

Abstract
Objective:
To assess whether Parkinson disease (PD) genes are somatically mutated in cutaneous melanoma (CM) tissue, because CM occurs in patients with PD at higher rates than in the general population and PD is more common than expected in CM cohorts.
Methods:
We cross-referenced somatic mutations in metastatic CM detected by whole-exome sequencing with the 15 known PD (PARK) genes. We computed the empirical distribution of the sum of mutations in each gene (Smut) and of the number of tissue samples in which a given gene was mutated at least once (SSampl) for each of the analyzable genes, determined the 90th and 95th percentiles of the empirical distributions of these sums, and verified the location of PARK genes in these distributions. Identical analyses were applied to adenocarcinoma of lung (ADENOCA-LUNG) and squamous cell carcinoma of lung (SQUAMCA-LUNG). We also analyzed the distribution of the number of mutated PARK genes in CM samples vs the 2 lung cancers.
Results:
Somatic CM mutation analysis (n = 246) detected 315,914 mutations in 18,758 genes. Somatic CM mutations were found in 14 of 15 PARK genes. Forty-eight percent of CM samples carried ≥1 PARK mutation and 25% carried multiple PARK mutations. PARK8 mutations occurred above the 95th percentile of the empirical distribution for SMut and SSampl. Significantly more CM samples harbored multiple PARK gene mutations compared with SQUAMCA-LUNG (p = 0.0026) and with ADENOCA-LUNG (p < 0.0001).
Conclusions:
The overrepresentation of somatic PARK mutations in CM suggests shared dysregulated pathways for CM and PD.
Epidemiologic evidence shows that cutaneous melanoma (CM) occurs 1.5–3.5 times more frequently among patients with Parkinson disease (PD) than in the general population.1 The CM-PD co-occurrence is also reported for first- and second-degree relatives of PD and CM patients.2–4 Alterations in the activity of melanin synthesis enzymes, impaired autophagy, and/or genetic predisposition for both diseases have been suggested as possible mechanisms.5 Although the risk for PD and for CM is higher in individuals with red hair color,6 an MC1R variant, a main genetic determinant of skin and hair color (R160W*MC1R -rs1805008), was reported to be associated with PD.7 It is of interest to note that some familial PD genes (PARK genes) play a role in regulating or maintaining the cell cycle, a key component in the malignant transformation process.8 Some PARK genes are tumor suppressors (e.g., PARK2-PRKN-PARKIN) and others are oncogenes (e.g., PARK7-DJ1).8,9 In this context, one plausible explanation underlying the CM-PD co-occurrence could be genes involved in both processes that co-segregate or are in linkage disequilibrium with each other. A study testing this notion reported no shared PD-related single-nucleotide polymorphisms (SNPs) in 2,297 melanoma cases and 6,651 controls.10 Moreover, no association was shown when cross-referencing 360 pigmentation or melanoma SNPs in 5,333 PD cases and 12,019 controls.11
Given the negative findings summarized above in cross-referencing germline DNA of CM and PD patients, we hypothesized that PARK genes may preferentially be mutated somatically in CM. The present study tested this notion.
METHODS
PARK genes.
To define PD predisposition genes and loci, OMIM (http://www.ncbi.nlm.nih.gov/omim) was searched with the words “Parkinson disease” and “genes” and “locus/loci.”
Data formatting.
The somatic CM mutation data used in this article merged melanoma exome/genome sequencing from different sources as described elsewhere.12,13 All mutational data from 6 different whole-exome/genome sources were collated from 4 published studies14–17 and unpublished data. The data were formatted so that all positional data were mapped to the same genome build. In this case, any data that were on hg18 were lifted over to hg19 using the Lift Genome Annotations tool available from UCSC (http://genome.ucsc.edu/cgi-bin/hgLiftOver, UC Santa Cruz Software, The Regents of the University of California, Santa Cruz, CA). In some cases, as data were merged, it became necessary to eliminate redundant information. For instance, with some samples, both a tumor and a cell line derived from it were sequenced and the overwhelming majority of mutations were shared. This is also true in the case of samples that were sequenced in more than one study15 and for multiple metastases extracted from the same patient in another study.17 When removing these duplicates and redundancies, all mutations were retained at a count of one and the sample name was merged into a single entry. This step was taken to ensure that the number of recurrent positions was not inflated in later analysis. Once the list of mutations was established, the positional data and changes were formatted to an oncotator input format and annotated using the web-based version of oncotator (http://www.broadinstitute.org/oncotator, Cambridge, MA). The next step taken was to remove any samples that were listed as acral, mucosal, or uveal melanoma subtypes, to ensure focusing on CM. In the final step, any samples in the initial publication that did not include a matched normal genotype were also removed.
The data were arranged in a table, where the rows represent genes and the columns are CM tissue samples. The entries are the number of somatic mutations per each combination of gene and tissue sample.
The list of somatic mutations was sorted by gene name. The somatic CM mutation platform was cross-referenced with the list of the defined PARK genes and loci (PARK1 to PARK20).
To assess CM-related specificity of the findings, identical analyses were performed for adenocarcinoma of lung (ADENOCA-LUNG) and squamous cell carcinoma of lung (SQUAMCA-LUNG), based on data derived from the COSMIC database (studies COSU417 and COSU418).18 The data format of the CM mutation data set was compatible with the COSMIC data sets. Splice variants are annotated in COSMIC and are used if a mutation arises in a region that maps to the coding domain of a gene and the predicted protein is different.18 No ethical approval was required for this study.
Statistical analysis.
The statistical analysis was performed in 2 steps. As an initial step, we analyzed each gene individually, and in the second step, we analyzed the PARK genes combined as a group.
Analysis by each PARK gene individually.
The question of interest was whether single PARK genes harbor a relatively high number of somatic mutations, when compared with other genes in CM tissue. To answer this question, 2 measures were defined: (1) the sum of mutations in each gene (Smut) and (2) the number of tissue samples in which a given gene was mutated at least once (SSampl). (Note that the difference between SMut and SSampl is that in SSampl, each sample is counted at most once.)
Analysis was performed by the following steps: (1) computation of the empirical distributions for SMut and SSampl for all analyzable genes, (2) determination of the percentiles (75th, 90th, and 95th) of the empirical distribution, and (3) determination of the location of the known PARK genes in the above empirical distributions.
For comparison, and to ensure CM specificity of the findings, identical analyses were applied for ADENOCA-LUNG and SQUAMCA-LUNG in COSMIC data sets of studies COSU417 and COSU418.18 Lung carcinomas were chosen as control tissues because the number of somatic mutations in lung cancer is roughly similar to that of CM (12–13/megabase in lung vs 10–18/megabase in CM).19–21
Analysis of the 15 PARK genes combined as a group.
Because some samples harbored mutations in more than 1 PARK gene, we analyzed the distribution of the number of mutated PARK genes in CM using 2 methods:
We compared the distribution of the number of mutated PARK genes in CM samples with the distribution in ADENOCA-LUNG and in SQUAMCA-LUNG samples using the Kolmogorov-Smirnov test.
Here, the question of interest was how often the 15 PARK genes are mutated as a group, relative to a randomly chosen group of 15 genes.
Using a bootstrap-like procedure, we generated the empirical distributions of the variables of interest (SMut, SSampl), aggregated over the 15 PARK genes. The aggregated variables are denoted by SMutGroup, which is the total number of mutations of the 15 PARK genes as a group in all samples, and SSamplGroup, which is the number of samples in which at least one gene from the group was mutated. The procedure was performed as follows:
Randomly select a group of 15 genes.
Compute SMutGroup and SSamplGroup for the selected group.
Repeat steps 1 and 2, ten thousand times.
Obtain the empirical distributions of SMutGroup and SSamplGroup.
We note that the 15 PARK genes were excluded from the pool of genes (18,758 − 15 = 18,743 genes). The same analysis was applied to each of the 3 cancer types.
Once the empirical distributions of these variables were generated for random gene groups, we verified the locations of the group of 15 PARK genes in these distributions, for SMutGroup and SSamplGroup and for each cancer type.
Statistical analyses used SAS (9.2 version) and MATLAB (r2014a version).
RESULTS
Analysis by each PARK gene individually.
A total of 15 PARK genes and 20 PARK-associated loci were identified (table 1, first column). Somatic CM analysis resulted in 315,914 mutations in 18,758 genes by whole-exome sequencing in 246 metastatic CM tissue samples.12 Fourteen of the 15 PARK genes were mutated in CM tissues. Table 1 summarizes the observed number of mutations and the number of samples harboring PARK gene mutations and the percentiles of the empirical distribution of mutations. As shown in the table, PARK8 (shown as straight bold numbers) was mutated in CM, as counted by Smut, 55 times, thus being above the 95th percentile of the empirical distribution (which is 49 times) and in SSampl = 40 CM samples, again above 36, the 95th percentile of number of samples with mutations.
Table 1.
PARK mutations in cutaneous melanoma, adenocarcinoma, and squamous cell carcinoma of lung
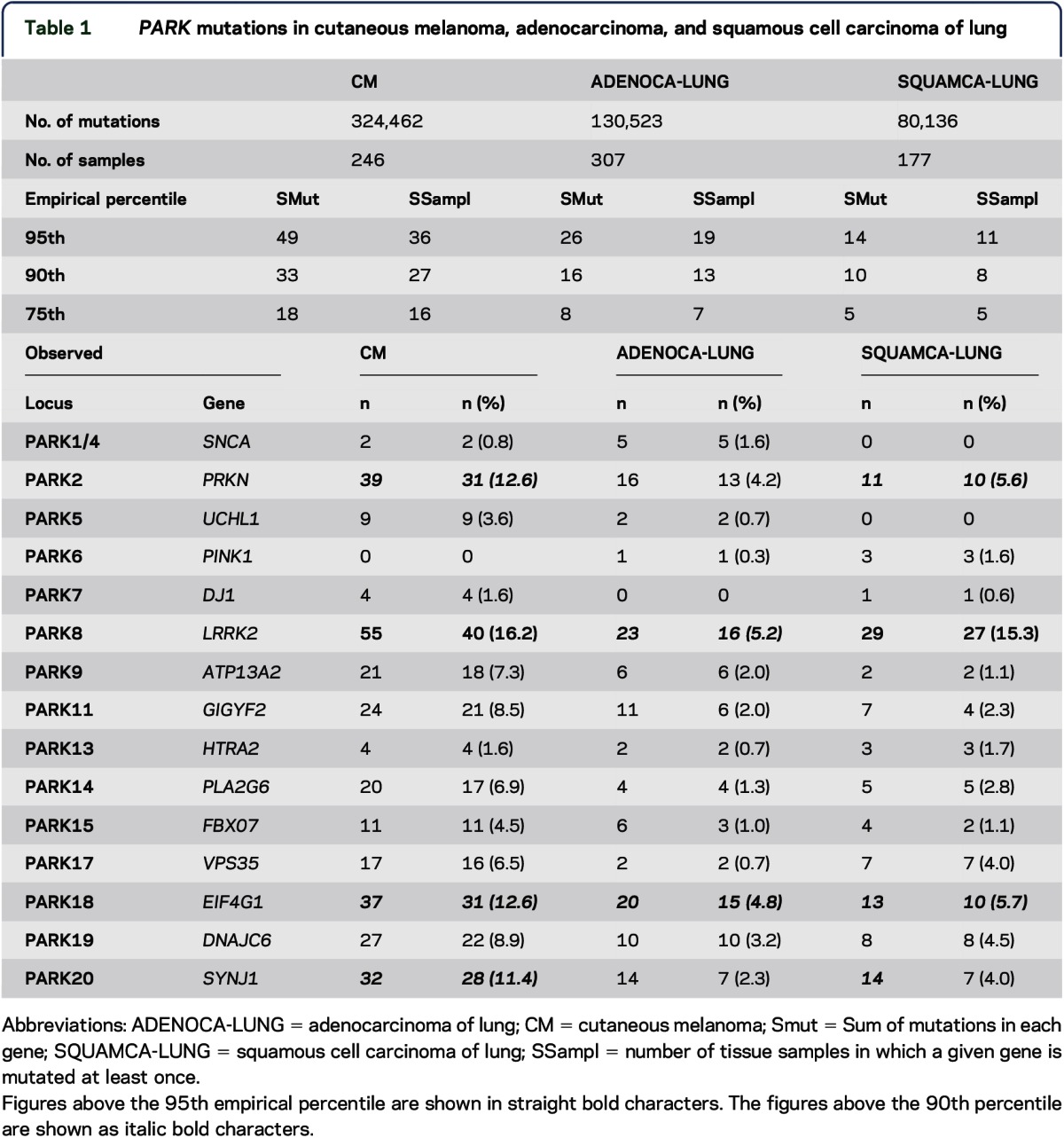
Three additional PARK genes (PARK2, PARK18, and PARK20, shown in table 1 as italic bold numbers) were mutated above the 90th percentile of the empirical distributions of SMut and SSampl.
Somatic PARK mutations were also detected in lung cancer tissue samples. Figure 1 summarizes PARK genes found above the 90th percentile of the empirical distribution of tissue samples (SSampl) in CM, ADENOCA-LUNG, and SQUAMCA-LUNG, and table 1 depicts the distributions of PARK gene mutations for the SMut and SSampl variables in the 3 cancer types.
Figure 1. PARK genes observed above the 90th percentile.
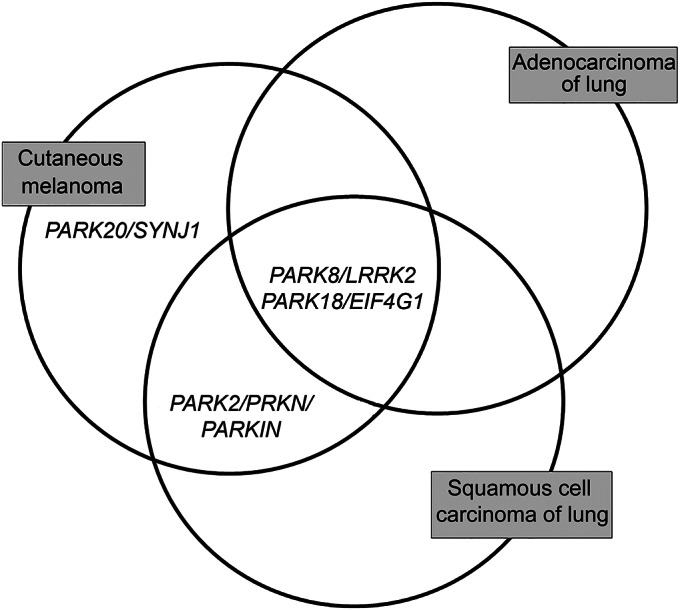
Venn diagram summarizing mutated PARK genes observed above the 90th percentile of the empirical distribution in melanoma, adenocarcinoma, and squamous cell carcinoma of lung.
Analysis of the 15 PARK genes as a group.
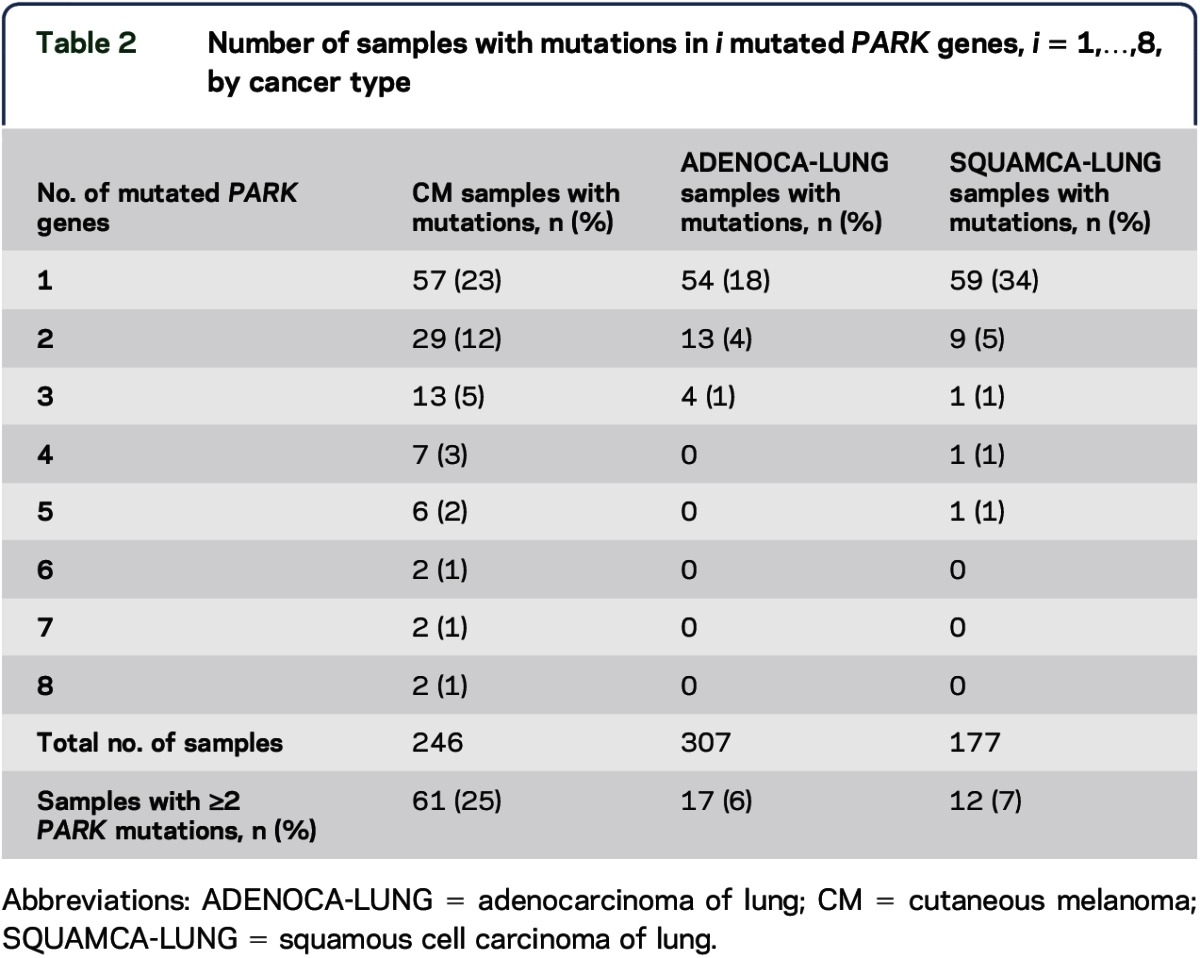
Overall, 48% of CM samples had a mutation in at least 1 PARK gene and 25% had mutations in multiple PARK genes (2–8 mutated genes) (table 2). Samples with mutations in multiple PARK genes (2–5 mutated genes) were found in only 6% of ADENOCA-LUNG and in 7% of SQUAMCA-LUNG samples. Comparison of the distribution of the number of mutated PARK genes in the 3 cancer types is shown in table 2. Significantly more CM samples harbored multiple PARK gene mutations compared with SQUAMCA-LUNG (p = 0.0026) and with ADENOCA-LUNG (p < 0.0001) (Kolmogorov-Smirnov test). This finding was significant after Bonferroni correction for multiple comparisons (required p value <0.025 = 0.05/2).
Table 2.
Number of samples with mutations in i mutated PARK genes, i = 1,…,8, by cancer type
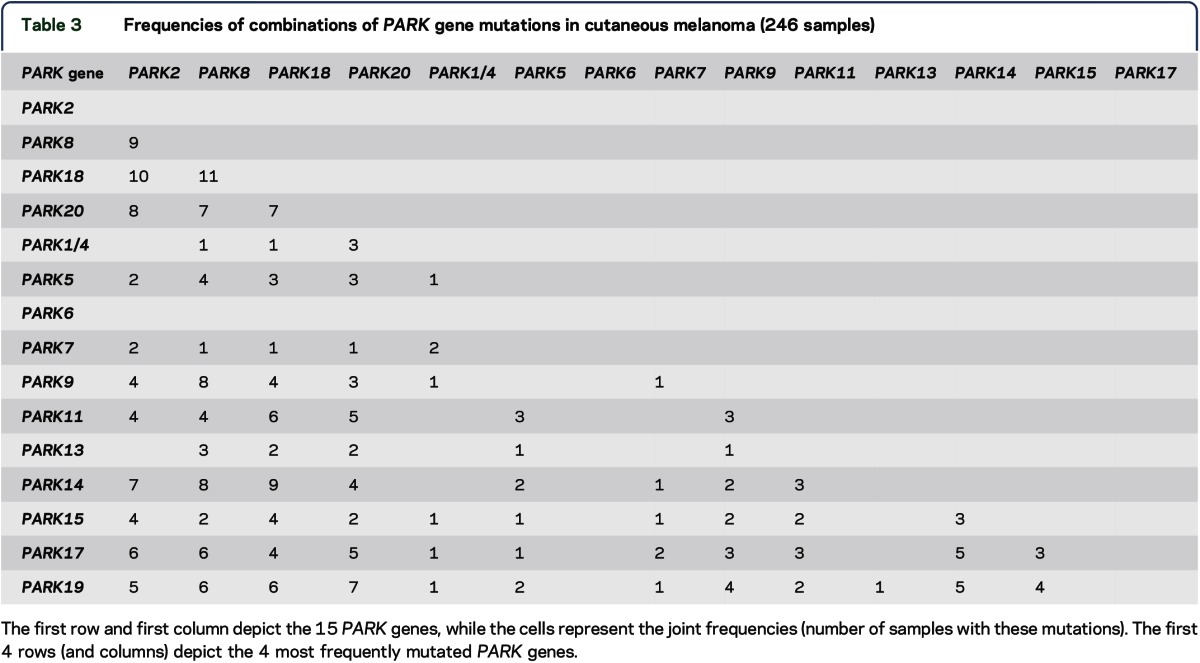
The number of samples carrying diverse combinations of multiple PARK gene mutations in CM samples is depicted in table 3. The higher frequencies of some of the combinations in CM samples (in particular, those involving PARK2, PARK8, PARK18, and PARK20, table 3) vs the paucity of such samples in ADENOCA-LUNG (table e-1 at Neurology.org/ng) and SQUAMCA-LUNG (table e-2) are clearly notable.
Table 3.
Frequencies of combinations of PARK gene mutations in cutaneous melanoma (246 samples)
To further study the PARK genes as a group, we used the bootstrap-like permutation method (see “Statistical analysis”). The results plotted as histograms are shown in figure 2, A–C: SMutGroup (upper panel) and SSamplGroup (lower panel). SMutGroup and SSamplGroup of PARK genes shown as the vertical lines appearing to be located in the upper end of the distribution of genes in CM (figure 2A) and SQUAMCA-LUNG (figure 2C) and not for ADENOCA-LUNG (figure 2B). For CM (figure 2A), the location of SMutGroup for the 15 PARK genes as a group was at the 83.5th percentile and that of SSamplGroup was at the 90.8th percentile. For SQUAMCA-LUNG (figure 2C), the location of SMutGroup was at the 92.8th percentile and that of SSamplGroup was at the 94.1th percentile. For ADENOCA-LUNG (figure 2B), the respective sums were located at the 76.3rd percentile for SMutGroup and at the 60.8th percentile for SSamplGroup.
Figure 2. Empirical distributions of the sums of mutations in groups of 15 genes in cutaneous melanoma, adenocarcinoma of lung, and squamous cell carcinoma of lung.
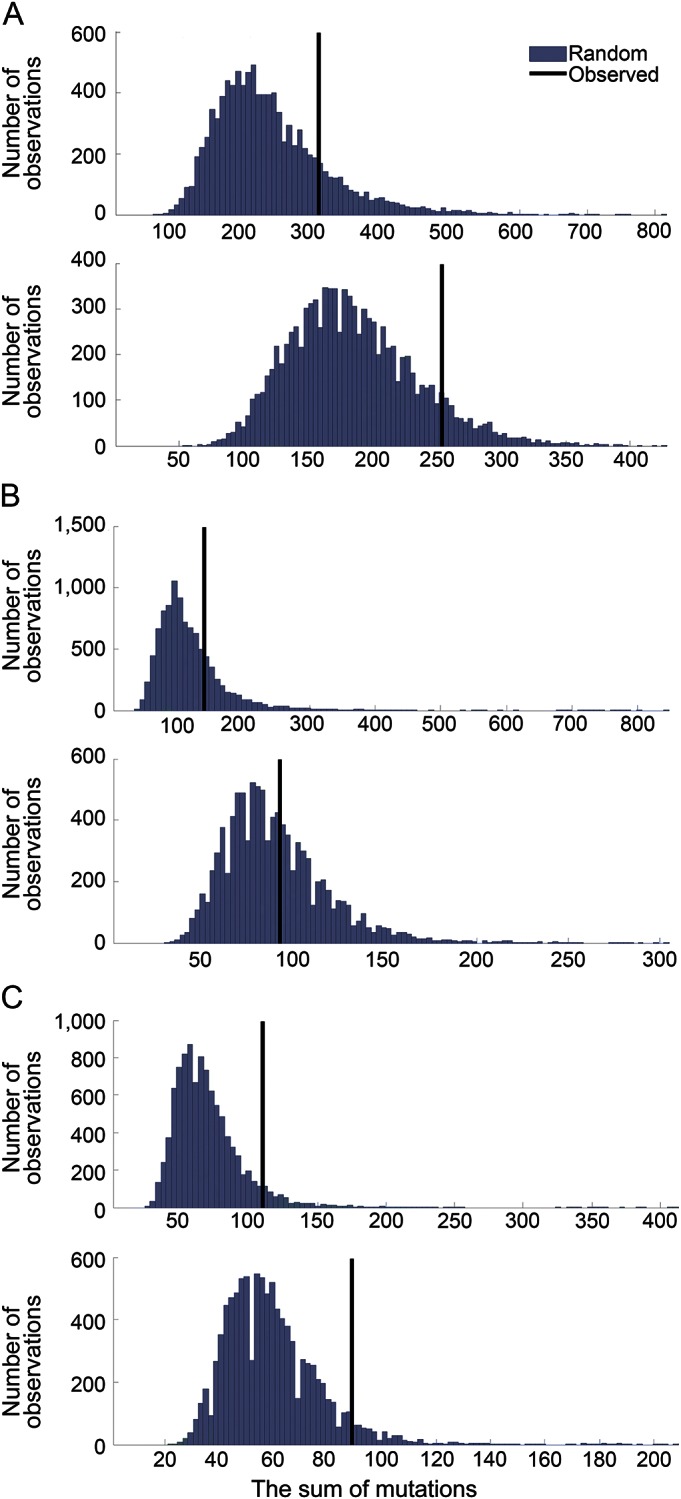
Histograms of the empirical distributions of the sums of mutations in groups of 15 genes, generated by randomly selecting 10,000 groups of 15 genes in (A) cutaneous melanoma, (B) adenocarcinoma of lung, and (C) squamous cell carcinoma of lung (SMutGroup—upper panel, SSamplGroup—lower panel). X axis: the sum of mutations; Y axis: number of observations. The vertical lines depict the locations of the sums of mutations (SMutGroup) in the PARK genes.
DISCUSSION
In the present study, we observed that genes associated with PD predisposition (PARK genes) are somatically mutated above random occurrence in CM. This overrepresentation suggests shared dysregulated functional pathways between CM and PD. It is possible that the co-occurrence of mutations in multiple PARK genes has a cumulative contribution beyond the influence of a single gene dysfunction.
The most commonly mutated PARK gene observed herein in CM (∼15% of tumors) was PARK8 or LRRK2, a gene that accounts for the most common autosomal dominant form in PD.22 LRRK2, leucine-rich repeat kinase 2, is a large protein displaying dual enzymatic functions, a protein kinase effector domain, and a ras-oncogene like GTPase domain, in addition to multiple protein interaction domains.23 The G2019S missense mutation, the most common LRRK2 germline mutation in PD, results in gain of function.23 A previous study reported that LRRK2 is overexpressed in papillary renal and thyroid cancers.24 Downregulation of LRRK2 in cultured renal and thyroid cancer cells compromised MET activation and reduced downstream signaling, suggesting that LRRK2 and MET cooperate in controlling tumor growth.24
Patients with Ashkenazi-Jewish PD, who carry the germline G2019S*LRRK2 mutation, were reportedly at higher risk for developing mainly hormone-related cancers, breast and prostate, but not for melanoma or any skin cancer compared with PD patients who do not carry this mutation.25–27 It is of interest to note that a new bioinformatics tool used for detecting somatic activating mutations identified the LRRK2 locus as a possible oncogene, albeit analyzed only in nonskin cancers, and not CM.28
The second most commonly mutated gene in our study was PARK2 encoding for parkin, an ubiquitin ligase 3. PARK2-inactivating germline mutations underlie the most common form of autosomal recessive early-onset PD.22 Several studies have shown that PARK2 is a potential tumor suppressor, inactivated in many cancers including glioblastoma, renal cell carcinoma, colon, lung, and pancreatic cancers.9,29,30 Germline PARK2-inactivating mutations were recently reported to be more frequently encountered in a CM cohort than in controls (odds ratio = 4.93).31 Furthermore, parkin was underexpressed in melanocytes whereas reexpression of parkin in melanoma cell lines resulted in reduction of cell proliferation rates.31 In that study, 43% of primary and 66% of metastatic CM cell lines harbored an inactivating PARK2 mutation.31
The third most frequently mutated gene in CM in the present study was PARK18 or EIF4G1.32 EIF4G1 is a member of the translation initiation complex eIF4F and plays a central role in eIF4F complex formation.33 The effect of eIF4G1 mutations on cap complex assembly and initiation of protein synthesis remains currently unknown. Notably, LRRK2 is involved in protein translation and cross-talks with elongations factors, one of which is encoded by EIF4G1.33 The possible association of this gene with carcinogenesis is indirectly inferred from abnormal expression patterns in cancers such as SQUAMCA-LUNG, inflammatory breast cancer, and nasopharyngeal cancer.34,35
The fourth most commonly mutated gene somatically in CM is PARK20 or SYNJ1, a gene whose biallelic mutations are associated with an uncommon form of autosomal recessive PD.22 Recently, functional protein-protein interactions of EIF4G1 with SYNJ1 were reported.36 SYNJ1 has not been shown to be involved in cancer pathogenesis, but its homolog, SYNJ2, was reportedly overexpressed in breast cancer.37
One caveat to these results and the putative specificity of PARK genes targeted somatically in CM is the fact that CM displays much higher somatic mutation rates than other cancer types: somatic mutation frequency per megabase is 2 in ovarian cancer, 3–4 in breast cancer, vs 10–18 in CM.19–21 To account for these different mutation rates, we chose to compute the relative location of PARK genes in CM, in comparison with the empirical distributions of the plethora of mutations in CM itself and were able to dissect PARK genes mutated above these distributions. In addition, we chose lung cancer, which has the closest mutation load (12–13/megabase) to CM as control tumor tissue19–21 and showed that CM has significantly more mutated PARK genes compared with ADENOCA-LUNG and SQUAMCA-LUNG.
The strength of our study is its approach focusing on all PARK genes and in the use of big data. There are inherent weaknesses that should be acknowledged. Our study relies exclusively on bioinformatics findings, and there is no assignment of the somatic sequence variants as pathogenic or nonpathogenic and there are no experimental data to prove (or disprove) the possible mechanisms of association of genetic variants with phenotypes. Additional concerns involve the use of stringent statistical nonparametric analysis that may only be able to detect high odds and omit moderate contributions of single genes or subsets and thus may leave true associations undetected.
The present study focused only on PARK genes that are somatically mutated in CM. Yet, the possible functional and genetic intersections between CM and PD are, in all likelihood, more complex. Notably, the reported list of somatic CM mutations encompasses several genes that are functionally associated with PD.20 For example, the alpha-synuclein gene (SNCA) is associated with 2 autosomal dominant inherited forms of PD (PARK1 and PARK4).22 SNCA accumulates in neurons and forms the main component of the Lewy bodies, the pathologic hallmark of familial and sporadic PD.22 SNCA is highly expressed in both primary and metastatic melanoma tissues, but not in nonmelanocytic cells.38 Another example is PLA2G6 (PARK14), a gene related to nevus count, whose germline mutations cause familial PD39 and whose variability has been associated with increased risk for developing melanoma.20
A common denominator between the cells affected in CM and PD is their shared neural crest origin. The initiation of CM malignant transformation process occurs presumably early in life and is associated in a number of cases with sunburn episodes during early childhood.40 It may be speculated that melanocytes derived from neuroectodermal stem cells with a germline “PD genotype” may be more prone to UV-induced mutations, increasing the likelihood of acquiring “driver” mutations during early childhood. This may be suggestive of some “genomic instability” that ultimately predisposes to PD and CM.
The present study highlights an association between somatic PD-related gene mutations and CM. Although the functional consequences of the mutations observed in shared genes in both diseases remain to be elucidated, our observation suggests shared pathways and offers one plausible explanation for the observed CM-PD relationship.
Supplementary Material
GLOSSARY
- ADENOCA-LUNG
adenocarcinoma of lung
- CM
cutaneous melanoma
- PD
Parkinson disease
- SNCA
alpha-synuclein gene
- SNPs
single-nucleotide polymorphisms
- SQUAMCA-LUNG
squamous cell carcinoma of lung
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Rivka Inzelberg: drafting/revising the manuscript, study concept or design, analysis or interpretation of data. Yardena Samuels: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, acquisition of data. Esther Azizi: drafting/revising the manuscript, study concept or design. Nouar Qutob: drafting/revising the manuscript, acquisition of data. Lilah Inzelberg: drafting/revising the manuscript, analysis or interpretation of data. Edna Schechtman: drafting/revising the manuscript, analysis or interpretation of data, statistical analysis. Eytan Domany: drafting/revising the manuscript, analysis or interpretation of data. Eitan Friedman: drafting/revising the manuscript, study concept or design, analysis or interpretation of data.
STUDY FUNDING
Y. Samuels is supported by the Israel Science Foundation grants 1604/13 and 877/13, the ERC (StG-335377), the Henry Chanoch Krenter Institute for Biomedical Imaging and Genomics, the estate of Alice Schwarz-Gardos, the estate of John Hunter, the Knell Family, the Peter and Patricia Gruber Award, and the Hamburger Family. E. Domany is supported by a grant from the Leir Charitable Foundation.
DISCLOSURE
Dr. Eytan Domany has served on the editorial board of BMC Bioinformatics Journal of Statistical Mechanics: Theory and Experiment, holds a patent for Serologic diagnosis of pemphigus vulgaris, has received research support from the Leir Charitable Foundation, has received royalty payments for Technology invention (Immunarray Co.), and holds stocks/stock options in Immunarray. The other authors report no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Liu R, Gao X, Lu Y, Chen H. Meta-analysis of the relationship between Parkinson disease and melanoma. Neurology 2011;76:2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kareus SA, Figueroa KP, Cannon-Albright LA, Pulst SM. Shared predispositions of parkinsonism and cancer: a population-based pedigree-linked study. Arch Neurol 2012;69:1572–1577. [DOI] [PubMed] [Google Scholar]
- 3.Gao X, Simon KC, Han J, Schwarzschild MA, Ascherio A. Family history of melanoma and Parkinson disease risk. Neurology 2009;73:1286–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsen JH, Jorgensen TL, Rugbjerg K, Friis S. Parkinson disease and malignant melanoma in first-degree relatives of patients with early-onset melanoma. Epidemiology 2011;22:109–112. [DOI] [PubMed] [Google Scholar]
- 5.Pan T, Li X, Jankovic J. The association between Parkinson's disease and melanoma. Int J Cancer 2011;128:2251–2260. [DOI] [PubMed] [Google Scholar]
- 6.Gao X, Simon KC, Han J, Schwarzschild MA, Ascherio A. Genetic determinants of hair color and Parkinson's disease risk. Ann Neurol 2009;65:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tell-Marti G, Puig-Butille JA, Potrony M, et al. The MC1R melanoma risk variant p.R160W is associated with Parkinson's disease. Ann Neurol 2015;77:889–894. [DOI] [PubMed] [Google Scholar]
- 8.Inzelberg R, Jankovic J. Are Parkinson disease patients protected from some but not all cancers? Neurology 2007;69:1542–1550. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y, Zack TI, Morris LG, et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 2014;46:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng S, Song F, Chen H, et al. No association between Parkinson disease alleles and the risk of melanoma. Cancer Epidemiol Biomarkers Prev 2012;21:243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong J, Gao J, Nalls M, et al. Susceptibility loci for pigmentation and melanoma in relation to Parkinson's disease. Neurobiol Aging 2014;35:1512.e5–1512.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dutton-Regester K, Gartner JJ, Emmanuel R, et al. A highly recurrent RPS27 5'UTR mutation in melanoma. Oncotarget 2014;5:2912–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet 2011;43:442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012;485:502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell 2012;150:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikolaev SI, Rimoldi D, Iseli C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet 2012;44:133–139. [DOI] [PubMed] [Google Scholar]
- 18.Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill VK, Gartner JJ, Samuels Y, Goldstein AM. The genetics of melanoma: recent advances. Annu Rev Genomics Hum Genet 2013;14:257–279. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonifati V. Genetics of Parkinson's disease—state of the art, 2013. Parkinsonism Relat Disord 2014;20(suppl 1):S23–S28. [DOI] [PubMed] [Google Scholar]
- 23.Martin I, Abalde-Atristain L, Kim JW, Dawson TM, Dawson VL. Abberant protein synthesis in G2019S LRRK2 Drosophila Parkinson disease-related phenotypes. Fly (Austin) 2014;8:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Looyenga BD, Furge KA, Dykema KJ, et al. Chromosomal amplification of leucine-rich repeat kinase-2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proc Natl Acad Sci USA 2011;108:1439–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agalliu I, San Luciano M, Mirelman A, et al. Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: a pooled analysis. JAMA Neurol 2015;72:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inzelberg R, Cohen OS, Aharon-Peretz J, et al. The LRRK2 G2019S mutation is associated with Parkinson disease and concomitant non-skin cancers. Neurology 2012;78:781–786. [DOI] [PubMed] [Google Scholar]
- 27.Saunders-Pullman R, Barrett MJ, Stanley KM, et al. LRRK2 G2019S mutations are associated with an increased cancer risk in Parkinson disease. Mov Disord 2010;25:2536–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye J, Pavlicek A, Lunney EA, Rejto PA, Teng CH. Statistical method on nonrandom clustering with application to somatic mutations in cancer. BMC Bioinformatics 2010;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuda S, Nakanishi A, Minami A, Wada Y, Kitagishi Y. Functions and characteristics of PINK1 and parkin in cancer. Front Biosci (Landmark Ed) 2015;20:491–501. [DOI] [PubMed] [Google Scholar]
- 30.Veeriah S, Taylor BS, Meng S, et al. Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 2010;42:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu HH, Kannengiesser C, Lesage S, et al. PARKIN inactivation links Parkinson's disease to melanoma. J Natl Cancer Inst 2016;108. [DOI] [PubMed] [Google Scholar]
- 32.Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 2011;89:398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorval V, Hebert SS. Lrrk2 in Transcription and translation regulation: Relevance for Parkinson's disease. Front Neurol 2012;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bauer C, Diesinger I, Brass N, Steinhart H, Iro H, Meese EU. Translation initiation factor eIF-4G is immunogenic, overexpressed, and amplified in patients with squamous cell lung carcinoma. Cancer 2001;92:822–829. [DOI] [PubMed] [Google Scholar]
- 35.Silvera D, Arju R, Darvishian F, et al. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol 2009;11:903–908. [DOI] [PubMed] [Google Scholar]
- 36.Dhungel N, Eleuteri S, Li LB, et al. Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on alpha-synuclein. Neuron 2015;85:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Chetrit N, Chetrit D, Russell R, et al. Synaptojanin 2 is a druggable mediator of metastasis and the gene is overexpressed and amplified in breast cancer. Sci Signal 2015;8:ra7. [DOI] [PubMed] [Google Scholar]
- 38.Matsuo Y, Kamitani T. Parkinson's disease-related protein, alpha-synuclein, in malignant melanoma. PLoS One 2010;5:e10481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshino H, Tomiyama H, Tachibana N, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 2010;75:1356–1361. [DOI] [PubMed] [Google Scholar]
- 40.Thomas NE, Edmiston SN, Alexander A, et al. Number of nevi and early-life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol Biomarkers Prev 2007;16:991–997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.