

Abstract
Objective:
To characterize the clinical and genetic features of cervical dystonia (CD).
Methods:
Participants enrolled in the Dystonia Coalition biorepository (NCT01373424) with initial manifestation as CD were included in this study (n = 1,000). Data intake included demographics, family history, and the Global Dystonia Rating Scale. Participants were screened for sequence variants (SVs) in GNAL, THAP1, and Exon 5 of TOR1A.
Results:
The majority of participants were Caucasian (95%) and female (75%). The mean age at onset and disease duration were 45.5 ± 13.6 and 14.6 ± 11.8 years, respectively. At the time of assessment, 68.5% had involvement limited to the neck, shoulder(s), and proximal arm(s), whereas 47.4% had dystonia limited to the neck. The remaining 31.5% of the individuals exhibited more extensive anatomical spread. A head tremor was noted in 62% of the patients. Head tremor and laryngeal dystonia were more common in females. Psychiatric comorbidities, mainly depression and anxiety, were reported by 32% of the participants and were more common in females. Family histories of dystonia, parkinsonian disorder, and tremor were present in 14%, 11%, and 29% of the patients, respectively. Pathogenic or likely pathogenic SVs in THAP1, TOR1A, and GNAL were identified in 8 participants (0.8%). Two individuals harbored novel missense SVs in Exon 5 of TOR1A. Synonymous and noncoding SVs in THAP1 and GNAL were identified in 4% of the cohort.
Conclusions:
Head tremor, laryngeal dystonia, and psychiatric comorbidities are more common in female participants with CD. Coding and noncoding variants in GNAL, THAP1, and TOR1A make small contributions to the pathogenesis of CD.
Cervical dystonia (CD) is the most common adult-onset isolated dystonia.1,2 The percentage of patients with CD with head tremor, neuropsychiatric comorbidities, and spread of dystonia to other anatomical segments is mostly unknown in large multicenter clinical series. Genetic factors are believed to be important given that most large clinical series report a positive family history in approximately 10% of probands.3–5 Although sequence variants (SVs) in THAP1, GNAL, and Exon 5 of TOR1A have been identified in small percentages of participants with isolated CD, the relative contributions of dystonia-associated genes to mainly adult-onset (>20 years)6 CD have not been fully characterized.3,5,7–14 In this study, we detail the clinical and genetic features of CD in a large multicenter cohort.
METHODS
Study design and patients.
The Dystonia Coalition (DC, www.rarediseasesnetwork.org/dystonia/) has developed a biospecimen repository (Coriell; Camden, NJ) and an associated clinical database to be used as a resource for dystonia research (http://clinicaltrials.gov/show/NCT01373424). Participants included in the analyses described herein were the first 1,000 with reported onset of dystonia in the nonlaryngeal, nonpharyngeal neck muscles. These participants were enrolled between January 5, 2011, and September 21, 2014. Data collection for the DC included the following: blood for the extraction of DNA and lymphocytes, Global Dystonia Rating Scale (GDRS), video examination, and clinical data elements (demographics, age at onset, site of onset, sensory tricks, anatomical distribution of dystonia, history of psychiatric disorders, and family history [dystonia, parkinsonism, and tremor]). Raters were also asked to indicate whether the neck was affected by tremor. The Dystonia Genetic Consortium is an international collaboration of neurologists interested in the genetics of dystonia and closely related neurogenetic disorders. Biological specimens acquired by the Dystonia Genetic Consortium are stored in the Department of Neurology at the University of Tennessee Health Science Center (UTHSC).
Standard protocol approvals, registrations, and patient consents.
The study was approved by the ethics committees of all participating centers. All participants gave written informed consent for participation in the study and inclusion in the biospecimen and video repositories.
Genetic screening.
Because of the limited amounts of DNA available for this project (2 μg per participant) and questions related to the pathogenicity of CIZ1 and ANO3 SVs in participants with isolated dystonia, a combination of high-resolution melting (HRM) and Sanger sequencing was restricted to coding and contiguous noncoding regions of THAP1, GNAL, and Exon 5 of TOR1A.3,5,8,10,15 Exon 5 of TOR1A harbors the classic DYT1 ΔGAG mutation, and cosegregating pathogenic SVs have not been identified in other exons of TOR1A.7,8,15,16 After the identification of 2 novel SVs in Exon 5 of TOR1A, an additional 1,160 DNA samples from the UTHSC Dystonia Genetics Consortium biorepository were also screened for these and other SVs within Exon 5 of TOR1A (table e-1 at Neurology.org/ng). Cases with known mutations in TOR1A and other dystonia-associated genes were excluded from this secondary screening effort.
In silico mutation prediction programs SIFT, MutationTaster, Polymorphism Phenotyping version 2 (PolyPhen-2), and Combined Annotation-Dependent Depletion (CADD) were used to evaluate the pathogenicity of novel and previously reported missense SVs.17–20 TargetScanHuman 6.2 (www.targetscan.org) was used to search for predicted microRNA (miRNA) targets within loci harboring SVs.21 Human Splicing Finder 3.0 (www.umd.be/HSF) was used to predict the effects of identified SVs on splicing motifs including acceptor and donor splice sites, and branch point and auxiliary sequences known to either enhance (exonic splicing enhancers [ESEs]) or repress (exonic splicing silencers [ESSs]) splicing.22 The allele frequency of the identified SVs was compared with the frequencies reported in Exome Variant Server (EVS, evs.gs.washington.edu/EVS/), 1000 Genomes Project (1KG, www.1000genomes.org), and Exome Aggregation Consortium (ExAC, exac.broadinstitute.org). American College of Medical Genetics and Genomics guidelines were used for the classification of SVs as “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” or “benign.”23 Details of HRM, Sanger sequencing, and variant analysis are included in appendix e-1 and table e-2.
Statistical analysis.
Fisher exact tests (2-tailed) were used to compare female-male frequencies. The χ2 statistic (2-tailed) was used to compare case-control genotype frequencies. Two-tailed t tests were used to determine whether age, age at onset, duration, and scores on rating scales differed between women and men. The Pearson correlation coefficient (r) was used to measure the strength of linear associations between GDRS scores and number of affected anatomical segments, dystonia duration, and dystonia age at onset. An alpha (α) of 0.05 was chosen for statistical significance.
RESULTS
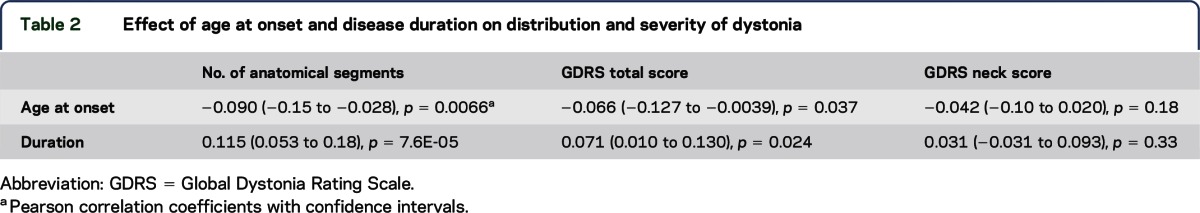
The majority (95%) of participants were Caucasian females (table 1). The mean age at onset and disease duration for the entire cohort of 1,000 participants were 45.5 and 14.6 years, respectively. A high percentage of the 1,000 participants with CD had a head tremor (62%), and head tremor was more common in females than in males. Total GDRS scores did not differ between males and females. The majority of participants (68.5%) had anatomical involvement limited to the neck ± shoulder(s) and proximal arm(s). Less than half of the patients enrolled in this cohort were judged to have isolated neck involvement (47.4%). Females with CD were more likely to manifest laryngeal dystonia. Otherwise, there were no significant effects of sex on patterns of anatomical involvement. Age at onset decreased with the number of affected anatomical segments and GDRS total score (table 2). Dystonia duration increased with the number of affected anatomical segments and GDRS total score.
Table 1.
Clinical features of the study population

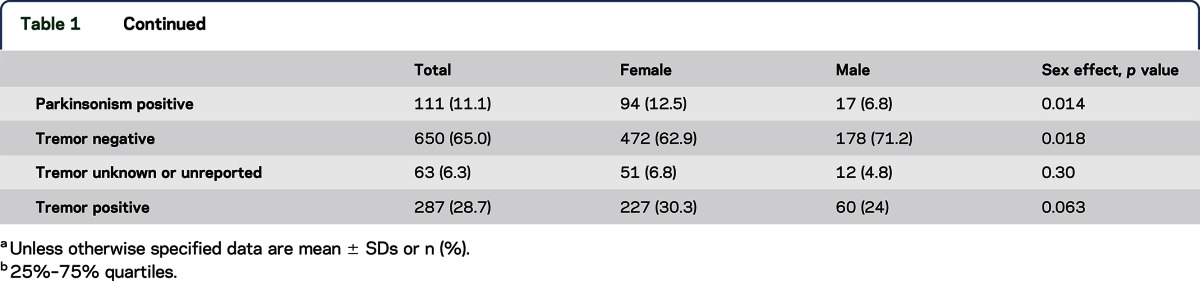
Table 2.
Effect of age at onset and disease duration on distribution and severity of dystonia
The majority of participants reported at least 1 sensory trick (71%), which did not differ between males and females. Psychiatric disorders were reported more commonly by females (35%) than males (23%) and included, in the order of frequency, isolated depression, combined anxiety and depression, and isolated anxiety. Although myoclonus (2%) and parkinsonism (1%) were reported in small numbers of participants, etiologic diagnosis was not part of the study protocol.
A family history of dystonia was reported by 14% of the participants (table 1). Among the participants with a family history, 2 participants reported that both their mother and father were affected, whereas 46 reported an affected mother and 27 reported an affected father. A family history of parkinsonism was reported by 11%. Parkinsonism was reported more commonly by females (13%) than males (7%). Overall, 29% of participants reported a family history of tremor and there was no significant difference between females (30%) and males (24%). The presence or absence of dystonia, parkinsonism, and tremor in family members was not confirmed by face-to-face examinations or telephone interviews.
Twelve coding SVs (tables 3 and 4, figure) predicted to be possibly pathogenic or deleterious by at least 1 of 4 (SIFT, MutationTaster, PolyPhen-2, and CADD) in silico prediction programs were identified in our cohort. In addition, the classic ΔGAG deletion mutation in TOR1A was identified in 1 participant (tables 3 and 4).
Table 3.
Bioinformatic analysis of SVs identified in participants with CD

Table 4.
Clinical features of probands harboring missense or indel SVs in coding regions
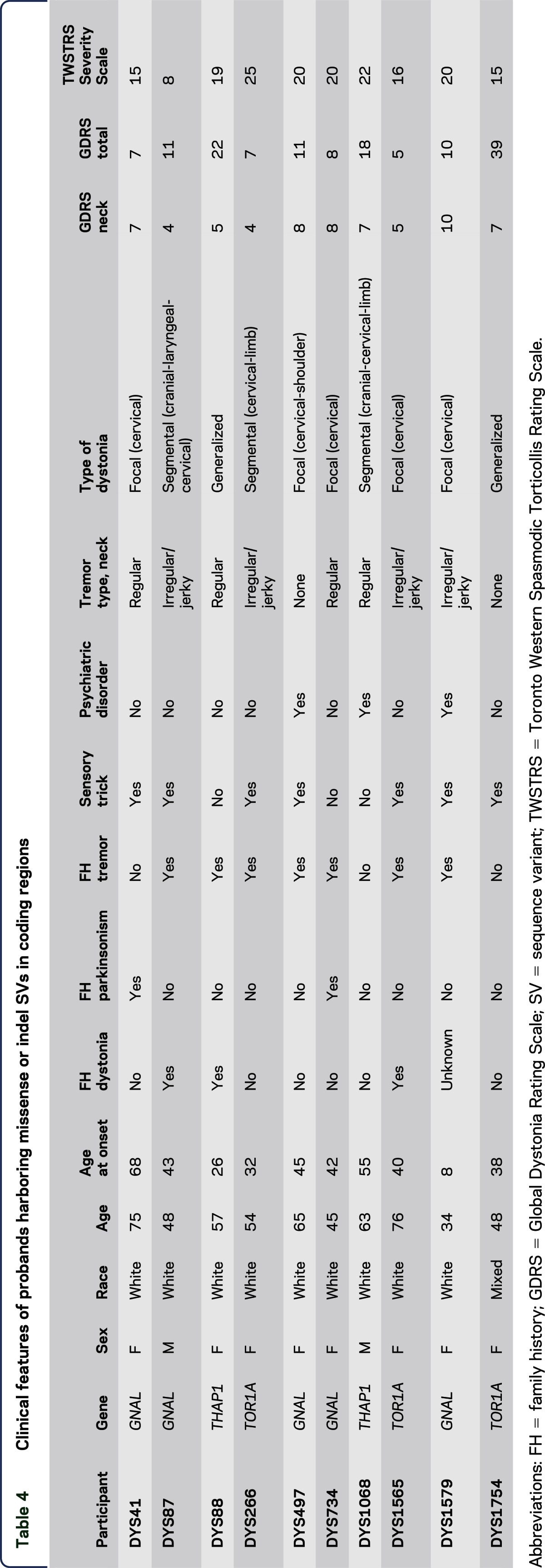
Figure. Pedigrees of probands with missense sequence variants in GNAL, THAP1, and TOR1A.

Arrows = probands; filled symbols = affected; half-filled symbols = possibly affected.
A definite family history of CD was reported by a single proband with a GNAL SV (DYS87, p.V354M). The mother of DYS87 (figure, II-A) had CD with age at onset of 43 years. Family history was inconclusive in another GNAL pedigree (DYS497). The mother of DYS497 (figure, I-A) had a low-frequency right-hand tremor with onset in her late 60s, approximately 5 years after an apparent episode of tardive dyskinesias due to neuroleptic medication treatment of a psychiatric disorder. Reportedly, the tremor did not improve with medical treatment. Gait was shuffling and hand writing was tremulous but not micrographic. Participant I-A from pedigree DYS497 was never formally diagnosed with drug-induced parkinsonism, Parkinson disease, or SWEDDs (scans without evidence of dopaminergic deficit).
Family histories of tremor were reported by 2 probands with TOR1A missense SVs (figure). The father of DYS266 had a late adult-onset tremor of historically unreliable anatomical distribution. The brother of DYS1565 was treated with deep brain stimulation for severe head and arm tremors. Two sisters (II-B and II-E) also had head tremors. Of note, participants I-A, II-B, and II-D had onset of tremor in their 40s and all 3 died of cancer, whereas participant II-E died after an automobile accident.
Among coding SVs, the THAP1 p.S51R SV had the highest CADD Phred score (27.1) and showed relatively high penetrance with multiple affected individuals in the pedigree (figure). The proband's sister (figure, III-D), also enrolled in the DC biorepository, harbors the same p.S51R SV but had initial onset in the distal arm-hand anatomical segment at 32 years of age that has now progressed to generalized dystonia. One of her 2 daughters (IV-F) has generalized dystonia with onset in the distal arm-hand at 7 years of age, and a granddaughter (V-I) has CD and possible lower extremity dystonia with onset at 15 years of age. Participant IV-E had onset of dystonia in the neck and shoulder in her mid-20s with spread to the right distal arm and hand by her early 30s. Participant IV-E is now 43 years old and has generalized dystonia with additional involvement of her left arm and leg. Her daughter, participant V-H, had onset of right-arm dystonia at 13 years of age. Participant III-B has spasmodic dysphonia and participant V-G has dystonia of unknown anatomical distribution.
Synonymous SVs were identified in GNAL (p.R22R, p.A338A) and THAP1 (p.P19P). The GNAL R22R SV was found in ExAC, 1KG, and EVS, and in 6 participants within the DC. GNAL R22R was not enriched in the DC cohort. However, the R22R variant had a CADD Phred score of 22.1 and is predicted to break an ESE site. GNAL A338A was identified in 33 patients in the DC cohort, all heterozygotes. In comparison with ExAC, GNAL A338A was enriched in the DC cohort (χ2 = 13.034, p = 0.0003). Moreover, this variant is predicted to be disease causing by MutationTaster and has a CADD Phred score of 19.2 and may generate a new ESS site. THAP1 P19P is categorized as disease causing by MutationTaster and was found in 1 participant in the DC cohort.
Noncoding SVs were identified in GNAL and THAP1. A c.218+19C>A SV in GNAL is predicted to be marginally deleterious by CADD but is found at high frequency within the ExAC database. The c.932-7T>G SV in GNAL is frequent in the DC, ExAC, and 1KG databases and benign (table 3). None of the coding or noncoding variants detected in this study are predicted to disrupt miRNA-binding domains.
Follow-up screening of TOR1A Exon 5 identified 3 patients with the classic DYT1 ΔGAG deletion (table e-1).7,8 In addition, 1 participant (DYS_85085, figure) was found to have the same c.962C>T (p.T321M) SV as DC DYS1565. DYS_85085 was initially evaluated at 53 years of age for mild gait dysfunction and found to have mild spasticity with bilateral Babinski responses and hyperreflexia of undetermined etiology. Despite mild foot drop, she was able to walk unaided at 66 years of age. At 59 years of age, she developed abnormal head posturing with subsequent spread of dystonia to her arms, worse in the left arm than in the right. On the most recent examination, she had CD with an irregular/jerky head tremor, along with bilateral arm tremors. Her deceased brother and father had a similar gait abnormality. DNA acquired from an alive and neurologically normal brother did not harbor the c.962C>T SV. The proband's deceased mother and maternal grandmother had jerky head tremors but normal gait.
DISCUSSION
We characterized the clinical features of CD and contributions of known isolated dystonia-associated genes to the etiopathogenesis of CD and established a platform for future clinical and genetic studies of dystonia. The demographics of our cohort are compatible with the previous epidemiologic studies of CD and other forms of dystonia.24 In particular, CD is more common in females than in males and, apparently, less common in African Americans than in Caucasians.1,25 Women with CD were more likely than men to exhibit a head tremor, develop laryngeal dystonia, and report a history of at least the onset of a psychiatric disorder. However, the effect of sex on the frequencies of psychiatric conditions should be interpreted with caution given that we did not use a control cohort for the nongenetic aspects of our study.
As suggested by our data set, the boundaries between focal and segmental dystonia and definition of CD are often imprecise.26 In the neck, for example, the trapezius and levator scapulae muscles insert on the scapula and cross the neck and shoulder anatomical regions. In our study, only 47% of participants were deemed to exhibit isolated neck involvement, whereas 69% were reported to manifest dystonia in the neck and shoulder(s). The current classification of dystonia states that CD is “considered a form of focal dystonia, although by convention the shoulder can be included as well as the neck.”6
Small percentages of participants were reported to have had myoclonus (2%) or parkinsonism (1%). Because of the limiting quantities of DNA available for this study, participants with myoclonus were not screened for mutations in SGCE, KCTD17, RELN, or CACNA1B. In addition, neurophysiologic studies were not performed to differentiate myoclonic from dystonic jerks and we did not formally diagnose the cause of parkinsonism in this small group of 10 patients or differentiate dystonic from nondystonic causes of SWEDDs.27
In our cohort, a considerable majority of participants had a head tremor. The presence of tremor in dystonia and dystonia in patients with various forms of tremor including essential tremor or “isolated tremor” has received increasing attention in the recent medical literature.28,29 The presence of either dystonia or isolated tremor in individuals within some pedigrees suggests that subtypes of isolated tremor may exist and share genetic etiologies with some forms of dystonia.30 Of note, the high family history of tremor (29%) in our cohort exceeded the combined family histories of parkinsonism and dystonia.
A role for genetic factors in the etiopathogenesis of CD has long been recognized.31,32 In a review of 266 patients with isolated CD from a single center, 12% had a positive family history of dystonia.32 Similar percentages of participants with first- or second-degree relatives with dystonia have been reported in large genetic screening studies of CD and other isolated dystonia cohorts.3,4,10 Therefore, the 14% of CD participants with a positive family history of dystonia is in line with the previous works.
A small but noteworthy percentage (11%) of participants in our CD cohort reported a positive family history of parkinsonism. In this regard, postmortem pathologic studies and animal models of dystonia suggest that mild damage to the dopaminergic nigrostriatal pathway may contribute to the pathogenesis of dystonia.33,34 However, isolated dystonia has not been shown to be a risk factor for Parkinson disease and vice versa, and family members were not accessible for neurologic examinations to diagnose SWEDDs, Parkinson disease, or other parkinsonian disorders.
Generally, the genetic contributions to CD identified in this study were small; patients with pathogenic or likely pathogenic missense SVs tended to show evidence of spread to contiguous anatomical segments over time, and among those genes interrogated, GNAL had the largest impact (tables 3 and 4). We identified several pathogenic or likely pathogenic GNAL missense variants in our cohort.23 Likely due to the focus on CD, the frequency of missense variants in GNAL reported herein was modestly higher than that reported in other screening studies.5,35 Perhaps most importantly, a synonymous variant of uncertain significance (p.A338A) was enriched in our CD cohort in comparison with the population controls contained in the ExAC database. Accordingly, A338A may increase the risk of developing CD.
Not unexpectedly, SVs in THAP1 were infrequent in our CD cohort. In previous screening studies, pathogenic or likely pathogenic THAP1 missense SVs were found in less than 1% of the patients with mainly adult-onset isolated dystonia.10,11 The synonymous THAP1 variant (p.P19P) identified in 1 participant in our cohort was previously reported in a patient with segmental craniocervical dystonia.10 The c.71+9C>A intronic variant was found in 1 participant in this study and previously reported as enriched in patients with dystonia in comparison with neurologically normal controls.36
Notably, our screening efforts exposed a non-ΔGAG SV in TOR1A (c.962C>T, p.T321M) in 2 pedigrees from different regions of the United States. Family history information acquired from the probands suggested that these pedigrees were unrelated. We also identified another SV in TOR1A (c.823A>G, p.K275E) of uncertain significance. Other TOR1A variants of uncertain significance have been described in single individuals with dystonia.37,38 Both p.K275E and p.T321M are contained at very low frequencies within the ExAC database, and all 3 probands harboring these TOR1A SVs reported a positive family history of dystonia. Studies of p.K275E and p.T321M torsinA in cellular systems and animal models should provide important and novel insights into the biology of dystonia. The classic TOR1A ΔGAG mutation is infrequent (2/1,319 patients = 0.15%) in participants with dystonia onset in the neck.
Given that our screenings were limited to THAP1, GNAL, and Exon 5 of TOR1A, the potential roles of CIZ1, ANO3, and Exons 1, 2, 3, and 4 of TOR1A in the pathogenesis of CD remain uncertain. In addition, we did not interrogate promoter and deep intronic regions for variants of possible pathogenic significance and cannot exclude the possibility that some participants in our cohort harbored large deletion mutations in these or other dystonia-associated genes. Conversely, those missense and noncoding variants reported herein were not subjected to cosegregation analyses or downstream molecular and cellular assays of deleteriousness.
Our data set provides a comprehensive presentation of CD phenotypes and rational overview of CD genetics. The majority of patients with CD have a head tremor and an important percentage exhibit spread to contiguous anatomical regions, have personal histories of psychiatric disorders, and have family histories of tremor and dystonia. Although coding and noncoding SVs in GNAL, THAP1, and TOR1A contribute to the development of CD, the yield of clinical genetic testing for pathogenic SVs in this collection of genes will be low in sporadic cases of CD, particularly in those participants without any evidence of spread to other anatomical segments, but may be considered in individuals from multiplex pedigrees with dystonia. The prevalence of pathogenic and likely pathogenic SVs in dystonia-associated genes reported herein should be considered a minimum because we did not exclude copy number variants or include GCH1, SGCE, KCTD17, RELN, CACNA1B, HPCA, and TAF1 in our screenings. Clinical genetic testing of TOR1A in patients with dystonia should not be limited to discovery of the classic ΔGAG mutation given that other TOR1A SVs may be pathogenic. Future studies into the molecular and cellular biology of CD should benefit from the identification of other genetic etiologies and must consider potential biological commonalities with Parkinson disease, isolated tremor, and psychiatric disorders, principally depression and anxiety.
Supplementary Material
GLOSSARY
- 1KG
1000 Genomes Project
- CADD
Combined Annotation-Dependent Depletion
- CD
cervical dystonia
- DC
Dystonia Coalition
- ESE
exonic splicing enhancer
- ESS
exonic splicing silencer
- EVS
Exome Variant Server
- ExAC
Exome Aggregation Consortium
- GDRS
Global Dystonia Rating Scale
- HRM
high-resolution melting
- miRNA
microRNA
- PolyPhen-2
Polymorphism Phenotyping version 2
- SV
sequence variant
- SWEDDs
scans without evidence of dopaminergic deficit
- UTHSC
University of Tennessee Health Science Center
Footnotes
Supplemental data at Neurology.org/ng
Contributor Information
Collaborators: Dystonia Coalition Investigators, Dystonia Genetic Consortium, Laura Marsh, Ludy Shih, Susan Bressman, Stewart Factor, Marie Vidailhet, Emmanuel Roze, Zoltan Mari, Charles Adler, Mark Hallett, Vesper F.M. L. Ramos, Stephen Grill, Christopher Goetz, Tanya Harlow, Susan Fox, David Standaert, Oksana Suchowersky, Fatta Nahab, Tao Xie, Alberto Espay, Michael Okun, Pedro Gonzalez-Alegre, Ergun Uc, Christine Klein, Sarah Pirio Richardson, Alfredo Berardelli, Giovanni Fabbrini, William Ondo, Claudia Testa, Alison Brashear, Ronald F. Pfeiffer, Monika Rudzińska, Zbigniew K. Wszolek, Ryan J. Uitti, Jay A. Van Gerpen, and Robert W. Bastian
AUTHOR CONTRIBUTIONS
Mark LeDoux conceived the study, performed all statistical analyses, and wrote the first draft. Satya Vemula contributed to generation of supplementary materials, tables, and figures. Joel Perlmutter and H.A. Jinnah managed activities of the Dystonia Coalition and supervised clinical data collection for this study. Satya Vemula, Jianfeng Xiao, and Mark LeDoux performed all the genetic and related bioinformatic analyses. Laura Wright, Ami Rosen, and Misty Thompson contributed to the collection and organization of clinical data. All authors contributed to data collection, data analysis, drafting, and/or revising the manuscript critically for important intellectual content, and approved the final version of the manuscript. All collaborators in the Dystonia Coalition and Dystonia Genetic Consortium contributed to data collection.
STUDY FUNDING
This study was supported by grants from the NIH (R01 NS069936, R01 NS082296, and Dystonia Coalition U54 NS065701) and the Dorothy/Daniel Gerwin Parkinson's Research Fund.
DISCLOSURE
Dr. LeDoux has served on the scientific advisory board of the National Spasmodic Torticollis Association and the Spastic Paraplegia Foundation; has received travel/speaker honoraria from Teva and Lundbeck; has served on the speakers' bureaus for Lundbeck, Teva Neuroscience, and UCB Pharma; is an employee of the University of Tennessee Health Science Center; has served as a consultant for Teva Neuroscience, US WorldMeds, Lundbeck, and the Mayo Clinic; has received research support from the NIH, NIH/National Institute of Neurological Disorders and Stroke, Auspex, Omeros, Teva, Accorda, US WorldMeds, Prana Biotechnology, the Benign Essential Blepharospasm Research Foundation, and CHDI; and has received royalty payments from Elsevier. Dr. Vemula, Dr. Xiao, and Dr. Thompson report no disclosures. Dr. Perlmutter has served on the scientific advisory boards of the American Parkinson Disease Association, the Dystonia Medical Research Foundation, the Greater St. Louis Chapter of the APDA, and ENROLL; has received travel/speaker honoraria from the University of Louisville, the Toronto Western Hospital, the University of Maryland, the Weil Cornell School of Medicine, the University of Saskatoon, Movement Disorders Society, the Society of Nuclear Medicine, the Rhode Island General Hospital, the Medical University of South Carolina, St. Lukes Hospital in St. Louis County, Emory University, the American Academy of Neurology, the Parkinson Disease Foundation, Columbia University, the Indiana Neurological Society, Beth Israel Hospital (Harvard Medical School), the University of Missouri (Columbia), the American Parkinson Disease Association, St. Louis University, and the University of Michigan; has served on the editorial boards of Neurology and Movement Disorders Journal; has been a consultant for Alberta Innovates, Pennsylvania State University, and NIH/National Institute of Neurological Disorders and Stroke; has been a part of the Parkinson Disease Study Group and Huntington Study Group; has received partial fellowship support from Medtronic Inc.; has received research support from Medtronic Inc., Express Scripts, NIH, the University of Toronto, the Huntington Disease Society of American Center of Excellence, the Bander Fund for Medical Business Ethics, the Fixel Foundation, the Michael J. Fox Foundation, CHDI, the McDonnell Center for Higher Brain Function, the Greater St. Louis Chapter of the American Parkinson Disease Association, and the American Parkinson Disease Association; and has been involved in legal proceedings regarding a patient's work-related injury. Dr. Wright reports no disclosures. Dr. Jinnah has served on the scientific advisory board of Medtronic Inc.; has received travel/speaker or honoraria from the Dystonia Medical Research Foundation and the Movement Disorders Society; has been a consultant for Medtronic Inc.; has performed botulinum toxin injections as a clinical service; and has received research support from Psyadon Pharmaceuticals, Merz Pharmaceuticals, IPSEN Pharmaceuticals, National Institute of Neurological Disorders and Stroke, Emory Neurosciences Initiative, Atlanta Clinical and Translational Institute, Emory University Research Council, the Dystonia Medical Research Foundation, the Lesch-Nyhan Syndrome Children's Research Foundation, Cure Dystonia Now, the Dystonia Coalition, and the Benign Essential Blepharospasm Research Foundation. Dr. Rosen reports no disclosures. Dr. Hedera has served on the editorial boards of the Journal of Parkinsonism and Restless Legs Syndrome, Neurology, and Parkinsonism and Related Disorders. Dr. Comella has served as a consultant for Acorda Therapeutics, Allergan Inc., Impax Pharmaceuticals, Ipsen Biopharmaceuticals, Lundbeck, Medtronic Inc., Merz Pharmaceuticals, Acadia Pharmaceuticals, Teva Neurosciences, Neurocrine Biosciences, Revance Therapeutic, and Ultragenyx Pharmaceuticals; has served on the editorial boards of Sleep Medicine and Clinical Neuropharmacology; has received research support from the NIH (R01 NS074343 and U54NS06570), Dystonia Medical Research Foundation, Parkinson's Disease Foundation, Allergan, Ipsen Biopharmaceuticals, Biotie, Revance, and Merz Pharmaceuticals; and has received royalties from Cambridge, Humana Press, and Wolters Kluwer. Dr. Weissbach and Dr. Junker report no disclosures. Dr. Jankovic has served on the scientific advisory board of Adamas Pharmaceuticals, Allergan, and Teva Pharmaceutical Industries Ltd; has received gifts and travel/speaker honoraria from Adamas Pharmaceuticals, Allergan, and Teva Pharmaceutical Industries Ltd; has served on the editorial boards of Acta Neurologica Scandinavica, the Journal of the Neurological Sciences, Neurology Medlink, Neurotherapeutics, Expert Review of Neurotherapeutics, the Journal of Parkinson's Disease, and Toxins; has received royalties from Cambridge, Elsevier, Future Science Group, Hodder Arnold, Lippincott Williams and Wilkins, and Wiley-Blackwell; and has received research support from Adamas Pharmaceuticals, Allergan, CHDI Foundation, Civitas/Acorda Therapeutics, Huntington Study Group, Ipsen Ltd, Kyowa Haako Kirin Pharma Inc., Lundbeck Inc, Medtronic, Merz Pharmaceuticals, the Michael J. Fox Foundation for Parkinson Research, NIH, the National Parkinson Foundation, Parkinson Study Group, Pfizer, Prothena Biosciences Inc, Psyadon Pharmaceuticals Inc., St. Jude Medical, and Teva Pharmaceutical Industries Ltd. Dr. Barbano has served on the scientific advisory board for Allergan; has served on the editorial board of Neurology Clinical Practice; has performed botulinum toxin injections at the University of Rochester; has received educational support from Allergan; has received research support from the Dystonia Coalition, Allergan, Vaccinex, Biotie, NIH, National Institute of Neurological Disorders and Stroke, ORDR, and Merz; holds stock/stock options in VisualDx; and has acted as an expert witness in legal proceedings including malpractice but not involving commercial entities. Dr. Reich has served on the editorial boards of Journal of Clinical Movement Disorders and Parkinsonism and Related Movement Disorders; has received royalties from Informa; and has received research support from National Institute of Neurological Disorders and Stroke. Dr. Rodriguez has served on the scientific advisory board of Teva Pharmaceuticals Ltd, Auspex Pharmaceuticals, Lundbeck, and Abbvie and has served on the speakers' bureaus of Abbvie and Allergan. Dr. Berman has served on the scientific advisory board of the Benign Essential Blepharospasm Research Foundation and the National Spasmodic Torticollis Association; has received travel/speaker honoraria from the Parkinson Study Group, the American Neurological Association, the Movement Disorder Society, the Dystonia Medical Research Foundation, and the Benign Essential Blepharospasm Research Foundation; has served on the editorial board of the Journal of Neurology and Neurophysiology; and has received research support from NIH, the Colorado Translational Research Imaging Center, the University of Colorado Center for Neuroscience, the Dana Foundation, the Benign Essential Blepharospasm Research Foundation Inc., and the Dystonia Medical Research Foundation. Dr. Chouinard has received travel/speaker honoraria from Allergan and Merz. Dr. Severt has served as a consultant for Allergan; has served on a scientific advisory board for Lundbeck; has received travel/speaker honoraria from Teva, Lundbeck, Allergan, and Impax; has served on the speakers' bureaus of Teva, Allergan, Lundbeck, and Impax; and has received research support from US Worldmeds. Dr. Agarwal has received grant support from TEVA, Lundbeck, US WorldMeds, Auspex, Amarantus, Adamas, Impax, Omeros, and CHDI; has received personal fees from Teva, Lundbeck, and US WorldMeds; has been a consultant for Cynapsus, US Worldmeds, UCB, Impax, and Lundbeck; has served on the speakers' bureaus of Teva, UCB, US Worldmeds, Impax, and Lundbeck; has received research support from Pfizer, US Worldmeds, PSG, HSG, Teva, Abbvie, Merck, Astellas, NIH, the Parkinson's support group, the Dystonia support group, and the Huntington's support group; and (spouse) holds stock/stock options in Exilexis, Pharmaathens, Atossa Genetics, Pain Therapeutics, Antares, MEI Pharma, Marina Biotech, and RXi Pharmaceuticals. Dr. Stover reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Defazio G, Jankovic J, Giel JL, Papapetropoulos S. Descriptive epidemiology of cervical dystonia. Tremor Other Hyperkinet Mov (NY) 2013;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Defazio G, Abbruzzese G, Livrea P, Berardelli A. Epidemiology of primary dystonia. Lancet Neurol 2004;3:673–678. [DOI] [PubMed] [Google Scholar]
- 3.Xiao J, Uitti RJ, Zhao Y, et al. Mutations in CIZ1 cause adult onset primary cervical dystonia. Ann Neurol 2012;71:458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao J, Zhao Y, Bastian RW, et al. The c.-237_236GA>TT THAP1 sequence variant does not increase risk for primary dystonia. Mov Disord 2011;26:549–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vemula SR, Puschmann A, Xiao J, et al. Role of Galpha(olf) in familial and sporadic adult-onset primary dystonia. Hum Mol Genet 2013;22:2510–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozelius LJ, Hewett JW, Page CE, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 1997;17:40–48. [DOI] [PubMed] [Google Scholar]
- 8.Xiao J, Bastian RW, Perlmutter JS, et al. High-throughput mutational analysis of TOR1A in primary dystonia. BMC Med Genet 2009;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuchs T, Gavarini S, Saunders-Pullman R, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet 2009;41:286–288. [DOI] [PubMed] [Google Scholar]
- 10.Xiao J, Zhao Y, Bastian RW, et al. Novel THAP1 sequence variants in primary dystonia. Neurology 2010;74:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeDoux MS, Xiao J, Rudzinska M, et al. Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat Disord 2012;18:414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchs T, Saunders-Pullman R, Masuho I, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet 2013;45:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dufke C, Hauser AK, Sturm M, et al. Mutations in CIZ1 are not a major cause for dystonia in Germany. Mov Disord 2015;30:740–743. [DOI] [PubMed] [Google Scholar]
- 14.Charlesworth G, Plagnol V, Holmstrom KM, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet 2012;91:1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao J, Vemula SR, LeDoux MS. Recent advances in the genetics of dystonia. Curr Neurol Neurosci Rep 2014;14:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LeDoux MS. The genetics of dystonias. Adv Genet 2012;79:35–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 2001;11:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575–576. [DOI] [PubMed] [Google Scholar]
- 21.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol 2011;18:1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jankovic J, Tsui J, Bergeron C. Prevalence of cervical dystonia and spasmodic torticollis in the United States general population. Parkinsonism Relat Disord 2007;13:411–416. [DOI] [PubMed] [Google Scholar]
- 25.Marras C, Van den Eeden SK, Fross RD, et al. Minimum incidence of primary cervical dystonia in a multiethnic health care population. Neurology 2007;69:676–680. [DOI] [PubMed] [Google Scholar]
- 26.LeDoux MS. Meige syndrome: what's in a name? Parkinsonism Relat Disord 2009;15:483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erro R, Schneider SA, Stamelou M, Quinn NP, Bhatia KP. What do patients with scans without evidence of dopaminergic deficit (SWEDD) have? New evidence and continuing controversies. J Neurol Neurosurg Psychiatry 2016;87:319–323. [DOI] [PubMed] [Google Scholar]
- 28.Defazio G, Conte A, Gigante AF, Fabbrini G, Berardelli A. Is tremor in dystonia a phenotypic feature of dystonia? Neurology 2015;84:1053–1059. [DOI] [PubMed] [Google Scholar]
- 29.Shaikh AG, Zee DS, Jinnah HA. Oscillatory head movements in cervical dystonia: dystonia, tremor, or both? Mov Disord 2015;30:834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedera P, Phibbs FT, Fang JY, Cooper MK, Charles PD, Davis TL. Clustering of dystonia in some pedigrees with autosomal dominant essential tremor suggests the existence of a distinct subtype of essential tremor. BMC Neurol 2010;10:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duane DD. Spasmodic torticollis: clinical and biologic features and their implications for focal dystonia. Adv Neurol 1988;50:473–492. [PubMed] [Google Scholar]
- 32.Chan J, Brin MF, Fahn S. Idiopathic cervical dystonia: clinical characteristics. Mov Disord 1991;6:119–126. [DOI] [PubMed] [Google Scholar]
- 33.Iacono D, Geraci-Erck M, Peng H, Rabin ML, Kurlan R. Reduced number of pigmented neurons in the substantia nigra of dystonia patients? Findings from extensive neuropathologic, immunohistochemistry, and quantitative analyses. Tremor Other Hyperkinet Mov (NY) 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neychev VK, Gross RE, Lehericy S, Hess EJ, Jinnah HA. The functional neuroanatomy of dystonia. Neurobiol Dis 2011;42:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma LY, Wang L, Yang YM, Feng T, Wan XH. Mutations in ANO3 and GNAL gene in thirty-three isolated dystonia families. Mov Disord 2015;30:743–744. [DOI] [PubMed] [Google Scholar]
- 36.Vemula SR, Xiao J, Zhao Y, et al. A rare sequence variant in intron 1 of THAP1 is associated with primary dystonia. Mol Genet Genomic Med 2014;2:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calakos N, Patel VD, Gottron M, et al. Functional evidence implicating a novel TOR1A mutation in idiopathic, late-onset focal dystonia. J Med Genet 2010;47:646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zirn B, Grundmann K, Huppke P, et al. Novel TOR1A mutation p.Arg288Gln in early-onset dystonia (DYT1). J Neurol Neurosurg Psychiatry 2008;79:1327–1330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.