

Abstract
Peptide-derived natural products are a class of metabolites that afford the producing organism a selective advantage over other organisms in their biological niche. While the polypeptide antibiotic produced by the non-ribosomal polypeptide synthetases (NRPS) are the most widely recognized, the ribosomally-synthesized and post-translationally-modified peptides (RiPPs) are an emerging group of natural products with diverse structures and biological functions. Both the NRPS derived peptides and the RiPPs undergo extensive post-translational modifications to produce structural diversity. Here we report the first characterization of the six cysteines in forty-five (SCIFF) [Haft, D.H. and Basu M.K. (2011) J Bacteriol 193, 2745–2755] peptide maturase Tte1186, which is a member of the radical SAM superfamily. Tte1186 catalyzes the formation of a thioether crosslink in the peptide Tte1186a encoded by an orf located upstream of the maturase, under reducing conditions in the presence of SAM. Tte1186 contains three [4Fe-4S] clusters that are indispensable for thioether crosslink formation; however, only one cluster catalyzes the reductive cleavage of SAM. Mechanistic imperatives for the reaction catalyzed by the thioether forming radical SAM maturases will be discussed.
Graphical Abstract
Microorganisms produce a wide variety of peptide-derived natural products that provide selective advantage to the producer in complex biological niches. The most widely recognized are the polypeptide antibiotics, such as vancomycin, which are produced by non-ribosomal polypeptide synthetases (NRPS) and undergo extensive modifications to include a wide variety of novel side chains.1,2 In contrast to NRPS natural products, the ribosomally-synthesized and post-translationally-modified peptides (RiPPs) are an emerging group of natural products that undergo extensive post-translational modifications to produce structural diversity. There is substantial interest in understanding the biosynthesis of these peptides because many have been shown to have biological activities.3 For example, the lanthipeptide nisin (Fig. 1A) is an extensively crosslinked RiPP that is commonly used as a food preservative.4 The gene encoding the RiPP precursors co-localize with the enzymes that catalyze the post-translational modification reactions.3 This co-localization is the basis for identification of large numbers of RiPPs in genome sequencing projects, the mechanisms of post-translational modifications for many of which are poorly understood.5–7
Figure 1.

(A) The mature nisin peptide has thioether crosslinks between Cys and dehydroalanine (Dha) and dehydrobutyrine (Dhb) residues. (B) The sactipeptide subtilosin A is shown, highlighting the three thioether crosslinks. In its fully mature form, subtilosin A is circularized. The stereochemistry at the three attachment sites is shown in red. The sactipeptides are distinct from the lanthipeptides in that the thioether crosslinks are formed to the Cα of the peptide. (C) Sequence logo for the SCIFF peptides showing the conserved C-terminal sequence. The figure was generated by aligning 100 SCIFF sequences selected from Interpro family IPR023975 using the Clustal Omega61 multiple sequence alignment and visualized by Weblogo.62,63
Sulfur-to-alpha carbon thioether crosslinked peptides (sactipeptides) are a group of RiPPs (Fig 1B). The thioether linkages in these natural products are distinct from the well-studied lanthipeptides, such as nisin, where the thioether crosslinks form between a Cys residue and a dehydrated Thr/Ser. Unlike the lanthipeptide systems, where crosslinks are introduced by a two-step redox neutral mechanism, sactipeptide maturase enzymes introduce chemically equivalent crosslinks by a radical-mediated pathway involving a 2-electron oxidation of the peptide substrate.
To date, six sactipeptides have been described.8–12 The genes encoding the precursor peptides for all six are co-localized in an operon with an orf encoding a member of the radical S-adenosyl-L-methionine (SAM) (RS) superfamily. The RS superfamily contains >100,000 members catalyzing a myriad of biologically important transformations.13 The enzymes within this superfamily generally contain the CxxxCxxC motif, identified as the radical SAM signature sequence.14 These conserved cysteines provide thiolate ligands to coordinate three iron atoms of a [4Fe-4S] cluster (referred to as the RS cluster) with the fourth iron of the cluster coordinated by the α-carboxylate and α-amino moieties of SAM (Fig. 2).
Figure 2.

All RS enzymes use the site differentiated [4Fe-4S] cluster to catalyze the reductive cleavage of SAM. Upon reduction from the +2 to the +1 oxidation state the [4Fe-4S] mediates an inner sphere electron transfer to SAM generating the dAdo•. This strong oxidant then abstracts a H-atom from substrate to initiate a radical-mediated transformation.
The catalytic cycles of RS enzymes can be divided into two phases. The first is reduction of the FeS cluster from the +2 to the +1 oxidation state. In many systems, NADPH and flavodoxin/flavodoxin reductase, or small molecule reductants such as dithionite often accomplish the task in vitro.15–24 The reduced cluster mediates the reductive cleavage of SAM to generate a 5′-deoxyadenosyl radical (dAdo•) (Fig. 2), which abstracts a H-atom from the cognate substrate to initiate catalysis. The reductive cleavage of SAM is a hallmark activity of all RS enzymes. A small amount of 5′-dAdo can form even in the absence of substrate in a so-called abortive cleavage reaction.25
Studies on the RS systems have revealed a subgroup that have additional FeS clusters26. With the exception of biotin synthase27 and lipoic acid synthase28, where the second cluster serves as a source of sulfur, and HydG, where the second cluster interacts with a “dangler” iron atom through L-cysteine that acts as the scaffold for the [Fe(CO)2(CN)] synthon, 29,30 the role(s) of these clusters is not known. There are limited data on these proteins, but available structural and functional data suggest that many of these so-called auxiliary clusters (AC) are coordinated by Twitch or SPASM domains within their respective proteins,31 and they may interact with the substrate.32–34 Interestingly several members of thioether crosslink forming maturases, including those involved in the biosynthesis of sactipeptides, have been shown to have one (and sometimes a second) auxiliary cluster (referred to as AC1 or AC2 hereafter), whose role(s) remain to be established.26
RS enzymes have been shown to catalyze thioether crosslinks in the biosynthesis of subtilosin A, sporulation killing factor, thurincin H, and the γ subunit of quinohemoprotein amine dehydrogenase.35–38 Additionally, the RS enzymes ThrC and ThrD were proposed to catalyze the formation of thioether crosslinks in the RiPPs Trnα and Trnβ, which together form the bacteriocin thuricin CD9. However, recent bioinformatic investigations have identified numerous instances where a gene encoding for a RS enzyme was clustered with a gene encoding for a putative RiPP precursor indicating that the enzyme may act as a RiPP maturase.5–7
In 2011 Haft and Basu identified a gene encoding a putative RS enzyme adjacent to a gene encoding for a peptide in the genomes of 128 different organisms, primarily in the Clostridia family.6 These two genes were observed clustered with the genes tgt, queA, yajC, secD, and secF. This genomic context led the authors to propose that the RS enzyme modifies the peptide and that the modified peptide has a housekeeping role. Sequence analysis of the peptides adjacent to the RS maturase showed that nearly all of them have a conserved C-terminal domain and a variable N-terminal region (Fig. 1C). The C-terminus contains a subsequence that is distinguished by the presence of six cysteines in a forty-five residue peptide (on average), leading to these peptides being designated SCIFF. The nature of the modification introduced in the SCIFF peptide, however has not been determined.
In this manuscript we characterize a SCIFF peptide/maturase pair and show that the enzyme introduces a thioether crosslink in the conserved C-terminal region of the peptide. Biochemical and spectroscopic investigations further demonstrate the presence of three [4Fe-4S] clusters in the maturase one of which, the RS cluster (RC), is required for reductive cleavage of SAM and for overall activity. The RC and both auxiliary clusters (AC1 and AC2) are shown to be required for the crosslink formation. Mutagenesis studies further allow the three clusters to be differentiated spectroscopically. The role(s) of these clusters in the activity of the SCIFF maturase is discussed in the context of the general mechanism of radical-mediated thioether crosslink formation.
EXPERIMENTAL PROCEDURES
Cloning and expression of tte1186a and tte1186
The genes encoding the peptide, Tte1186a, and corresponding maturase, Tte1186, from Caldanaerobacter subterraneus subsp. tengcongensis MB4 were obtained from Genscript (Piscataway, NJ). The genes, supplied in a pUC57 vector, were codon optimized for recombinant expression in Escherichia coli. Both tte1186a and tte1186 were excised from their pUC57 vector upon digestion with NdeI and XhoI and ligated into pET28a, digested with the same enzymes, for recombinant expression as N-terminal His6-tagged proteins. Sequences were verified by standard Sanger sequencing at the University of Michigan DNA Sequencing Core.
Tte1186a was expressed in E. coli BL21 (DE3) cells that were transformed with the tte1186a-pET28a vector. LB, 12 × 1 L in 2.8 L Fernbach flasks, supplemented with 34 μg/mL kanamycin was inoculated with 0.1 L of tte1186a-pET28a BL21 (DE3) overnight culture and grown at 37 ºC. Expression of Tte1186a was induced upon addition of IPTG to a final concentration of 0.1 mM when OD600nm reached ~ 0.6. Cells were grown at 37 ºC for 3–4 h, harvested via centrifugation at 5000×g, flash frozen in liquid nitrogen, and stored at −80 ºC.
Tte1186 was expressed in E. coli BL21 (DE3) cells that were co-transformed with tte1186-pET28a and pDB1282 to aid in cluster maturation.40,41 LB, 12 × 1 L in 2.8 L Fernbach flasks, supplemented with 34 μg/mL kanamycin and 100 μg/mL ampicillin was inoculated with 0.1 L of tte1186-pET28a/pDB1282 BL21 (DE3) overnight culture and grown at 37 ºC. At OD600nm 0.3–0.4, arabinose and iron (III) chloride hexahydrate were added to the LB to final concentrations of 0.05% (w/v) and 0.05 mM, respectively, to induce the isc and suf operons encoded by pDB1282. Tte1186 was induced upon addition of IPTG to a final concentration of 0.1 mM when OD600nm reached 0.6–0.8. Cells were grown at 37 ºC for 18–20 h, harvested via centrifugation at 5000×g, flash frozen in liquid nitrogen, and stored at −80 ºC.
Purification of Tte1186a
Cells (~30g) were suspended in 0.2 L of 0.05 M KPi (pH 7.4), 0.5 M KCl, 0.05 M imidazole, and 1 mM PMSF, and lysed using a Branson digital sonifier operated at 50% amplitude. The lysate was clarified via centrifugation at 18,000×g for 35 min at 4 ºC. The clarified lysate was loaded on a 1 mL HisTrap HP column (GE Healthcare), charged with nickel sulfate, equilibrated in 0.05 M KPi (pH 7.4), 0.5 M KCl, and 0.05 M imidazole. After loading, the column was washed with 8 column volumes of equilibration buffer and Tte1186a was eluted with a linear gradient from 0.05 – 0.5 M imidazole over 15 column volumes. Fractions containing Tte1186a identified by SDS-PAGE were pooled and desalted using a Sep-Pak C18 cartridge (Waters) as follows. The Sep-Pak cartridge was wetted with 5 mL acetonitrile containing 0.1% (v/v) TFA and equilibrated with 5 mL 0.1 % (v/v) TFA prior to use. Tte1186a fractions (2–3 mL) were loaded onto the cartridge, and the cartridge was subsequently washed with 5 mL 0.1%(v/v) TFA. Tte1186a was eluted from the cartridge with 5 mL acetonitrile containing 0.1% (v/v) TFA. The desalted Tte1186a was lyophilized to dryness.
Purification of Tte1186
Tte1186 was purified and reconstituted in an anaerobic environment (Coy Labs anaerobic chamber with 95% N2/5% H2 atmosphere). Cells (~30 g) were suspended in 0.2 L of 0.05 M KPi (pH 7.4), 0.5 M KCl, 0.05 M imidazole, and 1 mM PMSF, and lysed using a Branson digital sonifier operated at 50% amplitude. The lysate was clarified via centrifugation at 18,000×g for 35 min at 4 ºC. The clarified lysate was loaded on a 5 mL HisTrap HP column (GE Healthcare), which had been charged with nickel sulfate and equilibrated in 0.05 M KPi (pH 7.4), 0.5 M KCl, and 0.05 M imidazole. After loading, the column was washed with 10 column volumes of equilibration buffer and Tte1186 was eluted with a linear gradient from 0.05 – 0.5 M imidazole over 8 column volumes. Fractions containing Tte1186 were pooled and desalted to 0.05 M PIPES•NaOH (pH 7.4) and 10 mM DTT using BioGel P6 DG desalting gel 100–200 mesh (wet) (Bio-Rad).
The desalted, purified enzyme was reconstituted by adding 15 molar equivalents of iron (III) chloride hexahydrate and sodium sulfide nonahydrate to Tte1186. The reconstitution was incubated at room temperature for 4h and the reaction was clarified via centrifugation at 16,000×g for 5 min. The clarified reaction was desalted to 0.05 M PIPES•NaOH (pH 7.4) and 10 mM DTT using BioGel P6 DG desalting gel 100–200 mesh (wet) and concentrated to a minimal volume (~1 mL).
Concentrated reconstituted Tte1186 (1 mL) was loaded on a Sephacryl S-300 HR size exclusion column (16 cm x 60 cm, GE Healthcare) equilibrated with 0.05 M PIPES•NaOH (pH 7.4) and 10 mM DTT and eluted isocractically at 1 mL/min. Fractions containing Tte1186 were identified by SDS-PAGE, pooled, concentrated to a minimal volume (~0.5 mL), and flash frozen in liquid nitrogen. Protein concentration was determined by the Bradford method with BSA as the standard. All UV-visible spectra of Tte1186 were measured using an Agilent 8453 diode array spectrophotometer.
Construction and purification of Tte1186 Cys→Ala variants
Each of the six Cys to Ala variants was constructed by following the Stratagene QuikChange site-directed mutagenesis protocol using the primers in Table S1. All plasmids encoding for Tte1186 Cys → Ala variants were expressed and the proteins were purified as described for wild-type Tte1186.
Amino acid analysis
Tte1186a was dissolved in 0.05 M Tris•HCl (pH 8.0), 10 mM DTT to approximately 5 mg (crude powder)/mL. The concentration of Tte1186a was then determined by Amino Acid Analysis at the Molecular Structure Facility at the University of California-Davis.
Tte1186 was subjected to Amino Acid Analysis to determine the accuracy of concentration measured by the Bradford method. A 0.1 mL aliquot of concentrated Tte1186, purified as described above, was desalted to 10 mM NaOH using an Illustra NICK column (GE Healthcare). Tte1186 was eluted from the column using 0.4 mL 10 mM NaOH and the concentration of the eluent was determined via the Bradford method with BSA as the standard. The remaining eluent was flash frozen, and the protein concentration was analyzed at the Molecular Structure Facility at the University of California-Davis. This process was performed on three independent purifications of Tte1186.
Determination of iron and sulfide stoichiometery
Iron content of wild-type and all Tte1186 variants were determined by ICP-OES. Concentrated Tte1186 was diluted to 1 μM (based on corrected Bradford concentration) in 1% (v/v) trace-metals grade nitric acid. Analyses were conducted by the Analytical Facilities of the Department of Hydrology & Atmospheric Sciences at the University of Arizona. Acid-labile sulfide content of Tte1186 (wild-type and variants) was determined using the Beinert method.42
LC-MS analysis for reductive cleavage of SAM
Assays were set up under anaerobic conditions in a Coy Labs anaerobic chamber (95% N2/5% H2). Reactions (0.2 mL) contained 0.05 M Tris•HCl (pH 8.0), 10 mM DTT, 0.5 mM SAM (enzymatically synthesized and purified as previously described43), 0 or 10 mM dithionite, 0.02 mM Tte1186 (wild-type or cluster variants), and 0.017 mM Tte1186a. The reactions were initiated upon the addition of dithionite and allowed to proceed at room temperature. At 6 h after the addition of dithionite, reactions were quenched with 0.02 mL 30% (w/v) TCA.
The acid quenched time points from the assays were centrifuged at 16,000×g for 5 min to pellet the insoluble debris prior to LC-MS analysis using an LCQ DecaXP mass spectrometer (Thermo Fisher) connected to an Agilent 1100 HPLC equipped with a diode array detector. An aliquot (0.04 mL) of the supernatant was injected onto an Agilent Zorbax Eclipse C-18 column (4.6 mm × 250 mm) pre-equilibrated in 50 mM ammonium acetate (pH 6.0) (solution A) operated at 0.3 mL/min. Solution B was 40% acetonitrile/60% H2O. The analytes were separated using the following program: 0–3 min, 0% B; 3–4.4 min, 0–0.2% B; 4.4–5.8 min, 0.2–0.8% B; 5.8–7.2 min, 0.8–1.8% B; 7.2–8.6 min, 1.8–3.2% B; 8.6–10 min, 3.2–5% B; 10–25 min, 5–25% B; 25–40 min, 25–50% B; 40–49 min, 50–75% B; 49–52 min, 75% B. After each run the C18 column was washed with 11 mL of 100% acetonitrile and equilibrated with 10 mL of 50 mM ammonium acetate (pH 6.0). SAM (enzymatically synthesized and purified) and 5′-dAdo (Sigma) were injected as standards and separated as described above.
Direct infusion of Tte1186a on LTQ OrbiTrap XL
Purified Tte1186a was diluted to 0.0085 mM in 50% (v/v) Optima acetonitrile/49.9% (v/v) Optima H2O/0.1% (v/v) Optima TFA (Fisher) and directly infused on a LTQ OrbiTrap XL (Thermo Fisher) mass spectrometer at a flow rate of 5 μL/min. The LTQ OrbiTrap XL was operated in positive ion mode detecting with FT analyzer set to a resolution of 100,000, 1 microscan, and 200 ms maximum injection time. Data were recorded in LTQ Tune program (Thermo Fisher) and analyzed using Xcalibur (Thermo Fisher). The deconvoluted and deisotoped spectrum was generated from the raw data using the Xtract software (Thermo Fisher).
Mass spectrometry analysis for Tte1186a modification
Reactions (0.2 mL) contained 0.05 M Tris•HCl (pH 8.0), 10 mM DTT, 10 mM dithionite, 0 or 1 mM SAM, 0.02 mM Tte1186 (wild-type or cluster variants), and 0.034 mM Tte1186a. Reactions were initiated upon addition of dithionite and allowed to proceed under anaerobic conditions at room temperature. After 6 h, 0.1 mL of each reaction (with SAM and without SAM) was quenched with 0.01 mL of 30% (w/v) TCA. Iodoacetamide was added to the remaining 0.1 mL of the reaction to a final concentration of 0.02 M, and the reactions were incubated in the dark for 2 h. Afterwards, the iodoacetamide treated aliquot was quenched with 0.01 mL 30% (w/v) TCA. The acidified aliquots were centrifuged to remove the precipitate and cleaned up using C18 ZipTips (Millipore) as follows. The ZipTips were wetted with acetonitrile/0.1% (v/v) TFA and equilibrated with 0.1% (v/v) TFA. The supernatant from the acid quenched samples (both treated with and without iodoacetamide) were loaded on the ZipTip. The ZipTip was subsequently washed with 10% (v/v) acetonitrile/0.1% (v/v) TFA and the peptide was eluted with 0.1 mL 50% (v/v) acetonitrile/49.9% H2O/0.1% (v/v) TFA. The eluent was directly infused on a LTQ OrbiTrap XL mass spectrometer in the same manner as described above.
LC-MS analysis for peptide modification
Assays were setup as described above. The acidified reactions were centrifuged 16,000×g for 5 min to pellet the insoluble debris prior to LC-MS analysis using a LTQ OrbiTrap XL (Thermo Fisher) mass spectrometer connected to an Agilent 1100 HPLC equipped with a diode array detector. An aliquot (0.04 mL) of the supernatant was injected onto an Agilent Zorbax Eclipse C-18 column (4.6 mm × 250 mm) pre-equilibrated in 0.1 % (v/v) TFA in water (Buffer A). Buffer B was 0.1% (v/v) TFA in acetonitrile. The reaction components were eluted with a linear gradient from 0 to 70% B over 70 min running at 0.3 mL/min. After the gradient the column was washed with 100% B for 15 min with a flow rate of 1 mL/min and equilibrated with 0% B for 15 min with a flow rate of 1 mL/min. The LTQ OrbiTrap XL was operated in positive ion mode detecting with FT analyzer set to a resolution of 100,000, 1 microscan, and 200 ms maximum injection time.
CID fragmentation of unmodified and modified Tte1186a
The peak corresponding to the +8 charge state of both the unmodified and modified Tte1186a was isolated in the CID cell using an isolation width of 3.0 m/z, Q 0.25, and 30 ms activation time. The unmodified peptide was fragmented with 10% collision energy and the modified peptide was fragmented with 20% collision energy. The MS/MS experiments were detected using the FT analyzer set to a resolution of 100,000, 1 microscan, and 200 ms maximum injection time.
EPR sample preparation
All EPR samples were prepared in a Coy Labs anaerobic chamber with 95% N2/5% H2 atmosphere. All samples contained 0.05 M PIPES•NaOH (pH 7.4), 2 mM DTT, 10 mM dithionite, 0.5 mM wild-type or variant (ΔAC1, ΔAC2, ΔRC, ΔRC/ΔAC2, or ΔRC/ΔAC1) Tte1186, and either 0 mM or 5 mM SAM in 0.15 mL. The ΔAC1/ΔAC2 variant was set up in the same manner as the rest except 0.33 mM enzyme was used in the reaction mixture. In the absence of SAM, enzyme was incubated with dithionite for 10 min at room temp. Then, only in the reactions that contained SAM, SAM was added to the reactions and they were incubated for an additional 1 min then flash frozen in liquid nitrogen in the anaerobic chamber. All samples were prepared in 3.8 mm thin walled precision quartz EPR sample tubes (Wilmad Glass) and flash frozen in liquid nitrogen in the anaerobic chamber after the appropriate incubation time. Frozen samples were stored in liquid nitrogen.
EPR Spectroscopy
Samples for X-band (~9.4 GHz) EPR spectroscopy were measured at the CalEPR center at the University of California, Davis. Continuous wave (CW) spectra were collected using a Brüker Instruments Elexsys-II E500 CW EPR spectrometer (Brüker Corporation, Billerica, MA) equipped with an Oxford Instruments ESR900 liquid helium cryostat and Oxford Instruments ITC503 temperature and gas-flow controller. Samples were measured under non-saturating slow-passage conditions using a Super-High Q resonator (ER 4122SHQE). Spectra simulations were performed using the EasySpin 4.0 toolbox in Matlab.44
RESULTS
Identification of Tte1186a
Recent bioinformatics search of the SCIFF peptide (IPR023975) and the corresponding SCIFF RS maturase (IPR024025) families indicate that they have expanded to 584 and 726 sequences, respectively, and are primarily in genomes within the Clostridia family.39 The sizable difference between the number of maturase and SCIFF peptide homologs likely represents a tendency for the automated annotations to miss orfs corresponding to short peptides. This was also noted in the report that first identified the SCIFF family.6
To identify a SCIFF maturase/peptide pair that may be amenable for biochemical analysis, a subset of ~20 pairs were identified by bioinformatics search using the radical SAM protein CPF_2198 from Clostridium perfringens ATCC 13124 as the search query. The sequences were then analyzed by XtalPred,45 which provides an indication of how likely the protein is to crystalize. Of these, the Caldanaerobacter subterraneus subsp. tengcongensis MB4 putative SCIFF maturase homolog was chosen for further analysis. However, while the maturase had been annotated, the putative SCIFF peptide was not. A tBLASTn search of the sequence space surrounding the maturase using the highly conserved C-terminal sequence (GGCGECQTSCQSACKTSCTVGNQACE)6 (Fig 1C) led to the identification of a sequence encoding the conserved C-terminal sequence. The complete orf corresponding to the SCIFF peptide was identified by searching the same translation frame as the C-terminal piece to identify the starting Met residue. The resulting orf was subsequently termed tte1186a.
Purification of SCIFF peptide and corresponding radical SAM maturase
Genes corresponding to the maturase (tte1186) and its putative peptide substrate (tte1186a) were codon-optimized for optimal expression of N-terminal His6-tagged proteins in E. coli (see Fig. S1 for sequences). The recombinant peptide substrate was purified by His-trap affinity chromatography to >95% homogeneity, as judged by SDS-PAGE analysis (Fig. S2A). The exact mass of Tte1186a was obtained by high resolution MS analysis using a LTQ OrbiTrap XL mass spectrometer and was within 2.3 ppm of that expected based on the sequence of the protein minus the N-terminal Met (Fig. S2C–E).
The recombinant maturase Tte1186 was co-expressed together with pDB1282, a plasmid that contains the isc and suf operons involved in the maturation of nitrogenase from Azotobacter vinelandii, to aid in iron-sulfur cluster insertion into the protein.40,41 Tte1186 was purified by His-trap affinity chromatography under anaerobic conditions to obtain protein that was >95% pure, as judged by SDS-PAGE analysis (Fig. S2A). To obtain cofactor-replete enzyme the as-isolated protein was reconstituted with iron and sulfide under anaerobic conditions and subjected to size exclusion chromatography. The UV-visible feature at 420 nm was substantially increased upon reconstitution, consistent with increased FeS cluster content in Tte1186 (compare spectra for as-isolated and reconstituted proteins in Fig. S2B). Amino acid analysis of the purified and reconstituted protein revealed that the Bradford protein assay overestimates the concentration of Tte1186 by 1.8 ± 0.04-fold. This factor was used to correct the concentrations determined in all subsequent experiments.
Tte1186 contains three 4Fe-4S clusters
Iron and sulfide analysis of reconstituted Tte1186 revealed 14.5 ± 1.1 mol of iron and 10.3 ± 1.5 mol of sulfide per mol of Tte1186 monomer. Based on the Fe/S stoichiometry, our working hypothesis is that Tte1186 contains three [4Fe-4S] clusters.
To further delineate the stoichiometry and identify the Cys residues that are involved in binding the additional clusters, the three cysteine residues in the CxxxCxxC radical SAM signature motif that is common to nearly all members of the radical SAM superfamily were mutated to alanine. In Tte1186, Cys 104, Cys108, and Cys111 are the three residues observed in the signature motif and were subjected to site-directed mutagenesis to generate the C104A/C108A/C111A variant of Tte1186 (ΔRC). ΔRC was expressed and purified as described for wild-type (Fig S3). Iron and sulfide analysis for this variant showed that it contained 8.8 ± 1.4 mol of iron and 10.0 ± 2.3 mol sulfide per mol of Tte1186 monomer. While this variant contains the same amount of sulfide as the wild-type protein, it contains less iron suggesting that the ΔRC variant protein binds one fewer [4Fe-4S] cluster. It is possible that the excess sulfide in this variant may be adventitiously bound sulfide resulting from the reconstitution conditions.
The SPASM domain was first defined using the radical SAM enzymes AlbA, PqqE, and anSME.5,6,31 Each enzyme contains a Cys rich C-terminal domain that was observed to bind additional [4Fe-4S] clusters called auxiliary clusters.21,35,46,47 Since the characterization of these three enzymes, additional members of the RS superfamily, including QhpD, were observed to contain a Cys rich C-terminal domain that binds auxiliary [4Fe-4S] clusters.38 BLAST search of Tte1186 revealed that it was homologous to both anSME and AlbA, suggesting that it contains the C-terminal SPASM domain. Both the bioinformatic analysis of Tte1186 and the excess Fe and S observed in both the wild-type and ΔRC variant discussed above suggested that Tte1186, by analogy to PqqE, AlbA, QhpD, and anSME, contains at least one auxiliary cluster. Indeed, Tte1186 shares significant sequence similarity with QhpD (47%), AlbA (40%), and anSME (44%). Biochemical and structural studies have shown that anSME houses a RC and two auxiliary clusters (AC1 and AC2), all of which are required for activity.21,22,46 Seven Cys residues are conserved in the C-termini of all these proteins (Fig. S4) and have been shown to coordinate the two auxiliary clusters in anSME.46 Based on the seven conserved Cys residues identified in the sequence alignment we propose that Tte1186 contains two auxiliary clusters, AC1 and AC2. When taken in the context of the X-ray crystal structure of anSME46, we propose that AC1 and AC2 are coordinated by Cys344/Cys362/Cys413, and Cys400/Cys403/Cys409/Cys432 respectively.
The two AC variants of Tte1186 were generated using site-directed mutagenesis to determine if these groups of Cys residues coordinate auxiliary FeS clusters. Fe/S analysis shows that the ΔAC1 (C344A/C362A/C413A) variant contains 8.8 ± 1.3 Fe and 7.5 ± 0.8 S per monomer while the ΔAC2 (C400A/C403A/C409A) variant contains 7.6 ± 1.0 Fe and 5.5 ± 0.7 S per monomer (Fig. S3). These are consistent, in each case, with the loss of a single cluster. We also constructed a variant where six of the seven Cys residues in the SPASM domain are mutated to Ala (Fig. S3). Fe/S analysis shows that the ΔAC1/ΔAC2 variant contains 5.1 ± 1.2 Fe and 3.7 ± 1.1 S per monomer, which is consistent with the presence of only one [4Fe-4S] cluster, the RC. Variants of Tte1186 where residues that bind the RC and either AC1 or AC2 are deleted in pairs were also constructed (Fig. S3). Fe/S analysis of these the resulting reconstituted proteins reveal that ΔRC/ΔAC1 variant contains 5.6 ± 0.7 Fe and 3.9 ± 0.6 S per monomer, whereas the ΔRC/ΔAC2 variant contains 4.9 ± 0.3 Fe and 3.3 ± 0.3 S per monomer. Collectively, these data are consistent with the presence of three [4Fe-4S] clusters in Tte1186.
Tte1186 catalyzes reductive cleavage of SAM
The first step in the reactions catalyzed by nearly all RS enzymes entails reductive cleavage of SAM to generate dAdo• to initiate catalysis (Fig. 2). In the absence of substrate, the reductive cleavage gives rise to a so-called abortive cleavage cycle, whereby the dAdo• is quenched.25 Both SAM and 5′-dAdo are readily detected by LC-MS (Fig 3a and 3b). The reaction mixtures containing Tte1186, Tte1186a, and SAM accumulate 5′-dAdo in the presence of dithionite (Fig. 3d) but not in its absence (Fig. 3c). By contrast to the wild-type enzyme, no 5′-dAdo was detected when the ΔRC variant was incubated with SAM in the presence of dithionite (Fig. 3e). Additionally, no 5′-dAdo was observed when the double cluster variants of Tte1186 lacking the RC (ΔRC/ΔAC1 and ΔRC/ΔAC2) were incubated with SAM in the presence of dithionite (Fig. 3h and 3i). In the experiments where no 5′-dAdo was detected we could have observed as little as 45 pmoles of 5′-dAdo in reactions that typically contained 0.8 nmoles of the maturase. By contrast to the ΔRC variant, incubation of ΔAC1, ΔAC2, or ΔAC1/ΔAC2 with SAM under reducing conditions led to formation of 5′-dAdo (Fig 3f, 3g, and 3j). Collectively, these observations establish a role for the RC in Tte1186 in reductively cleaving SAM to form dAdo•, while the roles of AC1 and AC2 are presumably in peptide modification chemistry.
Figure 3.

Reductive cleavage of SAM activity assay for wildtype and Cys→Ala variants of Tte1186. SAM standard (a) elutes at ~ 25 min and 5′-dAdo standard (b) elutes at ~ 41 min. The mass spectrum of each standard is above the corresponding peak. No 5′-dAdo was observed when SAM was incubated with wild-type Tte1186 in the absence of dithionite (c), but it was observed when SAM and wild-type Tte1186 were incubated in the presence of dithionite (d). No 5′-dAdo was detected in all variants that lacked the RC (ΔRC, e; ΔRC/ΔAC1, h; and ΔRC/ΔAC2, i). Variants lacking AC1 and/or AC2 (ΔAC1, f; ΔAC2, g; and ΔAC1/ΔAC2, j) were still able to reductively cleave SAM. The elution of SAM and 5′-dAdo was monitored at 260 nm.
Tte1186 post-translationally modifies the SCIFF peptide
The co-localization of the genes encoding the peptide and the RS homolog Tte1186 suggests that the enzyme carried out a post-translational modification of the peptide. To determine the nature of the modification, if any, purified and reconstituted Tte1186 was incubated with Tte1186a under various conditions, and the products were analyzed by direct infusion on a high-resolution mass spectrometer. The mass spectra of the reaction mixtures that contain Tte1186a peptide and Tte1186 maturase ± SAM are shown in Fig. 4A. The mass spectrum in the absence of SAM was identical to the spectrum in the absence of dithionite or maturase (see Fig. 4A, black spectrum for a representative mass spectrum). Multiple charge states for the peptide were observed in the mass spectrum, which are within 1.4 ppm error of the calculated m/z for each [M+nH]n+ charge state of Tte1186a (Fig. 4A, black spectrum, see Table S2 for peak list). The envelope corresponding to the +4 charge state is shown to illustrate the resolution of the spectral envelope. By contrast to the control (black spectrum), the mass spectrum of Tte1186a isolated from the reaction that contained Tte1186, SAM, and dithionite (red spectrum), exhibits a shift in each [M+nH]n+ charge state consistent with the loss of two hydrogen atoms relative to the unmodified peptide (Fig 4A) This is clearly observed by a shift in each of the envelopes consistent with a 2.0 amu shift in overall mass. A comparison of the shift for the +4 charge state was shown in Fig. 4A. In this case, the [M+4H]4+ peak (red spectrum) observed for the peptide isolated from the reaction that contained dithionite, SAM and Tte1186 is 0.5087 m/z less relative to both the observed m/z (black spectrum) and the calculated m/z of the [M+4H]4+ peak for the fully reduced unmodified peptide in the negative controls (Figure 4A, see Table S2 for peak list and Fig S5A for deconvoluted and deisotoped mass spectra).
Figure 4.

Representative mass spectra of Tte1186a isolated after incubation with Tte1186 and dithionite in the absence (black spectrum) and presence (red spectrum) of SAM. The peaks corresponding to the charge states of Tte1186a are highlighted with the gray box with the respective charge above the corresponding peak. (A) Mass spectra of Tte1186a isolated from the reactions not quenched with iodoacetamide. The spectra on the right expands the peak for the +4 charge state to show that the Tte1186a isolated from the reaction containing SAM (red spectrum) is 2.0348 amu (4 × 0.5087) lighter than peptide isolated from the reaction lacking SAM (black spectrum). (B) Mass spectra of Tte1186a that was quenched with iodoacteamide after 6 hr incubation with Tte1186 and dithionite in either the presence or absence of SAM to alkylate all free cysteine thiolates. The spectrum on the right expands on the region corresponding to the +5 charge state isotope envelope to show that the peptide isolated from the reaction containing SAM (red spectrum) is 59.033 amu (5 × 11.8066) lighter than the peptide isolated from the reaction lacking SAM (black spectrum).
The Tte1186a peptide contains 6 Cys residues, two of which could potentially form a disulfide to result in a 2 amu shift in the mass of the peptide. To rule out a disulfide bond as the source of the 2 amu shift, the reactions were quenched with iodoacetamide to alkylate all free cysteine thiols. Control reactions show that all six Cys residues in the peptide could be alkylated by iodoacetamide when dithionite, SAM, or Tte1186 was omitted (see Fig. 4B, black spectrum for a representative mass spectrum). In these controls, the m/z values for the various charge states corresponding to [M+nH]n+ are within 4.7–8.2 ppm of the theoretical values (Fig. 4B, black spectrum, see Table S3 for peak list). By contrast, when SAM, Tte1186, and reductant are included only five Cys residues are alkylated by iodoacetamide (Fig 4B, red spectrum). In the mass spectrum, the envelope corresponding to each of the charge states shifts by 59.035/z to 59.0589/z, where z represents the charge state. The m/z values for the various charge states corresponding to [M+nH]n+ are within 3.2- 5.6 ppm of that calculated m/z for peptide with 5 alkylated Cys residues minus two hydrogens (see Table S3 for peak list and Fig S5B for deconvoluted and deisotoped mass spectra). Therefore, the thiol side chain on one of the six cysteine residues is chemically modified by Tte1186, such that it was not available to be alkylated by iodoacetamide.
To ensure that the crosslinked product observed here is the sole product of the reaction we carefully examined the LC-MS data of the reactions with Tte1186 and the Tte1186a peptide ±dithionite that were treated with iodoacetamide prior to analysis. The UV-visible traces (at 230 nm) are essentially identical, with the exception of the peak for dAdo, which only appears under reducing conditions (Fig S6Aa and S6Ba). Similarly, the total ion chromatogram exhibits a peak for dAdo in the presence of dithionite and not in its absence (Fig S6Ab, S6Ac, S6Bb, and S6Bc). The peptide/product peaks, which elute at 40 min, are broad and overlapping. However, when we examine the extracted ion chromatograms for either the substrate (Fig S6Ad and S6Bd) or the product (Fig S6Ae and S6Be), we observe that in the presence of dithionite, the ion corresponding to the substrate (at m/z of 1491) is reduced and replaced with one for the product (at m/z of 1479). The mass spectra corresponding to the peak ~40 min clearly show no product in the absence of dithionite and a mixture of the two in its presence, consistent with the total ion counts of the same peak ±dithionite (Fig S6Af and S6Bf). Finally, the deconvoluted and deisotoped spectra show a mixture of species (substrate+product) in the presence of dithionite and only substrate in its absence (Fig S6Ag and S6Bg). Therefore, the data show a loss of the substrate and a concomitant increase in a peak for a singly crosslinked product. While SAM is also required for the reaction, the stoichiometry of dAdo to the crosslinked could not be determined because of the abortive cleavage reactions discussed earlier.
The mass spectral analysis of the reaction catalyzed by Tte1186 shows that Tte1186 catalyzes a SAM and reductant dependent crosslinking of a Cys residue to form a thioether intramolecular linkage in the peptide. Thioether crosslinks have been observed previously in the maturation of subtilosin A, sporulation killing factor, thurincin H, and the γ subunit of quinohemoprotein amine dehydrogenase.35,36,37,38
Tte1186 catalyzes the crosslink between Cys32 and Thr37 in Tte1186a
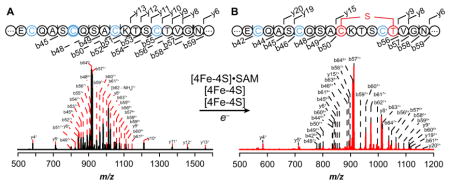
The nature of the modification introduced by Tte1186 on Tte1186a was further interrogated by MS/MS fragmentation experiments. To this end, the modification reactions containing Tte1186 and Tte1186a were quenched with iodoacetamide to block disulfide formation in the course of analysis and directly infused into the mass spectrometer. The peak corresponding to the +8 charge state of the modified and unmodified peptides was isolated, fragmented in the CID cell, and high resolution mass spectra of the corresponding fragments were obtained in the OrbiTrap analyzer.
Fragmentation of the unmodified peptide obtained from reactions where SAM was omitted is shown in Fig. 5A. We were readily able to observe and assign 18 b- and 14 y-ions primarily spanning the C-terminus of the peptide (see Table S4 for peak list). More importantly, b- and y-ions corresponding to fragments bearing each of the six conserved iodoacetamide-alkylated Cys residues (b45 through b62 and y4 through y62) are clearly present in the sample that did not contain SAM.
Figure 5.

CID MS/MS fragmentation spectrum of the +8 charge state peak of Tte1186a. (A) CID fragmentation spectrum of unmodified Tte1186a from reactions in which SAM was omitted. The four Cys residues in the sequences shown correspond to Cys 24, Cys 28, Cys 32, and Cys 36. Cys residues that were alkylated by iodoacetamide are shown in blue. The sequence shows the conserved C-terminus and that the observed b- and y-ion fragments covered majority of the C-terminus. (B) CID fragmentation spectrum of modified Tte1186a from reactions in which SAM was present. The three Cys residues (blue) correspond to Cys 24, Cys 28, and Cys 36 that are alkylated by iodoacetamide. The sequence shows the conserved C-terminus and that the observed b- and y-ion fragments covered majority of the C-terminus. No b- or y-ions were observed for the sequence between Cys 32 (red) and Thr 37 (red) due the cyclic structure imposed by a thioether crosslink. Furthermore, all b- and y-ions that contained Cys 32 through Thr 37 lacked two hydrogens and one carbamidomethyl group.
By contrast to the MS/MS spectrum of the unmodified Tte1186a, when Tte1186a isolated from reactions containing dithionite, SAM, and Tte1186 was subjected to CID fragmentation, only ions corresponding to b44 through b48 bearing iodoacetamide-alkylated C21, C24, C28, and y4 through y9 bearing IAM-alkylated C43 are observed (Fig. 5B, Table S4), and no b- or y-ions are observed for the region spanning C32 and T37. Finally, the b- and y-ions in which residues C32 through T37 are present (b56 through b64 and y15 through y61) lack one carbamidomethyl group and two hydrogen atoms. Taken together, these data are consistent with Tte1186 catalyzing the formation of a thioether crosslink between C32 and T37. Fragmentation at any point between C32 and T37 would not lead to release of any b- or y-ions because of the stable thioether linkage.
Tte11186 requires RC, AC1, and AC2 for thioether crosslink formation
We next examined which of the three FeS clusters in Tte1186 are required for thioether crosslink formation. In these experiments, the six cluster variants of Tte1186 (ΔRC, ΔAC1, ΔAC2, ΔRC/ΔAC1, ΔRC/ΔAC2, or ΔAC1/ΔAC2) were incubated with Tte1186a and SAM under reducing conditions. In the case of all of the variants, the mass spectrum of the peptide isolated from the reaction exhibits peaks that are similar to the mass spectrum of unmodified Tte1186a isolated from reactions with wild-type Tte1186 in the absence of dithionite (Fig. 6b–h and S7). A positive control reaction with wild-type Tte1186 carried out under the same conditions produced the same thioether crosslink that was described above, showing that the inability to observe any reaction was not due to any other factor (Fig. 6a and S7). Since we have shown that ΔAC1, ΔAC2, and ΔAC1/ΔAC2 Tte1186 variants are all able to carry out reductive cleavage of SAM to generate 5′-dAdo to initiate H-atom abstraction, we propose that AC1 and AC2 are required for thioether crosslink formation.
Figure 6.

Mass spectra of Tte1186a after incubation with wild-type Tte1186 and the six cluster variants. Incubation with wild-type Tte1186 and SAM in the presence (a) or absence (b) of dithionite served as the positive and negative controls. The spectra focus on the [M+4H]4+ charge state peak of the Tte1186a peptide. The mass spectra of Tte1186a isolated from reactions containing each Tte1186 cluster variant was identical to the mass spectrum of the peptide isolated from the wild-type Tte1186 negative control (ΔAC1, c; ΔAC2, d; ΔRC, e; ΔRC/ΔAC2, f; ΔRC/ΔAC1, g; ΔAC1/ΔAC2, h). Only wild-type Tte1186 was able to catalyze the formation of the crosslink in Tte1186a in the presence of SAM and dithionite, as indicated by the 0.5102 amu shift of spectrum a relative to the rest of the mass spectra.
EPR spectroscopy of Tte1186
To gain additional insights into the role of the three FeS clusters, wild-type Tte1186 and the various cluster variants (ΔRC, ΔAC1, ΔAC2, ΔRC/ΔAC1, ΔRC/ΔAC2, or ΔAC1/ΔAC2) were subjected to analysis by electron paramagnetic resonance under reducing conditions in the presence or absence of SAM. The spectrum of the wild-type protein is complex (Fig. 7Aa) with certain features appearing only when SAM is added (Fig. 7Ba). The spectra in the presence and absence of SAM are both consistent with the presence of multiple clusters with axial and/or rhombic g-tensors.
Figure 7.

Continuous wave electron paramagnetic resonance (EPR) spectroscopic analysis of the FeS clusters of wild-type Tte1186 and variants under reducing conditions in the presence (A) or absence (B) of SAM. The experimental spectra (black) represent Wild-type, a; ΔAC2, b; ΔAC1, c; ΔRC, d; ΔAC1/ΔAC2, e; ΔRC/ΔAC2, f; ΔRC/ΔAC1, g. Spectral simulations (red) were carried out using least squares fitting and a minimal set of components. In the case of ΔAC1/ΔAC2, a single component was used to simulate the RS cluster in the absence (Ae) or presence (Be) of SAM. The ΔRC/ΔAC2 and ΔRC/ΔAC1variants required two species each to simulate the spectra. Spectra were recorded at 10K, with a 9.4 GHz microwave frequency, modulation amplitude of 10 G, and 100 μW microwave power. The wild-type spectra in Aa and Ba were simulated using the g-values from the double cluster variants in the following compositions: Aa, 17% RC-SAM, 17% AC1a, 26% AC2a, and 39% AC2b; Ba, 10% RC-SAM, 50% RC+SAM, 8% AC1a, 10% AC2a, and 2% AC2b.
The EPR spectrum of wild-type Tte1186 exhibits peaks consistent with multiple clusters when RC, AC1, or AC2 are eliminated (see Fig. 7b, 7c, and 7d in both panel A and B), complicating assignment of the features to individual clusters. Therefore, to assign the features to individual clusters, we turned to the ΔRC/ΔAC1, ΔRC/ΔAC2, or ΔAC1/ΔAC2 variants, which retain AC2, AC1, and RC, respectively. With the ΔAC1/ΔAC2 variant, in the absence of SAM, we observe a weak signal, which is well simulated with the rhombic g-tensor of [2.04 1.92 1.91] (Fig. 7Ae, Table S5). In the presence of SAM the signal is significantly altered and is simulated with a g-tensor of [1.99 1.90 1.84] (Fig. 7Be, Table S5). The SAM-dependent appearance of this characteristic rhombic signal is consistent with other members of the radical SAM superfamily, where it is thought that binding of the cofactor induces an electronic change in the environment of the cluster leading to alteration of the redox potential and g-matrix.25,48 We assign this signal to the RS cluster.
The contributions of AC1 and AC2 to that of the wild-type sample were examined with the ΔRC/ΔAC2 and ΔRC/ΔAC1 variants of Tte1186, respectively. Both proteins exhibit similar signals ±SAM (see Fig. 7f and 7g in both panel A and B), as may be expected from the fact that they likely do not directly interact with the cofactor. The spectrum of each variant was well captured by using a mixture of two species, with the minor species likely the result of cluster degradation. The spectrum of the reduced AC1 was simulated with g-tensors of [2.06 1.93 1.88] and [2.02 1.95 1.90] for the major (AC1a, ~80%) and minor (AC1b, ~20%) species, respectively (Table S7, Fig. 7f and S8A). Similarly, the spectrum of AC2 could be simulated by g-tensors of [2.05, 1.93, 1.89] and [2.09, 1.94, 1.87] for the major (~60%) and minor (~40%) species, respectively (Table S5, Fig. 7g and S8B).
With the g-tensors for RC, AC1 and AC2 determined above, the EPR spectrum of the wild-type enzyme containing all three clusters could be simulated with a mixture of the relevant components. The wild-type spectrum in the absence of SAM (Fig. 7Aa) is well reproduced with the following composition: ~17% RC, ~17% AC1a, ~26%AC2a, and ~39% AC2b. In the presence of SAM (Fig. 7Ba) the spectrum is dominated by species attributed to the SAM binding cluster with SAM bound, making up ~50% of the observed spectrum. Also present is a small amount of the RC without SAM bound (~10%) and contributions from the auxiliary clusters AC1a (~8%), AC2a (~10%) and AC2b (~2%). We note that the spectra of the wild-type enzyme can be reproduced in the absence of the AC1b species and with minimal contribution of the AC2b species in the presence of SAM. Therefore, the assigned g-tensors for the three clusters adequately reproduce the complex spectrum obtained with wild-type protein in ±SAM.
As with the mutagenesis and activity studies discussed above, the EPR analysis is consistent with the presence of three [4Fe-4S] clusters in Tte1186. Moreover, the SAM-dependent appearance of the reduced RS cluster and the lack of observable changes in the spectrum of AC1 or AC2 are in-line with the biochemical observations that AC1 and AC2 are dispensable for the reductive cleavage of SAM.
DISCUSSION
The peptide maturase Tte1186 catalyzes the formation of a thioether crosslink in the Tte1186a peptide, which is encoded by an orf immediately upstream of the maturase. The results in this manuscript demonstrate that the protein accomplishes this remarkably simple reaction using three [4Fe-4S] clusters, one of which reductively activates SAM and two that are required for function. Tte1186 has been amenable to spectroscopic studies and we are able to deconvolute the complex EPR signal of the wild-type protein and assign the g-tensors for each of the three clusters.
While it may seem that the thioether crosslinks in the sactipeptides are reminiscent of the crosslinks found in the well-studied lanthipeptides, including nisin (see Fig. 1A), from a mechanistic perspective, lanthipeptides and sactipeptides are distinct. In nisin, the thioether crosslinks involve the addition of Cys residues to a dehydroalanine or dehydrobutyrine produced, from dehydration of Ser or Thr, respectively. In sactipeptides, the crosslinks are formed by a radical-mediated reaction that inserts the Cys thiol into the unactivated Cα-H of the acceptor site. In contrast to lanthipeptides, where studies by van der Donk and coworkers have led to extensive mechanistic insight,49 the mechanism of thioether crosslinks in sactipeptides remain unknown.
Prior to a discussion of the sactipeptides, it is instructive to first discuss in general terms the mechanism of biosynthesis of lanthipeptides (Fig. 8A). The key to these cyclization reactions is activation of the Thr or Ser residue for elimination of the hydroxyl group. This is accomplished by phosphorylation by an ATP-dependent reaction or glutamylation by a Glu-tRNA dependent reaction where the precursor Thr or Ser residue is phosphorylated/glutamylated, providing an activated center to facilitate elimination forming the dehydroresidue.50–52 Activation of the acceptor site to allow for Michael addition is key to the reaction. It is important to note that addition to both the re- and si-faces are observed, which is consistent with production of the dehydrated intermediate. Recent computational studies have suggested that the stereochemical outcome is governed by conformational restraints imposed by the substrate on the transition state.53
Figure 8.

Proposed mechanisms of thioether crosslink formation in lantipeptides and sactipeptides. (A) Mechanism of thioether crosslink formation in lanthipeptides. Substrate is activated to generate a reactive intermediate (Dha or Dhb), which undergoes Michael addition to generate the initial crosslink. (B) Proposed mechanism for radical-mediated thioether cross-links in sactipeptides. The distinguishing feature of this mechanism relative to those shown in Fig. S5 is formation of a reactive intermediate, such as a ketoimine, analogous to Dha or Dhb in the lantipeptide mechanism, which would be trapped from the re- or si-face to generate mixed regioselective outcomes.
Unlike the redox neutral mechanisms involved in the biosynthesis of lanthipeptides (Fig. 8A), formation of thioether crosslinks in sactipeptides entails an overall two-electron oxidation of the substrate. One can envision two possible mechanisms for the crosslinking reaction (see Fig. S9). The simplest mechanism (Fig. S9A) that one could propose would involve two parallel H-atom abstraction events by dAdo•, one at the α-carbon and another one at the Cys thiol, which is followed by the formation of the thioether crosslink. This would be unprecedented, as it requires the simultaneous production of two highly reactive intermediates, as well as the ability for the Aux cluster to reductively cleave SAM. This seems un-realistic, and in fact, in contrast to the RC, Aux clusters have never been shown to be required for dAdo production35,36 In Tte1186, only RC is shown to be able to reductively cleave SAM (see Fig 3). An alternative model (Fig. S9B) would generate dAdo• at the RC, which either directly or via an intervening enzyme-based radical, abstracts the H-atom from the α-carbon of the amino acid involved in the crosslink to form a substrate radical. The substrate radical would then combine with the Cys thiolate and electron transfer to the protein would complete the catalytic cycle. The recipient of the second electron could be the AC1, AC2, or alternately, the RC. It is premature to speculate on the precise recipient as no structural data are currently available for this protein, and the coordinating residues were inferred from sequence alignments. We note in passing that similar electron transfers have been postulated in other radical SAM enzymes with multiple FeS clusters32–38; but to date, there is no firm data on how this is accomplished in any of the systems. We cannot rule out a more direct role for one of the auxiliary clusters in activating the peptide and orchestrating the formation of the thioether crosslink.
While the second alternative above is reasonable, it is inconsistent with the stereochemical outcomes observed in thioether containing peptides. An interesting observation is that the structures of many sactipeptides that contain multiple thioether crosslinks (such as SboA, Trnα, and Trnβ) show that the linkage is introduced by the respective maturase (AlbA, ThrC, ThrD) by attack at either the si- or re-face of the initial radical intermediate to generate different stereochemical outcomes.54–56 As an example, examination of the structure of mature SboA shows that its maturase, AlbA, introduces both re- and si-adducts in the same peptide (see Fig. S9C). In the context of mechanisms shown in Fig. S9, these data suggest that sulfur insertion can occur at a position initially occupied by the α-proton, or from the opposite face. The latter would be reasonable, but the former requires the radical intermediate to be relatively flexible and able to rotate to expose the opposite face of the radical to the thiol for the crosslink to form on the same face as the abstracted α-proton, as that face would be occluded by the dAdo-H otherwise. Of course, if the peptide contained D-amino acids, we could rationalize the addition, but then would have to postulate that dAdo• can access both faces of the Cα, which would be unreasonable. We know that peptide substrates in all cases are ribosomally synthesized and contain all L-amino acids. While structural data on radical SAM enzymes are sparse, in all cases where data are available, the H-atom donor and acceptors are within VDW distance of one another and it is likely that radical intermediates are controlled in enzyme active sites by limiting their movement.57–59 The stereochemical outcomes of the reaction are not explained by the prevailing mechanisms in the literature,60 as summarized in Fig. S9.
Our alternative working model that can account for the observed regiochemistry is shown in Fig. 8B. We postulate that instead of direct coupling between the α-carbon radical and the thiol, the radical intermediate formed by dAdo• H-atom abstraction undergoes conversion to a more stable intermediate that can be trapped from either face by the thiol side chain. For example, the initially formed captodatively-stabilized radical intermediate may undergo an oxidation to a ketoimine, which is subsequently trapped by the thiol of the substrate on either face of the planar intermediate leading to stereoisomeric crosslinks in the product (Fig. 8B). Such a mechanism of action, would explain the curious regioselectivity of the sactipeptide maturases, as the preference for the re- or si- would then be dictated by binding determinants of the peptide to the maturase.
This new paradigm immediately suggests an intriguing mechanistic parallel between the sactipeptide and lanthipeptide biosynthetic reactions discussed earlier. As it will be recalled, the biosynthesis of the thioether crosslinks in lanthipeptides requires that the Ser or Thr residue be activated to a Dha or Dhb prior to being trapped by a Michael addition (see Fig. 8A). By analogy to the lanthipeptides, the 2-electron oxidation of unmodified sactipeptide substrate to a ketoimine would provide an activated intermediate for the reaction with Cys, which could then occur from either face to produce both possible stereoisomers. Presumably, the protein dictates which product is observed by using the conformation of the bound substrate to impose regioselectivity.
The Tte1186 maturase and peptide pair was identified bioinformatically and shown to be involved in a thioether crosslink formation. At present, the biological function of this modified product is not known. However, the methods used here to identify and characterize the reaction catalyzed by Tte1186 may be useful in studying the growing bioinformatically identified RiPP systems.5–7
Supplementary Material
Acknowledgments
Funding:
The work reported in this publication was supported by National Institutes of General Medical Sciences of the National Institutes of Health grant R01 GM72623 (awarded to VB) and GM104543 (awarded to R.D.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- NRPS
non-ribosomal polypeptide synthetases
- RiPPs
ribosomally-synthesized post-translationally-modified peptides
- SAM
S-adenosyl-L-methionine
- RS
radical SAM
- dAdo•
5′-deoxyadenosyl radical
- 5′-dAdo
5′-deoxyadenosine
- E. coli
Escherichia coli
- LC-MS
liquid chromatography-mass spectrometry
- HPLC
high-performance liquid chromatography
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- PIPES
1,4-piperazinediethanesulfonic acid
- PMSF
phenylmethanesulfonyl fluoride
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TCA
trichloroacetic acid
- Tris
tris(hydroxymethyl)aminomethane
- LB
Lennox broth
- EPR
electron paramagnetic resonance
- SPASM
subtilosin A, pyrroloquinonline quinone, anaerobic sulfatase, mycofactocin
- SCIFF
six-cysteines in forty-five
- RC
radical SAM cluster
- AC
auxiliary cluster
- KPi
potassium phosphate
- TFA
trifluoroacetic acid
- DTT
dithiothreitol
Footnotes
Notes: The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
N.A.B. and V.B. designed and carried out the biochemical experiments and prepared the EPR samples. J.W. and R.D.B. designed and carried out the EPR experiments. All authors contributed to writing the manuscript.
Tables S1 – S5 and Figures S1 – S9. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides1. Annu Rev Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 3.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Goransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, Link AJ, Liu W, Marahiel MA, Mitchell DA, Moll GN, Moore BS, Muller R, Nair SK, Nes IF, Norris GE, Olivera BM, Onaka H, Patchett ML, Piel J, Reaney MJ, Rebuffat S, Ross RP, Sahl HG, Schmidt EW, Selsted ME, Severinov K, Shen B, Sivonen K, Smith L, Stein T, Sussmuth RD, Tagg JR, Tang GL, Truman AW, Vederas JC, Walsh CT, Walton JD, Wenzel SC, Willey JM, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lubelski J, Rink R, Khusainov R, Moll GN, Kuipers OP. Biosynthesis, immunity, regulation, mode of action and engineering of the model lantibiotic nisin. Cell Mo Life Sci. 2007;65:455–476. doi: 10.1007/s00018-007-7171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haft DH. Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genomics. 2011;12:21. doi: 10.1186/1471-2164-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haft DH, Basu MK. Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J Bacteriol. 2011;193:2745–2755. doi: 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy K, O’Sullivan O, Rea MC, Cotter PD, Ross RP, Hill C. Genome mining for radical SAM protein determinants reveals multiple sactibiotic-like gene clusters. PloS One. 2011;6:e20852. doi: 10.1371/journal.pone.0020852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sit CS, van Belkum MJ, McKay RT, Worobo RW, Vederas JC. The 3D solution structure of thurincin H, a bacteriocin with four sulfur to alpha-carbon crosslinks. Angew Chem Int Ed Engl. 2011;50:8718–8721. doi: 10.1002/anie.201102527. [DOI] [PubMed] [Google Scholar]
- 9.Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci U S A. 2010;107:9352–9357. doi: 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu WT, Yang YL, Xu Y, Lamsa A, Haste NM, Yang JY, Ng J, Gonzalez D, Ellermeier CD, Straight PD, Pevzner PA, Pogliano J, Nizet V, Pogliano K, Dorrestein PC. Imaging mass spectrometry of intraspecies metabolic exchange revealed the cannibalistic factors of Bacillus subtilis. Proc Natl Acad Sci U S A. 2010;107:16286–16290. doi: 10.1073/pnas.1008368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, Mathews FS. Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking. Proc Natl Acad Sci U S A. 2001;98:14268–14273. doi: 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babasaki K, Takao T, Shimonishi Y, Kurahashi K. Subtilosin A, a new antibiotic peptide produced by Bacillus subtilis168: isolation, structural analysis, and biogenesis. J Biochem. 1985;98:585–603. doi: 10.1093/oxfordjournals.jbchem.a135315. [DOI] [PubMed] [Google Scholar]
- 13.Brown S, Babbitt P. Using the structure-function linkage database to characterize functional domains in enzymes. Current Protocols in Bioinformatics. 2014;48:2.10.1–2.10.16. doi: 10.1002/0471250953.bi0210s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harder J, Eliasson R, Pontis E, Ballinger MD, Reichard P. Activation of the anaerobic ribonucleotide reductase from Escherichia coli by S-adenosylmethionine. J Biol Chem. 1992;267:25548–25552. [PubMed] [Google Scholar]
- 16.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, Reichard P. Flavodoxin Is Required for the Activation of the Anaerobic Ribonucleotide Reductase. Biochem Biophy Res Comm. 1993;197:792–797. doi: 10.1006/bbrc.1993.2548. [DOI] [PubMed] [Google Scholar]
- 17.Ifuku O, Koga N, Haze SI, Kishimoto J, Wachi Y. Flavodoxin is Required for Conversion of Dethiobiotin to Biotin in Escherichia coli. Eur J Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 18.Layer G, Verfürth K, Mahlitz E, Jahn D. Oxygen-independent Coproporphyrinogen-III Oxidase HemN from Escherichia coli. J Biol Chem. 2002;277:34136–34142. doi: 10.1074/jbc.M205247200. [DOI] [PubMed] [Google Scholar]
- 19.Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Lipoyl Synthase Requires Two Equivalents of S-Adenosyl-L-Methionine To Synthesize One Equivalent of Lipoic Acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- 20.Brazeau BJ, Gort SJ, Jessen HJ, Andrew AJ, Liao HH. Enzymatic activation of lysine 2,3-aminomutase from Porphyromonas gingivalis. Appl Environ Microbiol. 2006;72:6402–6404. doi: 10.1128/AEM.01143-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grove TL, Lee KH, St Clair J, Krebs C, Booker SJ. In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grove TL, Ahlum JH, Qin RM, Lanz ND, Radle MI, Krebs C, Booker SJ. Further Characterization of Cys-Type and Ser-Type Anaerobic Sulfatase Maturating Enzymes Suggests a Commonality in the Mechanism of Catalysis. Biochemistry. 2013;52:2874–2887. doi: 10.1021/bi400136u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruender NA, Young AP, Bandarian V. Chemical and Biological Reduction of the Radical SAM Enzyme CPH4 Synthase. Biochemistry. 2015;54:2903–2910. doi: 10.1021/acs.biochem.5b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bianchi V, Reichard P, Eliasson R, Pontis E, Krook M, Jörnvall H, Haggård-Ljungquist E. Escherichia coli ferredoxin NADP+ reductase: activation of E. coli anaerobic ribonucleotide reduction, cloning of the gene (fpr), and overexpression of the protein. J Bacteriol. 1993;175:1590–1595. doi: 10.1128/jb.175.6.1590-1595.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-Adenosylmethionine Enzymes. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lanz ND, Booker SJ. Auxiliary iron-sulfur cofactors in radical SAM enzymes. Biochimica et biophysica acta. 2015;1853:1316–1334. doi: 10.1016/j.bbamcr.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Fugate CJ, Jarrett JT. Biotin synthase: insights into radical-mediated carbon-sulfur bond formation. Biochimica et biophysica acta. 2012;1824:1213–1222. doi: 10.1016/j.bbapap.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 28.Booker SJ, Cicchillo RM, Grove TL. Self-sacrifice in radical S-adenosylmethionine proteins. Curr Opin Chem Biol. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suess DLM, Pham CC, Bürstel I, Swartz JR, Cramer SP, Britt RD. The Radical SAM Enzyme HydG Requires Cysteine and a Dangler Iron for Generating an Organometallic Precursor to the [FeFe]-Hydrogenase H-Cluster. J Am Chem Soc. 2016;138:1146–1149. doi: 10.1021/jacs.5b12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suess DLM, Bürstel I, De La Paz L, Kuchenreuther JM, Pham CC, Cramer SP, Swartz JR, Britt RD. Cysteine as a ligand platform in the biosynthesis of the FeFe hydrogenase H cluster. Proc Natl Acad Sci U S A. 2015;112:11455–11460. doi: 10.1073/pnas.1508440112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grell TAJ, Goldman PJ, Drennan CL. SPASM and Twitch Domains in S-Adenosylmethionine (SAM) Radical Enzymes. J Biol Chem. 2015;290:3964–3971. doi: 10.1074/jbc.R114.581249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hänzelmann P, Schindelin H. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc Natl Acad Sci U S A. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hänzelmann P, Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc Natl Acad Sci U S A. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lees NS, Hanzelmann P, Hernandez HL, Subramanian S, Schindelin H, Johnson MK, Hoffman BM. ENDOR spectroscopy shows that guanine N1 binds to [4Fe-4S] cluster II of the S-adenosylmethionine-dependent enzyme MoaA: mechanistic implications. J Am Chem Soc. 2009;131:9184–9185. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fluhe L, Knappe TA, Gattner MJ, Schafer A, Burghaus O, Linne U, Marahiel MA. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol. 2012;8:350–357. doi: 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- 36.Flühe L, Burghaus O, Wieckowski BM, Giessen TW, Linne U, Marahiel MA. Two [4Fe-4S] Clusters Containing Radical SAM Enzyme SkfB Catalyze Thioether Bond Formation during the Maturation of the Sporulation Killing Factor. J Am Chem Soc. 2013;135:959–962. doi: 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- 37.Wieckowski BM, Hegemann JD, Mielcarek A, Boss L, Burghaus O, Marahiel MA. The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS lett. 2015;589:1802–1806. doi: 10.1016/j.febslet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 38.Nakai T, Ito H, Kobayashi K, Takahashi Y, Hori H, Tsubaki M, Tanizawa K, Okajima T. The Radical S-Adenosyl-L-methionine Enzyme QhpD Catalyzes Sequential Formation of Intra-protein Sulfur-to-Methylene Carbon Thioether Bonds. J Biol Chem. 2015;290:11144–11166. doi: 10.1074/jbc.M115.638320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, Yong SY, Bateman A, Punta M, Attwood TK, Sigrist CJA, Redaschi N, Rivoire C, Xenarios I, Kahn D, Guyot D, Bork P, Letunic I, Gough J, Oates M, Haft D, Huang H, Natale DA, Wu CH, Orengo C, Sillitoe I, Mi H, Thomas PD, Finn RD. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–21. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frazzon J, Fick JR, Dean DR. Biosynthesis of iron-sulphur clusters is a complex and highly conserved process. Biochem Soc Tran. 2001;30:680–685. doi: 10.1042/bst0300680. [DOI] [PubMed] [Google Scholar]
- 41.Frazzon J, Dean DR. Formation of iron-sulfur clusters in bacteria: an emerging field in bioinorganic chemistry. Curr Opin Chem Biol. 2003;7:166–173. doi: 10.1016/s1367-5931(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 42.Beinert H. Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 43.McCarty RM, Krebs C, Bandarian V. Spectroscopic, Steady-State Kinetic, and Mechanistic Characterization of the Radical SAM Enzyme QueE, Which Catalyzes a Complex Cyclization Reaction in the Biosynthesis of 7-Deazapurines. Biochemistry. 2013;52:188–198. doi: 10.1021/bi301156w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stoll S, Schweiger A. Easyspin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. Journal of Magnetic Resonance. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik A. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics. 2007;23:3403–3405. doi: 10.1093/bioinformatics/btm477. [DOI] [PubMed] [Google Scholar]
- 46.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, Drennan CL. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proc Natl Acad Sci U S A. 2013;110:8519–8524. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu SP, King D, Britt RD, Klinman JP. Pyrroloquinoline Quinone Biogenesis: Demonstration That PqqE from Klebsiella pneumoniaeIs a Radical S-Adenosyl- L-Methionine Enzyme. Biochemistry. 2009;48:10151–10161. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinckley GT, Frey PA. Cofactor dependence of reduction potentials for [4Fe-4S]2+/1+ in lysine 2,3-aminomutase. Biochemistry. 2006;45:3219–3225. doi: 10.1021/bi0519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, Zhang Q, van der Donk WA. Insights into the evolution of lanthipeptide biosynthesis. Protein Sci. 2013;22:1478–1489. doi: 10.1002/pro.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garg N, Salazar-Ocampo LMA, van der Donk WA. In vitro activity of the nisin dehydratase NisB. Proc Natl Acad Sci U S A. 2013;110:7258–7263. doi: 10.1073/pnas.1222488110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ortega MA, Hao Y, Zhang Q, Walker MC, van der Donk WA, Nair SK. Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature. 2015;517:509–512. doi: 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knerr PJ, van der Donk WA. Discovery, biosynthesis, and engineering of lantipeptides. Annual review of biochemistry. 2012;81:479–505. doi: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- 53.Tang W, Jiménez-Osés G, Houk KN, van der Donk WA. Substrate control in stereoselective lanthionine biosynthesis. Nature Chem. 2015;7:57–64. doi: 10.1038/nchem.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawulka KE, Sprules T, Diaper CM, Whittal RM, McKay RT, Mercier P, Zuber P, Vederas JC. Structure of subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to alpha-carbon cross-links: formation and reduction of alpha-thio-alpha-amino acid derivatives. Biochemistry. 2004;43:3385–3395. doi: 10.1021/bi0359527. [DOI] [PubMed] [Google Scholar]
- 55.Kawulka K, Sprules T, McKay RT, Mercier P, Diaper CM, Zuber P, Vederas JC. Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to alpha-carbons of phenylalanine and threonine. J Am Chem Soc. 2003;125:4726–4727. doi: 10.1021/ja029654t. [DOI] [PubMed] [Google Scholar]
- 56.Sit CS, McKay RT, Hill C, Ross RP, Vederas JC. The 3D structure of thuricin CD, a two-component bacteriocin with cysteine sulfur to alpha-carbon cross-links. J Am Chem Soc. 2011;133:7680–7683. doi: 10.1021/ja201802f. [DOI] [PubMed] [Google Scholar]
- 57.Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL. A locking mechanism preventing radical damage in the absence of substrate, as revealed by the x-ray structure of lysine 5,6-aminomutase. Proc Natl Acad Sci U S A. 2004;101:15870–15875. doi: 10.1073/pnas.0407074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dowling DP, Bruender NA, Young AP, McCarty RM, Bandarian V, Drennan CL. Radical SAM enzyme QueE defines a new minimal core fold and metal-dependent mechanism. Nat Chem Biol. 2013;10:106–112. doi: 10.1038/nchembio.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horitani M, Byer AS, Shisler KA, Chandra T, Broderick JB, Hoffman BM. Why Nature Uses Radical SAM Enzymes so Widely: Electron Nuclear Double Resonance Studies of Lysine 2,3-Aminomutase Show the 5′-dAdo• ‘Free Radical’ Is Never Free. J Am Chem Soc. 2015;137:7111–7121. doi: 10.1021/jacs.5b00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flühe L, Marahiel MA. Radical S-adenosylmethionine enzyme catalyzed thioether bond formation in sactipeptide biosynthesis. Curr Opin Chem Biol. 2013;17:605–612. doi: 10.1016/j.cbpa.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 61.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.