

Abstract
Background and Purpose
Activation of glucagon‐like peptide‐1 (GLP‐1) receptor exerts a range of cardioprotective effects. Geniposide is an agonist of GLP‐1 receptor, but its role in cardiac hypertrophy remains completely unknown. Here, we have investigated its protective effects and clarified the underlying molecular mechanisms.
Experimental Approach
The transverse aorta was constricted in C57/B6 mice and then geniposide was given orally for 7 weeks. Morphological changes, echocardiographic parameters, histological analyses and hypertrophic markers were used to evaluate hypertrophy.
Key Results
Geniposide inhibited the hypertrophic response induced by constriction of the transverse aorta or by isoprenaline. Activation of 5′‐AMP‐activated protein kinase‐α (AMPKα) and inhibition of mammalian target of rapamycin, ERK and endoplasmic reticulum stress were observed in hypertrophic hearts that were treated with geniposide. Furthermore, Compound C (CpC) or knock‐down of AMPKα restricted protection of geniposide against cell hypertrophy and activation of mammalian target of rapamycin and ERK induced by hypertrophic stimuli. CpC or shAMPKα also abolished the protection of geniposide against endoplasmic reticulum stress induced by thapsigargin or dihtiothreitol. The cardio‐protective effects of geniposide were ablated in mice subjected to CpC. GLP‐1receptor blockade counteracted the anti‐hypertrophic response and activation of AMPKα by geniposide. Knock‐down of GLP‐1 receptor also offset the inhibitory effects of geniposide on cardiac hypertrophy in vivo.
Conclusions and Implications
Geniposide protected against cardiac hypertrophy via activation of the GLP‐1 receptor/AMPKα pathway. Geniposide is a potential therapeutic drug for cardiac hypertrophy.
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide 1‐β‐D‐ribofuranoside
- AMPK
5′‐AMP‐activated protein kinase
- Ang
angiotensin
- ANP
atrial natriuretic peptide
- CpC
Compound C
- CSA
cross‐sectional area
- Ex
Exendin
- FS
fractional shortening
- GE
geniposide
- GLP
glucagon‐like peptide
- HW/BW
heart weight/body weight
- HW/TL
heart weight/tibia length
- LVIDd
left ventricular internal diastolic diameter
- mTOR
mammalian target of rapamycin
- TAC
constriction of the transverse aorta
- TG
thapsigargin
- β‐MHC
β‐myosin heavy chain
Tables of Links
| TARGETS |
|---|
| GPCRs a |
| GLP‐1 receptor |
| Enzymes b |
| Acetyl‐CoA carboxylase |
| AMPKα2 |
| ERK |
| mTOR, mammalian target of rapamycin |
| PERK |
| LIGANDS |
|---|
| Ang II, angiotensin II |
| ANP, atrial natriuretic peptide |
| Ex9‐39, exendin fragment 9‐39 |
| Exendin‐4, exenatide |
| GLP, glucagon‐like peptide |
| Isoprenaline |
| TG, thapsigargin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Cardiac hypertrophy is characterized by enlargement of the heart and is the response of the heart to a variety of stimuli (Tang et al., 2009). It can progress to heart failure, and ultimately leads to high rates of mortality and morbidity (Shah and Mann, 2011). Although considerable progress has been made in understanding the molecular mechanisms underlying hypertrophy, drugs that constrain these pathways are yet to be discovered.
One of the mechanisms that could promote cardiac hypertrophy is endoplasmic reticulum (ER) stress. Once ER stress is activated, branches of the protein response increase ROS and induce apotosis, contributing to the process of cardiac hypertrophy (McCullough et al., 2001; Harding et al., 2003). Therefore, it is of importance to detrermine the regulatory mechanisms of ER stress. 5′‐AMP‐activated protein kinase‐α (AMPKα), an important regulator of cardiac homeostasis known to limit protein synthesis by antagonizing the mammalian target of rapamycin (mTOR) kinase, has been shown to function as a physiological suppressor of ER stress(Inoki et al., 2003; Dong et al., 2010; Inoki et al., 2012). Moreover, activated AMPKα protected against cardiac ischaemia/reperfusion injury via the attenuation of ER stress (Tao et al., 2011). However, whether AMPKα is a pivotal modulator of ER stress in pressure overload‐induced heart disease has hitherto remained unaddressed.
Stimulation of glucagon‐like peptide‐1 (GLP‐1) receptor by a GLP‐1 analogue can activate AMPKα and suppress ER stress (Shao et al., 2015; Yang et al., 2015). Furthermore, there is considerable evidence for an important role of GLP‐1R in cardiovascular actions. An agonist of GLP‐1 receptor, exendin‐4, prevented cardiac remodelling in infarcted myocardium and ameliorated cardiac ischaemia/reperfusion injury (DeNicola et al., 2014; Tsutsumi et al., 2014). Geniposide has been isolated from the gardenia plant and has anti‐inflammatory and neuro‐protective properties (Fu et al., 2012; Lv et al., 2015). A previous study demonstrated that geniposide alleviated hepatic dyslipidemia by inhibiting ER stress in mice (Lee et al., 2013). The most striking finding, however, is that geniposide, although less potent than exenatide, is an agonist of GLP‐1 receptor (Gong et al., 2014).
These observations raised the possibility that geniposide would protect against cardiac hypertrophy. Here, we have shown that mice treated with geniposide reveal a suppressed hypertrophic response in hearts, induced by pressure overload. We also found that geniposide activated a GLP‐1 receptor /AMPKα pathway, and that blockade of the GLP‐1 receptor /AMPKα signalling prevented the anti‐hypertrophic effects of geniposide.
Methods
Animals and treatments
All animal care and experimental procedures complied with the Guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011) and the Guidelines for the Care and Use of Laboratory Animals of the Chinese Animal Welfare Committee and were approved by the Animal Use Committees of our hospital and our Institute. The animal studies follow the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male C57/B6 mice (8‐ to 10‐week‐old; body weight: 25.5 ± 2 g, NO: 0282970) were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). The animals were allowed free access to food and water and were maintained on a 12 h light/dark cycle in a controlled temperature (20–25°C) and humidity (50 ± 5%) environment for a period of 1 week before the study commenced. The transverse aortic constriction (TAC) surgery and subsequent analyses were performed in a blind fashion for all groups. Mice were anaesthetized i.p. using 3% sodium pentobarbital at a dose of 40 mg · kg−1. When mice lost the response to foot squeeze, TAC or a sham procedure was performed according to Jiang et al. (2014). To treat post‐operative pain, temgesic (0.1 mg · kg−1) was applied once daily for 6 days post‐surgery. Mice with TAC were grouped according to a random number table (n = 20 for every group). One week after the TAC or the sham procedure, mice were given geniposide (25 or 50 mg · kg−1) dissolved in sterile saline, intragastrically (09:00 h) for 7 weeks, and animals in the control group were given equal volumes of vehicle. Hypertrophy was also induced by i.p. injections of isoprenaline (50 mg · kg−1, 09:00 h) dissolved in sterile saline for 14 days (Belge et al., 2014), and meanwhile, mice were treated with geniposide (50 mg · kg−1, 09:00 h). In the inhibition experiment, mice (n = 12 for every group) were given repeated i.p. injections of Compound C (CpC; 20 mg · kg−1, 15:00 h, every other day) dissolved in 0.1% DMSO from 2 weeks after surgery to the study endpoint. Doppler analysis, as described previously (Zong et al., 2013) was carried out, without knowledge of the treatment, to confirm that hypertrophy was induced. At the end of the 7 week‐treatment, mice were killed with an overdose of sodium pentobarbital (200 mg · kg−1; i.p.) and the hearts and tibiae were collected to calculate the following ratios: heart weight (HW)/body weight (BW) (mg · g−1), HW/tibia length (TL) (mg · mm−1).
Morphological examination
The cardiac tissues were arrested in 10% KCl, fixed with 10% neutral formalin and then processed by standard histological protocol and stained with haematoxylin and eosin (HE). The sections (5 μm) were observed under light microscopy and photomicrographs were obtained by Photo Imaging System (Nikon (Tokyo, Japan), H550L). The slides were examined by two authors, without knowledge of the treatments. The cross‐sectional areas (CSA) of the myocytes were determined (image ‐pro plus 6.0 (Maryland, USA)). In each group, more than 200 myocytes (10 fields per animal, randomized five cardiomyocytes per field) were counted.
Recombinant adenoviral vectors and infection
To knock down AMPKα2 expression, which is the dominant and the more abundant catalytic subunit of AMPKα in the myocytes (Tian et al., 2001; Kahn et al., 2005), replication‐defective adenoviral vectors under the control of the U6 promoter were used. Three rat shAMPKα2 constructs were obtained from Sigma‐Genosys (Spring, TX, USA), and three Ad‐shAMPKα2 adenoviruses were generated by Vigene Bioscience (Rockville, MD, USA). The one that resulted in the most significant down‐regulation of endogenous AMPKα2 expression was selected for further experiments. Ad‐shRNA was the non‐targeting control. Next, neonatal rat cardiac myocytes (NRCM) were infected with Ad‐shAMPKα2.
Adeno‐associated virus (AAV) and injection
AAV‐shGLP‐1R and scrambled shRNA were previously generated by Vigene Bioscience. AAV9 can also infect pancreatic beta cells; therefore, mice were given an intramyocardial injection of 1 × 1011 viral genome particles of AAV9 in 30 μL PBS in five separate locations in the left ventricle free wall of the heart when the TAC surgery was performed. Eight weeks after injection, the level of mRNA for GLP‐1 receptor was determined.
Cell culture and surface area
H9c2 cardiomyocytes were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were seeded in DMEM (GIBCO (California, USA), C11995) supplemented with 10% FBS (GIBCO, 10099), penicillin (100 U · mL−1) and streptomycin (100 mg · mL−1) (GIBCO, 15140). Only cells below passage 15 were used. Cells were incubated with angiotensin II (Ang II; 1 μM) or isoprenaline (10 μM) for 24 h after starvation with DMEM medium containing 0.5% FBS overnight to induce hypertrophy. Meanwhile, geniposide dissolved in PBS was given. For AMPKα inhibition, H9c2 cells were incubated with CpC (20 μM) dissolved in 0.1% DMSO in the presence or absence of hypertrophic stimuli and geniposide (100 μM) for 24 h. AICAR (1 mM) was used to induce functional AMPKα. Cardiac myocytes were incubated with thapsigargin (TG; 1 μM) or DTT (0.5 mM) for 6 h to induce ER stress. For GLP‐1 receptor inhibition, H9c2 cells were incubated with Ex9‐39 (10 μM) dissolved in PBS in the presence or absence of hypertrophic stimuli and geniposide (100 μM) for 24 h. Neonatal rat cardiomyocytes were isolated as described previously (Li et al., 2005). To assess the CSA of the H9c2 cardiomyocytes, we used α‐actin to stain the cells. Cells were washed with PBS, fixed with 4% formaldehyde, permeabilized in 0.1% Triton X‐100 and stained with anti‐α‐actin at a dilution of 1:100 in 1% goat serum. Alexa Fluor 488‐goat anti‐mouse [Invitrogen (Carlsbad, CA, USA), A11017] was used to combine the first antibody, after which coverslips were mounted onto glass slides with SlowFade Gold antifade reagent with DAPI (Invitrogen, S36939). The slides were examined blind, and the results confirmed independently by two authors. The CSA were determined (image‐pro plus 6.0). In each group, more than 120 myocytes (eight fields per slide, randomized five cardiomyocytes per field) were outlined.
Quantitative real‐time PCR and Western blot analysis
Frozen pulverized left ventricles (50 mg of each sample) were homogenized in 1 mL TRIzol (Invitrogen, 15596‐026) on ice. The total RNA was reverse‐transcribed into cDNA using the Transcriptor First Strand cDNA Synthesis Kit [Roche (Basel, Switzerland), 04896866001]. The reactions were quantified by the LightCycler 480 SYBR Green 1 Master Mix (Roche, 04707516001). See Table S1 for all primer details. RIPA was used to extract the total protein from frozen heart tissues or iced cell lysates. The protein concentrations were detected using the BCA Protein Assay Kit. The proteins were loaded into 10% SDS‐PAGE gels and subsequently electrotransferred to a PVDF membrane (Millipore, IPVH00010). After incubation with primary antibodies overnight, the membrane was stained with IRDye®800CW‐conjugated secondary antibodies for 1 hour. The PVDF membrane containing targeted proteins was scanned using a two‐colour IR imaging system (Odyssey, LI‐COR). All the details of primary antibodies were provided in Table S2. The mRNA levels were normalized to GAPDH. Phosphorylation was quantified to the matched total protein.
Detection of ROS in cardiomyocytes
H9c2 myocytes were cultured in 96‐well plates and pretreated with geniposide and Ang II for the indicated times. ROS were then detected by DCFH‐DA. The cells were incubated with DCFH‐DA (10 μM) for 60 min in 37°C, and immunofluorescence was detected by a fluorescence microplate reader (excitation wavelength/emission wavelength: 485/525 mm).
Data and statistical analysis
These studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are expressed as mean ± SD. The group sizes of the in vivo experiments were estimated based on power analysis of HW/BW with an α error of 5% and a power of 80%, which is consistent with a published article (Puhl et al., 2016). To detect a 10% change in HW/BW with an expected SD of 5%, we needed 5 animals per group. In our study, n = 20 (the protection experiment) and n = 12 (the AMPK inhibition experiment and the GLP‐1 receptor knockdown experiment) obviously fulfilled the requirement. For the animal welfare, the group size in of n = 12 each group was chosen in the AMPK inhibition experiment and the GLP‐1 receptor knockdown experiment. Groups were compared by one‐way anova, followed by post hoc LSD test when anova found a significant value of F and no variance in homogeneity, otherwise, Tamhane's T2 post hoc test. Comparison between two groups was performed using an unpaired Student's t‐test. Statistical analysis of ROS was performed using a repeated measures anova. All in vivo, in vitro and imaging studies were performed blind. Statistical significance was assigned at P < 0.05.
Materials
Geniposide (#24512‐63‐8) was obtained from Shanghai Winherb Medical Science Co. (Shanghai, China, www.sh‐winherb.com/Zproduct.aspx?fn=2&key=geniposide). The purity of geniposide was above 98% determined by HPLC analysis. Isoprenaline (I5627), angiotensin II (Ang II, A9525), Compound C (CpC, P5499), 5‐aminoimidazole‐4‐carboxamide1‐β‐d‐ribofuranoside (AICAR, A9978), thapsigargin (TG, T9033), DTT (D9779), N‐acetyl‐l‐cysteine (NAC, A7250) and exendin fragment 9‐39 (Ex9‐39, E7269) were purchased from Sigma‐Aldrich (St . Louis, MO, USA). Anti‐α‐actin was purchased from Millipore (Massachusetts, USA). We also used IRDye®800CW‐conjugated secondary antibodies [LI‐COR Biosciences (Lincoln, NE, USA), for immunoblotting] in the study. 2, 7‐Dichlorofluorescin diacetate (DCFH‐DA) was obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Proteins were measured with assay kits obtained from Pierce (Pierce, 23225). All other chemicals were of analytical grade.
Results
Geniposide attenuated cardiac hypertrophy in vivo
In the present study, 25 or 50 mg · kg−1 of geniposide was given p.o. according to our preliminary experiments (Figure S1A). After 8 weeks of TAC, the mice had increased heart size, HW/BW, HW/TL and CSA (Figure 1A–D, P < 0.05). Mice with TAC and thus exposed to pressure‐load, also developed a decline in heart function with a decrease in ejection fraction and fractional shortening (FS) and had a significant increase in left ventricular internal diastolic diameter (LVIDd) (Figure S1B–D, P < 0.05). These morphological changes were dose‐dependently inhibited in mice given the treatment with geniposide (Figure S1B–D, P < 0.05). It also showed that 50 mg · kg−1 of geniposide attenuated TAC‐induced increases of atrial natriuretic peptide (ANP) and β‐myosin heavy chain (β‐MHC) (Figure 1E, P < 0.05). We next examined the potential effects of geniposide on hypertrophy induced by repeated i.p. isoprenaline injections, as described previously (Belge et al., 2014), and found that results were similar to those observed after treatment of TAC with geniposide (Figure 1F–H, Figure S1E, P < 0.05).
Figure 1.
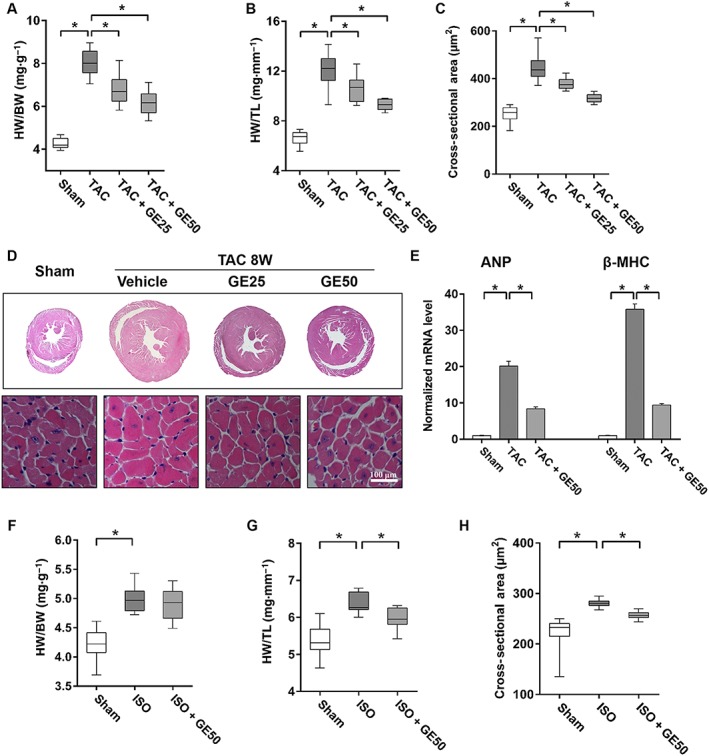
Geniposide (GE) prevented cardiac hypertrophy and dysfunction. (A–B) Summary data of (HW)/(BW) and HW/TL (n = 20). (C) The cross‐sectional areas of myocytes (n = 6). (D) Histology results from cross‐sections of whole hearts with haematoxylin and eosin staining. (E) Expression levels of the transcripts of ANP and β‐MHC (n = 5). (F–G) Statistical results of HW/BW and HW/TL after ISO injections (n = 12). (H) The cross‐sectional areas of myocytes (n = 5). Data shown in A, B, C, and in F,G,H are medians with first and third quartiles and ranges. * P < 0.05.
Geniposide activated AMPKα and inhibited mTOR, ERK and ER stress in hypertrophic heart
Previous studies have shown that geniposide suppressed palmitate‐induced ER stress (Lee et al., 2013). Here, we also found that geniposide (50 mg · kg−1) suppressed ER stress in hypertrophic hearts, characterized by reduced phosphorylated level of protein kinase dsRNA‐dependent‐like ER kinase and decreased expressions of glucose regulated protein 78 (GRP78), X‐box binding protein 1 (XBP‐1) and activating transcription factor 6 (ATF6) (Figure 2A, P < 0.05). Phosphorylated AMPKα and acetyl‐CoA carboxylase (ACC), a substrate of AMPKα, were elevated in mice given 50 mg · kg−1 geniposide compared with those in the TAC group without geniposide (Figure 2B, P < 0.05). Phosphorylation of mTOR and ERK induced by TAC, which are known as other downstream targets of AMPKα in its anti‐hypertrophic action (Chan et al., 2004; Li et al., 2007; Dolinsky et al., 2009), were also inhibited by geniposide (Figure 2B, P < 0.05). ER stress induced XBP‐1 mRNA splicing to generate a potent transcription factor XBP1s; therefore, the XBP‐1 assay was performed. As illustrated by Figure S2, TAC induced the production of XBP‐1 s, and geniposide treatment could attenuate the level of XBP‐1 s.
Figure 2.
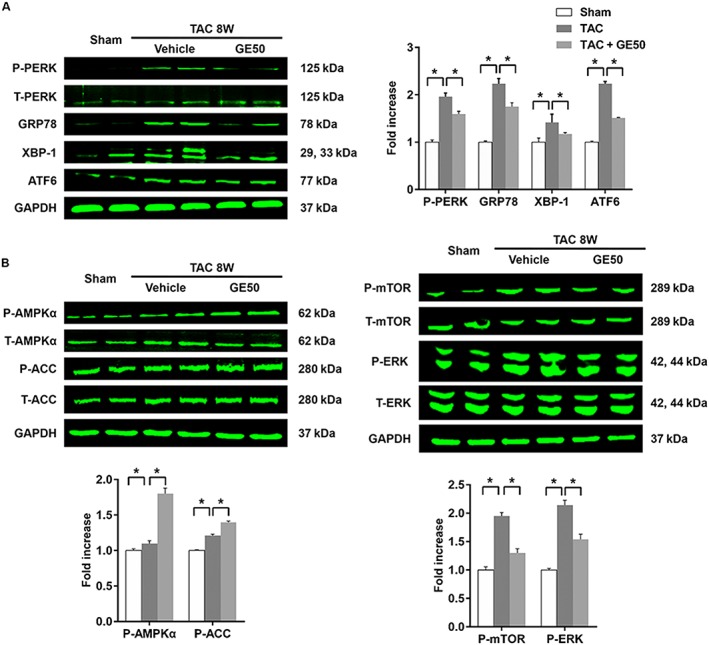
The effects of geniposide GE (50 mg · kg−1 for 7 weeks) on AMPKα and pro‐hypertrophic pathways. (A) The protein levels of phosphorylated protein kinase dsRNA‐dependent‐like ER kinase (PERK) and decreased expressions of glucose regulated protein 78 (GRP78), X‐box binding protein 1 (XBP‐1) and activating transcription factor 6 (ATF6) (n = 6). (B) The protein levels of phosphorylated AMPKα and the related targets (n = 6). * P < 0.05.
Geniposide protected against hypertrophy in an AMPKα‐dependent manner in vitro
Considering the protective role of AMPKα in cardiovascular diseases, we further confirmed the hypothesis that the protection of geniposide against hypertrophy was mediated by the activation of AMPKα. H9c2 cells treated with escalating doses of geniposide exhibited viability that was not statistically distinguishable from controls treated only with PBS (Figure S3A, P < 0.05). Geniposide also blocked Ang II (1 μM)‐induced hypertrophic response in a dose‐dependent manner, characterized by decreased ANP and CSA (Figure 3A–D, P < 0.05). In view of the activation of AMPKα in hypertrophy induced by geniposide (100 μM) (Figure S3E, P < 0.05), we incubated H9c2 cells with the AMPKα antagonist CpC and found that the effects of geniposide were blocked by CpC (Figure 3B–D, Figure S3E, P < 0.05). Given that CpC may have other actions, we knocked down AMPKα2 in neonatal rat myocytes (Viollet et al., 2010). As shown in Figure S3B, mRNA of AMPKα2 decreased after adenoviral infection, without a compensatory increase in AMPKα1. Knock‐down of AMPKα completely offset the protective effects of geniposide in neonatal rat myocytes (Figure S3C, Figure 3E–F, P < 0.05). Additionally, the synergistic effect of geniposide and AICAR, an AMPKα agonist, on Ang II‐induced hypertrophic response in H9c2 cells was also confirmed (Figure S3D, Figure 3G–H, P < 0.05). Considering that geniposide attenuated isoprenaline‐induced cardiac hypertrophy in vivo in our study and AMPKα was also implicated in isoprenaline‐induced hypertrophy (Zarrinpashneh et al., 2008), H9c2 cells were also incubated with isoprenaline (10 μM). Geniposide protected against isoprenaline‐induced cell hypertrophy in a dose‐dependent manner and, conversely, CpC co‐incubation with geniposide totally blocked these protective effects (Figure S4A–D, P < 0.05).
Figure 3.
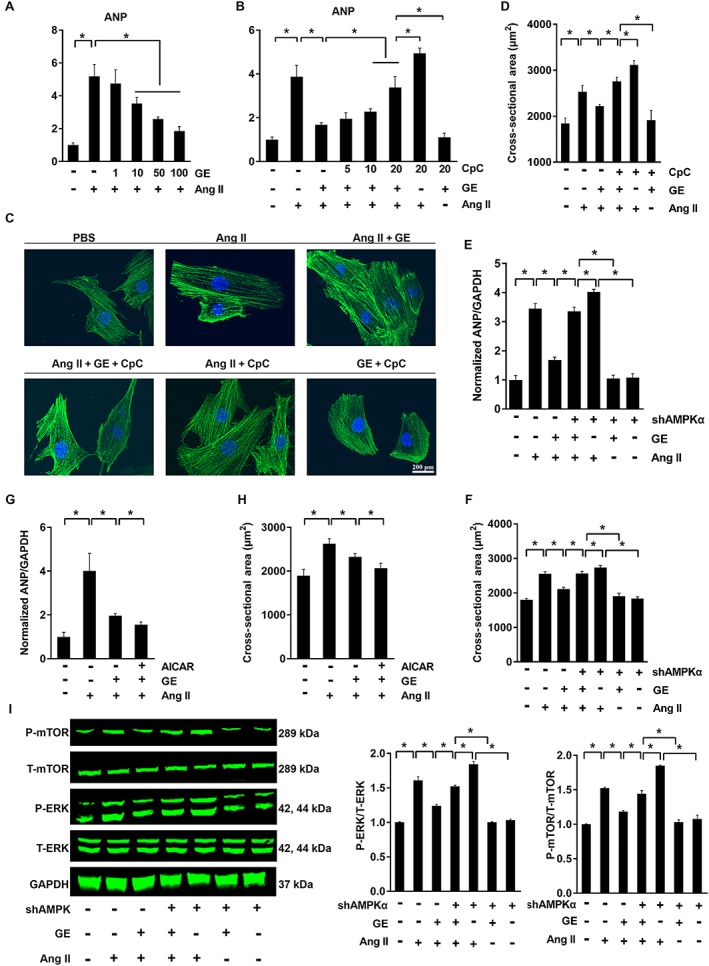
The effects of geniposide (GE; 100 μM for 24 h) on hypertrophy induced by Ang II (1 μM for 24 h) were blocked by CpC or shAMPKα in vitro. (A) The mRNA levels of ANP in H9c2 cells (n = 6). (B) CpC (5–20 μM for 24 h) reversed protection by geniposide in H9c2 cells (n = 6). (C–D) The immunofluorescence and CSA of H9c2 cells (n = 6). (E–F) ANP (n = 5) and CSA (n = 6) in neonatal rat cardiomyocytes after adenovirus infection. (G–H) ANP and CSA in group treated with AICAR in H9c2 cells (1 mmol · L−1 for 24 h) (n = 6). (I) Protein changes after infection of adenovirus in neonatal rat cardiomyocytes (n = 6). * P < 0.05.
Geniposide suppressed mTOR, ERK pathways and ER stress in an AMPK‐dependent manner in vitro
Our study also found that increased phosphorylation of mTOR and ERK induced by Ang II were attenuated after geniposide treatment in cardiomyocytes and that the actions of geniposide against phospho‐mTOR and phospho‐ERK were abolished by CpC or knock‐down in vitro, each of which could inhibit functional AMPKα (Figure S3E, Figure 3I, P < 0.05). We also confirmed our hypothesis that the inhibition of ER stress was mediated by the activation of AMPKα. The increased levels of GRP78 and XBP‐1 were reduced by geniposide, and CpC or shAMPKα blunted the protection of geniposide against the ER stress response (Figure 4A–B, G; Figure S4E, P < 0.05). Conversely, the combination of geniposide and AICAR further decreased the levels of ER stress (Figure 4C–D, P < 0.05). To further substantiate the effects of AMPKα on ER stress, we incubated cardiomyocytes with TG or DTT, which are potent inducers of ER stress. As expected, geniposide treatment reduced ER stress induced by TG or DTT and lost its inhibitory effects after CpC or shAMPKα treatment (Figure 4E–F, H, P < 0.05). Geniposide treatment also reduced the production of XBP‐1 s and this effect was lost after knock‐down of AMPKα (Figure S4F).
Figure 4.
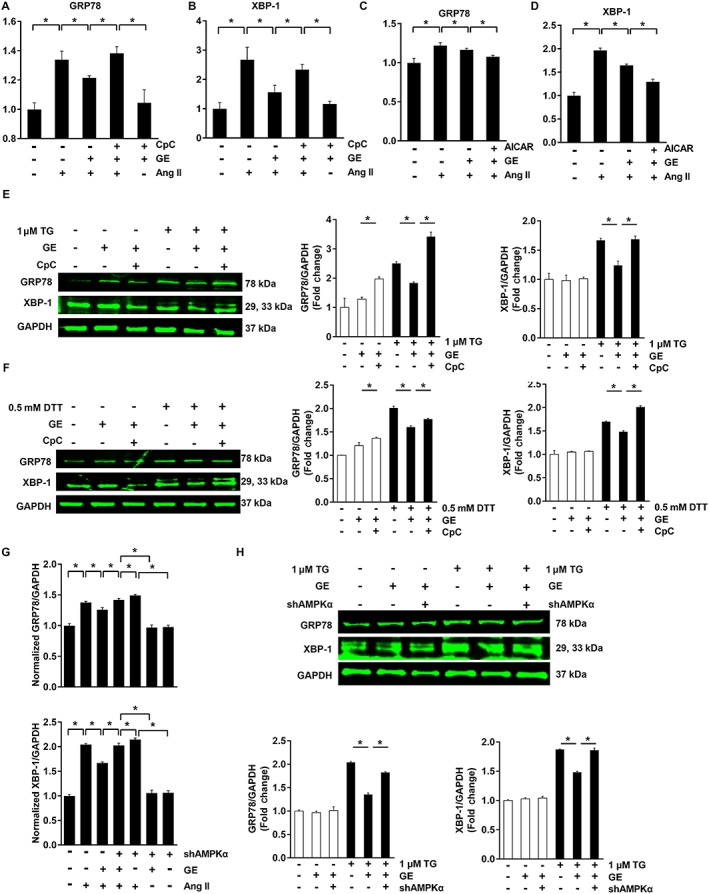
The effects of geniposide (GE) on ER stress were blunted by CpC or shAMPKα in vitro. (A–B) The effects of geniposide (100 μM for 6 h) on ER stress were abolished by CpC in H9c2 cells (20 μM for 6 h) (n = 6). (C–D) The synergistic effects of geniposide and AICAR on ER stress in H9c2 cells (1 mmol · L−1 for 24 h) (n = 6). (E–F) ER stress induced by TG (1 μM for 6 h) or DTT (1 μM for 6 h) (n = 10). (G) ER stress induced by angiotensin (Ang II, 1 μM for 24 h) was abolished by shAMPKα in neonatal rat cardiomyocytes (n = 5). (H) ER stress induced by TG was abolished by shAMPKα in neonatal rat cardiomyocytes (n = 6). * P < 0.05.
CpC counteracted protective effects of geniposide in vivo
Subsequently, we investigated whether mice treated with CpC after geniposide treatment showed reversal of the inverse morphological changes. We found that mice additionally treated with CpC exhibited an aggravated hypertrophic response illustrated by increased HW/BW and HW/TL, enlarged heart sizes, augmented CSA, expanded LVIDd, reduced FS and increased hypertrophic markers (Figure 5A–D, Figure S5A–B, P < 0.05). The activation of AMPKα and the protection against mTOR, ERK and ER stress produced by geniposide were abolished by CpC (Figure S5C, P < 0.05).
Figure 5.
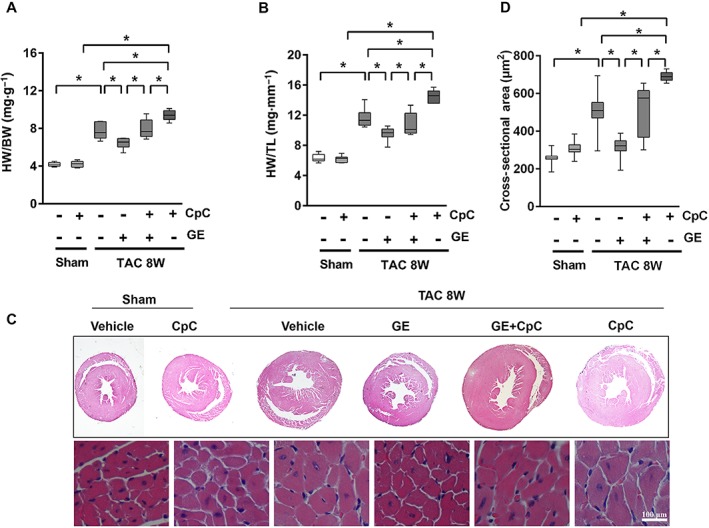
The effects of geniposide (GE; 50 mg · kg−1 for 7 weeks) were blocked by CpC (20 mg · kg−1, every other day, for 6 weeks) in vivo. (A–B) Summary data of HW/BW and HW/TL (n = 12). (C) Histology results from cross‐sections of whole hearts with haematoxylin and eosin staining. (D) The cross‐sectional areas of myocytes (n = 5). Data shown in A, B, D are medians with first and third quartiles and ranges. * P < 0.05.
ER stress contributed to the hypertrophic response via ROS
Elevated ROS were observed after TG or Ang II exposure and these changes were alleviated after geniposide treatment (Figure S6A–B, P < 0.05). NAC completely abolished the increase of ROS induced by TG or Ang II (Figure S6C–D, P < 0.05). Moreover, NAC markedly inhibited the hypertrophic response mediated by Ang II or TG treatment (Figure S6E–I, P < 0.05).
GE activated AMPKα via GLP‐1 receptor
It has been reported that geniposide is an agonist of GLP‐1 receptor (Gong et al., 2014). Our study found that the levels of mRNA for GLP‐1 receptor were increased in the group treated with geniposide (Figure 6A, P < 0.05). This was also the case in H9c2 cardiomyocytes that were incubated with geniposide and Ang II (Figure 6B, P < 0.05). However, after incubation with Ex9‐39 (a GLP‐1 receptor antagonist) combined with geniposide, the mRNA for GLP‐1 receptor was decreased (Figure 6B, P < 0.05). Intriguingly, Ex9‐39, which alone did not affect the hypertrophic response (Figure S7, P > 0.05), completely blocked the protective effects of geniposide against hypertrophy, with marked increases in CSA and ANP (Figure 6C–D, P < 0.05). However, when we reactivated AMPKα using AICAR (Figure 6F, P < 0.05), geniposide protection was restored (Figure 6C–D, P < 0.05). Neonatal rat cardiomyocytes were also incubated with Ex9‐39. Ex9‐39 counteracted the protective effects of geniposide against hypertrophy of neonatal rat cardiomyocytes, and activation of AMPKα via AICAR restored the anti‐hypertrophic effects of geniposide (Figure 6E, P < 0.05).
Figure 6.
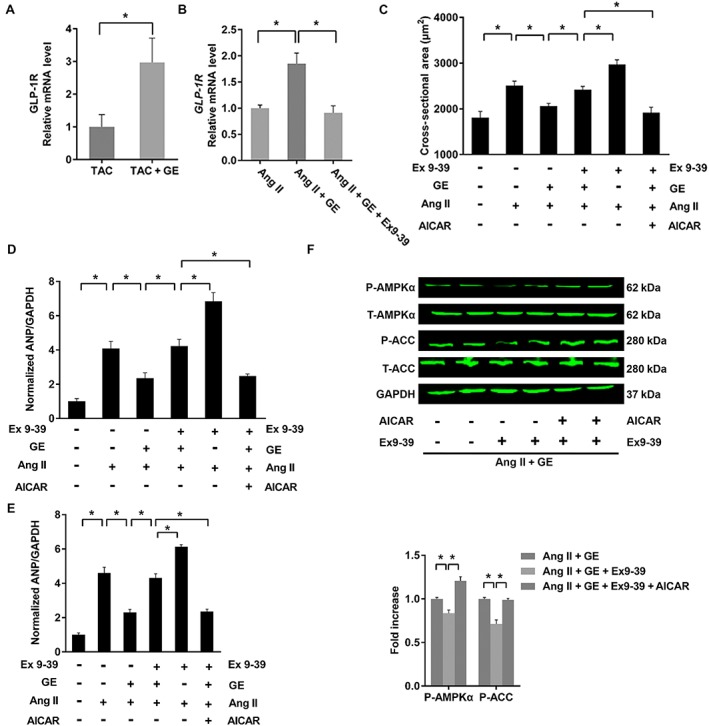
Protection by geniposide (GE; 100 μM for 24 h) was mediated by GLP‐1 receptor. (A) The mRNA levels of GLP‐1 receptor in the hypertrophic hearts (n = 5). (B) The mRNA levels of GLP‐1R in H9c2 cells (n = 5). (C) The cross‐sectional areas of H9c2 myocytes (n = 5). (D) Ex9‐39 (10 μM for 24 h) blocks protection by geniposide in H9c2 cells (n = 6). (E) Ex9‐39 blunts protection by geniposide in neonatal rat cardiomyocytes (n = 6). (F) Effects of Ex9‐39 on AMPKα and ACC (n = 6). * P < 0.05.
To further confirm the role of GLP‐1 receptor, mice were injected with AAV‐shGLP‐1R at the time of TAC surgery. Eight weeks after injection, the mRNA for these receptor was determined. We obtained a reduction of 86 ± 4% in mRNA for GLP‐1 receptor in mice after the AAV‐shGLP‐1R treatment (Figure S8, P < 0.05). Consistent with the results in vitro, GLP‐1R knock‐down abolished the protection by geniposide, which was characterized by the visible morphological changes, the deterioration in echocardiographic parameters and the increased hypertrophic markers (Figure 7A–F, P < 0.05).
Figure 7.
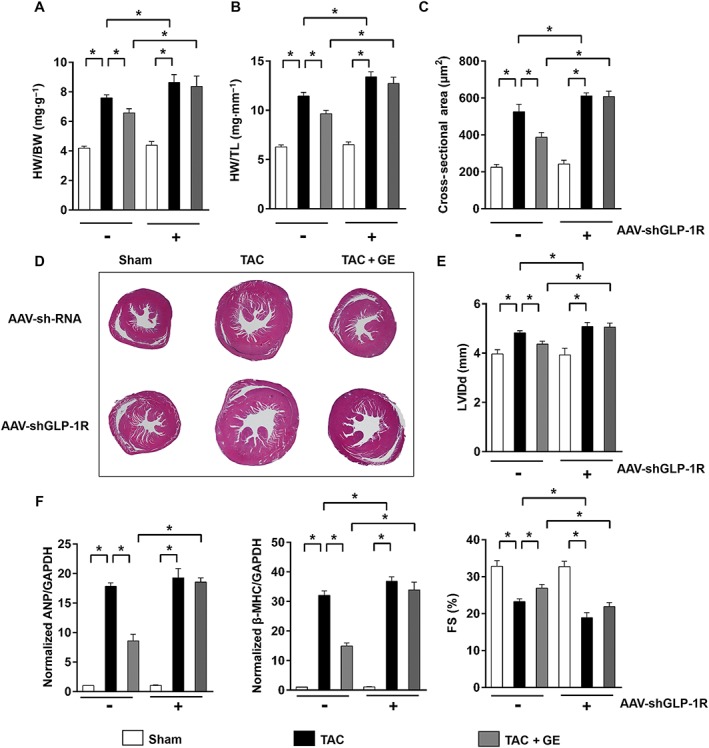
The effects of geniposide (GE; 50 mg kg−1 for 7 weeks) were abolished after knock‐down of GLP‐1 receptor in the heart. (A–B) Summary data of HW/BW and HW/TL (n = 12). (C) The cross‐sectional areas of myocytes (n = 6). (D) Histology results from cross‐sections of hearts with haematoxylin and eosin staining. (E) Echocardiographic parameters in the heart after knock down GLP‐1R (n = 10). (F) Expression levels of the transcripts of ANP and β‐MHC (n = 6). * P < 0.05.
Figure 8.
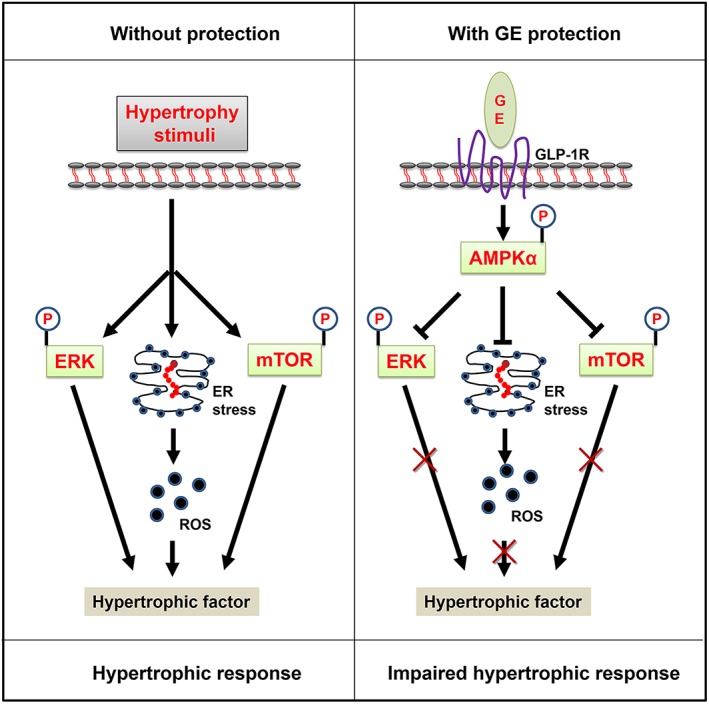
The proposed mechanism of geniposide (GE) protection against cardiac hypertrophy. Data in our study demonstrates that geniposide activated GLP‐1 receptor to increase phosphorylation of AMPKα in the heart. Increased AMPKα attenuated increased mTOR, ERK and ER stress to inhibit cardiac hypertrophy.
Geniposide exhibited activation of AMPKα at baseline
We also investigated the effects of geniposide in mice at baseline. No cardiac and hepatic changes were observed (Figure S9A–E, G, P > 0.05). However, mice given geniposide treatment for 7 weeks at baseline had increased phosphorylation of AMPKα (Figure S9F, P < 0.05).
Discussion and conclusions
Our study demonstrated that geniposide protected against cardiac hypertrophy in vivo and in vitro. To our knowledge, this is the first report to demonstrate that geniposide can block the cardiac hypertrophic response and activate AMPKα via GLP‐1 receptor. In addition, we showed that geniposide activated AMPKα to suppress ER stress in cardiac hypertrophy.
A previous study demonstrated that long‐term inhibition of AMPKα activity in transgenic mice expressing a kinase dead form of the enzyme, exacerbated post‐ischaemic cardiac dysfunction and aberrant apoptosis (Russell et al., 2004). AMPKα also negatively regulated protein synthesis and AMPKα ablation exacerbated TAC‐induced hypertrophy and dysfunction in mice (Zhang et al., 2008). Our data demonstrated that geniposide activated AMPKα and that cardioprotection was lost after AMPKα depletion, implying that AMPKα is the crucial mechanism that mediates the beneficial actions of geniposide. This is consistent with a previous report that adiponectin suppressed TAC‐induced hypertrophy in mice via activation of AMPKα (Belge et al., 2014). mTOR and ERK were downstream targets of AMPKα in its anti‐hypertrophic action. AMPKα directly phosphorylated raptor and suppressed the mTORC1 kinase complex (Zang et al., 2006). ERK, a member of the MAPK signalling pathways that directly modify transcription factors promoting cardiac gene expression and lead to cardiac hypertrophy, can also be suppressed by AMPKα (Shibata et al., 2004). Our study found that geniposide treatment attenuated phosphorylation of mTOR and ERK via functional AMPKα in the hypertrophic heart.
Our study also showed that geniposide suppressed induced ER stress. Overexpression of mutant Lys‐Asp‐Glu‐Leu (KDEL) receptor in transgenic mice activated ER stress and led to heart failure (Hamada et al., 2004). Consistent with a previous finding (Tu and Weissman, 2004), our study found that elevated levels of ER‐resident chaperones caused accumulation of ROS in cardiomyocytes. Moreover, the finding in our study that TG alone can induce the hypertrophic response was in agreement with a previous report (Zhang et al., 2010). Taken together, these facts indicate the important role of ER stress in hypertrophy. Functional AMPKα is associated with protection against hypoxic injury by down‐regulating ER stress in cardiomyocytes (Terai et al., 2005). Activation of AMPKα can also protect against cardiac ischaemia via attenuation of ER stress (Tao et al., 2011). The results from our study showed that geniposide‐impaired ER stress during the development of hypertrophy in vivo and in vitro was no longer inhibitory after ablation of AMPKα, indicating that the inhibitory effects of geniposide on hypertrophy were mediated by AMPKα dependent attenuation of ER stress.
Geniposide exerts its pharmacological effects as an agonist of GLP‐1 receptor (Gong et al., 2014). After activation of these receptor, production of cAMP was increased according to Lee and Jun (2014). Subsequently, the level of cAMP drops to basal levels resulting from its degradation to 5′‐AMP by PDEs, leading to an increase in the AMP/ATP ratio and activation of AMPKα (Omar et al., 2009; Nin et al., 2012). In addition, cAMP activates AMPKα in a PKA‐dependent manner (Fu et al., 2011; Hurtado et al., 2014). Correspondingly, a GLP‐1 analogue has been shown to reverse the molecular pathology and cardiac dysfunction in obese mice and diabetic rats by activating the AMPKα pathway (Inoue et al., 2015; Noyan‐Ashraf et al., 2013). A recent study reported that exendin‐4 ameliorated cardiac ischaemia/reperfusion injury via caveolae and caveolin (Tsutsumi et al., 2014), which are regulated by AMPKα (Zhang et al., 2014). Our study has also shown that geniposide could increase the level of GLP‐1 receptor, thereby activating AMPKα in cardiomyocytes and inhibiting cardiac hypertrophy. Although it has been reported that disruption of GLP‐1 signalling in mice did not influence ischaemia or doxorubicin‐induced cardiomyopathic injury (Ussher et al., 2014), the fact that geniposide lost its inhibitory effects on cardiac hypertrophy after knock‐down of GLP‐1 receptor in the heart was consistent with the available genetic evidence using GLP‐1 receptor knockout mice, which demonstrated that the key metabolic and cardiovascular actions of GLP‐1 receptor agonists were mediated through the known GLP‐1 receptor (Scrocchi et al., 1996; Kim et al., 2013; Noyan‐Ashraf et al., 2013; Tatarkiewicz et al., 2014). Moreover, activation of GLP‐1 receptor can modulate the ER stress response and prevent ROS production via activation of AMPK in cardiomyocytes (Balteau et al., 2014; Lee and Jun, 2014). Our study also found that geniposide could suppress ER stress via GLP‐1 receptor. However, how geniposide activates the GLP‐ receptor remains unclear. It is reported that geniposide‐induced GLP‐1 receptor activity is not altered by the removal of the free C4‐carboxyl group; hence, the structure–activity relationship remains to be elucidated in more detail (Gong et al., 2014).
Elevation of intracellular cAMP levels can also induce the activation of pro‐hypertrophic pathways such as PKA‐dependent activation of ERK and cAMP‐response element binding protein and PI3K‐dependent activation of mTOR. Conversely, following the stimulation of GLP‐1R, activation of AMPKα can suppress the phosphorylation of ERK and mTOR (Inoki et al., 2003; Li et al., 2007). In this study, we found that geniposide suppressed activation of mTOR and ERK, implying that functional AMPKα, rather than PKA and PI3K, was the primary regulator of ERK and mTOR in mice treated with geniposide. Further studies aimed at the potential interactions between GLP‐1 receptor and AMPKα will be of particular interest.
New low MW GLP‐1 receptor agonists still need to be discovered because exenatide and GLP‐1 are not active when taken orally. Our study demonstrated that geniposide protected against cardiac hypertrophy after oral dosing. Inchin‐ko‐to, mainly composed of an analogue of geniposide, is the ‘magic pill’ for jaundice and has long been clinically used in China and Japan (Shoda et al., 2004). With the dose selected in this study, mice treated with geniposide exhibited no obvious change in hepatic morphology despite hepatotoxicity being observed when rats received an overdose of geniposide (Wei et al., 2014). These data suggest that geniposide has the potential for development in clinical use.
In conclusion, we have demonstrated here that geniposide prevented cardiac hypertrophy induced by hypertrophic stimuli via GLP‐1 receptor and AMPKα. We also show that inhibition of AMPKα or knock‐down of GLP‐1 receptor results in loss of the protective effects of geniposide against hypertrophy. Our study provides novel insight into the development of cardiac hypertrophy and may have implications for the future treatment of cardiac hypertrophy through the application of geniposide.
Author contributions
Z.‐G.M., J.D. and W.B.Z. performed the research; Z.‐G.M. and Q.‐Z.T. designed the research study; Z.‐Y.B. and Y.Y. contributed essential reagents or tools; H.‐H.L. and N.Z. analysed the data; Q.‐Z.T. and Z.‐G.M. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1 Primer sequences used for RT‐PCR
Table S2 The information of the primary antibodies used in Western blots
Figure S1 Effects of geniposide (GE) on cardiac hypertrophy induced by pressure overload. HW/BW: heart weight/body weight. (A) The preliminary experiment indicated that geniposide (50–100 mg · kg−1) protected against cardiac hypertrophy (n = 5). (B–D) echocardiographic parameters in mice after 8 weeks of TAC (n = 14). (E) Left ventricular internal diastolic diameter (LVIDd) in mice subjected to isoprenaline (ISO) injection (n = 8). *P < 0.05 versus matched control.
Figure S2 Effects of geniposide (GE) suppressed the production of transcription factor XBP1s. M, marker.
Figure S3 Geniposide (GE) suppressed hypertrophy of myocytes in vitro. (A) Viability of H9c2 cells (n = 3). (B) mRNA of 5′‐AMP‐activated protein kinase‐α (AMPKα)1 and AMPKα2 after adenovirus infection in neonatal rat cardiac myocytes (n = 6). (C) Changes of AMPKα and acetyl‐CoA carboxylase (ACC) after adenovirus infection in neonatal rat cardiac myocytes (n = 6). (D) Changes of AMPKα and ACC after treatment with AICAR in H9c2 cells (n = 6). (E) Phosphorylated AMPKα and downstream proteins after Compound C (CpC) treatment in H9c2 cells (n = 6). *P < 0.05 versus matched control.
Figure S4 The effects of geniposide (GE) on hypertrophy induced by isoprenaline (ISO) (10 mmol · L−1 for 24 h) in H9c2 cell. (A) The levels of atrial natriuretic peptide (ANP) induced by isoprenaline in indicated groups (n = 5). (B) Compound C (CpC) (5–20 μM for 24 h) reversed hypertrophic response induced by isoprenaline (n = 5). (C) The cross‐sectional area of H9c2 myocytes (n = 5). (D) The protein levels of phosphorylated 5′‐AMP‐activated protein kinase‐α (AMPKα) in indicated groups (n = 6). (E) geniposide suppressed isoprenaline‐induced ER stress (n = 5). (F) shAMPKα offset the effect of geniposide against the production of transcription factor XBP1s. M, marker. *P < 0.05.
Figure S5 Compound C (CpC) offset effects of geniposide (GE) on hypertrophy in vivo. (A) Echocardiographic parameters in mice treated with CpC (n = 7). (B) The mRNA expressions of hypertrophic markers (n = 6). (C) CpC attenuates 5′‐AMP‐activated protein kinase‐α (AMPKα) and related targets (n = 6). *P < 0.05 versus matched control.
Figure S6 Endoplasmic stress induces accumulation of ROS. (A) Geniposide (GE) suppressed the accumulation of ROS induced by thapsigargin (TG). (B) Geniposide suppressed the accumulation of ROS induced by angiotensin II (Ang II). (C) N‐acetylcysteine (NAC) blunted the accumulation of ROS induced by TG. (D) NAC blunted the accumulation of ROS induced by Ang II. (E–F) NAC almost completely abolished the increase of atrial natriuretic peptide (ANP) and cross‐sectional area (n = 6). (G–H) NAC suppressed the increase of ANP and cross‐sectional area induced by TG (0.5 μM for 48 h) (n = 6). ROS were detected by DCFH‐DA in three experiments independently. Statistical analysis of ROS was performed using a repeated measures anova. *P < 0.05 versus matched control.
Figure S7 Atrial natriuretic peptide (ANP) and cross‐sectional area of myocytes induced by Ex9‐39 (n = 6).
Figure S8 The mRNA levels of glucagon‐like peptide 1 receptor (GLP‐1R) in the hypertrophic heart after infection (n = 8). *P < 0.05 versus matched control.
Figure S9 Effects of geniposide (GE) on the heart at baseline. (A–D) Statistical results of body weight (BW), heart weight (HW)/BW, HW/tibia length (TL) of the indicated groups (n = 6). (E) The cross‐sectional areas of myocytes (n = 6). (F) The protein levels of phosphorylated 5′‐AMP‐activated protein kinase‐α (AMPKα) in mice from indicated groups (n = 6). (G) Hepatic morphology after geniposide treatment (n = 5). *P < 0.05 versus sham.
Supporting info item
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81270303, 81470516, 81470402), the Fundamental Research Funds for the Central Universities of China (No. 2014302020202), and the Hubei Province's Outstanding Medical Academic Leader programme.
Ma, Z.‐G. , Dai, J. , Zhang, W.‐B. , Yuan, Y. , Liao, H.‐H. , Zhang, N. , Bian, Z.‐Y. , and Tang, Q.‐Z. (2016) Protection against cardiac hypertrophy by geniposide involves the GLP‐1 receptor / AMPKα signalling pathway. British Journal of Pharmacology, 173: 1502–1516. doi: 10.1111/bph.13449.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balteau M, Van Steenbergen A, Timmermans AD, Dessy C, Behets‐Wydemans G, Tajeddine N, et al. (2014). AMPK activation by glucagon‐like peptide‐1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am J Physiol Heart Circ Physiol 307: 1120–1133. [DOI] [PubMed] [Google Scholar]
- Belge C, Hammond J, Dubois‐Deruy E, Manoury B, Hamelet J, Beauloye C, et al. (2014). Enhanced expression of beta 3‐adrenoceptors in cardiac myocytes attenuates neurohormone‐induced hypertrophic remodeling through nitric oxide synthase. Circulation 129: 451–462. [DOI] [PubMed] [Google Scholar]
- Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR (2004). Activation of AMP‐activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem 279: 32771–32779. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola M, Du J, Wang Z, Yano N, Zhang L, Wang Y, et al. (2014). Stimulation of glucagon‐like peptide‐1 receptor through exendin‐4 preserves myocardial performance and prevents cardiac remodeling in infarcted myocardium. Am J Physiol Endocrinol Metab 307: E630–E643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky VW, Chan AY, Robillard FI, Light PE, Des Rosiers C, Dyck JR, et al. (2009). Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation 119: 1643–1652. [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, et al (2010). Reduction of AMP‐activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo . Circulation 121: 792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, Wakabayashi Y, Lippincott‐Schwartz J, Arias IM (2011). Bile acid stimulates hepatocyte polarization through a cAMP‐Epac‐MEK‐LKB1‐AMPK pathway. Proc Natl Acad Sci U S A 108: 1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Liu B, Liu J, Liu Z, Liang D, Li F, et al. (2012). Geniposide, from Gardenia jasminoides Ellis, inhibits the inflammatory response in the primary mouse macrophages and mouse models. Int Immunopharmacol 14: 792–798. [DOI] [PubMed] [Google Scholar]
- Gong N, Fan H, Ma AN, Xiao Q, Wang YX (2014). Geniposide and its iridoid analogs exhibit antinociception by acting at the spinal GLP‐1Rs. Neuropharmacology 84: 31–45. [DOI] [PubMed] [Google Scholar]
- Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, et al. (2004). Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol 24: 8007–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633. [DOI] [PubMed] [Google Scholar]
- Hurtado DLA, Martin‐Hidalgo D, Gil MC, Garcia‐Marin LJ, Bragado MJ (2014). The calcium/CaMKKalpha/beta and the cAMP/PKA pathways are essential upstream regulators of AMPK activity in boar spermatozoa. Biol Reprod 90: 1–10. [DOI] [PubMed] [Google Scholar]
- Inoki K, Kim J, Guan K (2012). AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol 52: 381–400. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590. [DOI] [PubMed] [Google Scholar]
- Inoue T, Inoguchi T, Sonoda N, Hendarto H, Makimura H, Sasaki S, et al. (2015). GLP‐1 analog liraglutide protects against cardiac steatosis, oxidative stress and apoptosis in streptozotocin‐induced diabetic rats. Atherosclerosis 240: 250–259. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG (2005). AMP‐activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE Guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Platt M, Shibasaki T, Quaggin S, Backx PH, Seino S, et al. (2013). GLP‐1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med 19: 567–575. [DOI] [PubMed] [Google Scholar]
- Lee HY, Lee GH, Lee MR, Kim HK, Kim NY, Kim SH, et al. (2013). Eucommia ulmoides Oliver extract, aucubin, and geniposide enhance lysosomal activity to regulate ER stress and hepatic lipid accumulation. PLoS One 8: e81349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Jun HS (2014). Anti‐diabetic actions of glucagon‐like peptide‐1 on pancreatic beta‐cells. Metabolism 63: 9–19. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Wei X, Zhang XF, Liu Y, Zhang Y, Chen K, et al. (2014). IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun 5: 3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Wang AB, Huang Y, Liu DP, Wei C, Williams GM, et al. (2005). Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free Radic Biol Med 38: 243–257. [DOI] [PubMed] [Google Scholar]
- Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, et al. (2007). Long‐term activation of adenosine monophosphate‐activated protein kinase attenuates pressure‐overload‐induced cardiac hypertrophy. J Cell Biochem 100: 1086–1099. [DOI] [PubMed] [Google Scholar]
- Lv C, Wang L, Liu X, Yan S, Yan SS, Wang Y, et al. (2015). Multi‐faced neuroprotective effects of geniposide depending on the RAGE‐mediated signaling in an Alzheimer mouse model. Neuropharmacology 89: 175–184. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ, et al. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down‐regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21: 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nin V, Escande C, Chini CC, Giri S, Camacho‐Pereira J, Matalonga J (2012). Role of deleted in. breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP‐activated protein kinase. J Biol Chem 287: 23489–23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyan‐Ashraf MH, Shikatani EA, Schuiki I, Mukovozov I, Wu J, Li RK, et al. (2013). A glucagon‐like peptide‐1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 127: 74–85. [DOI] [PubMed] [Google Scholar]
- Omar B, Zmuda‐Trzebiatowska E, Manganiello V, Goransson O, Degerman E (2009). Regulation of AMP‐activated protein kinase by cAMP in adipocytes: roles for phosphodiesterases, protein kinase B, protein kinase A, Epac and lipolysis. Cell Signal 21: 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SPH, Buneman OP, et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhl SL, Kazakov A, Müller A, Fries P, Wagner DR, Böhm M, et al. (2016). Adenosine A1 receptor activation attenuates cardiac hypertrophy and fibrosis in response to α1‐adrenoceptor stimulation in vivo . Br J Pharmacol 173: 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. (2004). AMP‐activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 114: 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrocchi LA, Brown TJ, MacLusky N, Brubaker PL, Auerbach AB, Joyner AL, et al. (1996). Glucose intolerance but normal satiety in mice with a null mutation in the glucagon‐like peptide receptor gene. Nat Med 2: 21254–21258. [DOI] [PubMed] [Google Scholar]
- Shah AM, Mann DL (2011). In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet 378: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Yuan G, Zhang J, Guo X (2015). Liraglutide reduces lipogenetic signals in visceral adipose of db/db mice with AMPK activation and Akt suppression. Drug Des Devel Ther 9: 1177–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, et al. (2004). Adiponectin‐mediated modulation of hypertrophic signals in the heart. Nat Med 10: 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoda J, Miura T, Utsunomiya H, Oda K, Yamamoto M, Kano M, et al. (2004). Genipin enhances Mrp2 (Abcc2)‐mediated bile formation and organic anion transport in rat liver. Hepatology 39: 167–178. [DOI] [PubMed] [Google Scholar]
- Tang Q, Cai J, Shen D, Bian Z, Yan L, Wang YX, et al. (2009). Lysosomal cysteine peptidase cathepsin L protects against cardiac hypertrophy through blocking AKT/GSK3beta signaling. J Mol Med (Berl) 87: 249–260. [DOI] [PubMed] [Google Scholar]
- Tao J, Zhu W, Li Y, Xin P, Li J, Liu M, et al (2011). Apelin‐13 protects the heart against ischemia–reperfusion injury through inhibition of ER‐dependent apoptotic pathways in a time‐dependent fashion. Am J Physiol Heart Circ Physiol 301: H1471–H1486. [DOI] [PubMed] [Google Scholar]
- Tatarkiewicz K, Sablan EJ, Polizzi CJ, Villescaz C, Parkes DG (2014). Long‐term metabolic benefits of exenatide in mice are mediated solely via the known glucagon‐like peptide 1 receptor. Am J Physiol‐Reg I 306: 490–498. [DOI] [PubMed] [Google Scholar]
- Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, et al. (2005). AMP‐activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 25: 9554–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Musi N, D'Agostino J, Hirshman MF, Goodyear LJ (2001). Increased adenosine monophosphate‐activated protein kinase activity in rat hearts with pressure‐overload hypertrophy. Circulation 104: 1664–1669. [DOI] [PubMed] [Google Scholar]
- Tsutsumi YM, Tsutsumi R, Hamaguchi E, Sakai Y, Kasai A, Ishikawa Y, et al. (2014). Exendin‐4 ameliorates cardiac ischemia/reperfusion injury via caveolae and caveolins‐3. Cardiovasc Diabetol 13: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Weissman JS (2004). Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164: 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussher JR, Baggio LL, Campbell JE, Mulvihill EE, Kim M, Kabir MG, et al. (2014). Inactivation of the cardiomyocyte glucagon‐like peptide‐1 receptor (GLP‐1R) unmasks cardiomyocyte‐independent GLP‐1R‐mediated cardioprotection. Mol Metab 3: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, et al. (2010). AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol 45: 276–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Zhang F, Zhang Y, Cao C, Li X, Li D, et al (2014). Proteomic investigation of signatures for geniposide‐induced hepatotoxicity. J Proteome Res 13: 5724–5733. [DOI] [PubMed] [Google Scholar]
- Yang J, Ao N, Du J, Wang X, He Y (2015). Protective effect of liraglutide against ER stress in the liver of high‐fat diet‐induced insulin‐resistant rats. Endocrine 49: 106–118. [DOI] [PubMed] [Google Scholar]
- Zang M, Xu S, Maitland‐Toolan KA, Zuccollo A, Hou X, Jiang B, et al. (2006). Polyphenols stimulate AMP‐activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor‐deficient mice. Diabetes 55: 2180–2191. [DOI] [PubMed] [Google Scholar]
- Zarrinpashneh E, Beauloye C, Ginion A, Pouleur AC, Havaux X, Hue L, et al. (2008). AMPKalpha2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem Biophys Res Commun 376: 677–681. [DOI] [PubMed] [Google Scholar]
- Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, et al. (2008). AMP activated protein kinase‐alpha2 deficiency exacerbates pressure‐overload‐induced left ventricular hypertrophy and dysfunction in mice. Hypertension 52: 918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang Q, Wu Y, Moriasi C, Liu Z, Dai X, et al. (2014). Endothelial cell‐specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo . Circulation 129: 1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZY, Liu XH, Hu WC, Rong F, Wu XD (2010). The calcineurin‐myocyte enhancer factor 2c pathway mediates cardiac hypertrophy induced by endoplasmic reticulum stress in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol 298: H1499–H1509. [DOI] [PubMed] [Google Scholar]
- Zong J, Deng W, Zhou H, Bian ZY, Dai J, Yuan Y, et al. (2013). 3,3′‐Diindolylmethane protects against cardiac hypertrophy via 5′‐adenosine monophosphate‐activated protein kinase‐alpha2. PLoS One 8: e53427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences used for RT‐PCR
Table S2 The information of the primary antibodies used in Western blots
Figure S1 Effects of geniposide (GE) on cardiac hypertrophy induced by pressure overload. HW/BW: heart weight/body weight. (A) The preliminary experiment indicated that geniposide (50–100 mg · kg−1) protected against cardiac hypertrophy (n = 5). (B–D) echocardiographic parameters in mice after 8 weeks of TAC (n = 14). (E) Left ventricular internal diastolic diameter (LVIDd) in mice subjected to isoprenaline (ISO) injection (n = 8). *P < 0.05 versus matched control.
Figure S2 Effects of geniposide (GE) suppressed the production of transcription factor XBP1s. M, marker.
Figure S3 Geniposide (GE) suppressed hypertrophy of myocytes in vitro. (A) Viability of H9c2 cells (n = 3). (B) mRNA of 5′‐AMP‐activated protein kinase‐α (AMPKα)1 and AMPKα2 after adenovirus infection in neonatal rat cardiac myocytes (n = 6). (C) Changes of AMPKα and acetyl‐CoA carboxylase (ACC) after adenovirus infection in neonatal rat cardiac myocytes (n = 6). (D) Changes of AMPKα and ACC after treatment with AICAR in H9c2 cells (n = 6). (E) Phosphorylated AMPKα and downstream proteins after Compound C (CpC) treatment in H9c2 cells (n = 6). *P < 0.05 versus matched control.
Figure S4 The effects of geniposide (GE) on hypertrophy induced by isoprenaline (ISO) (10 mmol · L−1 for 24 h) in H9c2 cell. (A) The levels of atrial natriuretic peptide (ANP) induced by isoprenaline in indicated groups (n = 5). (B) Compound C (CpC) (5–20 μM for 24 h) reversed hypertrophic response induced by isoprenaline (n = 5). (C) The cross‐sectional area of H9c2 myocytes (n = 5). (D) The protein levels of phosphorylated 5′‐AMP‐activated protein kinase‐α (AMPKα) in indicated groups (n = 6). (E) geniposide suppressed isoprenaline‐induced ER stress (n = 5). (F) shAMPKα offset the effect of geniposide against the production of transcription factor XBP1s. M, marker. *P < 0.05.
Figure S5 Compound C (CpC) offset effects of geniposide (GE) on hypertrophy in vivo. (A) Echocardiographic parameters in mice treated with CpC (n = 7). (B) The mRNA expressions of hypertrophic markers (n = 6). (C) CpC attenuates 5′‐AMP‐activated protein kinase‐α (AMPKα) and related targets (n = 6). *P < 0.05 versus matched control.
Figure S6 Endoplasmic stress induces accumulation of ROS. (A) Geniposide (GE) suppressed the accumulation of ROS induced by thapsigargin (TG). (B) Geniposide suppressed the accumulation of ROS induced by angiotensin II (Ang II). (C) N‐acetylcysteine (NAC) blunted the accumulation of ROS induced by TG. (D) NAC blunted the accumulation of ROS induced by Ang II. (E–F) NAC almost completely abolished the increase of atrial natriuretic peptide (ANP) and cross‐sectional area (n = 6). (G–H) NAC suppressed the increase of ANP and cross‐sectional area induced by TG (0.5 μM for 48 h) (n = 6). ROS were detected by DCFH‐DA in three experiments independently. Statistical analysis of ROS was performed using a repeated measures anova. *P < 0.05 versus matched control.
Figure S7 Atrial natriuretic peptide (ANP) and cross‐sectional area of myocytes induced by Ex9‐39 (n = 6).
Figure S8 The mRNA levels of glucagon‐like peptide 1 receptor (GLP‐1R) in the hypertrophic heart after infection (n = 8). *P < 0.05 versus matched control.
Figure S9 Effects of geniposide (GE) on the heart at baseline. (A–D) Statistical results of body weight (BW), heart weight (HW)/BW, HW/tibia length (TL) of the indicated groups (n = 6). (E) The cross‐sectional areas of myocytes (n = 6). (F) The protein levels of phosphorylated 5′‐AMP‐activated protein kinase‐α (AMPKα) in mice from indicated groups (n = 6). (G) Hepatic morphology after geniposide treatment (n = 5). *P < 0.05 versus sham.
Supporting info item