

Abstract
Ingenol derivatives with varying degrees of oxidation were prepared by two‐phase terpene synthesis. This strategy has allowed access to analogues that cannot be prepared by semisynthesis from natural ingenol. Complex ingenanes resulting from divergent C—H oxidation of a common intermediate were found to interact with protein kinase C in a manner that correlates well with the oxidation state of the ingenane core. Even though previous work on ingenanes has suggested a strong correlation between potential to activate PKCδ and induction of neutrophil oxidative burst, the current study shows that the potential to activate PKCβII is of key importance while interaction with PKCδ is dispensable. Thus, key modifications of the ingenane core allowed PKC isoform selectivity wherein PKCδ‐driven activation of keratinocytes is strongly reduced or even absent while PKCβII‐driven activation of neutrophils is retained.
Keywords: C—H oxidation, ingenol, natural products, protein kinase C
Protein kinase C (PKC) is a family of protein kinase enzymes that play a central role in controlling cell metabolism, growth, and apoptosis by phosphorylating serine and threonine residues of a large number of proteins involved in intracellular signal transduction.1 PKCs are categorized into three subfamilies: 1) classical/conventional PKCs (cPKCs) consisting of PKCα, PKCβI, PKCβII, and PKCγ isoforms, 2) novel PKCs (nPKCs) consisting of PKCδ, PKCε, PKCθ, and PKCη isoforms, and 3) atypical PKCs (aPKCs) consisting of PKCζ and PKCι isoforms, which share less sequence homology with the two other classes. For activation, the cPKCs require both the endogenous second messenger diacylglycerol (DAG) and Ca2+, whereas the nPKCs only require DAG to be activated. In contrast, neither DAG nor Ca2+ are required for the activation of aPKCs.2 DAG activates cPKCs and nPKCs by binding to the regulatory C1 domain. Interestingly, the C1 domain is also present in other proteins like RasGRPs, where it plays a critical role in the interaction with ingenol mebutate (2, Figure 1 A).3
Figure 1.
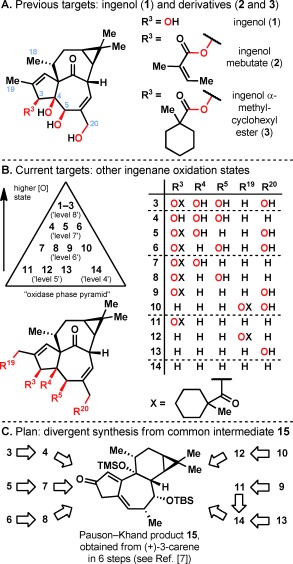
A) Previously synthesized ingenane targets: ingenol (1), ingenol mebutate (2; the active ingredient in Picato), and an ester derivative 3. B) Current ingenane targets with varying oxidation states. C) A synthetic plan that maximizes divergency from previously synthesized intermediate 15.
Ingenol mebutate (2) and several other structurally diverse natural products, including phorbol esters, bryostatins, prostratin, gnidimacrin, mezerein, and resiniferatoxin, act as potent DAG mimetics by binding to the C1 domain of cPKCs and nPKCs.4 However, while a variety of natural compounds activate PKCs, the outcome of PKC activation by a specific compound is highly context‐dependent, with the individual cellular targets (and off‐targets) and overall effects determined by the specific isozyme(s) involved, the timing of PKC activation, the cell type, and the signaling environment.5 This is reflected by comparing the biological activities of 12‐O‐tetradecanoylphorbol‐13‐acetate (TPA), bryostatin 1, and ingenol mebutate (2): the first is a strong tumor promoter, the second shows antiproliferative properties, and latter has antitumoral properties and represents the active ingredient in Picato, a marketed product for the topical treatment of actinic keratosis (solar keratosis).6
The two‐phase total synthesis of ingenol (1)7 and the semisyntheses of ingenol mebutate (2)8 and ingenol 3‐α‐methylcyclohexanecarboxylate (3)9 were reported previously (Figure 1 A). Two‐phase terpene synthesis can both enable scalable access to the parent natural product and constitute a template for preparation of analogues with deep‐seated changes.10 In the case of 1, the latter cannot be rapidly obtained through semisynthesis or synthetic biology. In this work, an exploratory oxidase phase (Figure 1 B) featuring 13 C—H oxidations is used to systematically evaluate the role of the four hydroxy groups in the biological activity of ingenol (1).
The decision to focus primarily on the oxidation pattern of the ingenanes was guided by the hypothesis that the alcohol functionalities have the greatest potential to fine‐tune interactions with PKCs. The work by Winkler and co‐workers demonstrated that simultaneous removal of the C18 and C19 methyl groups and the dimethylcyclopropane moiety resulted in a significant loss of affinity to PKCα,11 although the contribution to the loss of affinity from each group was not clarified. Analogues 4–14 therefore retain these motifs, with only a few C18 desmethyl derivatives synthesized over the course of this research program as an initial step to dissect the importance of these lipophilic groups (see below). Ingenol 3‐α‐methylcyclohexanecarboxylate (3) was chosen as the reference ester because 3 has previously been shown to be more chemically stable and more potent than ingenol mebutate (2).9
The synthesis of analogues 3–14 was accomplished by a two‐phase strategy that closely resembles our previous work on the total synthesis of ingenol (1).7 Since intermediate 15 (prepared from 3‐carene in 6 steps including an efficient Pauson–Khand reaction) is now being synthesized in greater than 100 gram batches at Kemxtree, it was the most logical point of divergence to access 3–14 in a minimal number of synthetic operations (Figure 1 C). The synthetic oxidase phase outlined below delivered novel hydroxylation patterns on the ingenane skeleton and necessitated chemo‐ and regioselective solutions to the complex problem of divergently functionalizing the ingenane core. Selective C—H oxidation was ultimately the enabling method to address this challenge, and a total of 13 C—H oxidation events were carried out in the synthesis of analogues 3–14 and 3′. To the best of our knowledge, this represents the most extensive use to date of C—H oxidation in the biological evaluation of a natural product family.12
To access many different oxidation levels of the ingenane skeleton, Pauson–Khand product 15 was first elaborated into the “more‐oxidized cyclase‐phase endpoint” 16 and “less‐oxidized cyclase‐phase endpoint” 17 in 4 steps and 3 steps, respectively (Figure 2; see the Supporting Information for details). Although cyclase‐phase endpoint 16 had been previously elaborated into ingenol (1; Path A),7 in this work, a critical modification was made wherein a stoichiometric osmium‐mediated dihydroxylation step was rendered catalytic (see the Supporting Information for details). Thereafter, in a diverging pathway, 16 was transformed into C4‐deoxy analogues 6 and 8 (Path B). The “allylic oxidation panel” previously developed during our synthesis of taxuyunnanine D13 was employed throughout this work. After extensive experimentation, a key Pd(OH)2/TBHP step14 (20→21) was found to be critical to the success of this reaction sequence, since the allylic oxidation at C3 was only possible using these conditions. The product of this oxidation reaction, 21, was structurally confirmed by X‐ray analysis to establish the relative stereochemistry of the generated analogues 6 and 8 (see the Supporting Information for details). Likewise, “less‐oxidized cyclase‐phase endpoint” 17 was treated with Pd(OH)2 and TBHP to give allylic alcohol 22, which was further oxidized to give analogues 5 and 7 (Path C).
Figure 2.
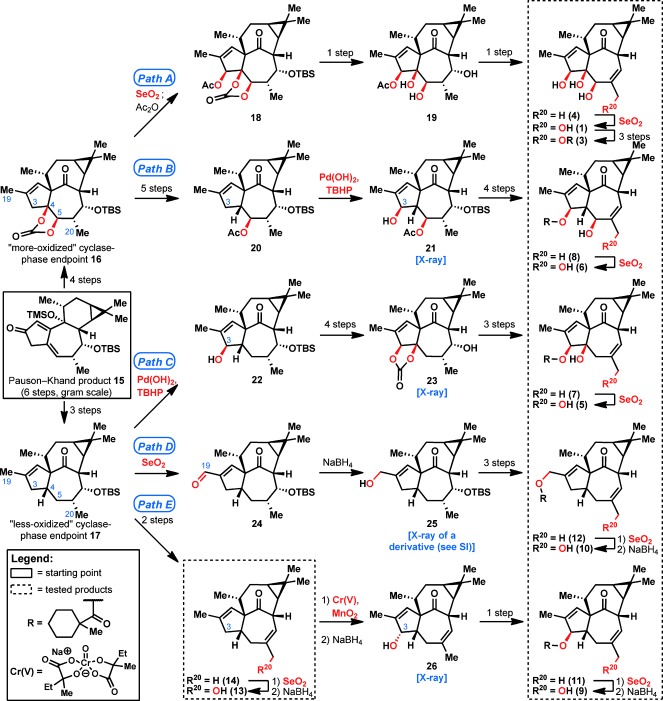
Overview of the synthetic sequence from Pauson–Khand product 15 to ingenol (1) and ingenol analogues 3–14 classified by oxidation level. Oxidation events are shown in red; other transformations have been abbreviated for overall clarity (see the Supporting Information). Ingenol analogues used for biological testing are placed within dotted boxes.
An interesting point of divergence occurred when using intermediate 17, since treatment of 17 with Pd(OH)2 and TBHP gave C3‐oxidized ingenane 22, but SeO2 oxidation gave C19‐oxidized product 24 (Path D). The lack of C3 oxidation when using SeO2 allowed access to analogues 10 and 12, which are devoid of the C3,C4,C5‐hydroxy triad that is so prominent in ingenol (1). Finally, transformation of cyclase‐phase endpoint 17 into ingenol analogue 14 led to another curious finding in oxidation chemistry (Path E): although 14 did not react under Pd(OH)2/TBHP conditions, it was successfully oxidized at C3 when using our recently developed CrV‐based allylic oxidation,13 giving ingenane 26 with an inverted stereocenter at C3. This inversion of stereochemistry did not obstruct the synthetic plan, since it 26 was easily converted into analogues 11 and 9 in 1 and 2 steps, respectively. As such, 10 new ingenol analogues (5–14) were synthesized, with most of these products containing an α‐methylcyclohexanecarboxylate ester for stability and potency. The feat of generating many derivatives with vastly different oxidation states was achieved by embracing the subtleties of C—H oxidation in complex‐molecule synthesis.15
Aside from these analogues with varying oxidation levels, some desmethyl analogues of ingenol (1) were also prepared (Figure 3). Much like in the previous route to 1,7 an 11‐desmethyl analogue of 15 (herein denoted as 15′) was prepared in 6 steps from 3‐carene (see the Supporting Information). Following the remaining steps in the previous synthesis of 1, analogues 4′ and 1′ were generated, along with α‐methylcyclohexanecarboxylate ester 3′. These desmethyl analogues surprisingly did not show reduced potency (see below).
Figure 3.
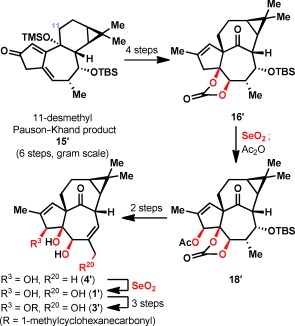
Synthesis of 11‐desmethyl ingenol analogues 1′, 3′, and 4′.
A previously described9 screening cascade was used to characterize immune response‐related effects of C20‐hydroxylated ingenol analogues 5, 6, 9, 10, 13 and desmethyl compound 3′. Thus, studies were conducted for in vitro activation of human recombinant PKCδ, stimulation of IL‐8 release in primary epidermal keratinocytes, and induction of oxidative burst (release of reactive oxygen species) in polymorphonuclear leukocytes (PMN; neutrophils). PKCδ has been shown to be critical for TPA‐induced keratinocyte differentiation.16 In this cell type, PKCδ also exerts pro‐apoptotic functions and is critical for the regulation of both intrinsic and extrinsic apoptosis pathways.17 The finding that PKCδ expression is low or even absent in some human squamous cell carcinomas suggests that it functions as a tumor suppressor in keratinocytes.18 In addition to its role in keratinocytes, PKCδ is also required for the attraction of neutrophils, which are immune cells that are an essential component of the antitumoral action of ingenol mebutate (2).19 This is supported by findings in cultured human endothelial cells, where 2 induces secretion of the neutrophil‐attracting chemokine IL‐8 and surface expression of the adhesion molecules E‐selectin and ICAM‐1 in a PKC‐dependent manner. Knockdown of PKCδ by small interfering RNA (siRNA) completely abolished neutrophil recruitment to endothelial cells subsequently treated with 2, thus proving the essential role of this specific isoform in the process.20
Comparison of the activity of ingenol analogues 5, 6, 9, 10, 13 with the reference ingenol ester 3 in the PKCδ activation and the keratinocyte IL‐8 release assay revealed a clear structure–activity relationship (Figure 4). Removal of a single hydroxy group at either position C5 or C4 (compounds 5 and 6, respectively) only resulted in a moderate loss of potency, and removal of both the C4‐ and C5‐hydroxy groups (9) resulted in a marked loss in activity: approximately 100‐fold in PKCδ activation and 16‐fold in IL‐8 induction when compared to 3. A near‐complete loss of activity was observed for analogue 10, in which the ester function was moved to the C19 position, and C3,C4,C5‐des‐hydroxy ingenol 13 was totally inactive. Surprisingly, despite previous literature data,11 removal of the C18‐methyl group (compound 3′) did not negatively affect the potency of 3.
Figure 4.
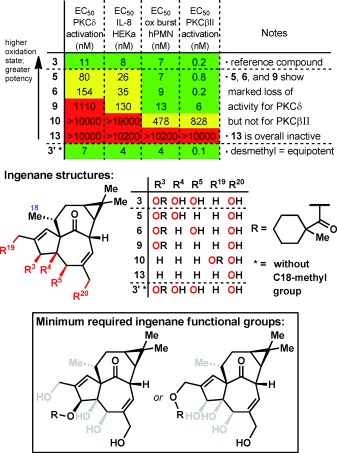
Biological testing of C20‐hydroxylated ingenol analogues 5, 6, 9, 10, 13, and desmethyl compound 3′ compared to reference compound 3, illuminating the minimally required functional groups on the ingenane core (N/A=not available; green=high potency; yellow=intermediate potency; red=low potency/inactive). For a full table including E max values, see the Supporting Information.
Remarkably, the data for neutrophil oxidative burst induction indicates that the C4,C5‐des‐hydroxyl analogue 9 is almost as potent as the reference ingenol ester 3 despite its low potency in activating PKCδ and inducing IL‐8 in keratinocytes. Furthermore, the C19‐ester 10 was established as an agonist of PKCδ with a sub‐micromolar half‐maximal effective concentration (EC50). This finding was unexpected because 10 was completely inactive in both the PKCδ and keratinocyte IL‐8 assay and also because earlier work on C3‐ingenol esters had shown a strong correlation between PKCδ activation and oxidative burst induction.21 Thus, it was hypothesized that a different PKC isoform was critical for oxidative burst induction in neutrophils. In order to identify relevant candidates, mRNA expression levels of classical and novel PKC isoforms were determined in primary human keratinocytes and neutrophils (see the Supporting Information). These data showed that PKCβ, which was barely detectable in keratinocytes, was clearly the dominating isoform in neutrophils, with expression levels more than tenfold higher than that of PKCδ and any of the other isoforms. A critical role of classical PKC isoforms and specifically PKCβ in neutrophils had been suggested earlier22 and is supported by studies showing approximately 50 % inhibition of phorbol‐ester‐induced oxidative burst in neutrophils isolated from PKCβ‐knockout mice and in primary human neutrophils treated with a small‐molecule PKCβ inhibitor.23 Since PKCβI and PKCβII are generated by splicing from a single gene and don’t differ in their regulatory (DAG, Ca2+) and catalytic domains,24 we decided to determine the effect of compounds 5, 6, 9, 10, 13, and 3’ on the activation of PKCβII because this isoform has been reported to be expressed at higher protein levels in human neutrophils25 (Figure 4). In line with our hypothesis, 9 and 10 activated PKCβII with EC50 values of 6 and 828 nm, respectively. These values are in the same range as those observed for the induction of oxidative burst in neutrophils, thus indicating that activation of PKCβII rather than PKCδ is critical for this process. Of note, the EC50 values for 5, 6 and 3’ were all in the sub‐nanomolar range.
In summation, the first systematic study of single‐ and multiple‐functional‐group alteration on ingenol (1) has been accomplished using two‐phase terpene synthesis. Enabled by over a dozen C—H activation steps, modifications of the ingenol core scaffold resulted in analogues with exquisite PKC isoform specificity, i.e., molecules that show low or even absent activity on PKCδ and keratinocytes but retain the potential to activate PKCβII and neutrophils. Beyond their utility as pharmacological tools, such compounds could be of use in therapeutic applications where one would like to avoid the activation of keratinocytes or endothelial cells but retain activity in neutrophils. This work highlights the power of two‐phase terpene synthesis and C—H oxidation to enable the preparation of rationally designed pharmacological probes to illuminate complex biological processes.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
Financial support for this work was provided by LEO Pharma and Bestsyn Technologies (postdoctoral fellowship to Y.J.). We thank Prof. A. L. Rheingold and Dr. C. E. Moore (UCSD) for X‐ray crystallography analysis, Dr. D.‐H. Huang, Dr. L. Pasternack (TSRI) for assistance with NMR spectroscopy, Dr. Y. Ishihara for assistance in the preparation of this manuscript, and Dr. M. Stahlhut and L. Rohde (LEO Pharma) for logistical and technical assistance, respectively.
References
- 1. Mochly‐Rosen D., Das K., Grimes K. V., Nat. Rev. Drug Discovery 2012, 11, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bosco R., Melloni E., Celeghini C., Rimondi E., Vaccarezza M., Zauli G., Mini‐Rev. Med. Chem. 2011, 11, 185. [DOI] [PubMed] [Google Scholar]
- 3. Song X., Lopez‐Campistrous A., Sun L., Dower N. A., Kedei N., Yang J., Kelsey J. S., Lewin N. E., Esch T. E., Blumberg P. M., Stone J. C., PLoS One 2013, 8, e72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Das J., Rahman G. M., Chem. Rev. 2014, 114, 12108. [DOI] [PubMed] [Google Scholar]
- 5. Black A. R., Black J. D., Front. Immunol. 2013, 3, 423.23335926 [Google Scholar]
- 6. Gupta A. K., Paquet M., J. Cutan. Med. Surg. 2013, 17, 173. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Jørgensen L., McKerrall S. J., Kuttruff C. A., Ungeheuer F., Felding J., Baran P. S., Science 2013, 341, 878; [DOI] [PubMed] [Google Scholar]
- 7b. McKerrall S. J., Jørgensen L., Kuttruff C. A., Ungeheuer F., Baran P. S., J. Am. Chem. Soc. 2014, 136, 5799. [DOI] [PubMed] [Google Scholar]
- 8. Liang X., Grue‐Sørensen G., Petersen A. K., Högberg T., Synlett 2012, 2647. [Google Scholar]
- 9. Liang X., Grue‐Sørensen G., Månsson K., Vedsø P., Soor A., Stahlhut M., Bertelsen M., Engell K. M., Högberg T., Bioorg. Med. Chem. Lett. 2013, 23, 5624. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Chen K., Baran P. S., Nature 2009, 459, 824; [DOI] [PubMed] [Google Scholar]
- 10b. Ishihara Y., Baran P. S., Synlett 2010, 1733. [Google Scholar]
- 11.
- 11a. Winkler J. D., Hong B.‐C., Bahador A., Kazanietz M. G., Blumberg P. M., Bioorg. Med. Chem. Lett. 1993, 3, 577; [Google Scholar]
- 11b. Winkler J. D., Kim S., Harrison S., Lewin N. E., Blumberg P. M., J. Am. Chem. Soc. 1999, 121, 296. [Google Scholar]
- 12.A recent example that describes the use of several C—H oxidation steps for medicinal chemistry investigations of a natural product family: Michaudel Q., Journot G., Regueiro‐Ren A., Goswami A., Guo Z., Tully T. P., Zou L., Ramabhadran R. O., Houk K. N., Baran P. S., Angew. Chem. Int. Ed. 2014, 53, 12091; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12287. [Google Scholar]
- 13. Wilde N. C., Isomura M., Mendoza A., Baran P. S., J. Am. Chem. Soc. 2014, 136, 4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu J.‐Q., Corey E. J., J. Am. Chem. Soc. 2003, 125, 3232. [DOI] [PubMed] [Google Scholar]
- 15. Gutekunst W. R., Baran P. S., Chem. Soc. Rev. 2011, 40, 1976.21298176 [Google Scholar]
- 16. Adhikary G., Chew Y. C., Reece E. A., Eckert R. L., J. Invest. Dermatol. 2010, 130, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 17. Zhao M., Xia L., Chen G. Q., Arch. Immunol. Ther. Exp. 2012, 60, 361. [DOI] [PubMed] [Google Scholar]
- 18. D’Costa M., Robinson J. K., Maududi T., Chaturvedi V., Nickoloff B. J., Denning M. F., Oncogene 2006, 25, 378. [DOI] [PubMed] [Google Scholar]
- 19. Challacombe J. M., Suhrbier A., Parsons P. G., Jones B., Hampson P., Kavanagh D., Rainger G. E., Morris M., Lord J. M., Le T. T., Hoang‐Le D., Ogbourne S. M., J. Immunol. 2006, 177, 8123. [DOI] [PubMed] [Google Scholar]
- 20. Hampson P., Kavanagh D., Smith E., Wang K., Lord J. M., Rainger G. E., Cancer Immunol. Immunother. 2008, 57, 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grue‐Sørensen G., Liang X., Månsson K., Vedsø P., Dahl Sørensen M., Soor A., Stahlhut M., Bertelsen M., Engell K. M., Högberg T., Bioorg. Med. Chem. Lett. 2014, 24, 54. [DOI] [PubMed] [Google Scholar]
- 22. Gray R. D., Lucas C. D., Mackellar A., Li F., Hiersemenzel K., Haslett C., Davidson D. J., Rossi A. G., J. Inflammation 2013, 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dekker L. V., Leitges M., Altschuler G., Mistry N., McDermott A., Roes J., Segal A. W., Biochem. J. 2000, 347, 285. [PMC free article] [PubMed] [Google Scholar]
- 24. Kawakami T., Kawakami Y., Kitaura J., J. Biochem. 2002, 132, 677. [DOI] [PubMed] [Google Scholar]
- 25. Balasubramanian N., Advani S. H., Zingde S. M., Leuk. Res. 2002, 26, 67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information