

Abstract
Mucopolysaccharidosis type VII (MPS VII) is a lysosomal storage disease caused by the deficiency of β-glucuronidase. In this study, we compared the changes relative to normal littermates in the proteome and transcriptome of the hippocampus in the C57Bl/6 mouse model of MPS VII, which has well-documented histopathological and neurodegenerative changes. A completely different set of significant changes between normal and MPS VII littermates were found in each assay. Nevertheless, the functional annotation terms generated by the two methods showed agreement in many of the processes, which also corresponded to known pathology associated with the disease. Additionally, assay-specific changes were found, which in the proteomic analysis included mitochondria, energy generation, and cytoskeletal differences in the mutant, while the transcriptome differences included immune, vesicular, and extracellular matrix changes. In addition, the transcriptomic changes in the mutant hippocampus were concordant with those in a MPS VII mouse caused by the same mutation but on a different background inbred strain.
Keywords: mucopolysaccharidosis type VII, MPS VII, lysosomal storage disease, transcriptome, proteomic analysis, mitochondria, β-glucuronidase, GUSB, neurodegeneration, hippocampus
1. Introduction
Mucopolysaccharidosis VII (MPS VII) is a monogenic disease caused by the lack of the enzyme β-glucuronidase (GUSB) and is known to affect intellectual abilities [1, 2]. Lysosomal storage lesions, the hallmark of the disease, and neurodegeneration are present in the hippocampus, which has been implicated in the disease [3-6]. However, the mechanisms by which these lesions lead to neurodegeneration are not known. Proteomics and transcriptomics have both been used to analyze molecular changes associated with disease. Proteome and transcriptome analyses have also been shown to complement each other in areas of overlap and extend the range of findings because of differences in methodology [7].
Transcriptomic analysis of MPS VII versus normal littermate mice on a C3H background has shown changes in pathways and processes common to all regions of the brain, as well as some region-specific alterations [8]. To further assess the changes associated with the MPS VII brain, the present study directly compared proteomics and transcriptomic analyses in the hippocampus from the MPS VII mouse on the C57Bl/6 (B6) strain background in which neurodegeneration has been studied [3, 9, 10]. The combined results of the proteome and transcriptome changes were in functional categories consistent with many of the known histopathology findings [3, 9, 10]. The analyses extended findings of pathological alterations in some non-overlapping areas and provided information on the molecular manifestation of MPS VII disease between strains of mice.
2. Materials and Methods
2.1 Animals
All animal procedures were performed according to protocols approved by the IACUC (Institutional Animal Care and Use Committee) of the Children's Hospital of Philadelphia (CHOP). MPS VII mice and normal controls were generated from the carrier strain B6.C-H2-Kbm1 /ByBir-Gusb mps/+/J [9] and were maintained in our breeding colony through carrier-carrier brother-sister mating. Identification of the MPS VII-affected mice was verified by PCR genotyping, as described previously [10]. Carriers were used for the normal animals and they have been shown to be equivalent to the wild type [8]. Four normal and four MPS VII animals 6 months of age were used for the transcriptomics assay. For the proteomics analysis, three normal and three MPS VII mice age matched to those used for the transcriptome assay were used.
2.2 Micro-dissection of brains
The mice were anesthetized with ketamine/xylazine and the brains were removed and placed immediately on ice. The hemispheres were separated along the medial longitudinal fissure and the hippocampi were dissected out separately from each hemisphere based on anatomical boundaries, as described in [8]. The pieces were immediately frozen in liquid nitrogen and stored at -80 C until used for RNA or protein isolation.
2.3 Protein isolation and analysis
2.3.1Protein extraction and trypsin hydrolysis
Frozen mouse hippocampi of 3 normal and 3 MPS VII mice were thawed in 0.3% SDS, 50mM Tris.HCl, pH 7.8, 0.5 mM MgCl2 (1 mL/50mg wet tissue) and disrupted in a small Dounce homogenizer. The mixture was heated for 5 min at 95C, cooled to room temperature and treated with benzonase to reduce viscosity by hydrolyzing nucleic acids. After centrifugation, a small aliquot of the clear supernatant was reserved for protein assay and the proteins were precipitated from the remainder by adding 20 ug linear polyacrylamide and 5 volumes of acetone and storing at -20C for two hours to overnight. The protein pellet was dissolved in 1× LDS sample buffer (Invitrogen), and resolved on NuPAGE 10% Bis-Tris gels (Invitrogen, Carlsbad, CA) by electrophoresis in MOPS running buffer until the dye front reached ∼3 cm. Proteins were visualized by staining for 10 min with colloidal Coomassie blue and each lane was cut into uniform (2 mm) slices using a MEF-1.5 Gel Cutter (The Gel Company, San Francisco, CA). Individual gel slices were cut into 1 mm cubes, destained, reduced with dithiothreitol, alkylated with iodoacetamide and hydrolyzed with trypsin as previously described [11].
2.3.2 LC-MS/MS analysis
Peptide digests were loaded directly onto a C18 capillary column (75 um × 100 mm; New Objective Proteoprep 2) isocratically in 2% Acetonitrile/0.1%FA at a flow rate of 1 uL per minute using an Eksigent 2D LC system. A linear gradient was then initiated at a flow rate of 300 nL per minute (3% - 40%B over 42 minutes; 40% - 100%B over 3 minutes; then 5 minutes at 100% B). Buffer A was 0.1% FA and Buffer B was 80% Acetonitrile/0.1%FA. Mass spectrometry was performed on a Thermo-Finnegan LTQ mass spectrometer in a data-dependent fashion as peptides were eluted off of the capillary column. A top 5 method was performed in which one survey scan was followed by MS/MS analysis of the 5 most intense ions. MS and MSn thresholds were set to 1500 and 500, respectively. A mass range of 300 – 1800 was implemented for all runs. A repeat count of 3 was selected such that after 3 MS/MS repeats this ion was placed onto an exclusion list for 0.5 minutes. An exclusion window was set to 0.5 below and 1.5 above the target m/z. MS/MS experiments were performed with an isolation width of 2, collision energy of 35, activation Q = 0.25, and an activation time of 30 msec.
2.3.3 Analysis of MS/MS data and database searching
Raw files were searched against the mouse-specific component of the Swiss-Prot database (fasta file created 24 March, 2009) using SEQUEST (Sorcerer2 platform, SageN) search engine to identify peptide MS2 spectral matches. Two missed cleavages were allowed. A fixed modification of S-carbamidomethylation for cysteine, and variable modification for methionine oxidation were used. A precursor mass window of 1.2 and a fragment tolerance of 0.7 Da were utilized for all ion trap–based searches. False discovery rate at the peptide and protein level was controlled using the Peptide Prophet and Protein Prophet algorithms [12, 13] as implemented in the Trans Proteomic Pipeline (TPP v4.0 JETSTREAM rev 2, Build 200902031524, Linux).
2.3.4 APEX quantification of LC-MS/MS datasets
APEX quantification of mouse brain proteins was performed using the APEX Quantitative Proteomics Tool [14] v.1.1 as described previously [15]. Using the interact.prot.xml file from TPP analysis, a training dataset ARFF file was constructed from the 100 most frequently identified proteins. The random forest classifier algorithm was applied to the training set and then to all the in silico-generated tryptic peptides from the mouse fasta file to allow calculation of the complete set of mouse protein observability index (Oi) values. Apex abundances for all the observed mouse brain proteins were finally calculated using the intertact.prot.xml files generated for each experiment by the TPP analysis of the SEQUEST search results.
2.4 RNA isolation and microarray analysis
2.4.1 RNA isolation
Total RNA was isolated from the right hippocampus. Frozen tissue was placed into TRIzol (Invitrogen) at 1 ml per sample and homogenized (Pellet Pestle Motor - Kontes, VWR) at maximum speed for 20-40 Sec. The RNA was further purified using the RNeasy Lipid Tissue mini kit (Qiagen) according to manufacturer's instructions. Total RNA quality was assessed by measuring the A260/280 ratio on a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). RNA integrity was verified by visualization of the 28S and 18S ribosomal rRNA bands, with no presence of smear, using a denaturing TAE- agarose gel.
2.4.2 Microarrays
1 μg RNA was used to prepare biotinylated aRNA samples using the MessageAMP II-biotin Enhanced Signal Round aRNA Amplification Kit (Ambion). Reverse transcription, in vitro transcription and fragmentation were performed according to manufacturer's instructions (Ambion). Samples of 10 μg aRNA were hybridized on Affymetrix mouse genome 430A 2.0 Gene Chips containing 22,690 oligonucleotide probe sets (www.affymetrix.com). Hybridization, staining and washing were performed on an Affymetrix Fluidics Station 400 at the Children's Hospital of Philadelphia Nucleic Acid Core facility according to Affymetrix protocols. Scanning was performed using the Affymetrix Gene Chip Scanner 3000 controlled by a GeneChip Operating software 1.4 (GCOS, Affymetrix).
2.4.3 Data normalization and analysis
Raw microarray image files were processed using the Affymetrix GCOS 1.4 software to calculate individual probe cell intensity data and generate CEL data files. The CEL files were imported into Partek Genomics Suite (v6.5, Partek Inc., St. Louis, MO) where RMA normalization was applied.
2.5 Statistics
2.5.1 Proteomics
Partek Genomics Suite (v6.5, Partek, Inc., St. Louis, MO) was used for statistical analysis. Proteins were considered significantly different at the non-corrected level of p<0.05. The software calculates p-values when one group has a protein detected in only one animal and the other group has that protein detected in multiple animals by assuming the variance of the more complete group. Statistical analysis of proteins undetected in one group but detected in all three animals of the other group and the issue of non-detected proteins is discussed in the results section.
2.5.2 Transcriptomics
Significant Analysis of Microarray (SAM) was used for the transcriptomics significance calculation because its false discovery rate calculation is optimized for microarray analysis [16]. Gene transcription was considered significantly different at the level of q<0.05 with a >1.5 fold change.
2.5.3 Statistics abbreviations
p-value (small p), probability test value for significance; P-value (large P), strength of association in Spearman's Rank Correlation [17] or enrichment value in DAVID analysis [18]; q-value, probability test with false discovery rate calculation [19].
2.5.4 Proteomic/transcriptomic comparison
UniProt protein accession numbers (www.uniprot.org) and Affymetrix probe IDs were converted to DAVID IDs (http://david.abcc.ncifcrf.gov/) for comparison.
2.5.5 Spearman rank order
Two-tailed Spearman rank order was calculated using the online calculator provided by http://www.vassarstats.net [17].
2.5.6 Functional annotation analysis
The significantly changed proteins (p<0.05) and gene transcription changes (q<0.05) were analyzed using Database for Annotation, Visualization and Integrated Discovery v6.7 (DAVID) (http://david.abcc.ncifcrf.gov) [20] for Gene Ontology (GO) terms [21] using the mus musculis background, Kyoto Encyclopedia of Genes and Genomes (KEGG) [22] and other database enrichment and functional clustering, or from literature-search generated gene lists as described in the results section. The Euler diagram of proportionality was generated by EulerAPE from http://www.eulerdiagrams.org/eulerAPE/#Downloads [23]. The functional groups used terms that defined cell processes or pathways and did not include terms that were broadly representative of all cells, such as cell membrane, cytoplasm or nucleus.
3. Results
3.1 Proteomic detection
We chose to analyze a subregion of the brain for proteomic analysis since a previous transcriptome study showed there were significant differences between brain regions in the alterations caused by the disease [8]. The hippocampus was selected because it has been studied in this model both for histopathology [3, 9, 10, 24] and behavioral abnormalities [4-6, 25]. The B6 strain of MPS VII mouse was chosen due to the severity of disease in order to maximize the differences between normal and MPS VII hippocampi [24]. The disease features in the C3H strain are essentially the same as the B6 background at the end-stage, but C3H has a significantly longer lifespan [24].
A total of 3268 independent proteins were detected in the hippocampus among all of the mice (Supplemental Data 1.xls), but not all proteins were detected in every animal, which is a common finding in gel based mass spectrometry analyses [26-28]. A total of 2989 unique proteins were detected among the normal mice and 2686 among the MPS VII mice. Significant differences between genotypes were found in 189 proteins (p <0.05).
A second group of protein changes were those detected in all mice of one genotype but in none of the other genotype, but a p-value and fold change could not be calculated because the actual level of the undetected group was unknown. The level of a protein that was not detected in one genotype was hypothesized to be less than the level in the genotype where the same protein was detected in all animals, and thus likely to represent a meaningful biological difference. To evaluate this assumption and determine if those protein changes could be included in the analysis, we used a “likelihood” approach that has been applied to non-detected proteins in proteomic analysis [28]. The average level for all the proteins detected in all three animals was compared to the average of those detected in any two animals and to the average of those detected in just one animal within the phenotype groups. The average level for all of the proteins detected in all three animals was 5-fold greater than the average level for those detected in just two animals (p<0.001) and was 10 fold greater than the average of all the proteins detected in any one animal (p<0.0001) (Supplemental Data 2.xlsx). This is consistent with the finding that proteomics favors the detection of the more abundant proteins [26, 27] and conversely, that non-detected proteins are likely to be associated with lower protein levels.
On the basis of this, it was concluded that the level of a protein not detected in any animal of one genotype was likely lower than when it was detected in all 3 of the other genotype. This group included 68 proteins, with 59 in the normal animals and 9 in the mutants. and were included in the analysis of mutant vs normal with only a direction of change (arrows in tables) assigned without p-value or fold change. This likelihood basis was also used to remove 12 outliers (6 increases, 6 decreases) where proteins detected in only one animal of a genotype had a higher level than the average for the other genotype with multiple detections. Thus the total number of proteins included in the analysis of changes in the MPS VII hippocampus was 245 (7% of the detected proteins) (Table 1). Of these, 174 proteins were decreased (71%) and 71 were increased (29%) in the MPS VII brain. These were then analyzed for changes in functional annotation terms, which require multiple changes per term, rather than individual gene products.
Table 1. Proteomic changes sorted by fold change in the MPS VII hippocampus.
Up and down arrows indicate direction of change when fold-change could not be calculated as discussed in the results section.
| Uniprot ID | Gene Name | p-value | Fold | Description |
|---|---|---|---|---|
| P10922 | H1f0 | ↑ | H1 histone family, member 0 | |
| P17047 | Lamp2 | ↑ | lysosomal-associated membrane protein 2 | |
| P97441 | Slc30a3 | ↑ | solute carrier family 30 (zinc transporter), member 3 | |
| Q61035 | Hars | ↑ | histidyl-tRNA synthetase | |
| Q68FD9 | Kiaa1549 | ↑ | RIKEN cDNA D630045J12 gene | |
| Q7TQ95 | Lnp | ↑ | limb and neural patterns | |
| Q924L1 | Letmd1 | ↑ | LETM1 domain containing 1 | |
| Q9DCP2 | Slc38a3 | ↑ | solute carrier family 38, member 3 | |
| Q9EPR4 | Slc23a2 | ↑ | solute carrier family 23 (nucleobase transporters), member 2 | |
| Q64310 | Surf4 | <0.001 | 5.73 | surfeit gene 4 |
| Q5DU25 | Iqsec2 | 0.010 | 4.02 | IQ motif and Sec7 domain 2 |
| A2RT62 | Fbxl16 | 0.016 | 3.65 | F-box and leucine-rich repeat protein 16 |
| P12367 | Prkar2a | 0.004 | 3.51 | protein kinase, cAMP dependent regulatory, type II alpha |
| O88712 | Ctbp1 | 0.012 | 3.12 | similar to CtBP1 protein; C-terminal binding protein 1 |
| P62881 | Gnb5 | 0.001 | 3.10 | guanine nucleotide binding protein (G protein), beta 5 |
| Q8BY89 | Slc44a2 | 0.024 | 2.85 | solute carrier family 44, member 2 |
| Q8CGF7 | Tcerg1 | 0.032 | 2.84 | transcription elongation regulator 1 (CA150) |
| Q8BTY2 | Slc4a7 | 0.006 | 2.84 | solute carrier family 4, sodium bicarbonate cotransporter, member 7 |
| Q8C0L0 | Txndc13 | 0.005 | 2.82 | thioredoxin-related transmembrane protein 4 |
| P14115 | Rpl27a | 0.002 | 2.77 | predicted gene 14439; predicted gene 8213; predicted gene 13981 |
| Q99020 | Hnrnpab | 0.038 | 2.73 | heterogeneous nuclear ribonucleoprotein A/B |
| Q8BIW1 | Prune | 0.031 | 2.71 | predicted gene 5217; prune homolog (Drosophila) |
| Q791T5 | Mtch1 | 0.044 | 2.70 | mitochondrial carrier homolog 1 (C. elegans) |
| Q8R5J9 | Arl6ip5 | 0.043 | 2.63 | ADP-ribosylation factor-like 6 interacting protein 5 |
| Q9D8E6 | Rpl4 | 0.003 | 2.58 | predicted gene 5835; ribosomal protein L4 |
| P61620 | Sec61a1 | 0.004 | 2.58 | Sec61 alpha 1 subunit (S. cerevisiae) |
| Q9CR95 | Necap1 | 0.016 | 2.57 | NECAP endocytosis associated 1 |
| P03893 | Mtnd2 | 0.028 | 2.45 | NADH-ubiquinone oxidoreductase chain 2 |
| Q9ESW4 | Agk | 0.004 | 2.38 | predicted gene 8546; acylglycerol kinase |
| Q8JZR6 | Slc4a8 | 0.015 | 2.35 | solute carrier family 4 (anion exchanger), member 8 |
| O35609 | Scamp3 | 0.043 | 2.34 | secretory carrier membrane protein 3 |
| P98086 | C1qa | 0.003 | 2.34 | complement component 1, q subcomponent, alpha polypeptide |
| Q9D832 | Dnajb4 | 0.041 | 2.23 | DnaJ (Hsp40) homolog, subfamily B, member 4 |
| P54285 | Cacnb1 | 0.021 | 2.15 | calcium channel, voltage-dependent, beta 3 subunit |
| Q8K2C9 | Ptplad1 | 0.013 | 2.14 | protein tyrosine phosphatase-like A domain containing 1 |
| Q9CY27 | Gpsn2 | 0.035 | 2.14 | predicted gene 4948; glycoprotein, synaptic 2 |
| Q8BVQ5 | Ppme1 | 0.039 | 2.08 | protein phosphatase methylesterase 1 |
| Q8BWQ6 | UPF0505 | 0.041 | 2.06 | RIKEN cDNA 9030624J02 gene |
| P63001 | Rac1 | 0.029 | 2.02 | RAS-related C3 botulinum substrate 1 |
| P05063 | Aldoc | 0.041 | 1.95 | aldolase C, fructose-bisphosphate |
| Q60668 | Hnrnpd | 0.035 | 1.91 | heterogeneous nuclear ribonucleoprotein D |
| Q9JKK7 | Tmod2 | 0.015 | 1.91 | tropomodulin 2 |
| Q00612 | G6pdx | 0.026 | 1.90 | glucose-6-phosphate dehydrogenase X-linked |
| Q3UVX5 | Grm5 | 0.048 | 1.88 | glutamate receptor, metabotropic 5 |
| Q9DB10 | UPF0466 | 0.039 | 1.87 | RIKEN cDNA 1500032L24 gene |
| Q07076 | Anxa7 | 0.014 | 1.85 | annexin A7 |
| Q8BI08 | Mal2 | 0.010 | 1.83 | mal, T-cell differentiation protein 2 |
| P14206 | Rpsa | 0.048 | 1.83 | ribosomal protein SA pseudogene |
| Q9ERD7 | Tubb3 | 0.043 | 1.80 | tubulin, beta 3; tubulin, beta 3, pseudogene 1 |
| Q3V0K9 | Pls1 | 0.032 | 1.80 | plastin 1 (I-isoform) |
| Q9JKD3 | Scamp5 | 0.004 | 1.79 | secretory carrier membrane protein 5 |
| P26638 | Sars | 0.039 | 1.74 | seryl-aminoacyl-tRNA synthetase |
| P32921 | Wars | 0.002 | 1.66 | tryptophanyl-tRNA synthetase; similar to tryptophanyl-tRNA synthetase |
| Q8CHH9 | Sept8 | 0.046 | 1.66 | septin 8 |
| Q9CPU4 | Mgst3 | 0.025 | 1.65 | microsomal glutathione S-transferase 3 |
| P54775 | Psmc4 | 0.020 | 1.63 | proteasome (prosome, macropain) 26S subunit, ATPase, 4 |
| P84091 | Ap2m1 | 0.029 | 1.62 | predicted gene 8717; adaptor protein complex AP-2, mu1 |
| Q9CZU6 | Cs | 0.032 | 1.57 | citrate synthase |
| Q640R3 | Hepacam | 0.010 | 1.55 | hepatocyte cell adhesion molecule |
| Q9CPR4 | Rpl17 | 0.019 | 1.55 | predicted gene 8081; similar to Ribosomal protein L17; |
| Q8BWF0 | Aldh5a1 | 0.025 | 1.54 | aldhehyde dehydrogenase family 5, subfamily A1 |
| P50396 | Gdi1 | 0.016 | 1.53 | guanosine diphosphate (GDP) dissociation inhibitor 1 |
| P05201 | Got1 | 0.046 | 1.47 | similar to Aspartate aminotransferase |
| Q9D051 | Pdhb | 0.035 | 1.41 | predicted gene 6123; pyruvate dehydrogenase (lipoamide) beta |
| P63080 | Gabrb3 | 0.026 | 1.38 | gamma-aminobutyric acid (GABA) A receptor, subunit beta 3 |
| P19246 | Nefh | 0.021 | 1.38 | similar to neurofilament protein; neurofilament, heavy polypeptide |
| P54071 | Idh2 | 0.015 | 1.29 | isocitrate dehydrogenase 2 (NADP+), mitochondrial |
| Q8BH59 | Slc25a12 | 0.019 | 1.28 | solute carrier family 25 (mitochondrial carrier, Aralar), member 12 |
| Q62277 | Syp | 0.047 | 1.25 | synaptophysin |
| Q99L43 | Cds2 | 0.026 | 1.24 | CDP-diacylglycerol synthase (phosphatidate cytidylyltransferase) 2 |
| P06745 | Gpi | 0.010 | 1.12 | glucose phosphate isomerase 1 |
| Q11011 | Npepps | 0.023 | -1.24 | aminopeptidase puromycin sensitive |
| Q8K310 | Matr3 | 0.032 | -1.32 | matrin 3; similar to Matrin 3 |
| Q9ERS2 | Ndufa13 | 0.034 | -1.49 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 13 |
| O54774 | Ap3d1 | 0.043 | -1.51 | adaptor-related protein complex 3, delta 1 subunit |
| P47802 | Mtx1 | 0.034 | -1.53 | metaxin 1 |
| P56382 | Atp5e | 0.038 | -1.54 | ATP synthase, H+ transporting, mitochondrial F1 complex, Ɛ subunit |
| Q60597 | Ogdh | 0.041 | -1.58 | oxoglutarate dehydrogenase (lipoamide) |
| P19096 | Fasn | 0.022 | -1.62 | fatty acid synthase |
| P52196 | Tst | 0.046 | -1.62 | thiosulfate sulfurtransferase, mitochondrial |
| Q9DCW4 | Etfb | 0.035 | -1.65 | similar to Electron transferring flavoprotein |
| Q9Z1B3 | Plcb1 | 0.028 | -1.67 | phospholipase C, beta 1 |
| Q9CQW1 | Ykt6 | 0.002 | -1.71 | YKT6 homolog (S. Cerevisiae) |
| Q61316 | Hspa4 | 0.002 | -1.77 | heat shock protein 4 |
| O35683 | Ndufa1 | 0.045 | -1.80 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 1 |
| P54227 | Stmn1 | 0.041 | -1.81 | stathmin 1; predicted gene 11223; predicted gene 6393 |
| Q80TR1 | Lphn1 | 0.038 | -1.81 | latrophilin 1 |
| Q8BKC5 | Ipo5 | 0.004 | -1.85 | hypothetical protein LOC100044315; importin 5 |
| Q9CPP6 | Ndufa5 | 0.035 | -1.85 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 5 |
| Q4KMM3 | Oxr1 | 0.019 | -1.87 | oxidation resistance 1 |
| P56375 | Acyp2 | 0.035 | -1.89 | acylphosphatase 2, muscle type |
| Q9CPW4 | Arpc5 | 0.044 | -1.90 | predicted gene 16372; actin related protein 2/3 complex, subunit 5 |
| Q8BIJ6 | Iars2 | 0.047 | -1.95 | isoleucine-tRNA synthetase 2, mitochondrial |
| Q8BHN3 | Ganab | 0.017 | -1.95 | alpha glucosidase 2 alpha neutral subunit |
| Q9QUH0 | Glrx | 0.041 | -1.97 | glutaredoxin |
| O08788 | Dctn1 | 0.042 | -1.98 | dynactin 1 |
| P97450 | Atp5j | 0.046 | -2.00 | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F pseudogene; similar to ATP synthase coupling factor 6, mitochondrial precursor (ATPase subunit F6); ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F |
| Q66GT5 | Ptpmt1 | 0.022 | -2.02 | protein tyrosine phosphatase, mitochondrial 1 |
| P48722 | Hspa4l | 0.026 | -2.04 | heat shock protein 4 like |
| A2AG50 | Map7d2 | 0.035 | -2.07 | MAP7 domain containing 2 |
| Q148V7 | Kiaa1468 | 0.021 | -2.07 | RIKEN cDNA 2310035C23 gene |
| Q9CQ60 | Pgls | 0.033 | -2.09 | 6-phosphogluconolactonase |
| P84089 | Erh | 0.049 | -2.11 | predicted gene 6941; enhancer of rudimentary homolog (Drosophila) |
| Q60902 | Eps15l1 | 0.033 | -2.12 | epidermal growth factor receptor pathway substrate 15-like 1 |
| Q8BGX2 | Q8BGX2 | 0.025 | -2.13 | predicted gene 5747; RIKEN cDNA 1810026J23 gene |
| Q9DB70 | Fundc1 | 0.046 | -2.16 | FUN14 domain containing 1 |
| P58281 | Opa1 | 0.026 | -2.17 | similar to optic atrophy 1; optic atrophy 1 homolog (human) |
| P70336 | Rock2 | 0.014 | -2.22 | Rho-associated coiled-coil containing protein kinase 2 |
| Q91V92 | Acly | <0.001 | -2.25 | ATP citrate lyase |
| Q80UJ7 | Rab3gap1 | 0.012 | -2.27 | RAB3 GTPase activating protein subunit 1 |
| Q9JI46 | Nudt3 | 0.012 | -2.31 | nudix (nucleotide diphosphate linked moiety X)-type motif 3; similar to diphosphoinositol polyphosphate phosphohydrolase |
| Q60865 | Caprin1 | 0.015 | -2.32 | cell cycle associated protein 1 |
| Q9D5V5 | Cul5 | 0.042 | -2.35 | cullin 5 |
| Q62446 | Fkbp3 | 0.024 | -2.40 | FK506 binding protein 3 |
| O08585 | Clta | 0.043 | -2.40 | clathrin, light polypeptide (Lca) |
| O54962 | Banf1 | 0.023 | -2.42 | barrier to autointegration factor 1 |
| P62627 | Dynlrb1 | 0.030 | -2.45 | dynein light chain roadblock-type 1 |
| Q0GNC1 | Inf2 | 0.004 | -2.45 | subacute ozone induced inflammation |
| Q05920 | Pc | 0.006 | -2.45 | pyruvate carboxylase |
| Q58A65 | Spag9 | 0.022 | -2.46 | sperm associated antigen 9 |
| Q8K3H0 | Appl1 | 0.049 | -2.47 | adaptor protein, phosphotyrosine interaction, PH domain and leucine zipper containing 1 |
| Q9D1C8 | Vps28 | 0.003 | -2.48 | vacuolar protein sorting 28 (yeast) |
| Q80ZJ1 | Rap2a | 0.005 | -2.48 | RAS related protein 2a |
| O88653 | Mapksp1 | 0.045 | -2.49 | similar to Mitogen-activated protein kinase kinase 1 interacting protein 1 (MEK binding partner 1) (Mp1); MAPK scaffold protein 1 |
| Q61699 | Hsph1 | 0.014 | -2.49 | heat shock 105kDa/110kDa protein 1 |
| Q9EPW0 | Inpp4a | 0.034 | -2.51 | inositol polyphosphate-4-phosphatase, type I |
| Q9Z2H5 | Epb41l1 | 0.028 | -2.53 | erythrocyte protein band 4.1-like 1 |
| P97493 | Txn2 | 0.046 | -2.55 | thioredoxin 2 |
| Q8BWR2 | Trp26 | 0.040 | -2.55 | RIKEN cDNA 1110049F12 gene |
| Q9CQI3 | Gmfb | 0.017 | -2.57 | glia maturation factor, beta |
| P61971 | Nutf2 | 0.022 | -2.58 | similar to Chain A, D92n,D94n Double Point Mutant Of Human Nuclear Transport Factor 2 (Ntf2); nuclear transport factor 2; |
| P84086 | Cplx2 | 0.015 | -2.58 | complexin 2 |
| Q8BGQ7 | Aars | 0.026 | -2.63 | alanyl-tRNA synthetase |
| Q8VCT3 | Rnpep | 0.020 | -2.65 | arginyl aminopeptidase (aminopeptidase B) |
| Q80Y14 | Glrx5 | 0.034 | -2.68 | glutaredoxin 5 homolog (S. cerevisiae) |
| Q9CQ85 | Timm22 | 0.017 | -2.69 | translocase of inner mitochondrial membrane 22 homolog (yeast) |
| Q811D0 | Dlg1 | 0.044 | -2.69 | discs, large homolog 1 (Drosophila); similar to Discs, large homolog 1 (Drosophila) |
| Q9CQ69 | Uqcrq | 0.019 | -2.70 | ubiquinol-cytochrome c reductase, complex III subunit VII |
| Q91VR8 | Brk1 | 0.030 | -2.70 | RIKEN cDNA 6720456B07 gene |
| O35127 | Grcc10 | 0.048 | -2.72 | gene rich cluster, C10 gene |
| O88851 | Rbbp9 | 0.024 | -2.76 | retinoblastoma binding protein 9; similar to Retinoblastoma-binding protein 9 (RBBP-9) (B5T overexpressed gene protein) (Bog protein) |
| Q8BGS2 | Bola2 | 0.024 | -2.78 | bolA-like 2 (E. coli) |
| P27546 | Map4 | 0.018 | -2.80 | microtubule-associated protein 4 |
| Q9EQ80 | Nif3l1 | 0.031 | -2.81 | Ngg1 interacting factor 3-like 1 (S. pombe) |
| Q61330 | Cntn2 | 0.020 | -2.82 | contactin 2 |
| Q9WVL0 | Gstz1 | 0.004 | -2.89 | glutathione transferase zeta 1 (maleylacetoacetate isomerase) |
| Q9D0R2 | Tars | 0.030 | -2.89 | threonyl-tRNA synthetase |
| Q9JKR6 | Hyou1 | 0.044 | -2.96 | hypoxia up-regulated 1 |
| Q9CZD3 | Gars | 0.009 | -3.06 | glycyl-tRNA synthetase |
| Q9CQX8 | Mrps36 | 0.028 | -3.13 | predicted gene 10078; predicted gene 3544; similar to mitochondrial ribosomal protein S36; mitochondrial ribosomal protein S36 |
| Q8VD37 | Sgip1 | 0.035 | -3.27 | SH3-domain GRB2-like (endophilin) interacting protein 1 |
| Q8BU30 | Iars | 0.046 | -3.28 | isoleucine-tRNA synthetase |
| P23116 | Eif3a | 0.013 | -3.38 | eukaryotic translation initiation factor 3, subunit A |
| Q8VBV7 | Cops8 | 0.031 | -3.44 | COP9 (constitutive photomorphogenic) homolog, subunit 8 |
| Q61301 | Ctnna2 | 0.048 | -3.44 | catenin (cadherin associated protein), alpha 2 |
| Q9D7X8 | Ggct | 0.046 | -3.46 | gamma-glutamyl cyclotransferase |
| Q80X50 | Ubap2l | 0.049 | -3.49 | ubiquitin associated protein 2-like |
| P28740 | Kif2a | 0.011 | -3.52 | kinesin family member 2A |
| P21619 | Lmnb2 | 0.042 | -3.60 | lamin B2 |
| P28271 | Aco1 | 0.020 | -3.61 | aconitase 1 |
| P60469 | Ppfia3 | 0.021 | -3.65 | protein tyrosine phosphatase, receptor type, f polypeptide (PTPRF), interacting protein (liprin), alpha 3 |
| Q8BRT1 | Clasp2 | 0.016 | -3.67 | CLIP associating protein 2 |
| Q9QYB5 | Add3 | 0.026 | -3.72 | adducin 3 (gamma) |
| Q80UG2 | Plxna4 | 0.004 | -3.76 | plexin A4 |
| Q9QWI6 | P140 | 0.017 | -3.89 | P140 gene |
| O70161 | Pip5k1c | 0.016 | -3.93 | phosphatidylinositol-4-phosphate 5-kinase, type 1 gamma |
| Q8CGY8 | Ogt | 0.014 | -4.02 | O-linked N-acetylglucosamine (GlcNAc) transferase (UDP-N-acetylglucosamine:polypeptide-N-acetylglucosaminyl transferase) |
| P61458 | Pcbd1 | 0.035 | -4.15 | pterin 4 alpha carbinolamine dehydratase/dimerization cofactor of hepatocyte nuclear factor 1 alpha (TCF1) 1 |
| Q9JMH9 | Myo18a | 0.022 | -4.30 | myosin XVIIIA |
| Q3U0V1 | Khsrp | 0.002 | -4.32 | KH-type splicing regulatory protein |
| P35123 | Usp11 | <0.001 | -4.40 | ubiquitin specific peptidase 4 (proto-oncogene) |
| Q64737 | Gart | 0.011 | -4.57 | phosphoribosylglycinamide formyltransferase |
| Q3UPL0 | Sec31a | 0.008 | -4.66 | Sec31 homolog A (S. cerevisiae) |
| Q9DBR7 | Ppp1r12a | 0.009 | -4.79 | protein phosphatase 1, regulatory (inhibitor) subunit 12A |
| Q9EQQ9 | Mgea5 | 0.011 | -5.00 | meningioma expressed antigen 5 (hyaluronidase) |
| Q9DB27 | Mcts1 | 0.034 | -5.03 | malignant T cell amplified sequence 1 |
| Q91YE6 | Ipo9 | 0.029 | -5.50 | importin 9 |
| Q9QXL2 | Kif21a | 0.022 | -5.61 | kinesin family member 21A |
| Q4ACU6 | Shank3 | 0.011 | -5.85 | SH3/ankyrin domain gene 3 |
| Q9Z130 | Hnrpdl | 0.035 | -6.04 | heterogeneous nuclear ribonucleoprotein D-like |
| Q9CXW3 | Cacybp | 0.018 | -6.74 | calcyclin binding protein |
| Q6P9K8 | Caskin1 | 0.043 | -6.95 | CASK interacting protein 1 |
| Q7TMB8 | Cyfip1 | 0.001 | -7.32 | cytoplasmic FMR1 interacting protein 1 |
| Q80U40 | Rimbp2 | 0.015 | -7.67 | RIMS binding protein 2 |
| P02468 | Lamc1 | 0.010 | -10.86 | laminin, gamma 1 |
| Q3UHD9 | Agap2 | <0.001 | -11.45 | ArfGAP with GTPase domain, ankyrin repeat and PH domain 2 |
| P08556 | Nras | ↓ | similar to neuroblastoma ras oncogene; neuroblastoma ras oncogene | |
| P36536 | Sar1a | ↓ | SAR1 gene homolog A (S. cerevisiae) | |
| P39054 | Dnm2 | ↓ | dynamin 2 | |
| P58059 | Mrps21 | ↓ | mitochondrial ribosomal protein S21; predicted gene 6686; predicted gene 6181 | |
| P59708 | Sf3b14 | ↓ | RIKEN cDNA 0610009D07 gene | |
| P61148 | Fgf1 | ↓ | fibroblast growth factor 1 | |
| P98203 | Arvcf | ↓ | armadillo repeat gene deleted in velo-cardio-facial syndrome | |
| Q03173 | Enah | ↓ | enabled homolog (Drosophila) | |
| Q05A62 | Dnal1 | ↓ | dynein, axonemal, light chain 1 | |
| Q3TDD9 | Klraq1 | ↓ | KLRAQ motif containing 1 | |
| Q3TES0 | Iqsec3 | ↓ | IQ motif and Sec7 domain 3 | |
| Q3U487 | Hectd3 | ↓ | HECT domain containing 3 | |
| Q3UH68 | Limch1 | ↓ | LIM and calponin homology domains 1 | |
| Q3UU96 | Cdc42bpa | ↓ | CDC42 binding protein kinase alpha | |
| Q5F2E8 | Taok1 | ↓ | TAO kinase 1 | |
| Q5SSM3 | Rich2 | ↓ | expressed sequence AU040829 | |
| Q5SXY1 | Specc1 | ↓ | cytospin B | |
| Q5XJV6 | Lmtk3 | ↓ | lemur tyrosine kinase 3 | |
| Q64152 | Btf3 | ↓ | predicted gene 9308; basic transcription factor 3; | |
| Q69ZW3 | Ehbp1 | ↓ | EH domain binding protein 1 | |
| Q6A065 | Cep170 | ↓ | centrosomal protein 170 | |
| Q6NVE8 | Wdr44 | ↓ | WD repeat domain 44 | |
| Q6PAR5 | Gapvd1 | ↓ | GTPase activating protein and VPS9 domains 1 | |
| Q6PDI5 | Ecm29 | ↓ | expressed sequence AI314180 | |
| Q6PFD5 | Dlgap3 | ↓ | discs, large (Drosophila) homolog-associated protein 3 | |
| Q6Y7W8 | Gigyf2 | ↓ | GRB10 interacting GYF protein 2 | |
| Q7SIG6 | Asap2 | ↓ | development and differentiation enhancing factor 2 | |
| Q7TSC1 | Bat2 | ↓ | HLA-B associated transcript 2 | |
| Q80U49 | Kiaa0284 | ↓ | expressed sequence AW555464 | |
| Q810B6 | Ankfy1 | ↓ | ankyrin repeat and FYVE domain containing 1 | |
| Q8BGR6 | Arl15 | ↓ | ADP-ribosylation factor-like 15 | |
| Q8BLY2 | Tarsl2 | ↓ | threonyl-tRNA synthetase-like 2 | |
| Q8BMI3 | Gga3 | ↓ | golgi associated, gamma adaptin ear containing, ARF binding protein 3 | |
| Q8BPM0 | Daam1 | ↓ | dishevelled associated activator of morphogenesis 1 | |
| Q8BY87 | Usp47 | ↓ | ubiquitin specific peptidase 47 | |
| Q8C1B1 | Camsap1l1 | ↓ | calmodulin regulated spectrin-associated protein 1-like 1 | |
| Q8CC88 | Kiaa0564 | ↓ | RIKEN cDNA 1300010F03 gene | |
| Q8CCN5 | Bcas3 | ↓ | breast carcinoma amplified sequence 3 | |
| Q8CDG3 | Vcpip1 | ↓ | valosin containing protein (p97)/p47 complex interacting protein 1 | |
| Q8K394 | Plcl2 | ↓ | phospholipase C-like 2 | |
| Q8QZZ7 | Tprkb | ↓ | Tp53rk binding protein | |
| Q8R4H2 | Arhgef12 | ↓ | similar to SP140 nuclear body protein (predicted); Rho guanine nucleotide exchange factor (GEF) 12 | |
| Q91WV0 | Dr1 | ↓ | down-regulator of transcription 1 | |
| Q923D5 | Wbp11 | ↓ | WW domain binding protein 11 | |
| Q9CPW2 | Fdx1l | ↓ | ferredoxin 1-like | |
| Q9CQV7 | Dnajc19 | ↓ | DnaJ (Hsp40) homolog, subfamily C, member 19 | |
| Q9CQZ1 | Hsbp1 | ↓ | heat shock factor binding protein 1 | |
| Q9D0L7 | Armc10 | ↓ | predicted gene 9209; armadillo repeat containing 10 | |
| Q9D1L0 | Chchd2 | ↓ | coiled-coil-helix-coiled-coil-helix domain containing 2; predicted gene 13202; similar to coiled-coil-helix-coiled-coil-helix domain containing 2; predicted gene 12350 | |
| Q9D8S9 | Bola1 | ↓ | bolA-like 1 (E. coli) | |
| Q9D8T7 | Slirp | ↓ | RIKEN cDNA 1810035L17 gene | |
| Q9ERG2 | Strn3 | ↓ | striatin, calmodulin binding protein 3 | |
| Q9ERU9 | Ranbp2 | ↓ | RAN binding protein 2 | |
| Q9JKL4 | Ndufaf3 | ↓ | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, assembly factor 3 | |
| Q9JL26 | Fmnl1 | ↓ | formin-like 1 | |
| Q9QWY8 | Asap1 | ↓ | similar to Development and differentiation enhancing; ArfGAP with SH# domain, ankyrin repeat and PH domain1 | |
| Q9R1Z7 | Pts | ↓ | 6-pyruvoyl-tetrahydropterin synthase | |
| Q9Z2I2 | Fkbp1b | ↓ | FK506 binding protein 1b | |
| Q9Z2V5 | Hdac6 | ↓ | histone deacetylase 6 |
3.2 Proteomic annotation enrichment analysis
The functional annotation terms were generated for the protein changes with DAVID enrichment analysis (http://david.abcc.ncifcrf.gov/) using the same analytical approach as our previous transcriptome analysis [8]. A minimum of 3 significantly changed proteins were required per annotation term. A total of 743 unique terms were generated (Supplemental Data 3.pdf) and related terms were consolidated into larger functional categories representing related biological processes (Table 2).
Table 2. Functional categories of terms from the proteomic and transcriptomic significant changes.
Functional categories are ranked by the % of the total proteomic terms.
| Proteomic | Transcriptomic | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Functional category | Terms | % of terms | Proteins | terms | % of terms | Genes |
| cytoskeleton | 62 | 8.4 | 66 | 3 | 1.3 | 3 |
| neuron | 59 | 8.0 | 41 | 3 | 1.3 | 4 |
| transcription | 48 | 6.5 | 47 | 14 | 6.1 | 4 |
| mitochondria | 47 | 6.3 | 80 | 4 | 1.7 | 8 |
| signaling | 37 | 5.0 | 47 | 5 | 2.2 | 5 |
| ion transport/channels | 35 | 4.7 | 34 | 7 | 3.0 | 8 |
| nucleus | 30 | 4.0 | 69 | 1 | 0.4 | 3 |
| vesicle | 30 | 4.0 | 37 | 24 | 10.4 | 14 |
| metabolism | 28 | 3.8 | 20 | 8 | 3.5 | 9 |
| transport | 25 | 3.4 | 51 | 1 | 0.4 | 7 |
| development | 13 | 1.8 | 12 | 4 | 1.7 | 5 |
| extracellular matrix/adhesion | 12 | 1.6 | 15 | 10 | 4.3 | 28 |
| apoptosis | 12 | 1.6 | 7 | 5 | 2.2 | 5 |
| immune | 8 | 1.1 | 12 | 66 | 28.7 | 45 |
| protein modification | 11 | 1.5 | 25 | |||
| neural disease | 10 | 1.3 | 13 | |||
| ubiquitin | 10 | 1.3 | 12 | |||
| cell cycle | 6 | 0.8 | 8 | |||
| unassigned terms | 259 | 34.9 | 75 | 32.6 | ||
The largest percentage of total detected proteins (14%) was the mitochondrial functional category, which included 42% of the mitochondrial-related genes in the MitoCarta database (http://www.broadinstitute.org/pubs/MitoCarta/) (Table 3). Among all the significantly changed proteins in the MPS VII brain, 80 (about 1/3) belonged to the mitochondrial category. Since mitochondrial protein alterations implicate the cellular energy generating system, a comparison to Palmfeldt's mitochondrial sub-categories list [29] (Table 4) showed: 1) the proteins associated with the canonical citric acid cycle enzymes were all increased (2 significantly), except alpha-ketoglutarate dehydrogenase (Oghd) which was significantly reduced; and 2) all 11 significant changes in the mitochondria localized respiratory chain were decreased (complex 2 is a component of the citric acid cycle and was grouped with the citric acid cycle proteins [30]). In addition, although cytosolic, 87% of the energy generating glycolytic enzymes were also increased, two significantly (Gpi and Aldoc) (Table 5). The increases in the citric acid cycle and glycolytic enzymes were disproportionate to the decreases in the overall proteome (60%), the mitochondrial proteins (65%), and the respiratory chain associated proteins (78%), which suggests an alteration in energy generation.
Table 3. Significant mitochondrial proteomic changes found in the MitoCarta database.
| uniprot # | Gene | p-value | fold | Description |
|---|---|---|---|---|
| Q924L1 | Letmd1 | ↑ | LETM1 domain containing 1 | |
| Q791T5 | Mtch1 | 0.044 | 2.70 | Mitochondrial carrier homolog 1 |
| P26638 | Sars | 0.039 | 1.74 | Seryl-tRNA synthetase, cytoplasmic |
| Q9CZU6 | Cs | 0.033 | 1.57 | Citrate synthase, mitochondrial |
| Q8BWF0 | Aldh5a1 | 0.025 | 1.54 | Succinate-semialdehyde dehydrogenase, mitochondrial |
| Q9D051 | Pdhb | 0.035 | 1.41 | Pyruvate dehydrogenase E1 component subunit beta, mitochondrial |
| P54071 | Idh2 | 0.016 | 1.29 | Isocitrate dehydrogenase [NADP], mitochondrial |
| Q8BH59 | Slc25a12 | 0.019 | 1.28 | Calcium-binding mitochondrial carrier protein Aralar1 |
| Q9ERS2 | Ndufa13 | 0.034 | -1.49 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 13 |
| P47802 | Mtx1 | 0.034 | -1.53 | Metaxin-1 |
| P56382 | Atp5e | 0.038 | -1.54 | ATP synthase subunit epsilon, mitochondrial |
| Q60597 | Ogdh | 0.040 | -1.58 | 2-oxoglutarate dehydrogenase E1 component, mitochondrial |
| P19096 | Fasn | 0.022 | -1.62 | Fatty acid synthase |
| P52196 | Tst | 0.046 | -1.62 | Thiosulfate sulfurtransferase |
| Q9DCW4 | Etfb | 0.036 | -1.65 | Electron transfer flavoprotein subunit beta |
| O35683 | Ndufa1 | 0.045 | -1.80 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 1 |
| Q9CPP6 | Ndufa5 | 0.035 | -1.85 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5 |
| Q4KMM3 | Oxr1 | 0.019 | -1.87 | Oxidation resistance protein 1 |
| P56375 | Acyp2 | 0.035 | -1.89 | Acylphosphatase-2 |
| Q8BIJ6 | Iars2 | 0.047 | -1.95 | Isoleucyl-tRNA synthetase, mitochondrial |
| Q9QUH0 | Glrx | 0.041 | -1.97 | Glutaredoxin-1 |
| P97450 | Atp5j | 0.046 | -2.00 | ATP synthase-coupling factor 6, mitochondrial |
| Q66GT5 | Ptpmt1 | 0.022 | -2.02 | Protein-tyrosine phosphatase mitochondrial 1 |
| P58281 | Opa1 | 0.027 | -2.17 | Dynamin-like 120 kDa protein, mitochondrial |
| Q91V92 | Acly | <0.0003 | -2.25 | ATP-citrate synthase |
| P97493 | Txn2 | 0.045 | -2.55 | Thioredoxin, mitochondrial |
| Q80Y14 | Glrx5 | 0.034 | -2.68 | Glutaredoxin-related protein 5 |
| Q9CQ85 | Timm22 | 0.017 | -2.69 | Mitochondrial import inner membrane translocase subunit Tim22 |
| Q9CQ69 | Uqcrq | 0.019 | -2.70 | Cytochrome b-c1 complex subunit 8 |
| Q9EQ80 | Nif3l1 | 0.031 | -2.81 | NIF3-like protein 1 |
| Q9CZD3 | Gars | 0.009 | -3.06 | Glycyl-tRNA synthetase |
| Q9CQX8 | Mrps36 | 0.028 | -3.13 | 28S ribosomal protein S36, mitochondrial |
| P28271 | Aco1 | 0.020 | -3.61 | Cytoplasmic aconitate hydratase |
| Q9D8S9 | Bola1 | ↓ | BolA-like protein 1 | |
| Q9R1Z7 | Pts | ↓ | 6-pyruvoyl tetrahydrobiopterin synthase | |
| P58059 | Mrps21 | ↓ | 28S ribosomal protein S21, mitochondrial | |
| Q9D1L0 | Chchd2 | ↓ | Coiled-coil-helix-domain-containing protein 2, mitochondrial | |
| Q9CQV7 | Dnajc19 | ↓ | Mitochondrial import inner membrane translocase subunit TIM14 |
Table 4. Mitochondrial proteins detected and changed by Palmfeld functional subcategories [28].
| Protein category | proteins | detected | changed | % detected | % significant |
|---|---|---|---|---|---|
| Citric acid cycle | 30 | 25 | 6 | 83% | 24% |
| Mitochondrial morphology | 12 | 6 | 1 | 50% | 17% |
| Mitochondrial translation | 99 | 44 | 7 | 44% | 16% |
| Antioxidant systems | 33 | 17 | 2 | 52% | 12% |
| Respiratory chain | 82 | 58 | 5 | 71% | 9% |
| Amino acid metabolism | 64 | 35 | 3 | 55% | 9% |
| Fatty acid metabolism | 21 | 18 | 1 | 86% | 6% |
| Protein quality control systems | 16 | 12 | 0 | 75% | 0% |
| Apoptosis | 7 | 3 | 0 | 43% | 0% |
| Known disease association (OMIM database) | 36 | 31 | 5 | 86% | 16% |
Table 5. Proteomic changes associated with energy generating categories.
| A. G lycolysis | C. Respiratory chain | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | p-value | fold | Gene | p-value | fold | Gene | p-value | fold | |
| Increases | Complex 1 | Complex 4 | |||||||
| Aldoc | 0.040 | 1.95 | 1 | Increases | Increases | ||||
| Aldoa | 0.207 | 1.47 | Ndufv1 | 0.080 | 1.43 | Cox6a1 | 0.877 | 1.06 | |
| Pgk1 | 0.204 | 1.36 | Ndufs2 | 0.311 | 1.29 | Decreases | |||
| Pkm2 | 0.168 | 1.32 | Ndufa8 | 0.069 | 1.20 | Cox4i1 | 0.950 | -1.01 | |
| Pgam1 | 0.389 | 1.31 | Ndufa9 | 0.100 | 1.18 | Cox6c | 0.859 | -1.08 | |
| Eno1 | 0.324 | 1.21 | Ndufb11 | 0.544 | 1.18 | Cox5b | 0.530 | -1.24 | |
| Eno3 | 0.349 | 1.2 | Ndufs3 | 0.430 | 1.15 | Cox6b1 | 0.271 | -1.26 | |
| Eno2 | 0.296 | 1.2 | Ndufs8 | 0.666 | 1.08 | Cox5a | 0.079 | -1.26 | |
| Pfkl | 0.567 | 1.12 | Decreases | Cox7a2 | 0.536 | -1.29 | |||
| Gpi | 0.010 | 1.12 | Ndufs1 | 0.735 | -1.04 | Cox7a2l | 0.275 | -1.95 | |
| Tpi1 | 0.807 | 1.08 | Ndufa6 | 0.791 | -1.06 | Cox7a1 | 0.168 | -2.11 | |
| Gapdh | 0.661 | 1.07 | Ndufa10 | 0.671 | -1.09 | ATP synthase | |||
| Pfkm | 0.918 | 1.02 | Ndufb5 | 0.506 | -1.11 | Increases | |||
| Decreases | Ndufa2 | 0.078 | -1.14 | Atp5a1 | 0.100 | 1.40 | |||
| Pfkp | 0.696 | -1.04 | Ndufb10 | 0.708 | -1.17 | Atp5b | 0.089 | 1.33 | |
| Hk1 | 0.241 | -1.14 | Ndufa7 | 0.811 | -1.18 | Atp5o | 0.188 | 1.07 | |
| Ndufs6 | 0.703 | -1.19 | Atp5j2 | 0.969 | 1.01 | ||||
| Ndufa12 | 0.464 | -1.21 | Decreases | ||||||
| Ndufa4 | 0.052 | -1.23 | Atp5h | 0.869 | -1.03 | ||||
| Ndufb6 | 0.600 | -1.24 | Atp5c1 | 0.852 | -1.06 | ||||
| B. Citric acid cycle | Ndufb7 | 0.361 | -1.25 | Atp5f1 | 0.447 | -1.11 | |||
| Gene | p-value | fold | Ndufs5 | 0.358 | -1.27 | Atp5l | 0.213 | -1.16 | |
| Increases | Ndufs7 | 0.385 | -1.30 | Atp5d | 0.489 | -1.35 | |||
| Dlst | 0.063 | 2.11 | Ndufc2 | 0.286 | -1.31 | Atp5e | 0.038 | -1.54 | |
| Clybl | 0.278 | 1.7 | Ndufb3 | 0.434 | -1.39 | Atp5i | 0.086 | -1.72 | |
| Cs | 0.033 | 1.57 | Ndufb8 | 0.148 | -1.43 | Atp5j | 0.046 | -2.00 | |
| Pdha1 | 0.090 | 1.57 | Ndufv2 | 0.327 | -1.48 | Atp5s | ↓ | ||
| Fh | 0.201 | 1.53 | Ndufa13 | 0.034 | -1.49 | Other | |||
| Dld | 0.075 | 1.46 | Ndufb9 | 0.081 | -1.60 | Increases | |||
| Idh3a | 0.130 | 1.43 | Ndufv3 | 0.477 | -1.62 | Etfdh | 0.386 | 1.91 | |
| Mdh2 | 0.126 | 1.43 | Ndufa1 | 0.045 | -1.80 | Etfa | 0.597 | 1.26 | |
| Pdhb | 0.035 | 1.41 | Ndufs4 | 0.287 | -1.81 | Cyb5b | 0.686 | 1.14 | |
| Dlat | 0.092 | 1.39 | Ndufa5 | 0.035 | -1.85 | Decreases | |||
| Sucla2 | 0.425 | 1.31 | Ndufb2 | 0.272 | -1.89 | Cyc1 | 0.776 | -1.09 | |
| Sdhb | 0.449 | 1.3 | 2 | Ndufab1 | 0.089 | -2.44 | Etfb | 0.036 | -1.65 |
| Idh2 | 0.016 | 1.29 | Ndufa3 | ↓ | Txnl1 | 0.360 | -1.94 | ||
| Sdhc | 0.386 | 1.26 | 2 | Complex 3 | Fdx1 | ↓ | |||
| Suclg1 | 0.629 | 1.25 | Increases | Fdx1l | ↓ | ||||
| Pdk1 | 0.731 | 1.2 | Uqcrc1 | 0.066 | 1.81 | ||||
| Sdha | 0.557 | 1.16 | 2 | Uqcrc2 | 0.145 | 1.53 | |||
| Sdhd | 0.790 | 1.09 | 2 | Decreases | |||||
| Idh3g | 0.819 | 1.07 | Uqcrfs1 | 0.902 | -1.02 | ||||
| Aco2 | 0.878 | 1.03 | Uqcr10 | 0.155 | -1.30 | ||||
| Decreases | Uqcrh | 0.085 | -1.88 | ||||||
| Ogdh | 0.040 | -1.58 | Uqcrq | 0.019 | -2.70 | ||||
Changes in bold detected at p<0.05 or exclusively in one group; 2 Complex 2 (Sdh) proteins included with the citric acid cycle proteins
The greatest number of functional terms (62) were in the cytoskeleton category, which were generated from 66 significantly changed proteins (Table 6). Most of these were decreased in the mutant (74%), with all classes of motor proteins (kinesins, dyneins, and myosins) showing significant reductions in MPS VII. Also of interest are several protein changes associated with the cytoskeletal-membrane linking ankyrin proteins such as: Shank3 whose superfamily Shank proteins play a role in synapse formation and dendritic spine maturation [31] and Asap1, Asap2, Agap2, and Ankfy1, which are all involved with endocytosis.
Table 6.
Significant cytoskeletal protein changes (p<0.05 or exclusively in one group).
| Uniprot # | Gene Name | p-value | fold | Description |
|---|---|---|---|---|
| P10922 | H1f0 | ↑ | Histone H1.0 | |
| Q9EPR4 | Slc23a2 | ↑ | Solute carrier family 23 member 2 | |
| Q8BTY2 | Slc4a7 | 0.006 | 2.84 | Sodium bicarbonate cotransporter 3 |
| P14115 | Rpl27a | 0.002 | 2.77 | 60S ribosomal protein L27a |
| Q8BIW1 | Prune | 0.031 | 2.71 | Protein prune homolog |
| Q8R5J9 | Arl6ip5 | 0.043 | 2.63 | PRA1 family protein 3 |
| Q9D8E6 | Rpl4 | 0.003 | 2.58 | 60S ribosomal protein L4 |
| P54285 | Cacnb1 | 0.021 | 2.15 | Voltage-dependent L-type calcium channel subunit beta-1 |
| P63001 | Rac1 | 0.029 | 2.02 | Ras-related C3 botulinum toxin substrate 1 |
| Q60668 | Hnrnpd | 0.036 | 1.91 | Heterogeneous nuclear ribonucleoprotein D0 |
| Q9JKK7 | Tmod2 | 0.015 | 1.91 | Tropomodulin-2 |
| P14206 | Rpsa | 0.048 | 1.83 | 40S ribosomal protein SA |
| Q9ERD7 | Tubb3 | 0.043 | 1.80 | Tubulin beta-3 chain |
| Q3V0K9 | Pls1 | 0.032 | 1.80 | Plastin-1 |
| Q8CHH9 | sept8 | 0.046 | 1.66 | Septin-8 |
| Q9CPR4 | Rpl17 | 0.020 | 1.55 | 60S ribosomal protein L17 |
| P19246 | Nefh | 0.021 | 1.38 | Neurofilament heavy polypeptide |
| Q9ERS2 | Ndufa13 | 0.034 | -1.49 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 13 |
| P54227 | Stmn1 | 0.040 | -1.81 | Stathmin |
| Q4KMM3 | Oxr1 | 0.019 | -1.87 | Oxidation resistance protein 1 |
| Q9CPW4 | Arpc5 | 0.044 | -1.90 | Actin-related protein 2/3 complex subunit 5 |
| O08788 | Dctn1 | 0.042 | -1.98 | Dynactin subunit 1 |
| P70336 | Rock2 | 0.014 | -2.22 | Rho-associated protein kinase 2 |
| O54962 | Banf1 | 0.023 | -2.42 | Barrier-to-autointegration factor |
| Q0GNC1 | Inf2 | 0.004 | -2.45 | Inverted formin-2 |
| P62627 | Dynlrb1 | 0.030 | -2.45 | Dynein light chain roadblock-type 1 |
| Q58A65 | Spag9 | 0.022 | -2.46 | C-jun-amino-terminal kinase-interacting protein 4 |
| Q9Z2H5 | Epb41l1 | 0.027 | -2.53 | Band 4.1-like protein 1 |
| Q9CQI3 | Gmfb | 0.017 | -2.57 | Glia maturation factor beta |
| Q811D0 | Dlg1 | 0.044 | -2.69 | Disks large homolog 1 |
| Q91VR8 | Brick1 | 0.030 | -2.70 | Probable protein BRICK1 |
| P27546 | Map4 | 0.018 | -2.80 | Microtubule-associated protein 4 |
| Q9CQX8 | Mrps36 | 0.028 | -3.13 | 28S ribosomal protein S36, mitochondrial |
| P23116 | Eif3a | 0.012 | -3.38 | Eukaryotic translation initiation factor 3 subunit A |
| Q61301 | Ctnna2 | 0.048 | -3.44 | Catenin alpha-2 |
| P28740 | Kif2a | 0.011 | -3.52 | Kinesin-like protein KIF2A |
| P21619 | Lmnb2 | 0.041 | -3.60 | Lamin-B2 |
| Q8BRT1 | Clasp2 | 0.016 | -3.67 | CLIP-associating protein 2 |
| Q9QYB5 | Add3 | 0.026 | -3.72 | Gamma-adducin |
| Q9QWI6 | P140 | 0.017 | -3.89 | p130Cas-associated protein |
| O70161 | Pip5k1c | 0.016 | -3.93 | Phosphatidylinositol-4-phosphate 5-kinase type-1 gamma |
| Q9JMH9 | Myo18a | 0.023 | -4.30 | Myosin-XVIIIa |
| Q9DBR7 | Ppp1r12a | 0.009 | -4.79 | Protein phosphatase 1 regulatory subunit 12A |
| Q9QXL2 | Kif21a | 0.022 | -5.61 | Kinesin-like protein KIF21A |
| Q4ACU6 | Shank3 | 0.012 | -5.85 | SH3 and multiple ankyrin repeat domains protein 3 |
| Q6P9K8 | Caskin1 | 0.042 | -6.95 | Caskin-1 |
| Q7TMB8 | Cyfip1 | 0.001 | -7.32 | Cytoplasmic FMR1-interacting protein 1 |
| Q80U40 | Rimbp2 | 0.015 | -7.67 | RIMS-binding protein 2 |
| Q3UHD9 | Agap2 | <0.000003 | -11.45 | Arf-GAP, GTPase, ANK repeat and PH domain-containing protein 2 |
| P08556 | Nras | ↓ | GTPase NRas | |
| Q9QWY8 | Asap1 | ↓ | Arf-GAP with SH3 domain, ANK repeat and PH domain-containing protein 1 | |
| P58059 | Mrps21 | ↓ | 28S ribosomal protein S21, mitochondrial | |
| Q03173 | Enah | ↓ | Protein enabled homolog | |
| Q6PFD5 | Dlgap3 | ↓ | Disks large-associated protein 3 | |
| Q923D5 | Wbp11 | ↓ | WW domain-binding protein 11 | |
| Q80U49 | Kiaa0284 | ↓ | Protein KIAA0284 | |
| Q6A065 | Cep170 | ↓ | Centrosomal protein of 170 kDa | |
| Q6PDI5 | Ecm29 | ↓ | Proteasome-associated protein ECM29 homolog | |
| Q8C1B1 | Camsap1l1 | ↓ | Calmodulin-regulated spectrin-associated protein 1-like protein 1 | |
| P39054 | Dnm2 | ↓ | Dynamin-2 | |
| Q3UH68 | Limch1 | ↓ | LIM and calponin homology domains-containing protein 1 | |
| Q3UU96 | Cdc42bpa | ↓ | Serine/threonine-protein kinase MRCK alpha | |
| Q7SIG6 | Asap2 | ↓ | Arf-GAP with SH3 domain, ANK repeat and PH domain-containing protein 2 | |
| Q810B6 | Ankfy1 | ↓ | Ankyrin repeat and FYVE domain-containing protein 1 | |
| Q8BPM0 | Daam1 | ↓ | Disheveled-associated activator of morphogenesis 1 | |
| Q9JL26 | Fmnl1 | ↓ | Formin-like protein 1 |
3.3 Transcriptomic annotation enrichment analysis
A transcriptome analysis of BL6 MPS VII versus normal hippocampus was performed to compare to the proteomic results (Supplemental Data 4.xlsx). The number of mice was chosen to be in the same range as the proteome cohort, which was limited due to the nature of the assay. This experiment was also designed to focus on the most prominent differences between the normal and diseased brains. The transcriptomic data was analyzed using the Significance Analysis of Microarrays (SAM) [16] program. Significant differences (q< 0.05 and fold >1.5) were found in the expression of 81 probe sets accounting for 69 genes, of which 66 were up-regulated and 3 were down-regulated (Table 7).
Table 7.
Significant transcriptomic changes in the Bl6 hippocampus (q< 0.05 and fold >1.5).
| Probeset ID | Gene Symbol | Gene Title | Fold | q-value |
|---|---|---|---|---|
| 1420699_at | Clec7a | C-type lectin domain family 7, member a | 11.2 | <0.01 |
| 1419202_at | Cst7 | cystatin F (leukocystatin) | 10.6 | <0.01 |
| 1448303_at | Gpnmb | glycoprotein (transmembrane) nmb | 9.8 | <0.01 |
| 1423547_at | Lyz2 | lysozyme 2 | 7.4 | <0.01 |
| 1436996_x_at | Lyz1 | lysozyme 1 | 7.0 | <0.01 |
| 1439426_x_at | Lyz1 | lysozyme 1 | 7.0 | <0.01 |
| 1426509_s_at | Gfap | glial fibrillary acidic protein | 5.9 | <0.01 |
| 1418021_at | C4b | complement component 4B (Childo blood group) | 5.7 | <0.01 |
| 1426808_at | Lgals3 | lectin, galactose binding, soluble 3 | 5.5 | <0.01 |
| 1420394_s_at | Gp49a /// Lilrb4 | glycoprotein 49 A /// leukocyte immunoglobulin-like receptor, subfamily B, member 4 | 4.9 | <0.01 |
| 1426508_at | Gfap | glial fibrillary acidic protein | 4.9 | <0.01 |
| 1435477_s_at | Fcgr2b | Fc receptor, IgG, low affinity IIb | 4.3 | <0.01 |
| 1427076_at | Mpeg1 | macrophage expressed gene 1 | 4.2 | <0.01 |
| 1424754_at | Ms4a7 | membrane-spanning 4-domains, subfamily A, member 7 | 4.1 | <0.01 |
| 1449164_at | Cd68 | CD68 antigen | 4.0 | <0.01 |
| 1419004_s_at | Bcl2a1a /// Bcl2a1b /// Bcl2a1d | B-cell leukemia/lymphoma 2 related protein A1a /// B-cell leukemia/lymphoma 2 related p | 4.0 | <0.01 |
| 1418808_at | Rdh5 | retinol dehydrogenase 5 | 4.0 | 0.027 |
| 1419100_at | Serpina3n | serine (or cysteine) peptidase inhibitor, clade A, member 3N | 3.8 | 0.014 |
| 1427301_at | Cd48 | CD48 antigen | 3.6 | <0.01 |
| 1419561_at | Ccl3 | chemokine (C-C motif) ligand 3 | 3.4 | <0.01 |
| 1437540_at | Mcoln3 | mucolipin 3 | 3.2 | <0.01 |
| 1448021_at | Fam46c | family with sequence similarity 46, member C | 3.0 | <0.01 |
| 1422875_at | Cd84 | CD84 antigen | 2.9 | <0.01 |
| 1421792_s_at | Trem2 | triggering receptor expressed on myeloid cells 2 | 2.9 | <0.01 |
| 1452352_at | Ctla2b | cytotoxic T lymphocyte-associated protein 2 beta | 2.8 | 0.014 |
| 1451941_a_at | Fcgr2b | Fc receptor, IgG, low affinity IIb | 2.7 | <0.01 |
| 1428114_at | Slc14a1 | solute carrier family 14 (urea transporter), member 1 | 2.7 | 0.041 |
| 1427221_at | Slc6a20a | solute carrier family 6 (neurotransmitter transporter), member 20A | 2.7 | 0.014 |
| 1449401_at | C1qc | complement component 1, q subcomponent, C chain | 2.7 | <0.01 |
| 1455332_x_at | Fcgr2b | Fc receptor, IgG, low affinity IIb | 2.6 | <0.01 |
| 1419598_at | Ms4a6d | membrane-spanning 4-domains, subfamily A, member 6D | 2.6 | <0.01 |
| 1454268_a_at | Cyba | cytochrome b-245, alpha polypeptide | 2.6 | 0.041 |
| 1419298_at | Pon3 | paraoxonase 3 | 2.5 | 0.041 |
| 1448710_at | Cxcr4 | chemokine (C-X-C motif) receptor 4 | 2.5 | <0.01 |
| 1418028_at | Dct | dopachrome tautomerase | 2.4 | 0.041 |
| 1419482_at | C3ar1 | complement component 3a receptor 1 | 2.4 | <0.01 |
| 1437726_x_at | C1qb | complement component 1, q subcomponent, beta polypeptide | 2.4 | 0.014 |
| 1419599_s_at | Ms4a6d | membrane-spanning 4-domains, subfamily A, member 6D | 2.3 | <0.01 |
| 1416612_at | Cyp1b1 | cytochrome P450, family 1, subfamily b, polypeptide 1 | 2.3 | <0.01 |
| 1452014_a_at | Igf1 | insulin-like growth factor 1 | 2.3 | <0.01 |
| 1434366_x_at | C1qb | complement component 1, q subcomponent, beta polypeptide | 2.2 | 0.027 |
| 1424067_at | Icam1 | intercellular adhesion molecule 1 | 2.2 | 0.014 |
| 1451161_a_at | Emr1 | EGF-like module containing, mucin-like, hormone receptor-like sequence 1 | 2.2 | <0.01 |
| 1449156_at | Ly9 | lymphocyte antigen 9 | 2.2 | <0.01 |
| 1448380_at | Lgals3bp | lectin, galactoside-binding, soluble, 3 binding protein | 2.2 | <0.01 |
| 1419483_at | C3ar1 | complement component 3a receptor 1 | 2.1 | <0.01 |
| 1419128_at | Itgax | integrin alpha X | 2.0 | <0.01 |
| 1422660_at | Rbm3 | RNA binding motif protein 3 | 2.0 | 0.027 |
| 1421223_a_at | Anxa4 | annexin A4 | 2.0 | <0.01 |
| 1448148_at | Grn | granulin | 2.0 | 0.014 |
| 1420361_at | Slc11a1 | solute carrier family 11 (proton-coupled divalent metal ion transporters), member 1 | 1.9 | <0.01 |
| 1417963_at | Pltp | phospholipid transfer protein | 1.9 | 0.014 |
| 1451784_x_at | H2-D1 | histocompatibility 2, D region locus 1 | 1.9 | 0.038 |
| 1425545_x_at | H2-D1 | histocompatibility 2, D region locus 1 | 1.9 | 0.014 |
| 1451683_x_at | H2-D1 | histocompatibility 2, D region locus 1 | 1.9 | 0.041 |
| 1419315_at | Slamf9 | SLAM family member 9 | 1.9 | 0.014 |
| 1419132_at | Tlr2 | toll-like receptor 2 | 1.8 | <0.01 |
| 1417870_x_at | Ctsz | cathepsin Z | 1.8 | <0.01 |
| 1448640_at | Slc14a1 | solute carrier family 14 (urea transporter), member 1 | 1.8 | 0.032 |
| 1417868_a_at | Ctsz | cathepsin Z | 1.8 | <0.01 |
| 1448591_at | Ctss | cathepsin S | 1.8 | 0.014 |
| 1448891_at | Fcrls | Fc receptor-like S, scavenger receptor | 1.8 | <0.01 |
| 1435903_at | Cd300a | CD300A antigen | 1.7 | 0.027 |
| 1419455_at | Il10rb | interleukin 10 receptor, beta | 1.7 | <0.01 |
| 1416527_at | Rab32 | RAB32, member RAS oncogene family | 1.7 | 0.014 |
| 1426025_s_at | Laptm5 | lysosomal-associated protein transmembrane 5 | 1.7 | 0.014 |
| 1425025_at | Tmem106a | transmembrane protein 106A | 1.7 | 0.014 |
| 1418826_at | Ms4a6b | membrane-spanning 4-domains, subfamily A, member 6B | 1.7 | <0.01 |
| 1418910_at | Bmp7 | bone morphogenetic protein 7 | 1.7 | <0.01 |
| 1428018_a_at | AF251705 | cDNA sequence AF251705 | 1.7 | 0.014 |
| 1460248_at | Cpxm2 | carboxypeptidase X 2 (M14 family) | 1.6 | 0.014 |
| 1436890_at | Uap1l1 | UDP-N-acteylglucosamine pyrophosphorylase 1-like 1 | 1.6 | 0.014 |
| 1456567_x_at | Grn | granulin | 1.6 | <0.01 |
| 1438910_a_at | Stom | stomatin | 1.5 | 0.014 |
| 1418825_at | Irgm1 | immunity-related GTPase family M member 1 | 1.5 | 0.014 |
| 1449195_s_at | Cxcl16 | chemokine (C-X-C motif) ligand 16 | 1.5 | 0.014 |
| 1416340_a_at | Man2b1 | mannosidase 2, alpha B1 | 1.5 | 0.014 |
| 1421812_at | Tapbp | TAP binding protein | 1.5 | 0.027 |
| 1448606_at | Lpar1 | lysophosphatidic acid receptor 1 | -2.1 | <0.01 |
| 1455965_at | Adamts4 | a disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 mo | -2.1 | 0.041 |
| 1419064_a_at | Ugt8a | UDP galactosyltransferase 8A | -2.6 | 0.041 |
DAVID functional annotation terms were generated from the list of significantly altered gene transcript changes in the same manner as the proteome. This generated 229 unique annotation terms which grouped into 29 clusters (Supplemental Data 5.pdf). As with the proteome, the gene expression changes and terms were compared to the functional categories previously assigned in the analysis of the C3H mouse [8]. Of the significant gene expression changes in the BL6 mouse in the present study, 40 of the changes (58%) were the same as in the C3H mouse and 100% of the shared changes were in the same direction (Figure 1). Furthermore, 94% of the annotation terms generated from this BL6 mouse study were present in the terms generated in the C3H mouse hippocampus [8]. Thus the overall pattern of altered processes between strains was highly consistent, which also correlates with clinical and pathological findings [3]. Functional categories were assigned similarly to the proteomic assignments (Table 2). Four of the categories generated by the proteome (protein modification, neural disease, ubiquitin and cell cycle) were not represented in the BL6 transcriptome (Table 2).
Figure 1.
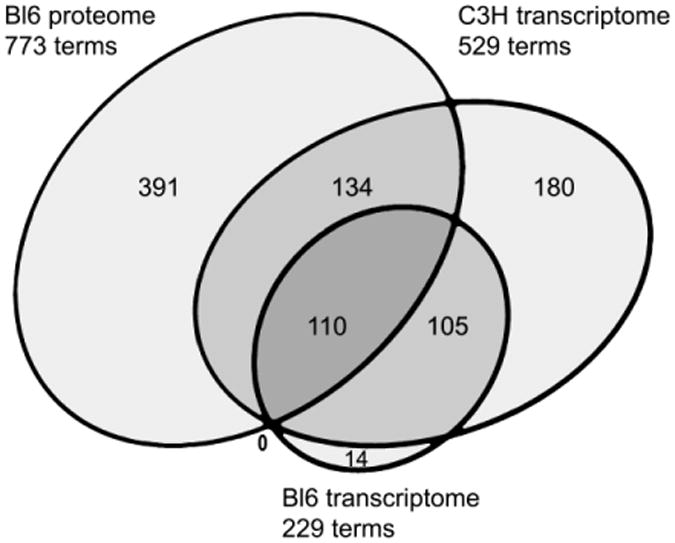
Proportional Euler diagram of shared DAVID functional terms from diseased versus normal mice between Bl6 proteomic and Bl6 and C3H transcriptomic analyses.
3.4 Comparison of Proteomic and Transcriptomic analyses
The results of the two assays were first compared using the UniProt tissue annotation enrichment tool [32], which ascertains if a list of genes or proteins is statistically over-represented (enriched) for specific tissue annotations, as a method of validating the detected proteins. There was a high degree of enrichment for the tissue annotation terms “brain” (P≈10-187), “hippocampus” (P≈10-174), and “brain cortex” (P≈10-114) (Supplemental Data 6.pdf). To ascertain that systematic database bias [29] was not responsible for this result, and since mitochondrial proteins were the largest category of proteins detected, the entire MitoCarta list was submitted and the enrichment score for “hippocampus” and “brain” were much lower (P≈10-23, P≈10-2 respectively).
Tissue annotation of the transcriptome was accomplished by analyzing the 3200 highest expressing probe sets which was about 12% of the probes on the chipset and about the number of proteins detected. This also showed that “brain,” “hippocampus,” and “brain cortex” were enriched at P≈10-100 (Supplemental Data 6.pdf). To test for systematic bias, 2 random sets of 3200 probes were analyzed which showed that the top tissue annotations were non-neural (Supplemental Data 6.pdf) and the top neural tissue, “hippocampus,” was enriched only to P≈10-9. The proteomic and transcriptomic tissue annotations were compared by Spearman rank order correlation and there was greater correlation to each other than to the random probe sets.
Since we did not find an analysis of this type in the literature, we explored what would happen using an iterative analysis of varying numbers of top expressing probes. With up to 200 probes, “hippocampus” was the top annotation, with “brain” second. Between 200-3200 probes, the term “brain” was the top annotation followed by “hippocampus.” At more than 3200 terms, the annotation for “hippocampus” began to decrease, the annotation for “brain” began to level off, and miscellaneous other annotations began to rapidly rise (Figure 2).
Figure 2.
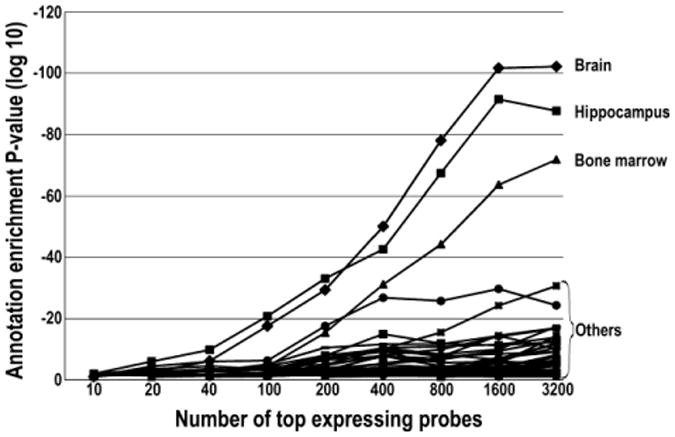
Transcriptomic tissue annotation profile from increasing numbers of top expressing probes showing the ranking of tissue annotation for the top expressing probes at increasing depth.
One of the problems with integrated proteomic and transcriptomic analysis using the methods here is that many probes present on the microarray are not detected in the proteome and not all of the detected proteins have corresponding probes on the microarray. To compare the results of the two methods we converted the Uniprot Identifiers and the Affymetrix probe set IDs to DAVID ID numbers using the DAVID program. Of the detected proteins, 86% had a matching probeset in the microarray (cognate pairs of transcripts and proteins), which represented about one-fifth of all the microarray probes. Of the 245 proteins that were significantly different in the MPS VII brain, 82% had a cognate mRNA transcript represented on the microarray and 43 of the changed proteins were not represented in the microarray. However, the annotation terms they generated were largely redundant with the terms already found, thus their absence had little effect on the characterization of the pathologic changes. Conversely, only 10 (14%) of the 69 significant gene transcription changes had cognates among the detected proteins and none of them were changed between normal and diseased brains.
Overall, the changes in the proteome and transcriptome were in opposite directions, with 71% of the significant proteomic changes being decreases and 96% of the significant transcriptomic changes being increases. While it is known that mRNA and protein levels correlate poorly, this discrepancy was explored by examining cognate pairs from the two assays for their direction of change. When the cognate changes were filtered for successively smaller p- and q-values, they became increasingly more likely to be in the same direction (Supplemental Data 7.pdf). This is consistent with studies that have shown: 1) as noise is reduced, the direction of change is more consistent between proteins and transcripts [33]; and 2) that gross proteome and transcriptome changes can be in the opposite directions in an integrated analysis [34]. Thus, the difference in the overall directions-of-change was consistent with what others have seen.
Despite the fact that there were no mutually significant cognate changes between the two assays, 110 of the 229 transcriptomic annotation terms (48%) were among the 773 annotation terms generated in the proteomic analysis (Figure 1). The shared terms were grouped by their assigned functional categories (Table 8). Although fewer in number, the transcriptomic terms generated in the BL6 strain in the present study were very similar to those identified for the hippocampus in the C3H strain, which carries the same mutation in GUSB [8, 24]. Additionally, the proportion of transcriptome terms that were shared with the proteome was similar for both strains (Figure 1).
Table 8. Shared transcriptomic and proteomic terms grouped by assigned functional category.
| Immune | Mitochondria |
|---|---|
| GO:0001775-cell activation | GO:0005739-mitochondrion |
| GO:0002252-immune effector process | GO:0031090-organelle membrane |
| GO:0002443-leukocyte mediated immunity | GO:0055114-oxidation reduction |
| GO:0002684-positive regulation of immune system process | Development |
|
|
|
| GO:0006955-immune response | GO:0035239-tube morphogenesis |
| GO:0007186-G-protein coupled receptor protein signaling pathway | GO:0035295-tube development |
| GO:0010033-response to organic substance | nucleus |
| GO:0016192-vesicle-mediated transport | GO:0000166-nucleotide binding |
| GO:0045321-leukocyte activation | Transcription |
|
|
|
| IPR013783:Immunoglobulin-like fold | GO:0045449-regulation of transcription |
| transducer | Transport |
|
|
|
| Vesicle | transport |
|
|
|
| cytoplasmic vesicle | Not assigned |
|
|
|
| GO:0006897-endocytosis | acetylation |
| GO:0010324-membrane invagination | alternative splicing |
| GO:0016023-cytoplasmic membrane-bounded vesicle | cell membrane |
| GO:0016044-membrane organization | cytoplasm |
| GO:0031410-cytoplasmic vesicle | disulfide bond |
| GO:0031982-vesicle | glycoprotein |
| GO:0031988-membrane-bounded vesicle | glycosylation site:N-linked (GlcNAc…) |
| GO:0051130-positive regulation of cellular component organization | GO:0000267-cell fraction |
| mmu04142:Lysosome | GO:0005506-iron ion binding |
| Extracellular matrix/Adhesion | GO:0005615-extracellular space |
|
|
|
| cell adhesion | GO:0005624-membrane fraction |
| GO:0005576-extracellular region | GO:0005626-insoluble fraction |
| GO:0007155-cell adhesion | GO:0005783-endoplasmic reticulum |
| GO:0022610-biological adhesion | GO:0005886-plasma membrane |
| GO:0044421-extracellular region part | GO:0006355-regulation of transcription, DNA-dependent |
| Secreted | GO:0007166-cell surface receptor linked signal transduction |
| Apoptosis | GO:0007242-intracellular signaling cascade |
|
|
|
| GO:0008219-cell death | GO:0008270-zinc ion binding |
| GO:0010941-regulation of cell death | GO:0008283-cell proliferation |
| GO:0016265-death | GO:0010604-positive regulation of macromolecule metabolic process |
| GO:0042981-regulation of apoptosis | GO:0016021-integral to membrane |
| GO:0043067-regulation of programmed cell death | GO:0031224-intrinsic to membrane |
| Metabolism | GO:0042127-regulation of cell proliferation |
|
|
|
| GO:0006508-proteolysis | GO:0042470-melanosome |
| GO:0008233-peptidase activity | GO:0043167-ion binding |
| GO:0008234-cysteine-type peptidase activity | GO:0043169-cation binding |
| GO:0070011-peptidase activity, acting on L-amino acid peptides | GO:0044459-plasma membrane part |
| Protease | GO:0046872-metal ion binding |
| Signaling | GO:0046914-transition metal ion binding |
|
|
|
| GO:0019220-regulation of phosphate metabolic process | GO:0048770-pigment granule |
| GO:0042325-regulation of phosphorylation | GO:0051252-regulation of RNA metabolic process |
| GO:0044093-positive regulation of molecular function | hydrolase |
| GO:0051174-regulation of phosphorus metabolic process | lipoprotein |
| Ion transport/Channels | membrane |
|
|
|
| calcium | metal-binding |
| GO:0005509-calcium ion binding | mmu04062:Chemokine signaling pathway |
| GO:0042592-homeostatic process | mmu04670:Leukocyte transendothelial migration |
| GO:0048878-chemical homeostasis | mutagenesis site |
| Neuron | oxidoreductase |
|
|
|
| GO:0007610-behavior | phosphoprotein |
| GO:0030030-cell projection organization | receptor |
| GO:0030182-neuron differentiation | sequence variant |
| GO:0050877-neurological system process | signal |
| Cytoskeleton | signal peptide |
|
|
|
| GO:0005856-cytoskeleton | splice variant |
| GO:0043228-non-membrane-bounded organelle | topological domain:Cytoplasmic |
| GO:0043232-intracellular non-membrane-bounded organelle | topological domain:Extracellular |
| transmembrane | |
| transmembrane region zinc |
4. Discussion
The present study was undertaken to assess the changes associated with diseased brain tissue in a widely studied model of lysosomal storage disease, MPS VII, which is a genetic model of mental retardation. We focused the analysis on a single region because transcriptomic analysis in this model showed not only numerous changes between normal and diseased brains, but also significant differences in the changes between brain regions [8]. We chose the hippocampus because of the extensive histopathology associated with this region [3, 9, 10].
Proteomic and transcriptomic analytical methods each provide meaningful insights into the study of normal and diseased states [7]. Global changes in mRNA levels have been found to not correlate well with the translated cognate proteins [35, 36] due to post-transcriptional regulatory processes, mRNA stability, and protein stability [37-39]. Proteomic and transcriptomic analysis both give rise to noisy data, use very different experimental methodologies, which complicates comparisons [40], and can even show an overall opposite directions of change by the two assay systems [34], as we observed. Nevertheless, they are complementary in their findings [7] as we also observed.
Our long-term goal is to obtain understanding of the complex pathological processes that arise in this single gene disease affecting the CNS. However, the changes in either protein abundance or transcript expression from individual genes give an incomplete picture of the pathogenic mechanisms. In contrast, analyzing the changes for functional annotation terms and broader categories, which require multiple changes, provides a common set of descriptors for the disease-associated alterations that can occur even in the absence of cognate changes.
None of the statistically significant changes in either protein or mRNA levels between normal and MPS VII hippocampus involved the same gene product. Limited studies of non-inherited diseases have also found few overlapping changes between the two assays [41-43]. Despite this discordance, the annotation analysis of the changes implicated many of the same pathologic processes, with about a third of the functional annotation terms from the two assays being the same. A number of these terms were for processes involved in known pathology in the MPS VII brain, such as those involving the hallmark lysosome/vesicular system, neuronal functions, and inflammatory/immune processes [3, 9, 10]. Thus, functional annotation analysis implicated many of the same areas of pathology even though the specific gene products had no overlap.
While “omic” assays are unbiased for comparison within each assay system, comparisons between assay systems have biases for technological and biological reasons. For example, both assays showed changes in complement cascade components, with significantly changed mRNA levels where no protein was detected, and protein changes where no corresponding mRNA probe was available on the microarray. The complement changes are interesting in light of the fact that complement component C1q has been found to bind to and be spatially associated with chondroitin sulfate proteoglycans in another lysosomal storage disease, Niemann-Pick C [44], and chondroitin sulfate is the main storage product of MPS VII. Furthermore, complement components have a role in brain development and are altered in a number of neurodegenerative diseases [45].
Comparison of the disease changes between the two assays was constrained by the limits of the proteomic assay since most of the altered proteins had probes on the microarray chip, but only a sixth of the significantly altered mRNAs had their cognate proteins detected. For example, CD68, a prominent marker of microglia activation, was one of the most significant gene expression changes in both the Bl6 and C3H mouse, yet was not detected in the proteomic assay. Nevertheless, the protein increase is readily detectable by immunohistochemistry in the MPS VII diseased brain and is a good indicator of early neuropathology and therapeutic correction [9]. This indicates that the amount of protein needed for immunofluorescence detection of a difference is below the level needed to for detection in the proteomic assay.
Proteomic analysis is also constrained due to its detection of the more abundant proteins, such as mitochondrial and cytoskeletal proteins, which resulted in those being the majority of protein differences between the MPS VII and normal brains. Mitochondrial abnormalities have been identified in several MPS diseases [46], including accumulation of defective mitochondria through the impairment of autophagy [47] and may be responsible for the energy imbalances seen in MPS VII [48].
The proteomic changes in the major energy generating pathways are consistent with Warburg's finding of an increase in aerobic glycolysis in tumors [49], which can also occur in inflammatory conditions [50]. Evidence of this alteration in the MPS VII brain is that most of the glycolytic and citric acid cycle enzymes were increased. Furthermore, the two glycolytic enzymes that were decreased (Hk1 and Pfkp) are involved in the ATP investment phase of glycolysis and have been shown in tumor cells to consistently decrease when the other glycolytic enzymes are increased [51]. In the citric acid cycle, the only decrease was in α-ketoglutarate dehydrogenase (Ogdh), which is believed to be a regulator of flux through the citric acid cycle [52]. An impaired function of this enzyme is characteristic of several neurodegenerative diseases, where it is decreased [52]. In contrast, in the respiratory chain, all 11 of the significantly changed proteins were decreases, which have been associated with neurodegeneration [53]. Thus, the alterations in mitochondrial and other energy system proteins in the brain are consistent with altered energy balance, and thus are likely to have a role in neurodegeneration.
Despite the differences between the two assays, the overlapping annotation terms were consistent with known histopathologic alterations such as the classic lysosomal alterations, while others suggest new directions to investigate such as the role of complement and energy generation in this monogenic disease.
Supplementary Material
Acknowledgments
We wish to thank T. Clarke, for technical assistance; E. Rappaport for help with microarray processing; and J. Tobias for bioinformatics advice. This work was supported by the NIH NINDS (R01-NS038690, R01-NS088667), the Ethel Foerderer Foundation; and an Intellectual and Developmental Disabilities Research Center from the NICHD (U54-HD086984).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta glucuronidase deficiency: report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82:249–257. doi: 10.1016/s0022-3476(73)80162-3. [DOI] [PubMed] [Google Scholar]
- 2.Neufeld E, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic & Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3421–3452. [Google Scholar]
- 3.Heuer GG, Passini MA, Jiang K, Parente MK, Lee VM, Trojanowski JQ, Wolfe JH. Selective neurodegeneration in murine mucopolysaccharidosis VII is progressive and reversible. Ann Neurol. 2002;52:762–770. doi: 10.1002/ana.10373. [DOI] [PubMed] [Google Scholar]
- 4.Brooks AI, Stein CS, Hughes SM, Heth J, McCray PM, Jr, Sauter SL, Johnston JC, Cory-Slechta DA, Federoff HJ, Davidson BL. Functional correction of established central nervous system deficits in an animal model of lysosomal storage disease with feline immunodeficiency virus-based vectors. Proc Natl Acad Sci U S A. 2002;99:6216–6221. doi: 10.1073/pnas.082011999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frisella W, O'Connor L, Vogler C, Roberts M, Walkley S, Levy B, Daly T, Sands M. Intracranial injection of recombinant adeno-associated virus improves cognitive function in a murine model of mucopolysaccharidosis type VII. Mol Ther. 2001:351–358. doi: 10.1006/mthe.2001.0274. [DOI] [PubMed] [Google Scholar]
- 6.O'Connor LH, Erway LC, Vogler CA, Sly WS, Nicholes A, Grubb J, Holmberg SW, Levy B, Sands MS. Enzyme replacement therapy for murine mucopolysaccharidosis type VII leads to improvements in behavior and auditory function. J Clin Invest. 1998;101:1394–1400. doi: 10.1172/JCI1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang W, Li F, Nie L. Integrating multiple ‘omics’ analysis for microbial biology: application and methodologies. Microbiology. 2010;156:287–301. doi: 10.1099/mic.0.034793-0. [DOI] [PubMed] [Google Scholar]
- 8.Parente MK, Rozen R, Cearley CN, Wolfe JH. Dysregulation of Gene Expression in a Lysosomal Storage Disease Varies between Brain Regions Implicating Unexpected Mechanisms of Neuropathology. PLoS One. 2012;7:e32419. doi: 10.1371/journal.pone.0032419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffin TA, Anderson HC, Wolfe JH. Ex Vivo Gene Therapy Using Patient iPSC-Derived NSCs Reverses Pathology in the Brain of a Homologous Mouse Model. Stem Cell Reports. 2015 doi: 10.1016/j.stemcr.2015.02.022. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy B, Galvin N, Vogler C, Birkenmeier EH, Sly WS. Neuropathology of murine mucopolysaccharidosis type VII. Acta Neuropathol. 1996;92:562–568. doi: 10.1007/s004010050562. [DOI] [PubMed] [Google Scholar]
- 11.Spodik B, Seeholzer SH, Coleman TR. Using Xenopus egg extracts to modify recombinant proteins. In: Coleman EA, editor. Protein-Protein Interactions. Cold Spring Harbor Lab. Press; Cold Spring Harbor: 2002. pp. 355–374. [Google Scholar]
- 12.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 13.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 14.Braisted JC, Kuntumalla S, Vogel C, Marcotte EM, Rodrigues AR, Wang R, Huang ST, Ferlanti ES, Saeed AI, Fleischmann RD, Peterson SN, Pieper R. The APEX Quantitative Proteomics Tool: generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics. 2008;9:529. doi: 10.1186/1471-2105-9-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuntumalla S, Braisted JC, Huang ST, Parmar PP, Clark DJ, Alami H, Zhang Q, Donohue-Rolfe A, Tzipori S, Fleischmann RD, Peterson SN, Pieper R. Comparison of two label-free global quantitation methods, APEX and 2D gel electrophoresis, applied to the Shigella dysenteriae proteome. Proteome Sci. 2009;7:22. doi: 10.1186/1477-5956-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowry R. Vassar Stats: Website for Statistical Computation. 2015 http://www.vassarstats.net/corr_rank.html.
- 18.Gene_Ontology_Consortium. GO Enrichment Analysis. 2015 http://geneontology.org/page/go-enrichment-analysis.
- 19.Storey JD. A direct approach to false discovery rates. J R Statist Soc B. 2002;64:478–498. [Google Scholar]
- 20.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic AcidsRes. 2010;38:D355–360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R P, Chow S. Constructing area-proportional Venn and Euler diagrams with three circles [Abstract] INRIA Rocquencourt. Euler Diagrams Workshop; Paris: 2005. [Google Scholar]
- 24.Casal ML, Wolfe JH. Variant clinical course of mucopolysaccharidosis type VII in two groups of mice carrying the same mutation. Lab Invest. 1998;78:1575–1581. [PubMed] [Google Scholar]
- 25.Liu W, Griffin G, Clarke T, Parente MK, Valentino RJ, Wolfe JH, Fraser NW. Bilateral single-site intracerebral injection of a nonpathogenic herpes simplex virus-1 vector decreases anxiogenic behavior MPS VII mice, Molecular therapy. Methods & clinical development. 2015;2:14059. doi: 10.1038/mtm.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor SL, Leiserowitz GS, Kim K. Accounting for undetected compounds in statistical analyses of mass spectrometry 'omic studies. Stat Appl Genet Mol Biol. 2013;12:703–722. doi: 10.1515/sagmb-2013-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albrecht D, Kniemeyer O, Brakhage AA, Guthke R. Missing values in gel-based proteomics. Proteomics. 2010;10:1202–1211. doi: 10.1002/pmic.200800576. [DOI] [PubMed] [Google Scholar]
- 28.Wood J, White IR, Cutler P. A likelihood-based approach to defining statistical significance in proteomic analysis where missing data cannot be disregarded. Signal Processing. 2004;84:1777–1788. [Google Scholar]
- 29.Palmfeldt J, Vang S, Stenbroen V, Pedersen CB, Christensen JH, Bross P, Gregersen N. Mitochondrial proteomics on human fibroblasts for identification of metabolic imbalance and cellular stress. Proteome Sci. 2009;7:20. doi: 10.1186/1477-5956-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaban Y, Boekema EJ, Dudkina NV. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim Biophys Acta. 2014;1837:418–426. doi: 10.1016/j.bbabio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic AcidsRes. 2007;35:D61–65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.UniProt_Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N, Park CC, Wen PZ, Brewer H, Weitz K, Camp DG, 2nd, Pan C, Yordanova R, Neuhaus I, Tilford C, Siemers N, Gargalovic P, Eskin E, Kirchgessner T, Smith DJ, Smith RD, Lusis AJ. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011;7:e1001393. doi: 10.1371/journal.pgen.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho YE, Singh TS, Lee HC, Moon PG, Lee JE, Lee MH, Choi EC, Chen YJ, Kim SH, Baek MC. In-depth identification of pathways related to cisplatin-induced hepatotoxicity through an integrative method based on an informatics-assisted label-free protein quantitation and microarray gene expression approach. Mol Cell Proteomics. 2012;11:M111 010884. doi: 10.1074/mcp.M111.010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waters KM, Pounds JG, Thrall BD. Data merging for integrated microarray and proteomic analysis, Briefings. Functional Genomics And Proteomics. 2006;5:261–272. doi: 10.1093/bfgp/ell019. [DOI] [PubMed] [Google Scholar]
- 36.Haider S, Pal R. Integrated analysis of transcriptomic and proteomic data. Curr Genomics. 2013;14:91–110. doi: 10.2174/1389202911314020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogel C, Marcotte EM. Calculating absolute and relative protein abundance from mass spectrometry-based protein expression data. Nat Protoc. 2008;3:1444–1451. doi: 10.1038/nport.2008.132. [DOI] [PubMed] [Google Scholar]
- 38.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat RevGenet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 40.Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- 41.Chin MH, Qian WJ, Wang H, Petyuk VA, Bloom JS, Sforza DM, Lacan G, Liu D, Khan AH, Cantor RM, Bigelow DJ, Melega WP, Camp DG, 2nd, Smith RD, Smith DJ. Mitochondrial dysfunction, oxidative stress, and apoptosis revealed by proteomic and transcriptomic analyses of the striata in two mouse models of Parkinson's disease. J Proteome Res. 2008;7:666–677. doi: 10.1021/pr070546l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritter SY, Subbaiah R, Bebek G, Crish J, Scanzello CR, Krastins B, Sarracino D, Lopez MF, Crow MK, Aigner T, Goldring MB, Goldring SR, Lee DM, Gobezie R, Aliprantis AO. Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheum. 2013;65:981–992. doi: 10.1002/art.37823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kjellqvist S, Maleki S, Olsson T, Chwastyniak M, Branca RM, Lehtio J, Pinet F, Franco-Cereceda A, Eriksson P. A combined proteomic and transcriptomic approach shows diverging molecular mechanisms in thoracic aortic aneurysm development in patients with tricuspid- and bicuspid aortic valve. Mol Cell Proteomics. 2013;12:407–425. doi: 10.1074/mcp.M112.021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez ME, Klein AD, Scott MP. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J Neuroinflammation. 2012;9:216. doi: 10.1186/1742-2094-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 46.Kiselyov K, Muallem S. Mitochondrial Ca2+ homeostasis in lysosomal storage diseases. Cell Calcium. 2008;44:103–111. doi: 10.1016/j.ceca.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- 48.Woloszynek JC, Coleman T, Semenkovich CF, Sands MS. Lysosomal dysfunction results in altered energy balance. J Biol Chem. 2007;282:35765–35771. doi: 10.1074/jbc.M705124200. [DOI] [PubMed] [Google Scholar]
- 49.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 50.Wen H, Ting JP, O'Neill LA. A role for the NLRP3 inflammasome in metabolic diseases--did Warburg miss inflammation? Nat Immunol. 2012;13:352–357. doi: 10.1038/ni.2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koopman WJ, Distelmaier F, Smeitink JA, Willems PH. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.