorta, mild developmental delay | 0, Alive, awaiting HSCT |
| P14 (III-1) | F | Germany (no known consanguinity) | 128 | Maturation arrest | n/a | c. 35_43del CCGACGGCA p.Thr12_Gly14del |
Pneumonia (none after HSCT), bronchiectasis | None | 25, G-CSF non-responder, HSCT at age 20 because of secondary acute myeloid leukemia (AML), alive and well |
A SNP array-based genetic linkage analysis was performed as described previously15 and identified a single interval of perfect segregation spanning 9.52Mb to 11.04Mb on chromosome 3 of NCBI’s human genome build 36.3 (Figure 1a) and containing 30 genes (Supplementary Table 1), with a multi-marker LOD score of at least 4.5, or 6.0 if the sibships are assumed to have a common ancestor (OnlineMethods).
We performed a literature search to prioritize genes in the linkage interval (Supplementary Table 1) for Sanger sequencing. Since aberrant ER function had been previously documented in patients with mutations in ELANE4,5 and G6PC36,16, JAGN1, encoding an ER-resident protein originally characterized in Drosophila17, was an attractive candidate. Sanger sequencing revealed one homozygous mutation that segregates perfectly with the disease in both sibships of Family A. This mutation, c.3G>A in exon 1 of the JAGN1 gene, leads to disruption of the defined start of translation (Supplementary Fig. 2). As confirmatory evidence, exome sequencing of P2 was performed. JAGN1 was the sole gene in Table 1 having a variant meeting our filtering criteria (Online Methods and Supplementary Table 2).
Thereafter, we assessed a cohort of 74 patients with SCN for mutations in JAGN1 and identified 9 additional patients from 8 families bearing homozygous mutations in JAGN1 (Figure 1b, Table 1). The majority were missense mutations; however, one patient had a nonsense mutation (Table 1, Figure 1b, Supplementary Fig. 3 and Supplementary Table 3). Immunoblot analysis on patient EBV-immortalized B cell lines using antibodies recognizing the N-terminus of JAGN1 showed that different mutations resulted in decreased or non-detectable protein in some patients (P5, P7 and P13) and expression of a mutant and likely non-functional protein (P9, P12) (Figure 1c). Immunoblot analysis in patient fibroblast cell lines confirmed the findings for P12 and P13 and illustrated that the nonframeshift deletion mutation in P14 leads to decreased expression of a JAGN1 protein variant of slightly reduced molecular weight (Supplementary Figure 4).
The clinical findings (Table 1) did not show an obvious distinction between SCN patients with JAGN1 mutations and those with ELANE mutations3 or HAX1 mutations7 of the non-syndromic type, but abnormalities in bone, or pancreas, or teeth were observed occasionally (Table 1). Serial blood counts from several patients did not reveal a genotype-phenotype relationship (Supplementary Table 4). Overall, variability in the level of neutropenia cannot easily be associated with the distinct genotypes. For instance, all patients in family A have the same mutation, but exhibit different disease severities. This is reminiscent of ELANE18- or G6PC3-mutated6,19 SCN where no clear association between genotype and phenotype has been observed, suggesting that modifier genes or environmental factors may influence neutrophil counts. Heterozygous JAGN1 mutation carriers were found to have normal differential blood counts (data not shown). Interestingly, response to rhG-CSF treatment was poor in several JAGN1-deficient patients (Supplementary Table 4), and in some patients severe bone pain limited the use of rhG-CSF.
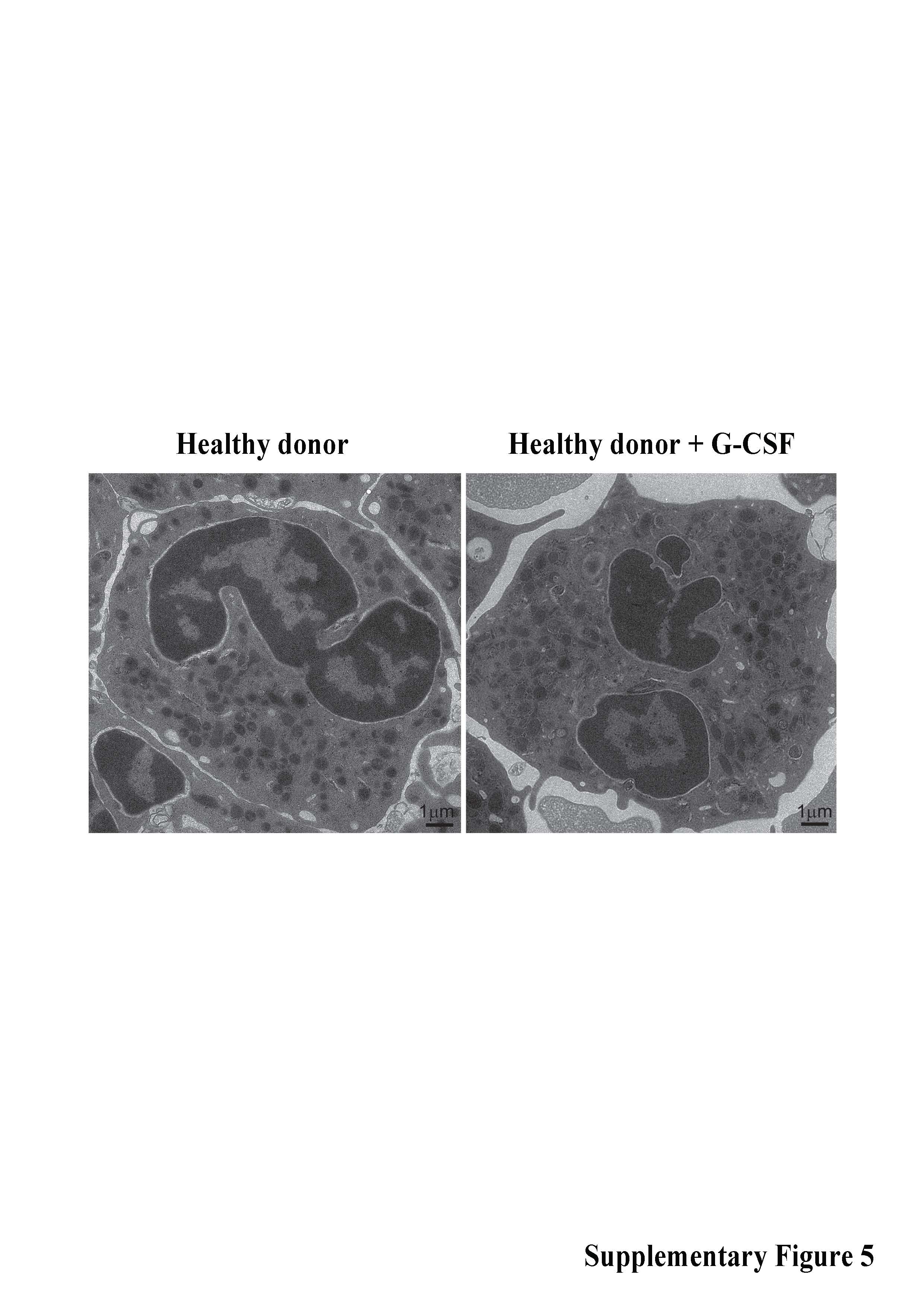
JAGN1 orchestrates the concentration of ER into subcortical clusters during the vitellogenesis stage of Drosophila oogenesis17. We performed transmission electron microscopy (TEM) studies to assess the ultrastructure of the ER and granules in neutrophils. In contrast to healthy myeloid progenitor cells, the ER appeared enlarged and granules were almost completely absent in JAGN1-mutant cells (Figure 2a). TEM analysis of peripheral blood neutrophils isolated from a healthy donor before injection with rhG-CSF to those isolated the following day revealed no marked difference in the ultrastructure of the cells (Supplementary Figure 5), suggesting that G-CSF treatment is not primarily responsible for the altered ultrastructure observed in JAGN1-deficient myeloid cells. Consistent with increased ER-stress, protein levels of binding immunoglobulin protein (BIP, encoded by the gene HSPA5) were elevated in JAGN1-mutant granulocytes (Figure 2b).
Figure 2.
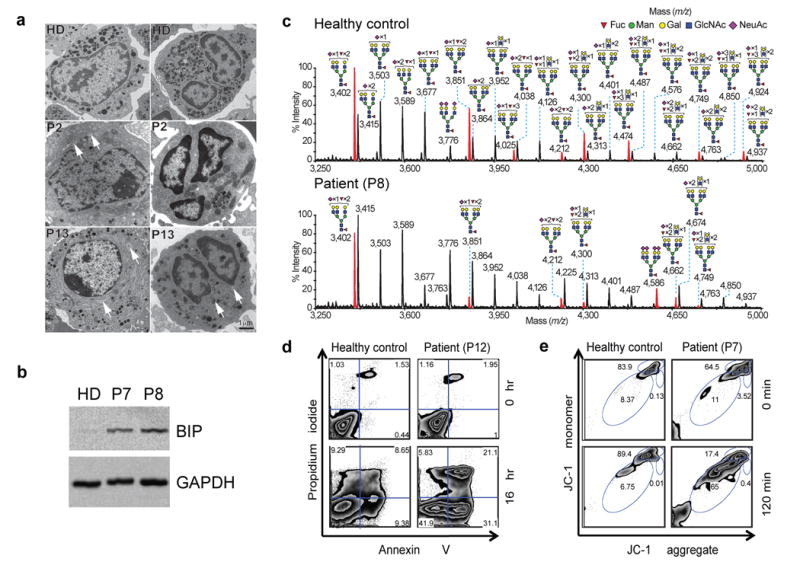
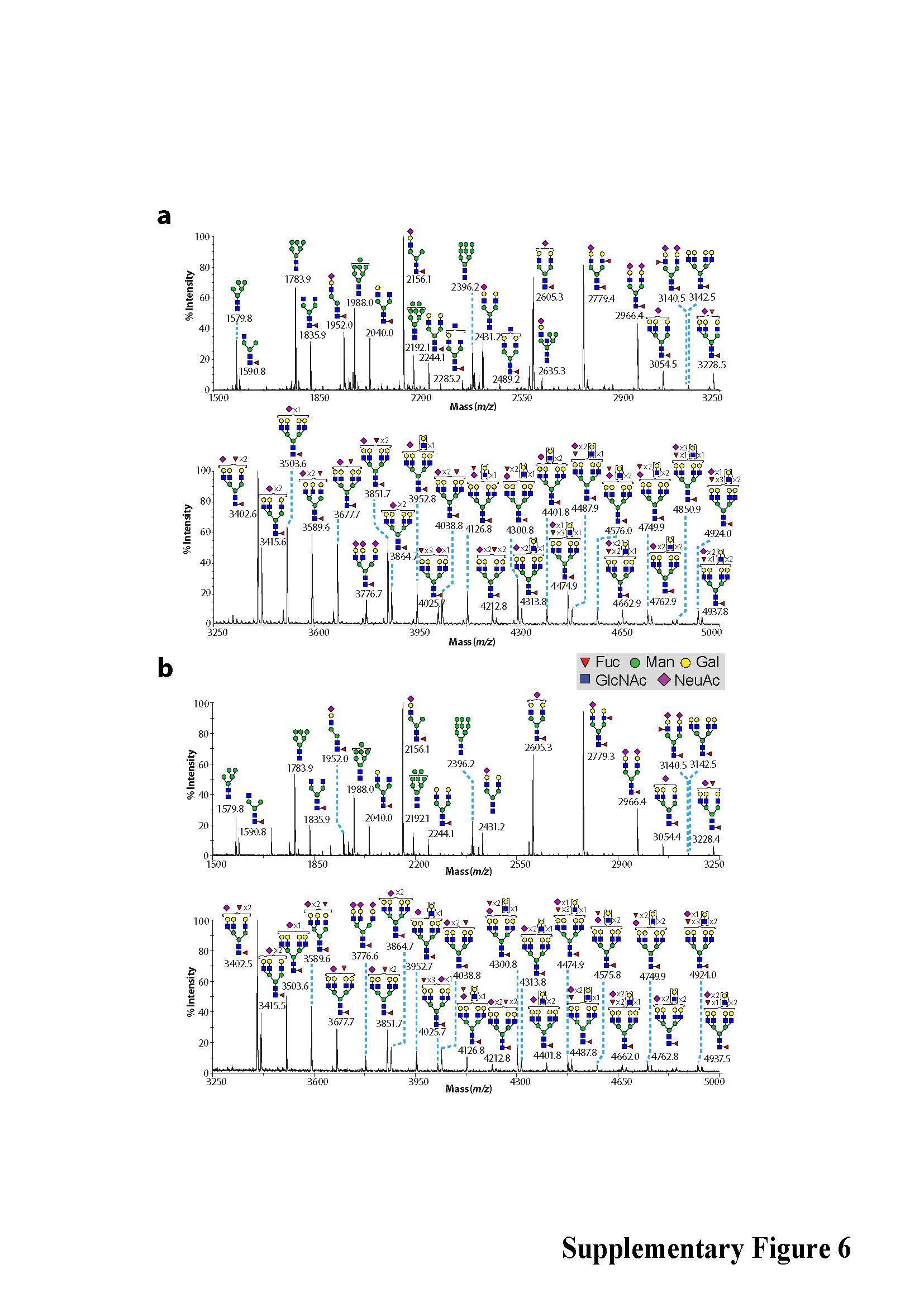
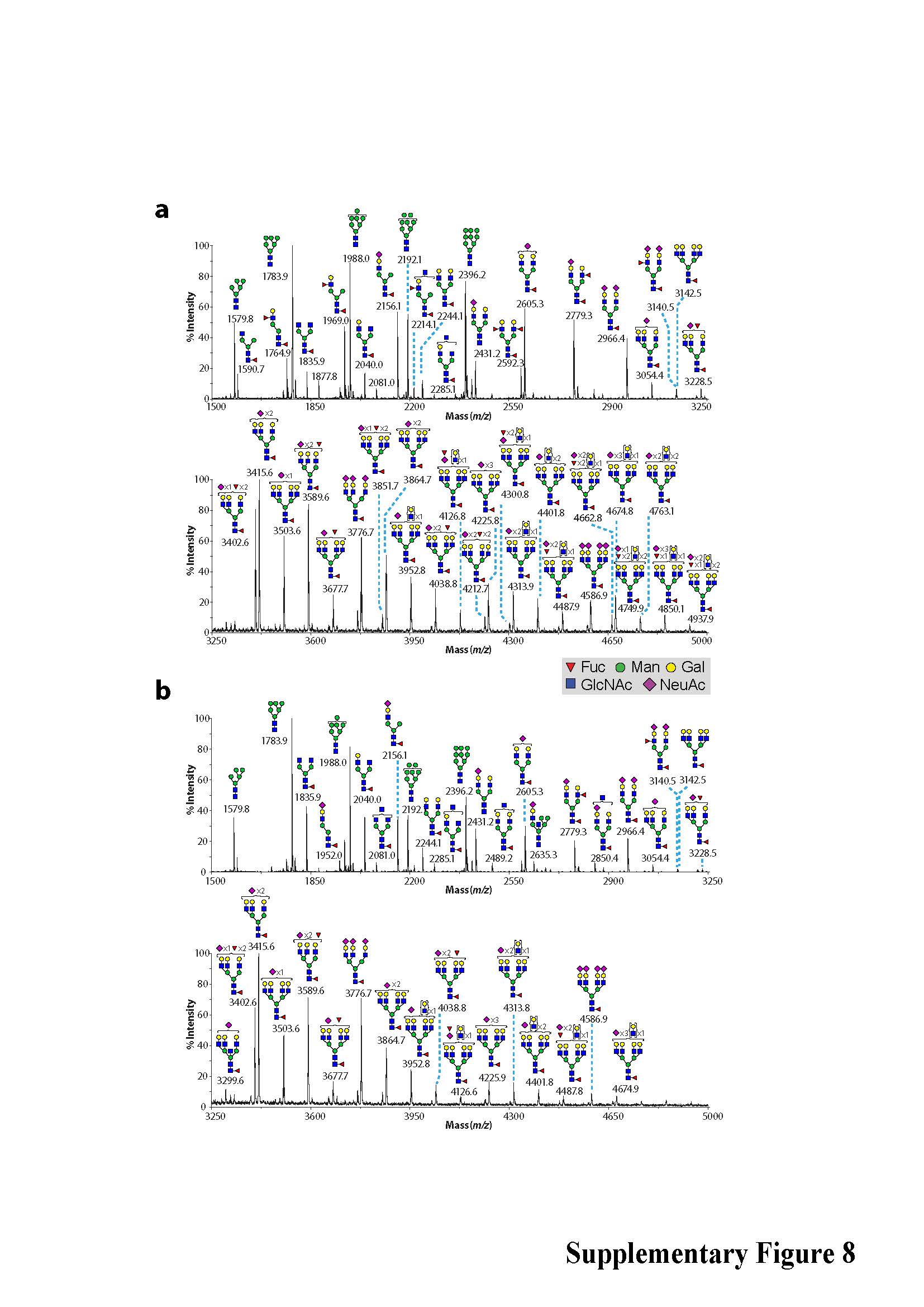
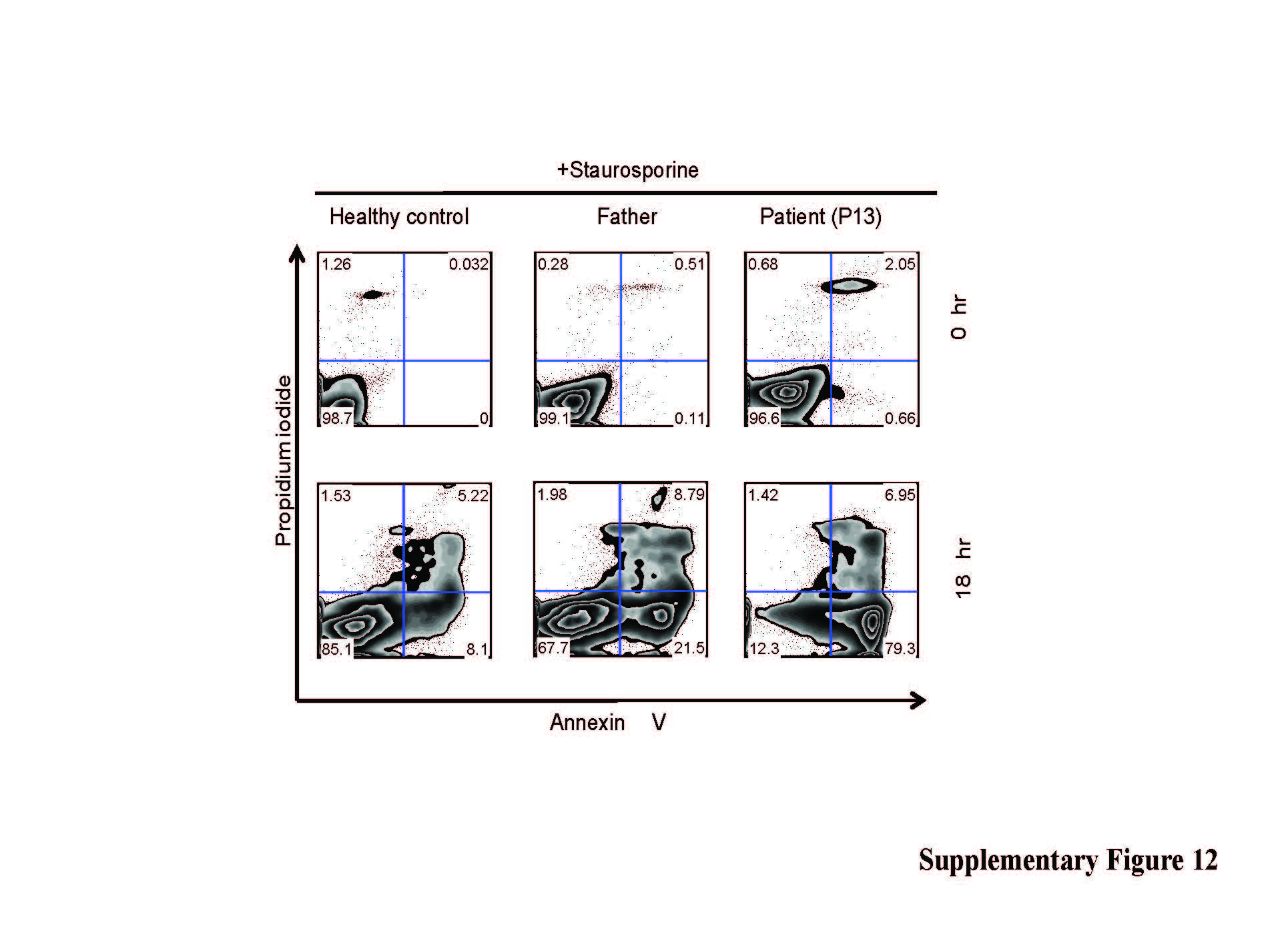
Phenotype of JAGN1-mutant neutrophils. (a) Transmission electron microscopy (TEM) of the bone marrow. Myelocytes (left panels) from patients showed aberrant, enlarged endoplasmic reticulum structures (arrows in left panels) in contrast to myelocytes from a healthy donor (HD). Differentiated granulocytes from patients (right panels) showed a paucity of typical granules (arrows in the right panel). (b) Increased protein expression of BIP in JAGN1-mutated granulocytes from patients P7 and P8. (c) MALDI-TOF MS partial spectra of permethylated N-glycans isolated from a heterozygous carrier (healthy control; mother) and patient P8 with JAGN1 mutation. The structures detected at m/z 3,402, 3,851, 4,212, 4,300 and 4,749 were found in reduced abundance in the patients compared to healthy controls, while those detected at m/z 4,025, 4,474 and 4,924 in healthy controls were almost undetectable in the patients. Red peaks depict the glycans whose abundance is markedly altered in the patient. For clarity, black peaks in the patient’s spectrum are not annotated. See the upper panel for annotations. Structure assignments are based on composition, tandem MS, and biosynthetic knowledge. (d) Increased apoptosis in JAGN1-mutant neutrophils (P12) in response to staurosporine as determined by flow cytometric analysis using Annexin-V and propidium iodide. (e) JAGN1-mutated neutrophils (P7) show rapid loss of the mitochondrial membrane potential (Δψmt) upon stimulation with valinomycin, as determined by flow cytometric analysis using the lipophilic cation JC-1 as dye.
In light of differences observed in ER structure in JAGN1-mutant granulocytes, we performed comparative global glycomic analyses of peripheral blood neutrophils. The N-glycomes of neutrophils from the clinically healthy mother (Figure 2c) and father (Supplementary Figure 6) resembled N-glycomes of healthy control samples analyzed in earlier studies20,21. We treated an unrelated healthy donor with rhG-CSF one day prior to neutrophil isolation and analyzed the N-glycosylation pattern. No qualitative changes in N-glycan content or in abundance of the multi-fucosylated N-glycan structures were detected (Supplementary Figure 7). In contrast, JAGN1-mutant neutrophils of patients P7 and P8 exhibited anomalous N-glycomic profiles, characterized by a marked reduction in fucosylation of all their multi-antennary glycans (Figure 2c and Supplementary Figure 8). Although immature N-glycans were similar in abundance to the healthy control samples (Supplementary Figure 6), their high mass N-glycomes showed a decrease in abundance of multi-LeX constituents (Figure 2c). Studies on unrelated patients P3 and P12 revealed similar findings with even more pronounced defects (Supplementary Figure 9). In contrast to N-glycan structures, O-glycosylation patterns were similar between patient and control cells (Supplementary Figure 10).
Since defective expression of neutrophil elastase (ELANE) has been implicated in the pathomechanism of SCN22,23, we assessed whether JAGN1 has an effect on the expression, glycosylation, or localization of ELANE. We analyzed ELANE protein expression by Western blot and subcellular localization using immunofluorescence in cells in which JAGN1 had been knocked down using siRNAs. No difference was seen (Supplementary Figure 11). Furthermore, the N-glycosylation profile of ELANE did not depend on JAGN1, as documented by treatment of cellular extracts with PNGase and EndoH (Supplementary Figure 11).
We assessed whether JAGN1 deficiency was associated with increased apoptosis in neutrophils, as seen in individuals with mutations in ELANE4,5, HAX17, or G6PC36. Peripheral blood neutrophils were exposed to staurosporine. A higher percentage of JAGN1-deficient neutrophils underwent apoptosis as compared to neutrophils from healthy individuals (Figure 2d and Supplementary Fig. 12). To assess whether the intrinsic pathway of apoptosis is involved, neutrophils were treated with valinomycin, a potassium-specific transporter leading to dissipation of the mitochondrial membrane potential Δψmt. JAGN1-deficient neutrophils had a propensity to rapidly depolarize their Δψmt (Figure 2e and Supplementary Fig. 13).
Next, we determined the subcellular localization of JAGN1 in mammalian cells. Using a JAGN1-specific polyclonal antibody, we found that JAGN1 was predominantly localized to the ER in HeLa cells (Figure 3a). This was confirmed in immunofluorescence microscopy studies using GFP-tagged JAGN1 constructs in fibroblasts and HeLa cells, respectively (Supplementary Figure 14).
Figure 3.
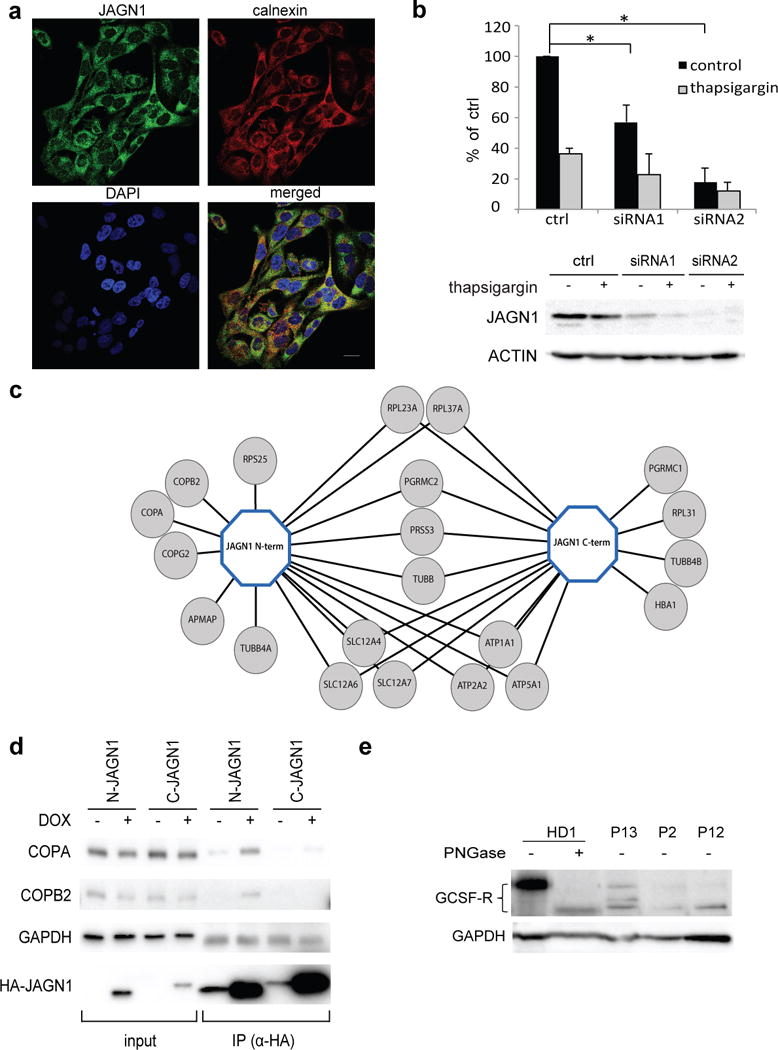
A role of JAGN1 in the secretory pathway. (a) Endogenous JAGN1 co-localized mainly with endoplasmic reticulum protein calnexin. HeLa cells were immunostained with anti-JAGN1 antibody (green) and anti-calnexin antibody for visualisation of ER (red). DAPI staining was used to visualize nuclei (blue). Images were acquired by a confocal laser scanning microscope fitted with a 63x objective. Scale bar, 20μm. (b) siRNA-mediated knockdown of JAGN1 leads to significantly (p <0.05) decreased secretion of Gaussia luciferase in comparison to control. For control, scrambled siRNA was used in this experiment. Thapsigargin was used as a positive control for secretion as it induces ER stress. The result shows a ratio between samples averaged over three independent experiments, statistical significance was calculated using a two-tailed Student’s t-test. (c) Determination of JAGN1-protein complex. Tandem affinity purification of N- and C-terminally STREP-HA-tagged JAGN1 protein complexes followed by liquid chromatography mass spectrometry identified several interacting proteins, including members of the COPI complex. (d) Immunoprecipitation of COPA and COPB2 using N- and C-terminally STREP-HA-tagged JAGN1 was performed and confirmed physical interaction between these proteins, consistent with the data shown in (c). (e) Western blotting to study glycosylation pattern of G-CSF-R in patient neutrophils. Neutrophils were isolated from blood from a healthy donor and patients of three families (P13, P2 and P12, respectively). Neutrophils from a healthy donor were also treated with PNGase F to remove glycans. GAPDH or ACTIN served as loading controls in panels (d) and (e), respectively.
We sought to determine whether JAGN1 deficiency affects the secretory pathway. We measured secretion of Gaussia luciferase in HeLa cells in which JAGN1 expression was reduced by RNA-mediated interference. In siRNA-JAGN1-knockdown cells, secretion of Gaussia luciferase was significantly reduced (Figure 3b). Secretion of proteins, however, was not globally affected; JAGN1-knockdown cells were not different from controls with respect to secretion of horseradish peroxidase (HRP, data not shown).
Since it was unknown why JAGN1 may be necessary for the secretory pathway, we performed affinity purification followed by mass spectrometry to identify interaction partners of JAGN1. N- and C-terminal JAGN1 streptavidin binding peptide haemagluttinin (STREP-HA) constructs were generated in HEK293T cells and tandem affinity purifications performed as previously described24. Three members of the Coat Protein I (COPI) complex (COPA, COPB2, and COPG2) were identified (Figure 3c and Supplementary Data). Co-immunoprecipitation experiments confirmed the interaction for both COPA and COPB2, respectively (Figure 3d). The COPI complex is known to play a central role in vesicular trafficking from the Golgi complex to the ER25.
From clinical trials of rhG-CSF, it had been known that 5–10% of both congenital and acquired neutropenia patients respond poorly to rhG-CSF26, but no predictive biomarker or molecular mechanism had been found. The majority of JAGN1-deficient patients showed no or a poor therapeutic response to rhG-CSF. Its cognate interaction partner GCSF-R is heavily glycosylated27 and relevant for development of neutrophils28. Global assessment of glycosylation (Figure 2c and Supplementary Figures 6–10) implicated a defect of N-glycosylation in patient neutrophils. We hypothesized that inadequate G-CSF receptor (G-CSF-R)-mediated signaling may offer an explanation for defective neutrophil differentiation. Compared to neutrophils isolated from healthy individuals, JAGN1-deficient patient neutrophils displayed decreased abundance and reduced molecular weight of G-CSF-R (Figure 3e). Upon treatment with peptide-N-glycosidase F (PNGase F), no difference in molecular weight could be determined, suggesting that a defect in the N-glycosylation accounts for the aberrant molecular weight in the G-CSF-R in patients (Figure 3e). It remains difficult to determine the relative contributions of aberrant G-CSF-R signaling and increased apoptosis to the neutropenia.
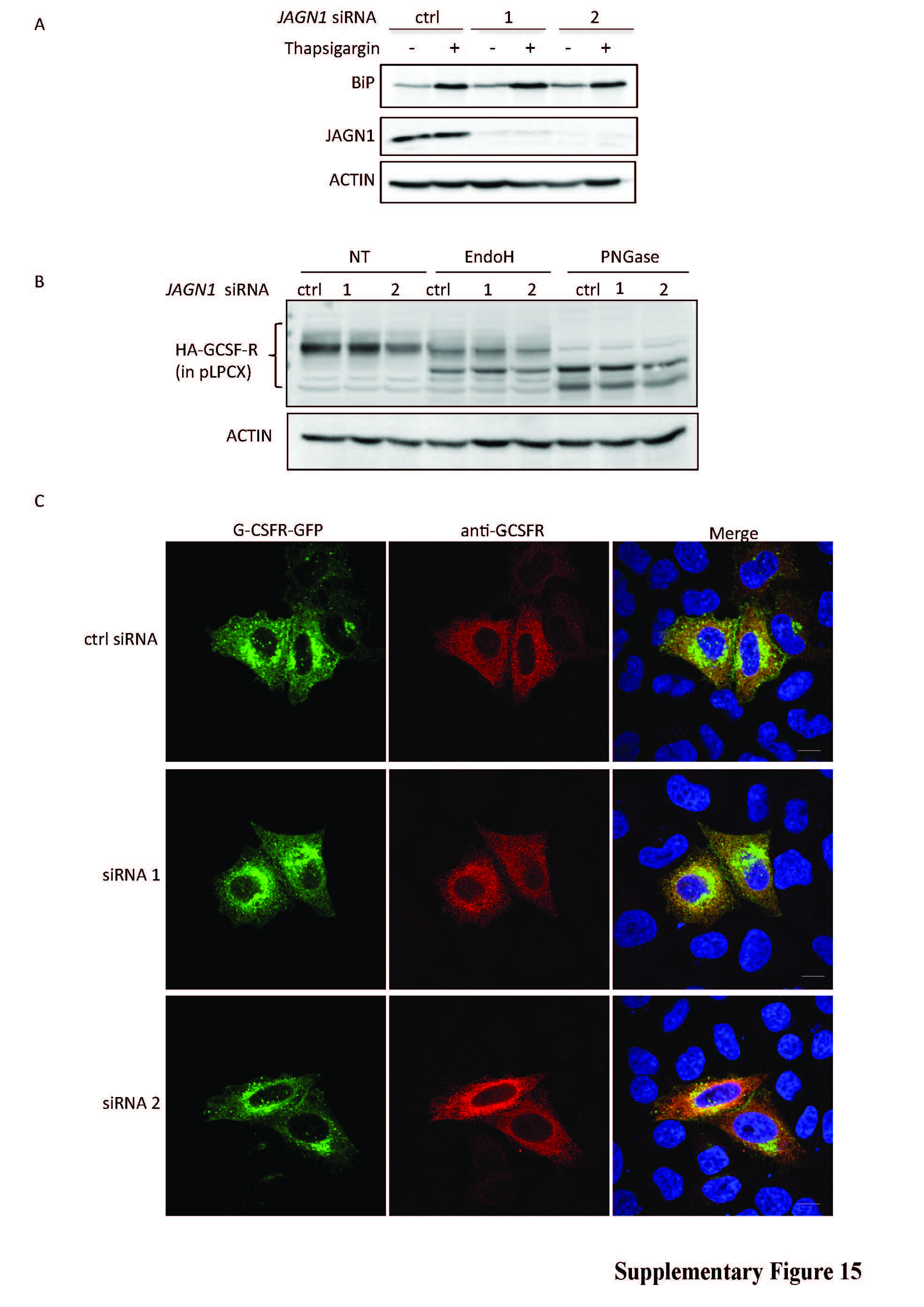
We attempted to model this effect using siRNA-mediated knockdown of JAGN1 in HeLa cells. However, HeLa cells neither recapitulated increased ER stress upon JAGN1 knockdown (Supplementary Figure 15a) nor easily detectable differences in G-CSF-R N-glycosylation (Supplementary Figure 15b), pointing to cell-specific differences between primary neutrophils and HeLa cells. Localization of G-CSF-R in these experiments did not show any differences (Supplementary Figure 15c). Next, we expressed G-CSFR in HeLa cells and studied STAT3 phosphorylation upon exposure to rhG-CSF in cells transfected with JAGN1-specific siRNA. In contrast to cells treated with control siRNA, JAGN1-knockdown HeLa cells had reduced phosphorylation of STAT3 upon exposure to rhG-CSF (Supplementary Figure 15d). Collectively, these data suggest decreased G-CSF-R-dependent signaling in the absence of JAGN1. Due to discrepancies between HeLa cells and primary neutrophils, we cannot prove precise molecular mechanisms.
By TEM, biochemical studies, and analysis of the N-glycome, JAGN1-deficiency is associated with alterations in the ER and specific granules in human neutrophils. Additionally JAGN1-deficiency is associated with aberrant neutrophil N-glycosylation. Most striking is a substantial reduction in antennae fucosylation (Figure 2c and Supplementary Figure 8).
Other human monogenic diseases have both SCN and aberrant membrane trafficking. Mutations in AP3B1, encoding the beta subunit of the adaptor protein 3 (AP3) complex involved in protein sorting in the lysosomal compartment and related organelles, are the cause of Hermansky-Pudlak-syndrome type II, a syndromic disorder including congenital neutropenia8,30. Reduced expression of the endosomal adaptor protein p14/LAMTOR2 causes a congenital neutropenia syndrome associated with aberrant lysosomal function and defective G-CSF receptor signaling9. Cohen syndrome is caused by mutations in VPS13B10, which encodes peripheral Golgi membrane protein with critical function to maintain integrity of the Golgi ribbon31. The SEC1P/MUNC18-like protein VPS45, cycling on and off membranes during vesicle transport in yeast32, is essential for SNARE-mediated membrane trafficking33. VPS45 mutations have been identified in SCN patients with extramedullary hematopoiesis, suggesting a role for viability and migration of neutrophils11,12. All diseases mentioned in this paragraph are recessive.
Even though JAGN1 is ubiquitously expressed, the only phenotype of JAGN1 deficiency seen in all patients is congenital neutropenia. This cell specificity could be either because some other protein carries out the function of JAGN1 in other cell types (as G6PC1 can replace G6PC3, but not in hematopoietic cells6) or because JAGN1 is essential for the secretion/membrane localization of proteins necessary for neutrophil function. In line with the latter hypothesis, we could demonstrate aberrant expression of G-CSFR, a critical cytokine-receptor governing differentiation of neutrophils.
In an accompanying paper, Wirnsberger et al. confirm the relevance of Jagn1 for neutrophil granulocytes in a murine knockout model34. Even though, in contrast to human patients, Jagn1-deficient mice do not show neutropenia, they are characterized by increased susceptibility to fungal infections due to defective killing capacity of neutrophil granulocytes. Interestingly, GM-CSF but not G-CSF rescues this functional defect in mice34, potentially opening a new therapeutic option for affected patients. In summary, by identifying patients with JAGN1 deficiency, we define a role for JAGN1 in the early secretory pathway required for physiological differentiation and viability of neutrophils.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Subjects
All material from patients and healthy donors was obtained with informed assent/consent in accordance with the Declaration of Helsinki. The study was approved by the institutional review board at Hannover Medical School and the Ludwig Maximilians University Munich.
Genome-wide genetic linkage analysis
For family A, DNA samples from five affected individuals, four parents, and one unaffected sibling were genotyped using the Affymetrix (Santa Clara, CA, USA) 250k StyI SNP mapping array (GEO Platform GPL3718), following the procedures in35 and recommended by Affymetrix.
Genotypes were analyzed using the software findhomoz15 to identify intervals where the affected individuals are homozygous for the same genotypes at consecutive markers, and the unaffected individuals have different genotypes for some markers in the interval. The same or a similar homozygosity mapping procedure was used for Families B, C, D, E, F and G to identify an interval on 3p consistent with linkage that included JAGN1. For families E and F, we used another Affymetrix (GEO Platform GPL6801) chip for genotyping.
For family A, we computed LOD scores using the software Superlink36 for some of the 257 SNPs in a 3Mbp region on chromosome 3 that includes the minimal linkage region and >500kb extra on each side. Twenty of the 257 SNPs have perfect and fully informative segregation.
To model the likely consanguinity in the pedigree, we added hypothetical ancestors to the two nuclear family pedigrees. The parents on the left (I-1, I-2) were assumed to have a pair of great grandparents in common and the parents on the right (I-3, I-4) were assumed to have a different pair of great grandparents in common. First, the pedigrees are kept separate and the LOD scores are the sum over the two pedigrees. In a model of the unproven shared ancestry of the carrier parents, one great grandparent on the left was assumed to share a pair of great grandparents with a great grandparent on the right, so that the putative founder pair is six generations up from the carrier parents. The assumption of common great grandparents to represent likely consanguinity has been used previously13,37.
The disease is modeled as fully penetrant recessive with a disease allele frequency of 0.001. The LOD scores depend on the frequencies of the disease-associated marker alleles. After identifying the 20 SNPs that segregate perfectly, we chose seven SNPs (Figure 1A) at which to compute single-marker LOD scores, and obtained their affected allele frequencies from HapMap38. These seven SNPs were selected as described below so that no two have extreme linkage disequilibrium. The affected allele frequency of each SNP was taken to be the largest (worst) frequency among the four populations in HapMap. The multipoint LOD scores reported in Results were computed using the seven selected SNPs.
HapMap has linkage disequilibrium data for four populations. We considered two SNPs to be in extreme linkage disequilibrium with each other if HapMap explicitly lists an r2-value of at least 0.8 between the SNPs in any of the four populations, or if there is a third SNP with which we consider the first and second SNPs are in extreme linkage disequilibrium. This means that if there is a chain of SNPs with r2-value of at least 0.8 connecting the first and second SNP, where the r2-value is the maximum of the four populations, then the SNPs were considered to be in extreme linkage disequilibrium.
Exome sequencing
Exome sequencing of patient 2 was performed using 50ng of genomic DNA. DNA was fragmented including simultaneous adaptor ligation (tagmentation) using the Nextera transposome. Subsequently, adaptor ligated genomic DNA was enriched for exonic regions during PCR amplification. Clusters were generated using the Illumina cBot Cluster Generation System following the TruSeq PE Cluster Kit v3 Reagent Preparation Guide. Sequencing was performed in a multiplexed pool of 12 samples distributed on four lanes of the flow cell. Reads were demultiplexed and aligned using Burrows-Wheeler Aligner (BWA) software39 to the human genome 19. Insertion/deletion realignment was performed as well as Genome Analysis Toolkit (GATK)40 based quality score recalibration. For single nucleotide variants (SNVs) and Deletions/Insertions variants (DIVs) calling, Unified Genotyper and GATK Variant quality score recalibration were performed as described previously41 with minor modifications. Generation of lists of SNVs and DIVs mentioned were annotated with ANNOVAR42 using dbSNP build 137. Variants present in 1000 Genomes (in February 2012) and/or dbSNP build 137 with minor allele frequency (MAF) ≥ 0.01 were excluded from further analyses; lists were filtered to include only nonsense, missense and splice-site variants present in the shared homozygous region on Chromosome 3 within 9.52–11.04Mb.
Sequence analysis
We used SIFT43, PolyPhen-244, and Consurf45 to analyze the effect of amino acid substitutions on JAGN1 (see Table S3). We gathered sequences for homologs to JAGN1 (NP_115881.3) by running three rounds of PSI-BLAST46 with default settings against NCBI’s non-redundant database current on Jan 12, 2009. We kept only sequences that aligned with E-value of at most 10−4 after three rounds of PSI-BLAST. We eliminated sequences that aligned with more than 90% identity with human JAGN1 or with each other, retaining a total of 41 homologs, including human JAGN1. We created a multiple alignment using ClustalW 1.8347 with default settings. We ran SIFT43, Consurf45, and PolyPhen-244 on Feb 14, 2013, using the web interfaces. For SIFT and Consurf, we provided the alignment produced by ClustalW.
Immunoblot analyses
Immunoblotting for expression levels of BIP and GAPDH was performed as described previously6. Total protein was extracted from Epstein-Barr virus (EBV)-immortalized B cell lines and primary fibroblasts from the patients and controls. Primary antibodies used to detect JAGN1 were derived from peptides in the mouse sequence NP_080641. Polyclonal rabbit anti-JAGN1 antibodies were raised in a rabbit and affinity purified against KLH coupled peptides ASRAGPRAAGTDGSDFQHR (positions 2–20 of NP_080641). As a loading control, monoclonal mouse anti-GAPDH (Santa Cruz Biotechnology, sc-32233) was used.
Electron microscopy
Sample preparation for transmission electron microscopy was performed as described previously6. Samples were assessed using a Philips electron microscope 301 (Fei, Hillsboro, OR, USA).
Apoptosis assays
Neutrophil granulocytes were isolated from peripheral blood and exposed to staurosporine (2.5 μg/ml; Sigma-Aldrich, St. Louis, MO, USA) to induce apoptosis. Neutrophils were isolated from a healthy donor one day after an injection of rh-G-CSF. Cells were stained with Annexin-V (Life Technologies, Paisley, UK) and propidium iodide (Sigma-Aldrich) and analyzed by flow cytometry, similar to previous studies6. Flow cytometry data were analyzed using FlowJo version 9.0.1 (FlowJo, Ashland, OR, USA).
Measurement of mitochondrial membrane potential in neutrophil granulocytes
Neutrophil granulocytes were treated with valinomycin (Sigma-Aldrich), a potassium-specific transporter leading to dissipation of the mitochondrial membrane potential Δψmt. Flow cytometry using the lipophilic cation JC-1 (Life Technologies) was done as described previously7 to measure the relative percentage of cells with low Δψmt.
Cloning of JAGN1
Two fusion constructs were generated: EGFP was cloned N-terminally (EGFP-JAGN1) or C-terminally (JAGN1-EGFP) to human JAGN1. The full-length JAGN1 open reading frame was amplified from cDNA. PCR primers for EGFP-JAGN1 contained a BglII or SalI restriction site, respectively (Supplementary Table 5). PCR primers for JAGN1-EGFP contained a NheI or BamHI restriction site (Supplementary Table 5). Both PCR products were subcloned into the pGEM-T® Vector System I (Promega, Madison, WI, USA). BglII/SalI digestion sites were used for insertion into the pEGFP-C1 plasmid (Clontech Laboratories Inc, Mountain View, CA, USA) to create EGFP-JAGN1. NheI/BamHI digestion sites and the pEGFP-N1 plasmid (Clontech Laboratories Inc.) were used to create JAGN1-EGFP. The Kozak consensus sequence was integrated upstream of the start codon of JAGN1-EGFP. Sequences were verified by digestion and sequencing. Vectors from Clontech Laboratories Inc. were provided by Dr. Axel Schambach (Hannover Medical School, Germany).
Cell culture
Cells were cultured at +37°C in a humidified atmosphere with 5% CO2 in glucose-rich DMEM (Dulbecco′s modified Eagle′s medium, PAA, Pasching, Austria) containing 10% (v/v) fetal calf serum (FCS, Life Technologies, Gibco) for HeLa cells and 20% (v/v) FCS for fibroblasts, respectively, 50 units/ml penicillin, 50 μg/ml streptomycin and 292 μg/ml L-Glutamin (all from Gibco).
Immunofluorescence-based studies to assess JAGN1 intracellular localization
To generate Figure 3a, HeLa cells were seeded onto glass coverslips (Thermo Fisher Scientific, Waltham, MA, USA) and cultivated for at least 24 hours. After washing with phosphate-buffered saline (PBS), cells were fixed in 3.7% paraformaldehyde for 35 min, and permeabilized with PBS containing 0.1% Triton X-100. Cells were blocked with PBS containing 5% BSA at room temperature for 40 min and subsequently incubated with polyclonal rabbit anti-JAGN1 (see above) and a mouse monoclonal anti-human Calnexin antibody (BD, Franklin Lakes, NJ) for 1 h. Cells were then washed in PBS and incubated for 1 h either with Alexa Fluor® 488-conjugated goat anti-rabbit IgG or Alexa Fluor® 594-conjugated goat anti-mouse IgG secondary antibodies (Life Technologies). Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) for 15 min at room temperature. Coverslips were mounted in fluorescent mounting medium (Dako, Hamburg, Germany). Images were acquired by an Olympus FV1000 confocal laser scanning microscope (Tokyo, Japan) using a 63× objective, and analyzed using Image J48.
To generate the data in Supplementary Figure 14, HeLa cells and fibroblasts were cultured on coverslips (Thermo Fisher Scientific). After 24–36 h, transient transfection was performed with Lipofectamine 2000 (Life Technologies) according to the manufacturer′s instructions. HeLa cells and fibroblasts were transfected with JAGN1-EGFP or EGFP-JAGN1. After 36–48 h cells were fixed with 4% (w/v) paraformaldehyde in PBS for 25 min and then permeabilized with 0.2% (w/v) saponin in PBS for 5 min. All the following steps were performed with 0.5% saponin. After quenching with 50 mM NH4Cl in PBS for 10 min, blocking of the non-specific binding sites followed with 10% goat serum in PBS for 30 min. To stain the ER, monoclonal mouse-anti-Calnexin (BD) and to stain Golgi apparatus, monoclonal mouse-anti-Golgin-97 antibodies (1:200 Molecular Probes®, Life Technologies) were used. Alexa Fluor® 555-conjugated goat anti-mouse IgG (1:1000, Molecular Probes®, Life Technologies, A21424) was used as an secondary antibody. For nuclear staining, cells were stained with Hoechst (33342, Molecular Probes®, Life Technologies) and coverslips were mounted in Fluorescence Mounting Medium (Dako). Between all steps, coverslips were washed with PBS. Mitochondria staining was performed with MitoTracker Red CMXRos (150 nm, 30 min, Molecular Probes®, Life Technologies), according to the manufacturer′s instructions. Images were acquired using a Leica DM IRB inverted confocal microscope (Leica Microsystems, Wetzlar, Germany) equipped with a TCS SP2 AOBS scanning head and analyzed using the Leica software LCS and ImageJ software.
Processing of neutrophils to obtain N- and O-glycans for subsequent glycomic analysis
For glycomics experiments, each human sample (~3 million cells) was subjected to sonication in the presence of detergent (CHAPS) and treated as described previously49.
Mass spectrometric N- and O-glycomic analyses
Permethylated N- and O-glycans prepared as described above were analyzed on a 4800 MALDI-TOF/TOF (Applied Biosystems, Life Technologies) mass spectrometer, as described previously21.
Gaussia luciferase secretion assay
To perform the Gaussia luciferase secretion assay, HeLa cells were first seeded on 6-well plates a day prior to the transfection. To silence JAGN1, the cells were transfected with either negative control siRNA (#4404021) or siRNAs 1 and 2 (#s39100 or #s39101, Life Technologies) against JAGN1 by using Lipofectamine RNAi/Max kit (Life Technologies) according to the manufacturer’s instructions. The medium was changed the following day. On the fourth day, the cells were transfected with pCMV-Gluc (New England Biolabs, Ipswich, MA, USA) using the JetPEI transfection reagent (PolyPlus Transfection). The medium was replaced on the following morning, and the cells were either treated with thapsigargin (1μg/ml, Applichem, Darmstadt, Germany) or left untreated. Samples were collected eight hours after change of the medium. Luciferase activity was measured using coelenterazine as a substrate (Biolux Gaussia luciferase assay kit, New England Biolabs). The result shows a ratio between samples averaged over three independent experiments.
To verify the knockdown of JAGN1, cells were lysed with buffer containing 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 5 mm NaF, 1 mm orthovanadate, 10% glycerol, 1% Triton X-100, 0.5% Nonidet P-40 and protease inhibitors before samples were used for Western blot analysis. The knockdown of JAGN1 was detected by using an anti-JAGN1 antibody (see above), and actin (Santa Cruz Technologies, Texas, USA) was used as a loading control.
Tandem affinity purification and mass spectrometry
Human JAGN1 cDNA was cloned into C- and N-terminal pTO-STREP-HA-GW vectors (Life Technologies). The plasmids were transfected into HEK293 Flp-In-TREx cells (Life Technologies), to express JAGN1 protein upon administration of doxycycline. Generation of JAGN1 HEK293 Flp-In TREx cells and subsequent tandem affinity purification coupled to mass spectrometry (TAP-MS) were performed as described previously50.
Five confluent 15 cm dishes were stimulated with 1 μg/mL doxycycline (Thermo Fisher Scientific) for 24 h. Cells were harvested and lysed in TNN-HS buffer (50 mM HEPES pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, protease inhibitors from Sigma-Aldrich). STREP-HA-JAGN1 was purified using strepTactin sepharose beads (IBA, Goettingen, Germany) followed by elution with biotin (Alfa Aesar, Karlsruhe, Germany). The biotin eluate was then immunopurified with anti-HA–agarose beads (Sigma-Aldrich). The bound material was eluted with 100 mM formic acid (Suprapur, Merck Millipore, Darmstadt, Germany) and neutralized with triethylammonium bicarbonate (TEAB) (Sigma-Aldrich). The samples were digested with trypsin (Promega) and 2 × 5% v/v of the resultant peptides were analyzed by LC–MSMS on a linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) coupled to an 1200 series high-performance liquid chromatography system (Agilent Technologies, Santa Clara, CA, USA)24.
The acquired raw MS data files were converted into MASCOT generic format (mgf) files, and the resultant peak lists were queried against the human SwissProt database (v. 2010.09) using the search engines MASCOT (v2.3.02, MatrixScience, London, UK) and Phenyx (v2.6, GeneBio, Geneva, Switzerland)51. One missed tryptic cleavage site was allowed. Carbamidomethyl cysteine was set as a fixed modification, and oxidized methionine was set as a variable modification. The peptides which were independently identified by the two algorithms were combined by discarding all the spectra identifying different peptides, and grouping the remaining spectra into protein groups sharing peptides. The peptide score thresholds of both search engines were set such that the combined results achieved a protein group false detection rate (FDR) of <1%, as determined by applying the whole procedure against a reversed database. This resulted in imposing a Phenyx z-score >4.75 for single peptide identifications or >4.2 for multiple peptide identifications, and Mascot single peptide identifications ion score >40, multiple peptide identifications ion score >14. Individual peptides selected by this procedure featured a false positive rate of <0.1%.
In parallel, TAP-MS of N- and C-terminal STREP-HA-GFP Flp-In-TREx cells were performed. All proteins identified from the STREP-HA-GFP experiments were removed from the JAGN-1 interactors list. The final JAGN1 interaction network was drawn using Cytoscape (version 2.8.1).
Immunoprecipitation assay
For the immunoprecipitation assay, JAGN1 HEK293 Flp-In TREx cell lines expressing N- and C-terminally tagged versions of JAGN1 were used. Doxycycline stimulation and lysate isolation were performed as for the tandem affinity purification, and 10% of the input material was used. The lysates were pre-cleared with Protein G-sepharose beads (GE Healthcare, Wauwatosa, WI, USA) and immunopurified with anti-HA-agarose beads (Sigma-Aldrich). Input and immunoprecipitation samples were separated by sodium dodecyl sulphate-polyacrylamide-gel electrophoresis (SDS-PAGE), blotted and stained with antibodies against COPA (Sigma-Aldrich), COPB2 (Sigma-Aldrich), GAPDH (Santa Cruz Biotechnology) and HA-tag (Sigma-Aldrich).
G-CSF-R studies in healthy donor and in patient neutrophil granulocytes
Granulocytes were isolated from peripheral blood of a healthy donor and three patients from distinct families A (P2), G (P12), and H (P13) using density gradient centrifugation (Ficoll-Histopaque, Amersham Biosciences, Piscataway, NJ). Cells were lysed as described above in the paragraph “Gaussia luciferase secretion assay” and the healthy donor sample was either left untreated or treated with PNGase F (New England Biolabs) according to the manufacturer’s instructions. G-CSF-R was detected in Western blotting by using an antibody from Santa Cruz Biotechnology (H-176, sc-9173). Actin (Santa Cruz Biotechnology) was used as loading control.
G-CSF stimulation assay in HeLa cells
HeLa cells were seeded on 6-well plate and a day after the cells were transfected with JAGN1-specific siRNA oligos (described above). On the fourth day, the cells were transfected with wildtype (wt) CSF3R-GFP in pMMP vector using JetPEI (PolyPlus Transfection) according to the manufacturer’s instructions. Day after the transfection, starvation was started with the medium containing 0.5% serum. The following day cells were stimulated with 100 ng/ml G-CSF (Amgen, Thousand Oaks, CA, USA) for the indicated times and cells were lysed as described above in the paragraph “Gaussia luciferase secretion assay”. Phosphorylated STAT3 (Y705, Cell Signaling, Danvers, MA, USA), total STAT3 (BD Biosciences, Franklin Lakes, NJ, USA), G-CSF-R (Santa Cruz Biotechnology, H-176, sc-9173) and JAGN1 (see above) were detected by Western blotting. Actin (Santa Cruz Biotechnology) was used as a loading control.
Immunofluorescent and glycosylation studies of G-CSF-R and neutrophil elastase in JAGN1–silenced HeLa-cells
HeLa cells were seeded on coverslips on 6-well plates and transfected using reverse transfection with either control siRNA or JAGN1-specific siRNAs 1 or 2 (described above) using Lipofectamine RNAiMax-kit (Life Technologies). The medium was changed the following day. Two days after siRNA-transfection, samples were again transfected using JetPEI transfection reagent (PolyPlus Transfection) either with HA-tagged wt ELANE in pLPCX-vector (Clontech Laboratories Inc.) or with wt CSF3R-GFP in pMMP-vector. Cover slips were fixed with 4% paraformaldehyde 36–48 hours later and stained following the first immunofluorescence staining protocol described above. Primary antibodies used were polyclonal goat anti-neutrophil elastase (Santa Cruz, clone C-17) and rabbit anti-G-CSF-R (Santa Cruz, clone H-176). Secondary antibodies used were Alexa Fluor® 488-conjugated donkey anti-goat IgG and Alexa Fluor® 594-conjugated goat anti-rabbit IgG. Images were acquired by Olympus FV1000 confocal laser scanning microscope (Tokyo, Japan) and a 63x objective.
For glycosylation studies, the cells were lysed as described above in the paragraph “Gaussia luciferase secretion assay” 36–48 hours post plasmid transfection and the cleared protein lysate was either non-treated or treated with EndoH or PNGase F (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s protocol. Samples were subjected to Western blot. Primary antibodies used were rabbit anti-JAGN1 N-terminal antibody (see above), rabbit anti-G-CSF-R (Santa Cruz H-176) and rabbit anti-neutrophil elastase (Santa Cruz (C-17). Actin (Santa Cruz Technologies, Texas, USA) was used as a loading control.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the patients and their families for their participation in this study. The help of all contributing medical, technical and administrative staff is greatly appreciated. The authors thank Jacques Colinge for bioinformatic analysis of mass spectrometry data. The authors thank Guy Leverger, Hubert Ducou Lepointe, Judy Levin, the association IRIS, and Mrs. Grosjean for their support. This study was supported by the CeMM intramural funds and the FWF START Programme (to KB) and grants from the European Research Council (Advanced Grants to CK and JP, Starting Grant to KB), the European program on rare diseases (E-RARE Neutro-Net), and the German Research Foundation (SFB914 and Gottfried-Wilhelm-Leibniz Program). The Imperial College work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC, grants BB/F0083091 and BB/K016164/1). The French SCN registry is supported by grants from Amgen SAS, Chugai SA, GIS Maladies Rares, Institut de veille sanitaire, Inserm, the Association Sportive de Saint Quentin Fallavier, CEREDIH and the Société d’Hémato Immunologie Pédiatrique. This research is supported in part by the Intramural Research Program of the NIH, NLM, the DZIF (German Center for Infection Research) and the Care-for-Rare Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
K.B. identified the JAGN1 mutation in the index family and the majority of the other families reported and performed experiments together with P.M.J., E.S., S.M., W.G., L.S., Ja.Di., G.A., J.v.B. B.L., M.A., K.W., R.S., J.v.W.t.B., N.R., A.E., Je.Do., C.B.-C., and C.K. took care of and enrolled patients into the study. T.R., E.M.G., A.A.S., J.P., D.P. were responsible for genome-wide analyses and bioinformatic analysis. A.A., S.M.H., A.D. performed glycoprotein analysis. G.B. performed TEM analyses. E.S., R.G., J.W.B., C.D.C., G.S.-F., and K.L.B. performed the JAGN1-interactome experiments and data analysis. U.E. and J.M.P. generated and characterized polyclonal antibodies and gave critical advice. C.K. designed and coordinated the investigations. The manuscript was written by C.K. and K.B. with help from E.M.G., A.A.S., and P.M.J. The final version was approved by all authors.
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
References
- 1.Kostmann R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr. 1956;45(Suppl 105):1–78. [PubMed] [Google Scholar]
- 2.Klein C. Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol. 2011;29:399–413. doi: 10.1146/annurev-immunol-030409-101259. [DOI] [PubMed] [Google Scholar]
- 3.Dale DC, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–22. [PubMed] [Google Scholar]
- 4.Grenda DS, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110:4179–87. doi: 10.1182/blood-2006-11-057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köllner I, et al. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006;108:493–500. doi: 10.1182/blood-2005-11-4689. [DOI] [PubMed] [Google Scholar]
- 6.Boztug K, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360:32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein C, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nat Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 8.Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- 9.Bohn G, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2006;13:38–45. doi: 10.1038/nm1528. [DOI] [PubMed] [Google Scholar]
- 10.Kolehmainen J, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003;72:1359–1362. doi: 10.1086/375454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stepensky P, et al. The Thr224Asn mutation in the VPS45 gene is associated with congenital neutropenia and primary myelofibrosis of infancy. Blood. 2013;121:5078–87. doi: 10.1182/blood-2012-12-475566. [DOI] [PubMed] [Google Scholar]
- 12.Vilboux T, et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. N Engl J Med. 2013;369:54–65. doi: 10.1056/NEJMoa1301296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Person RE, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003;34:308–12. doi: 10.1038/ng1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devriendt K, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001;27:313–7. doi: 10.1038/85886. [DOI] [PubMed] [Google Scholar]
- 15.Glocker EO, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–35. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jun HS, et al. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-β-deficient neutrophils in a congenital neutropenia syndrome. Blood. 2010;116:2783–92. doi: 10.1182/blood-2009-12-258491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Cooley L. Jagunal is required for reorganizing the endoplasmic reticulum during Drosophila oogenesis. J CellBiol. 2007;176:941–52. doi: 10.1083/jcb.200701048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horwitz MS, et al. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109:1817–24. doi: 10.1182/blood-2006-08-019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boztug K, et al. Extended spectrum of human glucose-6-phosphatase catalytic subunit 3 deficiency: novel genotypes and phenotypic variability in severe congenital neutropenia. J Pediatr. 2012;160:679–683 e2. doi: 10.1016/j.jpeds.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 20.Babu P, et al. Structural characterisation of neutrophil glycans by ultra sensitive mass spectrometric glycomics methodology. Glycoconj J. 2009;26:975–86. doi: 10.1007/s10719-008-9146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayee B, et al. G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction. Glycobiology. 2011;21:914–24. doi: 10.1093/glycob/cwr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horwitz MS, Corey SJ, Grimes HL, Tidwell T. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am. 2013;27:19–41. doi: 10.1016/j.hoc.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tidwell T, et al. Neutropenia-associated ELANE mutations disrupting translation initiation produce novel neutrophil elastase isoforms. Blood. 2014;123:562–9. doi: 10.1182/blood-2013-07-513242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudashevskaya EL, et al. A method to resolve the composition of heterogeneous affinity-purified protein complexes assembled around a common protein by chemical cross-linking, gel electrophoresis and mass spectrometry. Nat Protoc. 2013;8:75–97. doi: 10.1038/nprot.2012.133. [DOI] [PubMed] [Google Scholar]
- 25.Duden R. ER-to-Golgi transport: COP I and COP II function. Mol Membr Biol. 2003;20:197–207. doi: 10.1080/0968768031000122548. [DOI] [PubMed] [Google Scholar]
- 26.Dale DC, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496–502. [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Sartorelli AC. Evidence for the glycosylation of the granulocyte colony-stimulating factor receptor. Biochem Biophys Res Commun. 1994;205:238–44. doi: 10.1006/bbrc.1994.2655. [DOI] [PubMed] [Google Scholar]
- 28.Welte K, Zeidler C, Dale DC. Severe congenital neutropenia. Semin Hematol. 2006;43:189–195. doi: 10.1053/j.seminhematol.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 29.de Koning JP, et al. The membrane-distal cytoplasmic region of human granulocyte colony-stimulating factor receptor is required for STAT3 but not STAT1 homodimer formation. Blood. 1996;87:1335–42. [PubMed] [Google Scholar]
- 30.Jung J, et al. Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood. 2006;108:362–9. doi: 10.1182/blood-2005-11-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seifert W, et al. Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J Biol Chem. 2006;286:37665–75. doi: 10.1074/jbc.M111.267971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bryant NJ, James DE. The Sec1p/Munc18 (SM) protein, Vps45p, cycles on and off membranes during vesicle transport. J Cell Biol. 2003;161:691–6. doi: 10.1083/jcb.200212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ. The Sec1p/Munc18 protein Vps45p binds its cognate SNARE proteins via two distinct modes. J Cell Biol. 2006;173:927–36. doi: 10.1083/jcb.200512024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirnsberger G, et al. Jagunal-homolog 1 is a critical regulator of neutrophil function in fungal host defense. Nat Genet. doi: 10.1038/ng.3070. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfeifer D, et al. The hyper-IgE syndrome is not caused by a microdeletion syndrome. Immunogenetics. 2007;59:913–26. doi: 10.1007/s00251-007-0257-z. [DOI] [PubMed] [Google Scholar]
- 36.Fishelson M, Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–98. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 37.Hamada T, et al. Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1) Hum Mol Genet. 2002;11:833–40. doi: 10.1093/hmg/11.7.833. [DOI] [PubMed] [Google Scholar]
- 38.The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salzer E, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica. 2013;98:473–8. doi: 10.3324/haematol.2012.068791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–46. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Meth. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–33. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 48.Girish V, Vijayalakshmi A. Affordable image analysis using NIH Image/ImageJ. Indian J Cancer. 2004;41:47. [PubMed] [Google Scholar]
- 49.Jang-Lee J, et al. Glycomic profiling of cells and tissues by mass spectrometry: fingerprinting and sequencing methodologies. Methods Enzymol. 2006;415:59–86. doi: 10.1016/S0076-6879(06)15005-3. [DOI] [PubMed] [Google Scholar]
- 50.Pichlmair A, et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature. 2012;487:486–90. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 51.Colinge J, Masselot A, Giron M, Dessingy T, Magnin J. OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics. 2003;3:1454–63. doi: 10.1002/pmic.200300485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.