

Abstract
The replacement of a carboxylic acid with a surrogate structure, or (bio)-isostere, is a classical strategy in medicinal chemistry. The general underlying principle is that by maintaining the features of the carboxylic acid critical for biological activity, but appropriately modifying the physicochemical properties, improved analogs may result. In this context, a systematic assessment of the physicochemical properties of carboxylic acid isosteres would be desirable to enable more informed decisions of potential replacements to be used for analog design. Herein we report the structure–property relationships (SPR) of 35 phenylpropionic acid derivatives, in which the carboxylic acid moiety is replaced with a series of known isosteres. The data set generated provides an assessment of the relative impact on the physicochemical properties that these replacements may have compared to the carboxylic acid analog. As such, this study presents a framework for how to rationally apply isosteric replacements of the carboxylic acid functional group.
Introduction
The replacement of an atom, or group of atoms, or even an entire scaffold of a biologically active compound with a surrogate structure that exhibits broadly similar biological properties is a fundamental strategy of medicinal chemistry, known as isosteric or bioisosteric replacement.1−3 In general, for this approach to be successful, similarities must exist between at least some of the properties of the isostere (we use the term ”isostere” in the broadest sense to include both classical and nonclassical isosteres, as well as bioisosteres) and those of the fragment being replaced, such that the new analogs retain the biological activities of the parent compound. At the same time, however, the isosteric replacement must produce changes in the physicochemical properties or susceptibility to metabolism compared to the parent compound in order to lead to improved derivatives. The success of any isosteric replacement is invariably context-dependent, however, and depends on the particular molecular environment of the biological target (e.g., the ability of the target to accommodate surrogate structures), and whether those surrogate structures will also provide the desired improvements in properties. For this reason, a screening of a series of alternative structures is almost always necessary.4 In this situation, the availability of experimental data detailing the structure–property relationships (SPR) of the existing palette of isosteres is most desirable. However, when these data are not available, the selection/prioritization of potential replacements is typically based on a variety of factors, such as the historical success rate of specific isosteres, calculated physicochemical properties, chemical/medicinal chemistry intuition, as well as synthetic accessibility.
The importance of the carboxylic acid functional group in drug design is illustrated by the fact that >450 marketed drugs are carboxylic acid containing molecules.5 However, the presence of this functional group in a drug or a drug candidate can be responsible for undesired consequences, such as limited permeability across biological membranes, metabolic instability, and potential idiosyncratic toxicities. To circumvent one or more of these shortcomings, medicinal chemists typically resort to prodrug (e.g., ester prodrugs) or isosteric replacement strategies. As part of our continued interest in the area of isosteric replacements of the carboxylic acid functional group,4,6−9 we set out to define the SPR of a number of acidic moieties that are frequently used as replacements of the carboxylic acid in drug design. In this particular context, the most important physicochemical parameters are arguably the acidity and lipophilicity, as well as the effect that the isosteric replacements may have on compound permeability. Since these three parameters are interrelated, even partial/incomplete information [e.g., just pKa or distribution coefficient (logD7.4) values] can be valuable in anticipating the physiochemical outcome of an isosteric replacement. However, a systematic SPR study enabling an accurate ranking of specific properties of carboxylic acid isosteres would provide a rational basis on which design decisions could be made. Somewhat surprisingly, thus far, such a study has never been reported. Although comparison of experimentally determined physicochemical properties of a number of compounds bearing carboxylic acid isosteres to the parent acid is available in the literature,4 these data are often incomplete or fragmented. Furthermore, these data sets are not readily comparable given that different isosteres are frequently found in completely unrelated molecules and often different methods are used to define the experimental values. Thus, to enable a more rigorous side-by-side comparison, we have assembled and characterized a library of 35 model compounds that are derivatives of phenylpropionic acid. Each compound was then evaluated experimentally for water solubility, acidity, lipophilicity, and passive permeability in a Parallel Artificial Membrane Permeability Assay (PAMPA). The effect of each of the carboxylic acid replacements on plasma protein binding was also evaluated. The data generated in these studies permit an assessment of the SPR of the most commonly incorporated carboxylic acid isosteres, which may be useful in the selection and prioritization of potential replacements of the carboxylic acid moiety to be used in analog design.
Results
Library Design
The design of the library scaffold took multiple factors into consideration. To ensure that the physicochemical properties of each member of the library provided accurate representation of the properties of the corresponding carboxylic acid isostere, it was desirable that each model compound be only minimally functionalized. At the same time, given that the assays employed to determine the physicochemical properties would rely upon either UV- or mass spectrometry-based detection methods, the model compounds should both contain a chromophore and exhibit sufficient molecular weight (i.e., MW ≥ 150 Da). Finally, a short aliphatic spacer separating the chromophore from the acidic moiety was viewed as desirable to avoid possible interference caused by conjugation that might affect the intrinsic physicochemical properties of the carboxylic acid surrogate. Based on these considerations, phenylpropionic acid (1, Table 1) was selected as the reference compound and each of the compounds in the library was designed as an analog thereof. In addition to the technical reasons mentioned above, the phenylpropionic acid is also an attractive template, as this substructure is found in a wide range of biologically active compounds, thus making the compound library directly relevant to medicinal chemistry.
Table 1. Calculated and Experimental Properties of Test Compounds.


Kinetic solubility in aqueous phosphate buffer (pH 7.4) determined by LC/MS after 24 h of incubation (experiment run by Analyza).
Distribution coefficient between n-octanol and aqueous buffer (pH 7.4) determined by LC/MS (experiment run by WuXi AppTech).
Calculated values using ChemAxon.10
Effective permeability (PAMPA assay run by Analyza).
Membrane retention.
Log of the apparent permeability coefficient.
pKa values determined by capillary electrophoresis; for diprotic compounds, the second equivalence point is indicated in brackets (experiment run by Analyza).
Plasma protein binding, fraction unbound (fu) determined by equilibrium dialysis.
The X-ray crystal structure revealing the H-bond pattern is shown in the Supporting Information.
The permeability value could not be calculated, as the concentration of test compound in the acceptor plate was below the limit of quantitation (<LOQ).
Compound precipitated in donor plate.
Compound appeared to exhibit high nonspecific binding.
Compound appeared to be unstable during the 24 h incubation time of the assay; ND = not determined.
The library of model compounds (36 entries) shown in Table 1 include examples from 24 different classes of carboxylic acid isosteres for which there is evidence of successful applications in drug design.4 These examples were chosen to represent as wide a range of carboxylic acid surrogates as possible with respect to structure and properties. They include noncarbon acyclic acids such as phosphonic and sulfonic acids and sulfonamides, modified carbon-based acids such as hydroxamic acids and acylurea, heterocycles like tetrazole and thiazolidinedione, and carbacycles such as phenols and squaric acid derivatives. For some of the classes of carboxylic acid isosteres that present multiple, nonequivalent points of attachment or derivatization (e.g., sulfonamides, acyl sulfonamides), a minimal set of representative congeners was prepared recognizing, however, that the physicochemical properties of these particular classes of isosteres could be effectively modulated by varying the nature of the substituents. Compounds 1, 6, 8, and 10 were commercially available, while all other analogs were synthesized based on literature precedent (see Experimental Section). In addition to the structures being characterized by standard methods (1H- and 13C-NMR, IR, HRMS), X-ray structures of compounds 1–3, 5, 6, 8, 10–20, 22, 26, 28, 32, 35, and 36 were also obtained (see Supporting Information).
Determination of Physicochemical Properties
The compound library was initially evaluated for kinetic solubility in aqueous buffer (pH 7.4). These results, summarized in Table 1, reveal that the vast majority of compounds exhibit aqueous solubility that is either comparable or greater than that of the parent acid (1). The only exceptions were compounds 21 (which is significantly less soluble than 1) and 34, for which a determination of aqueous solubility was not possible due to the fact that this compound was not detected after the 24 h incubation in aqueous buffer. With the knowledge that solubility would not limit the subsequent property evaluation of the remaining members, each test compound was evaluated for: (a) lipophilicity, by determining the distribution coefficient between n-octanol and aqueous buffer at pH 7.4 (i.e., logD7.4) via the shake-flask method; (b) acidity, by determining the pKa values via capillary electrophoresis; (c) permeability, using a PAMPA assay; and (d) plasma protein binding, by determining the fraction unbound (fu) via equilibrium dialysis. Calculated logD7.4 and pKa values were also obtained for comparison employing ChemAxon.10Table 1 assembles the accumulated data from these studies.
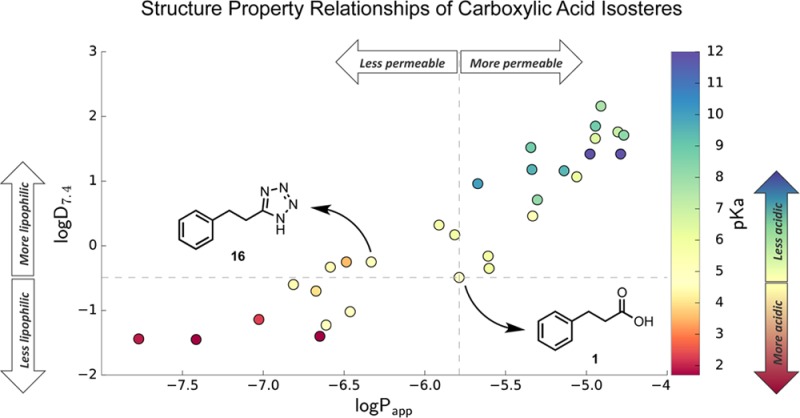
To highlight how the properties of each carboxylic acid isostere change in comparison to acid 1, Figures 1 and 2 illustrate the distribution of each of the measured values relative to 1. As shown in Figure 1, the replacement of the carboxylic acid with these surrogate structures can lead to a considerable variation of physicochemical properties relative to the reference compound 1: pKa values span over approximately 10 pKa units (2–12), with most isosteres being either equally acidic or less acidic than the parent acid. Only a handful of acidic moieties (e.g., noncarbon acids 6–9, oxadiazol-5(4H)-thione 22, as well as squaric acid derivatives 30 and 31) had measured pKa values below 4. The modulation of the pKa of a target molecule is often an important aspect in compound optimization. The influence of pKa on compound permeability (vide infra) as well as binding to the pharmacological target can be profound; therefore, a ranking of carboxylic acid isosteres based on their intrinsic acidity provides a starting point for prioritizing which moieties are most likely to impart pKa values within the desired range. In addition, these data could be helpful in determining whether neighboring group substitution may be required to modulate the pKa values of specific isosteres of interest if the intrinsic acidity falls outside of the optimal range.
Figure 1.

Plot showing lipophilicity (i.e., logD7.4), acidity (i.e., pKa), and permeability (i.e., logPapp) of test compounds, relative to the carboxylic acid compound 1; logD7.4 and logPapp values for compounds 1, 16, 14, and 17 are the averages obtained from three independent experiments; (**) p < 0.01 by two-tailed t test confirming statistical significant difference in membrane permeability between 16 and 1; NS indicates that the difference in logD7.4 values between 16 and 1 does not reach statistical significance.
Figure 2.

Plot of compound lipophilicity (i.e., logD7.4), plasma protein binding (i.e., fu), and acidity (i.e., pKa) relative to the carboxylic acid compound 1; fu values are the averages obtained from three independent experiments; logD7.4 values for compounds 1, 16, 14, and 17 are the averages obtained from three independent experiments.
In contrast to the wide range of pKa values, lipophilicity (i.e., logD7.4), and permeability coefficient (Papp) values in the PAMPA assay appear to be more narrowly distributed within approximately ∼3 log units, with a near equal number of isosteres leading to a relative increase and a decrease in lipophilicity and permeability. Examples of isosteres with logD7.4 values closest to the acid include tetrazole 16, oxazolidinedione 18, oxadiazol-5(4H)-thione 22, tetramic acid 24, and cyclopentane-1,3-diones 25–27. Permeability coefficients (Papp) were obtained for 33 of the 36 test compounds. The data for the oxathiadiazole 21, and the fluorophenols derivatives 32 and 33 were considered unreliable due to high nonspecific binding (32) or compound precipitation (21 and 33) on the donor plate during the assay (Table 1). Carboxylic acid isosteres with relatively high membrane permeability in the PAMPA assay (i.e., log Papp > −5.8) included acylurea 15, sulfonamide 11, thiazolidinedione 17, thiadiazol-5(4H)-one 20, cyclopentane-1,2-diones 28 and 29, and substituted phenol 35 and 36.
Similar to the results of pKa determination, the effect of isosteric replacements on plasma protein binding was pronounced (Figure 2). Some derivatives displayed very high fraction unbound (fu) values, including the acylurea 15 (77%), hydroxamic esters 4 and 5 (68% and 64%, respectively), and sulfonamide 10 (61%), while others [oxadiazol-5(4H)-thione 22, isoxazole 23, and substituted phenols 32 & 33] were virtually completely bound (i.e., fu < 1%).
Since many medicinal chemistry optimization projects rely on computational chemistry programs to predict specific properties to prioritize compounds for synthesis, we employed programs in ChemAxon to calculate logD7.4 and pKa values for all analogs. From the data depicted in Table 1, a comparison of experimental and calculated logD7.4 or pKa values reveals that the calculated values for the carboxylic acid and most isosteres appear to correlate well with experimental results; however, there are notable exceptions in which the discrepancy can be as large as 2 to 3 log units (see Figure 3A/B and Figure 4A/B). This variance is especially notable for the oxadiazol-5(4H)-thione (22) and conjugated 1,3-dicarbonyl systems, giving rise to vinylogous acids (e.g., 24–27, 30, and 31). As such, caution should be exercised when employing computational chemistry programs to predict the properties of these structural types.
Figure 3.
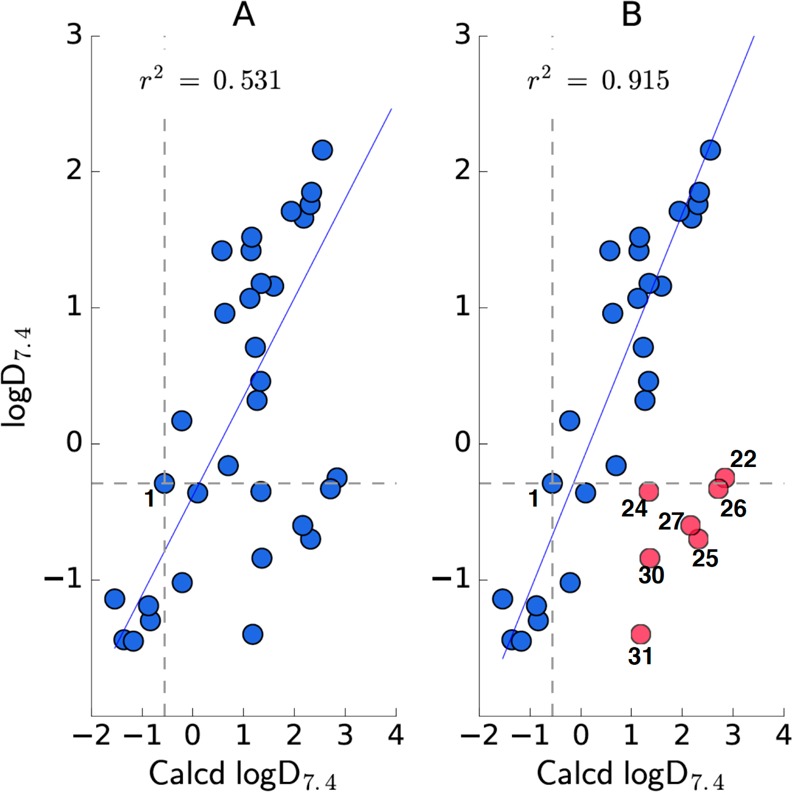
Comparison between experimental and calculated logD7.4 values; (A) the linear regression for the entire data set; (B) the linear regression when major outliers (i.e., discrepancy >1 log unit, shown in red) are excluded.
Figure 4.
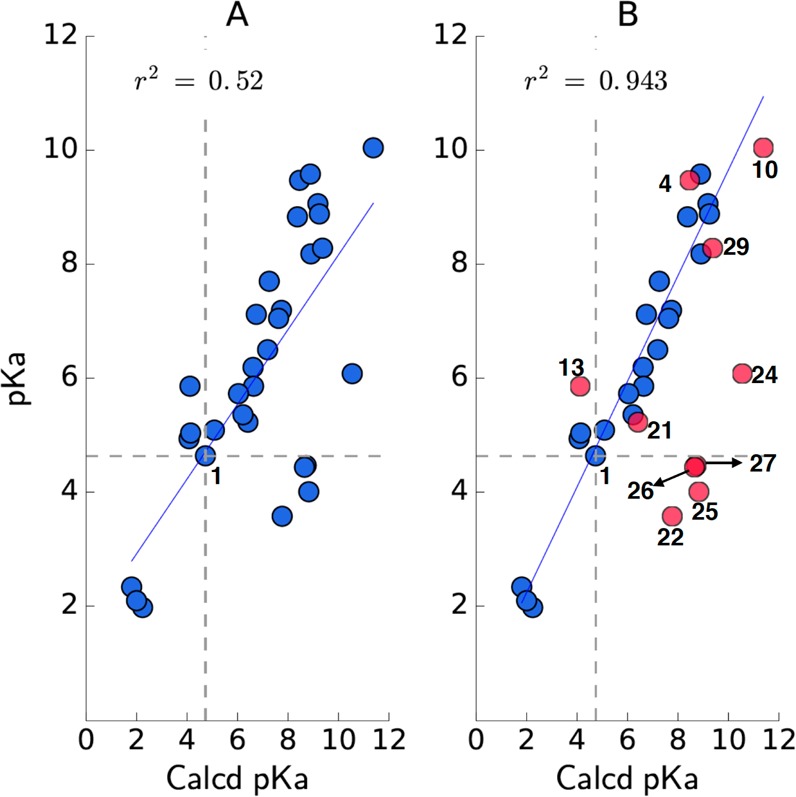
Comparison between experimental and calculated pKa values; (A) the linear regression for the entire data set; (B) the linear regression when major outliers (i.e., discrepancy >1 log unit, shown in red) are excluded.
While the specific value for each of the individual properties is useful, the correlations one can draw between the data sets are more helpful. As stated above, most of the measured properties are interrelated and one property certainly influences the others. Although no overt correlation was evident between the fu and logD7.4 values (Figure 2), overall there appears to be a relationship linking the ionization state of the acidic moiety with the fu. Thus, in most cases, the least acidic isosteres (i.e., pKa > 8), such as the hydroxamic acids (i.e., 2 and 3) and esters (i.e., 4 and 5), sulfonamides (i.e., 10 and 11), and acyl-urea (i.e., 15) had higher fu values (>29%) relative to the carboxylic acid and most other isosteres that were predominantly negatively charged at pH 7.4 (Table 1).
Evaluation of the data also revealed a reasonably good correlation between the lipophilicity and permeability data (r2 = 0.828; Figure 1). However, notably, some compounds with very similar acidity and lipophilicity exhibited significantly different permeability coefficients in the PAMPA assay. For example, comparison of the amino squaric acid (31) with the phosphinic acid (7) derivative suggests that the latter may be significantly less permeable than the former, in spite of comparable acidity and lipophilicity (Table 1 and Figure 1). Relatively large differences in permeability coefficients between compounds that are essentially isometric with respect to lipophilicity and acidity are also observed when comparing carboxylic acid 1 with other isosteres, such as the cyclopentane-1,3-dione (25–27), or the oxadiazol-5(4H)-thione (20), or the tetrazole (16). In particular, the difference in permeability between 1 and 16 was confirmed in three independent experiments (p < 0.01).
This observation indicated that, in addition to acidity and lipophilicity, other factors can influence compound permeability in the PAMPA assay. One possibility is that the desolvation energy may be significantly different with different acidic moieties, even in those cases where acidity and lipophilicity are comparable. Since the solvation/desolvation energy is determined in large part by hydrogen-bond (HB) interactions, we conducted further studies to compare specifically the HB capacity of tetrazole 16 and carboxylic acid 1. For this endeavor, we employed a colorimetric assay11 in which the H-bonding between a fluorescent pyrazinone sensor (HB acceptor) and the analyte HB donor (i.e., 1 or 16) was monitored by following the characteristic blue-shift of the maximum wavelength (λmax) of the sensor in the UV spectrum (Figure 5). Given that the change in λmax of the sensor depends on the strength of the HB interaction with the analyte, stronger HB donors will cause comparatively larger shifts than weaker HB donors. Interestingly, these titration experiments revealed that the tetrazole derivative engages in significantly stronger HB interactions than the corresponding carboxylic acid (Figure 5). These results suggest that if similar differences exist in the interaction with water, then a tighter solvation and correspondingly higher desolvation energy would be expected for 16 compared to 1, leading to less permeability in a PAMPA assay. Further evaluation of this possibility will require additional studies to compare 16 and 1 in the deprotonated forms (i.e., tetrazolate and carboxylate) as HB acceptors.
Figure 5.
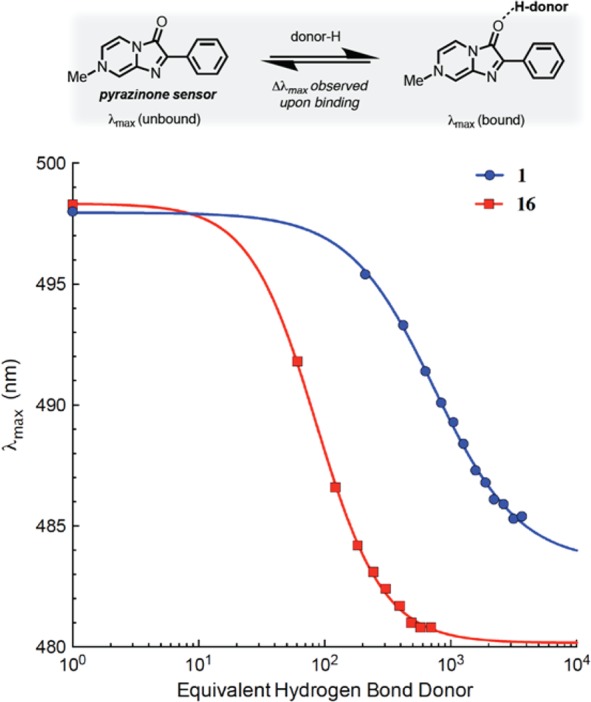
Titration curves from a colorimetric assay that monitors the blue-shift in λmax of the pyrazinone sensor upon complexation with the HB donor analyte (1 or 16).
Discussion
The carboxylic acid is one of the most frequent functional groups in small molecules that bind to protein targets and is considered a privileged substructure.12 The acidity, combined with the ability to establish relatively strong electrostatic interactions and hydrogen-bonds, as well as the ability to participate in interactions with dipoles, make this functional group highly versatile, and in turn able to engage in molecular interactions with a wide array of complementary functional groups. These properties also imply that the carboxylic acid moiety can impart relatively high water solubility, which is an important attribute for a drug-like molecule. On occasion, these same properties can also contribute to some of the deficits of carboxylic acids as drug candidates, including, for example, relatively poor permeability across biological membranes. Additionally, other potential liabilities that have been associated with the carboxylic acid functional group in drugs or drug candidates include a relatively rapid metabolism mediated by uridine 5′-diphospho-glucuronosyl-transferase (UGTs).13 This phase 2 metabolic process can be responsible for limited compound half-life and for the formation of reactive acyl-glucuronides that can cause covalent modifications of proteins and potentially serious adverse side effects.14 The replacement of the carboxylic acid with appropriate isosteres can be especially useful to circumvent these possible drawbacks while maintaining the positive aspects of the carboxylic acid functional group. Furthermore, in addition to these specific applications, carboxylic acid isosteres can be employed more broadly in analog design to explore the effects of diversification of structure and physicochemical properties of compounds of interest. The utility of this strategy is evident from the fact that several carboxylic acid isosteres, such as tetrazole, thiazolidinedione, sulfonamides, and acyl sulfonamides have ultimately led to the clinical development of entire classes of important therapeutic agents (e.g., see Figure 6).
Figure 6.

Representative examples of drugs/drug candidates and drug classes containing carboxylic acid bioisosteres.
Based on the importance of acid isosteres to the field of medicinal chemistry and the critical role that these fragments can play in tuning the properties of acidic, biologically active compounds, we assembled a set of 35 carboxylic acid isosteres on the same scaffold and measured a focused set of key physicochemical properties, such as acidity, lipophilicity, and permeability. Furthermore, given that molecules possessing the carboxylic acid functionality are often found to exhibit a high degree of plasma protein binding,15,16 the effect of these isosteric replacements on the fraction unbound was also investigated. Finally, using computational chemistry programs, we calculated pKa and logD7.4 values to compare with experimental values. The data set generated in this study provides an assessment of the effect that such isosteres can have on the physicochemical properties of the corresponding carboxylic acid. In addition, our results highlight some correlations between properties that can be used during the selection/prioritization of isosteres for analog design.
In agreement with previous studies,17 our results indicate that overall there is a clear correlation between the experimental logD7.4 values and the apparent permeability coefficients in the PAMPA assay (Figure 1). Generally, more lipophilic and less acidic compounds exhibit higher rates of passive diffusion. However, in selected cases the impact that isosteric replacements produce on compound permeability appears to deviate significantly from this relationship. These deviations are most effectively illustrated by the comparison between tetrazole 16 and carboxylic acid 1. The tetrazole, which among the carboxylic acid isosteres is one that has been the focus of intense studies,18,19 is generally considered to be more lipophilic20 and thus potentially more permeable than the carboxylic acid.21 However, our comparative studies with model compounds reveal that while the two acidic moieties exhibit similar pKa and logD7.4 values, the tetrazole is significantly less permeable in the PAMPA assay. Interestingly, previous Caco2 bidirectional permeability studies with a series of carboxylic acids and corresponding 1H-tetrazoles derivatives found that several tetrazoles were less permeable than the carboxylic acids due to active efflux mechanisms.22,23 However, in our studies, given the artificial nature of the PAMPA assay, active transport systems, metabolism, and protein binding are excluded from consideration. Therefore, any differences in the rate of transport should ultimately result from specific differences in physicochemical properties between the acidic moieties. Our studies found that the model compound bearing the tetrazole moiety can establish considerably stronger HB interactions than the corresponding compound bearing the carboxylic acid. This result is consistent with previous reports in which other 1H-tetrazoles were found to be relatively strong HB donors24 and suggests that the tetrazole derivative may be more tightly solvated in water than the carboxylic acid, leading to correspondingly higher desolvation energies. As the desolvation energy is known to affect compound permeability across biological membranes,25,26 the different rates of diffusion that have been observed for 16 and 1 in the PAMPA assay may be related, at least in part, to the difference in HB strength between the tetrazole and the carboxylic acid. In this regard, it should be noted that the lack of equivalence between the hydrogen-bonding and the acidity scale is not unexpected as this phenomenon has been documented.24,27
In addition to the differences in the permeability rates in the PAMPA assay, 16 and 1 were also found to exhibit significant differences in the plasma protein-binding assay, with the tetrazole derivative being more tightly bound than the corresponding carboxylic acid compound. A similar observation was reported when comparing the plasma protein-binding of other match paired tetrazole and carboxylic acid analogs.28 Also consistent with previous reports illustrating the importance of molecular ionization states on plasma protein binding,15,16 a broader examination of the plasma protein-binding data from all carboxylic acid isosteres evaluated in this study suggests that derivatives that are predominantly neutral at pH 7.4 generally exhibit larger fu values; however, there seems to be no clear correlation between the plasma protein binding and the lipophilicity of our test compounds. Although this outcome appears to be in disagreement with previous studies that reported a relatively good correlation between the calculated logD7.4 values and plasma protein binding of a series of carboxylic acid molecules,29 it is possible that such a correlation can be established only within specific families of closely related compounds and not between different classes of carboxylic acid isosteres. When we compared the fu values of 1 and 25–27 with the corresponding, but more lipophilic congeners that were brominated in the para position, we found that within each of the matching pairs the fu was considerably lower for the brominated derivative (see Supporting Information).
Finally, discrepancies between the calculated and the experimental logD7.4 and pKa values that are evident for all vinylogous acids tested likely arise from these or similar vinylogous systems being absent or underrepresented in the training set used by the software. As such, the predictive ability of the software could be readily improved by simply updating the training set with the data provided in this study. Our results underscore the importance of experimental values and suggest that caution should be exercised when calculated physicochemical properties are used to prioritize carboxylic acid isosteres for analog design.
Conclusions
Taken together, the data generated in this study permit an assessment of the relative differences in physicochemical properties of carboxylic acid isosteres, compared to the corresponding carboxylic acid. This data set comprises a benchmark that may be useful to compare the physicochemical properties of carboxylic acid isosteres and ultimately permit more informed and effective prioritizations of isosteric replacements in medicinal chemistry for analog design.
Experimental Section
The synthesis of test compounds is highlighted in Schemes 1–11. The hydroxamic acids 2(30) and 3(31) (Scheme 1) were prepared starting respectively from carboxylic acid 1 and O-benzylhydroxylamine.32 Hydroxamic ester derivative 4(33) was obtained via 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDCI) mediated coupling of 1 with O-methyl hydroxylamine, whereas reverse hydroxamic ester 5(34) was obtained by O-alkylating acetohydroxamic acid, following procedures described by Wrigglesworth and co-workers.35
Scheme 1. Synthesis of Hydroxamic Acid and Ester Derivatives.

Reagents and Reaction Conditions: (a) CDI, hydroxylamine, CH3CN, r.t., 14 h, (70%); (b) N-methylmorpholine, EDCI·HCl, O-methyl hydroxylamine, r.t., 16 h, (83%); (c) (i) K2CO3, Na2CO3, MeOH/H2O, 50 °C, 10 min; (ii) (2-bromoethyl)benzene, 50 °C, 16 h, (32%); (d) Boc2O, THF/H2O, NEt3, r.t., 2 h, (45%); (e) (i) NaH, DMF, r.t., 30 min; (ii) (2-bromoethyl)benzene, r.t., 16 h, (53%); (f) (i) TFA/CH2Cl2, r.t., 18 h, (85%); (ii) Ac2O, DMAP, pyridine, CH2Cl2, r.t., 2 h, (79%); (g) H2 (1 atm), Pd/C, MeOH, r.t., 3 h, (77%).
Scheme 11. Synthesis of Substituted Phenol Derivatives.

Reagents and Reaction Conditions: (a) NBS, DMF, 0 °C to r.t., 40 h, (86%); (b) (i) K2CO3, acetone, 70 °C, 1 h; (ii) benzyl bromide, 70 °C, 3 h then r.t., 14 h (99%); (c) ZnBr2, benzylmagnesium chloride, PEPPSI-IPr, THF, r.t., 16 h, (74%); (d) H2 (1 atm), Pd/C, MeOH or EtOH, r.t., 1 h (97–99%); (e) (i) n-BuLi, THF, –50 °C, 20 min; (ii) B(O-i-Pr)3, −50 °C to r.t., 1 h; (f) (PPh3)2Pd(Br)(Succ), benzyl bromide, Na2CO3, THF/H2O, 60 °C, 14 h, (42%); (g) (i) KHCO3, DMF, r.t., 5 min; (ii) benzyl bromide, r.t., 16 h, (93%); (h) AcOH, UHP, r.t., 20 h, (98%); (j) N-methylmorpholine N-oxide, K2OsO4·2H2O, CH2Cl2, r.t., 20 h, (95%).
With respect to phosphinic (736) and sulfinic acid (937) derivatives (Scheme 2), the former was obtained by reacting diethylchlorophosphite with phenethylmagnesium bromide, while the latter was prepared in two steps starting from 2-phenylethanethiol, via an oxidation–reduction sequence.
Scheme 2. Synthesis of Phosphinic and Sulfinic Acid Derivatives.

Reagents and Reaction Conditions: (a) Phenethylmagnesium bromide, Et2O, 5 °C to r.t., 16 h, (32%); (b) NCS, CH2Cl2/H2O, r.t., 3.5 h, (90%); (c) Na2SO3, NaHCO3, CH3CN/H2O, r.t., 24 h, (75%).
The model compounds bearing the sulfonamide (1138) and acylurea (1539) functional groups were synthesized starting respectively from 3-phenylpropan-1-amine and benzyl urea as previously described (Scheme 3). The acylsulfonamide derivatives 12 and 13 (Scheme 3) were obtained by coupling 1 with the appropriate sulfonamides, whereas sulfonylurea 14 was obtained by reacting methylsulfonamide with benzyl-carbamoylimidazole, which can be readily prepared from benzylamine hydrochloride and 1,1′-carbonyldiimidazole (CDI).40
Scheme 3. Synthesis of Sulfonamide, Acylsulfonamide, Sulfonylurea, and Acylurea Derivatives.

Reagents and Reaction Conditions: (a) MsCl, NEt3, CH2Cl2, 4 °C, 24 h, (88%); (b) methanesulfonamide, EDCI·HCl, DMAP, CH2Cl2, 45 °C, 8 h, then r.t., 48 h, (70%); (c) (i) 4 M HCl in dioxane, r.t., 5 min; (ii) CDI, DMF, CH3CN, r.t., 2 h; (iii) NaH, methanesulfonamide, DMF, r.t., 16 h, (11%); (d) (i) TBTU, DIEA, CH2Cl2, r.t., 25 min; (ii) N,N-dimethylsulfamide, r.t., 14 h, (81%); (e) Ac2O, CH2Cl2, 50°C, 24 h, then r.t., 48 h, (75%).
The tetrazole derivative 16(41) (Scheme 4) was prepared from nitrile 41 and sodium azide,42 or alternatively, from the cyanoalkylamide 42 and TMS-azide, under Mitsunobu conditions, followed by elimination of the acrylonitrile.43
Scheme 4. Synthesis of Tetrazole Derivative.

Reagents and Reaction Conditions: (a) EDCI·HCl, HOBt, DMF, DIEA, 3-aminopropionitrile, r.t., 15 h, (95%); (b) (i) PPh3, DIAD, TMSN3, 0 °C, 30 min, then r.t., 2 h, then 50 °C, 17 h; (ii) DBU, CH2Cl2, r.t., 6 h, (63%); (c) NH2OH·HCl, DMSO, 90 °C, 3.5 h, (61%); (d) NaN3, ZnBr2, H2O, 150 °C, 24 h, (20%).
In the case of thiazolidine-2,4-dione (1744) and oxazolidine-2,4-dione (18) derivatives, these model compounds were obtained by reacting the appropriately lithiated heterocycle with benzyl bromide (Scheme 5).
Scheme 5. Synthesis of Thiazolidine-2,4-dione and Oxazolidine-2,4-dione derivatives.

Reagents and Reaction conditions: (a) (i) n-BuLi, THF, –78 °C, 10 min (ii) benzyl bromide, –78 °C to r.t, 1.5 h, (53%); (b) (i) t-BuOK, diethyl carbonate, MeOH, 70 °C, 19 h, then r.t., 24 h; (ii) n-BuLi, THF, –78 to 0 °C, 30 min; (iii) benzyl bromide, –78 °C to r.t., 2 h, (5%).
For the synthesis of the oxadiazole derivatives (19 and 22) as well as related thiadiazole (20) and oxathiadiazole (21) compounds (Scheme 6), common precursor N′-hydroxy-3-phenylpropanimidamide (43) was reacted with the appropriate acylating agent according to conditions reported by Kohara and co-workers.45
Scheme 6. Synthesis of 5-Oxo-1,2,4-oxadiazole and Related Thiadiazole Derivatives.

Reagents and Reaction Conditions: (a) NH2OH, H2O, EtOH, 75 °C, 5.5 h, (92%); (b) (i) isobutyl chloroformate, pyridine, DMF, 0 °C to r.t., 16 h; (ii) toluene, 120 °C, 2 h, then 140 °C, 24 h, (59%); (c) (i) 1,1′-thiocarbonyldiimidazole, THF, r.t., 40 min; (ii) BF3·OEt2, THF, r.t., 3 h, (28%); (d) SOCl2, THF, CH2Cl2, 0 °C, 1 h, (<10%); (e) DBU, 1,1′-thiocarbonyldiimidazole, CH3CN, r.t., 24 h, (43%).
The synthesis of known 3-hydroxyisoxazol 23(46) was conducted starting from Meldrum’s acid, as highlighted in Scheme 7.
Scheme 7. Synthesis of Isoxazol-3-ol Derivative.

Reagents and Reaction Conditions: (a) (i) pyridine, CH2Cl2, 0 °C, 10 min; (ii) phenylacetyl chloride, 0 °C, 4 h, (91%); (b) N,O-di-Boc-hydroxylamine, toluene, 65 °C, 16 h, (quant.); (c) 4 M HCl, MeOH, r.t., 16 h, (10%).
The synthesis of a tetramic acid derivative (2447) was accomplished according to known procedures in three steps, starting from N-Boc protected phenylalanine and Meldrum’s acid, while structurally related cyclopentane-1,3-diones (25 and 26) were obtained via chemoselective benzylation of the appropriate isobutyl-protected cyclopentane-1,3-dione with benzyl bromide under conditions reported by Koreeda and co-workers,48 followed by hydrolysis of the vinylogous ester (Scheme 8). The 1,3-dione derivative 27 was prepared in one step from cyclopentane-1,3-dione and benzaldehyde under reductive alkylation conditions.49
Scheme 8. Synthesis of Tetramic Acid and Cyclopentane-1,3-Dione Derivatives.

Reagents and Reaction Conditions: (a) (i) Meldrum’s acid, DMAP, CH2Cl2, EDCI·HCl, 0 °C to r.t., 15 h; (ii) EtOAc, reflux, 30 min; (iii) TFA/CH2Cl2, r.t., 15 min, (91%); (b) (i) LDA, THF, –78 °C, 20 min; (ii) benzyl bromide, −78 °C to r.t., 3 h, (27–45%); (iii) acetone, HCl (2 M), r.t., 16 h, (42–67%); (c) Hantzsch ester, L-proline, benzaldehyde, CH2Cl2, r.t., 16 h, (60%).
The synthesis of cyclopentane-1,2-dione derivative 28(50) (Scheme 9) was accomplished in two steps by reacting known dipotassium salt 45(51) with benzyl bromide to give intermediate 46, followed by a debenzylation/decarboxylation sequence of the latter to obtain the desired 1,2-dione. The 1,2-dione 29(52) was synthesized via magnesium methoxide induced cyclization of α,β-unsaturated dione 47.
Scheme 9. Synthesis of Cyclopentane-1,2-dione Derivatives.

Reagents and Reaction Conditions: (a) benzyl bromide, DMF, 120 °C, 2 h, (47%); (b) AcOH, HCl (37%), 130 °C, 3 h, (71%); (c) (i) benzaldehyde, NaOH, MeOH, r.t., 24 h; (ii) TsOH·H2O, acetone, r.t., 38 h (73%); (d) Mg(OMe)2, MeOH, reflux, 90 min, (53%).
The cyclobutane-1,2,3-trione derivative 30 (Scheme 10) was obtained by reacting diethyl squarate with benzylmagnesium bromide, followed by acid-mediated hydrolysis. Squaric acid monoamide derivative 31(53) (Scheme 10) was prepared by direct condensation of benzylamine with squaric acid under microwave assisted conditions.
Scheme 10. Synthesis of Squaric Acid Derivatives.

Reagents and Reaction Conditions: (a) benzylmagnesium bromide, THF, 0 °C to r.t., 40 min, (82%); (b) 3 M HCl, acetone, r.t., 3 h (66%); (c) benzylamine, 200 °C (microwave irradiation), 20 min, (22%).
The synthesis of fluorophenol derivative 32 (Scheme 11) was completed in 4 steps starting from 2,6-difluorophenol. Thus, after bromination and protection of the phenol as benzyl-ether, Negishi cross-coupling conditions were employed to obtain 51, which was finally deprotected by hydrogenolysis to give model compound 32. The corresponding meta-benzylated isomer 33 was obtained via selective ortho-borylation of protected phenol 52, followed by a Suzuki-Miyaura cross-coupling reaction with benzyl bromide,54 and final deprotection.55 Finally, the synthesis of substituted phenols 34–36 was accomplished starting from 2-hydroxythiophenol via benzylation reaction (34), followed by S-oxidation to the corresponding sulfoxide (35) and sulfone (36).
Materials and Methods
All solvents were reagent grade. All reagents were purchased from Aldrich or Fisher Scientific and used as received. Thin layer chromatography (TLC) was performed with 0.25 mm E. Merck precoated silica gel plates. Flash chromatography was performed with silica gel 60 (particle size 0.040–0.062 mm) supplied by Silicycle and Sorbent Technologies. TLC spots were detected by viewing under a UV light, or using KMnO4 or ceric ammonium molybdate stains. Melting points (mp) were acquired on a Thomas-Hoover apparatus and are uncorrected. Infrared (IR) spectra were recorded on a Jasco Model FT/IR-480 Plus spectrometer. Proton (1H) and carbon (13C) NMR spectra were recorded on a Bruker AMX-500 spectrometer. Chemical shifts were reported relative to the residual solvent’s peak. High-resolution mass spectra were measured at the University of Pennsylvania Mass Spectrometry Center on either a VG Micromass 70/70H or VG ZAB-E spectrometer. Single-crystal X-ray structure determinations were performed at the University of Pennsylvania with an Enraf Nonius CAD-4 automated diffractometer. Analytical reverse-phased (Sunfire C18; 4.6 × 50 mm, 5 mL) high-performance liquid chromatography (HPLC) was performed with a Waters binary gradient module 2525 equipped with Waters 2996 PDA and Waters micromass ZQ. All samples were analyzed employing a linear gradient from 10% to 90% of CH3CN in H2O over 8 min and flow rate of 1 mL/min, and unless otherwise stated, the purity level was >95%. Preparative reverse-phase HPLC purifications were performed on a Gilson instrument (i.e., Gilson 333 pumps, a 215 liquid handler, 845Z injection module, and PDA detector) employing Waters SunFire preparative C18 OBD columns (5 μm 19 × 50 or 19 × 100 mm). Unless otherwise stated, HPLC purifications were carried out employing a linear gradient from 10% to 90% of CH3CN in H2O for 15 min with a flow rate of 20 mL/min. Unless otherwise stated, all final compounds were found to be >95% as determined by HPLC/MS and NMR.
3-Phenylpropionic acid (1)
Commercially available. X-ray quality crystals were obtained by slow evaporation from a CH2Cl2 solution (see Supporting Information): mp (CH2Cl2) 49–51 °C.
N-(Hydroxy)-3-phenylpropanamide (2)
To a stirred solution of 1 (1.00 g, 6.66 mmol, 1.00 equiv) in anhydrous CH3CN (22 mL) at r.t. under N2, CDI (1.30 g, 7.99 mmol, 1.20 equiv) was added in one portion, and the resulting solution was stirred for 35 min. A solution of hydroxylamine (50 wt % in H2O, 0.450 mL, 7.32 mmol, 1.10 equiv) was then added dropwise, and the solution was stirred for 14 h at r.t.. The reaction was quenched by careful addition of 1 M KHSO4 (until pH 1–2), then extracted with EtOAc (×3). The combined organic extracts were washed with phosphate buffer (pH 7), H2O, brine, then dried over Na2SO4, filtered, and concentrated in vacuo. Recrystallization from EtOAc/hexanes provided the title product as a colorless to off-white solid (0.774 g, 4.69 mmol, 70%). X-ray quality crystals were obtained by layer diffusion of cyclohexane into a CH2Cl2 solution (see Supporting Information): mp (CH2Cl2/cyclohexane) 73–74 °C; 1H NMR (500 MHz, CDCl3) δ 9.08 (br s, 2H), 7.27–7.14 (m, 5H), 2.90 (t, J = 7.8 Hz, 2H), 2.40 (t, J = 7.8 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.3, 140.3, 128.8, 128.5, 126.6, 34.9, 31.5 ppm; IR (KBr) ν 3204, 3059, 3025, 2925, 2861, 1642, 1496, 1452, 1382 cm–1; HRMS (ES+) calculated for C9H11NNaO2 [M + Na]+ 188.0687, found 188.0688.
N-Hydroxy-N-phenethylacetamide (3)
To a solution of 39 (0.183 g, 0.679 mmol, 1.00 equiv) in MeOH (3.40 mL), Pd/C (10 wt % (wet), 0.018 g) was added. The suspension was stirred under an H2 atmosphere (balloon) for 3 h. The reaction mixture was then filtered through a pad of Celite, washed with EtOAc, and concentrated in vacuo. Purification by a short silica gel column chromatography (20% EtOAc in hexanes) afforded the title compound (0.083 g, 0.46 mmol, 68%). X-ray quality crystals were obtained by slow evaporation from a CH2Cl2/hexanes solution (see Supporting Information): mp (CH2Cl2/hexanes) 49–51 °C; 1H NMR (500 MHz, CDCl3) mixture of E/Z isomers δ 7.44–7.08 (m, 3H), 3.97–3.71 (m, 2H), 2.98 (dt, J = 32.7, 6.7 Hz, 1H), 2.10 (s, 1H), 1.61 (s, 1H) ppm; 13C NMR (125 MHz, CDCl3) mixture of E/Z isomers δ 172.3, 165.2, 138.7, 138.0, 129.1, 128.9, 128.6, 127.1, 126.5, 51.6, 49.7, 33.5, 33.1, 20.5, 18.4 ppm; IR (KBr) ν 3172, 2922, 2848, 1614 cm–1; HRMS (ES+) calculated for C10H14NO2 [M + H]+ 180.1025, found 180.1020.
N-Methoxy-3-phenylpropanamide (4)
To a solution of 1 (0.210 g, 1.40 mmol, 1.00 equiv) in CH2Cl2 at r.t. under N2, N-methylmorpholine (0.190 mL, 1.70 mmol, 1.20 equiv) was added dropwise, and the reaction mixture was stirred for 15 min. EDCI·HCl (0.328 g, 1.70 mmol, 1.20 equiv) was added, and the suspension was stirred until a clear solution was obtained. O-methylhydroxylamine hydrochloride (0.146 g, 1.70 mmol, 1.20 equiv) was then added and the solution was stirred for 16 h at r.t.. The reaction was quenched with satd. aq. NH4Cl, and extracted with CH2Cl2 (×3). The combined organic extracts were washed with H2O, brine, then dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (2–10% MeOH in CH2Cl2) afforded the title compound (0.210 g, 1.17 mmol, 83%) as a clear oil: 1H NMR (500 MHz, CDCl3) δ 10.05 (s, 1H), 7.28–7.21 (m, 5H), 3.66 (s, 3H), 2.98 (t, J = 7.8 Hz, 2H), 2.45 (app t, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 170.3, 140.5, 128.6, 128.4, 126.4, 64.0, 34.8, 31.5 ppm; IR (KBr) ν 3182, 3000, 2974, 2936, 1659, 1497, 1453 cm–1; HRMS (ES+) calculated for C10H13NNaO2 [M + Na]+ 202.0844, found 202.0838.
N-Phenethoxyacetamide (5)
Acetohydroxamic acid (0.276 g, 3.67 mmol, 1.00 equiv) and K2CO3 (0.508 g, 3.67 mmol, 1.00 equiv) were dissolved in MeOH/H2O (1:1, 17 mL) and the resulting solution was stirred at r.t. for 10 min. Na2CO3 (0.428 g, 4.04 mmol, 1.10 equiv) was added in one portion at r.t., then the reaction was stirred at 50 °C for 10 min. Phenethyl bromide (0.500 mL, 3.67 mmol, 1.00 equiv) was added dropwise, and the reaction mixture was vigorously stirred for 16 h at the same temperature. After cooling to r.t., the reaction mixture was concentrated in vacuo, then diluted with 1 M HCl (until pH 2) and extracted with CH2Cl2 (×4). The combined organic extracts were washed with brine, then dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (40–100% EtOAc in hexanes) provided the title compound (0.208 g, 1.16 mmol, 32%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from an Et2O/toluene solution (see Supporting Information): mp (Et2O/toluene) 87–87.5 °C; Lit.34 (benzene) 91–93 °C; 1H NMR (500 MHz, MeOH-d4) δ 7.28–7.17 (m, 5H), 4.89 (s, 1H), 4.02 (t, J = 7.1 Hz, 2H), 2.94 (t, J = 7.1 Hz, 2H), 1.85 (s, 3H) ppm; 13C NMR (125 MHz, MeOH-d4) δ 168.7, 137.9, 128.5, 128.1, 126.0, 100.2, 76.6, 34.0, 18.0 ppm.
Phenethylphosphonic acid (6)
Commercially available. X-ray quality crystals were obtained by slow diffusion of ligroin into an i-PrOH solution (see Supporting Information): mp (i-PrOH/ligroin) 135–136 °C.
Phenethylphosphinic acid (7)
Diethylchlorophosphite (1.50 g, 9.58 mmol, 1.00 equiv) in anhydrous Et2O (10 mL) was cooled at 5 °C under N2. Phenethylmagnesium chloride (1.0 M in THF, 10.0 mL, 10.0 mmol, 1.04 equiv) was added dropwise while maintaining the internal temperature between 0–10 °C. The reaction mixture was stirred at r.t. for 16 h, then filtered under N2. The filtrate was concentrated in vacuo to give a clear, colorless liquid. The liquid was dissolved in H2O (1.5 mL) then concentrated HCl (37 wt %, 0.050 mL) was added. The resulting mixture was stirred for 15 min at r.t., and then extracted with EtOAc (×3). The combined organic extract were washed with brine, then dried over MgSO4, filtered and concentrated in vacuo. The clear liquid was diluted in 2 M aq. NaOH (4.0 mL) at r.t. and the solution was stirred for 1 h, then washed once with Et2O. The aqueous layer was acidified with concentrated HCl (37 wt %, until pH 1) and extracted with EtOAc (×3). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo to afford the title compound (0.520 g, 3.06 mmol, 32%) as a pale yellow oil: 1H NMR (500 MHz, DMSO-d6) δ 1.98–1.88 (m, 2H), 2.83–2.73 (m, 2H), 6.98 (d, J = 522.9 Hz, 1H), 7.22–7.15 (m, 1H), 7.32–7.22 (m, 4H), 9.77 (s, 1H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 27.3, 31.9 (d, J = 90.0 Hz), 126.7, 128.7, 129.0, 141.7 (d, J = 16.3 Hz) ppm; 31P NMR (146 MHz, CDCl3) δ 29.0 ppm; IR (KBr) ν 3027, 2920, 2861, 2367, 1497, 1455 cm–1; HRMS (ES–) calculated for C8H10O2P [M–H]− 169.0418, found 169.0448.
Sodium 2-phenylethane-1-sulfonate (8)
Commercially available. X-ray quality crystals were obtained by slow vapor diffusion of Et2O into a MeOH solution (see Supporting Information): mp (Et2O/MeOH) > 150 °C.
Sodium 2-phenylethane-1-sulfinate (9)
A solution of 40 (0.205 g, 1.00 mmol, 1.00 equiv) in CH3CN (1.2 mL) was added dropwise to a stirred solution of Na2SO3 (0.189 g, 1.50 mmol, 1.50 equiv) and NaHCO3 (0.252 g, 3.00 mmol, 3.00 equiv) in H2O (1.4 mL) at r.t., and the resulting solution was vigorously stirred for 24 h. The solvents were removed in vacuo, and the colorless solid residue was suspended in hot EtOH, then filtered through a short pad of Celite (rinsed three times with hot EtOH). Evaporation of the solvents in vacuo provided the title compound (0.144 g, 0.749 mmol, 75%) as a colorless solid. An analytically pure sample was obtained by recrystallization from MeOH/Et2O: mp (Et2O/MeOH) > 150 °C; 1H NMR (500 MHz, MeOH-d4) δ 7.27–7.22 (m, 4H), 7.17–7.13 (m, 1H), 2.91–2.88 (m, 2H), 2.54–2.51 (m, 2H) ppm; 13C NMR (125 MHz, MeOH-d4) δ 129.4, 126.9, 64.6, 29.7 ppm; IR (KBr) ν 3443, 3085, 3061, 3026, 2961, 2927, 2854, 1602, 1214, 1180 cm–1.
2-Phenylethane-1-sulfonamide (10)
Commercially available. X-ray quality crystals were obtained by slow evaporation from EtOAc (see Supporting Information): mp (EtOAc) 121–122 °C.
N-(3-Phenylpropyl)methanesulfonamide (11)
To a solution of 3-phenylpropylamine (1.00 mL, 7.03 mmol, 1.00 equiv) in CH2Cl2 (20 mL) at 0 °C under N2, NEt3 (3.00 mL, 21.5 mmol, 3.06 equiv) was added. The solution was stirred at the same temperature for 5 min, then methanesulfonyl chloride (0.550 mL, 7.06 mmol, 1.00 equiv) was added dropwise, then the flask was placed in a fridge (approximately 4 °C) for 24 h. The solvents were removed in vacuo, and the residue was diluted with satd. aq. NaHCO3 and extracted with EtOAc (×3). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (10–90% EtOAc in hexanes) afforded the title compound (1.32 g, 6.19 mmol, 88%) as a colorless solid. An analytically pure sample was obtained by recrystallization from cold Et2O. X-ray quality crystals were obtained from a mixture of CH3CN/H2O (see Supporting Information): mp (Et2O) 45–46 °C; 1H NMR (500 MHz, CDCl3) δ 7.29 (t, J = 7.4 Hz, 2H), 7.24–7.15 (m, 3H), 4.86 (t, J = 5.8 Hz, 1H), 3.13 (t, J = 6.8 Hz, 2H), 2.93 (s, 3H), 2.75–2.60 (m, 2H), 1.90 (t, J = 7.2 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 140.9, 128.6, 128.4, 126.2, 42.7, 40.1, 32.8, 31.6 ppm; IR (KBr) ν 3458, 2923, 1326, 1161 cm–1; HRMS (ES+) calculated for C10H15NNaO2S [M + Na]+ 236.0721, found 236.0711.
N-(Methylsulfonyl)-3-phenylpropanamide (12)
A solution of 1 (0.546 g, 3.63 mmol, 1.00 equiv), methanesulfonamide (0.346 g, 3.63 mmol, 1.00 equiv), EDCI·HCl (0.836 g, 4.36 mmol, 1.20 equiv), and DMAP (0.533 g, 4.36 mmol, 1.20 equiv) in CH2Cl2 (20 mL) was stirred at 45 °C for 8 h, then at r.t. for 48 h. The reaction was quenched with H2O, then extracted with CH2Cl2 (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC afforded the title compound (0.574 g, 2.52 mmol, 70%) as a colorless solid. X-ray quality crystals were obtained from a mixture of CH3CN/H2O (see Supporting Information): mp (CH3CN/H2O) 105.5–106.5 °C; 1H NMR (500 MHz, CDCl3) δ 8.49 (s, 1H), 7.30 (t, J = 7.4 Hz, 2H), 7.21 (dd, J = 18.3, 7.3 Hz, 3H), 3.21 (s, 3H), 2.97 (t, J = 7.6 Hz, 2H), 2.63 (t, J = 7.6 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.4, 139.6, 128.9, 128.5, 126.8, 41.6, 38.3, 30.5 ppm; IR (KBr) ν 3439, 2919, 1339, 1144 cm–1; HRMS (ES+) calculated for C10H13NNaO3S [M + Na]+ 250.0514, found 250.0511.
N-(N,N-Dimethylsulfamoyl)-3-phenylpropanamide (13)
To a suspension of 1 (0.052 g, 0.244 mmol, 1.00 equiv) and TBTU (0.133 g, 0.413 mmol, 1.00 equiv) in anhydrous CH2Cl2 (2.3 mL) at r.t. under N2, DIEA (0.300 mL, 1.72 mmol, 5.00 equiv) was added dropwise, and the resulting suspension was stirred at r.t. for 25 min. N,N-dimethylsulfamide (0.051 g, 0.293 mmol, 1.20 equiv) was then added in one portion, and the resulting solution was stirred at r.t. for 14 h. The reaction was quenched by addition of 1 M aq. KHSO4, then extracted with EtOAc (×3). The combined organic extracts were washed with brine, then dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (7–40% EtOAc in hexanes) provided the title compound (0.071 g, 0.277 mmol, 81%) as a colorless crystalline solid. X-ray quality crystals were obtained by slow evaporation from EtOAc/hexanes (see Supporting Information): mp (EtOAc/hexanes) 103–104 °C; 1H NMR (500 MHz, CDCl3) δ 8.61 (s, 1H), 7.30–7.26 (m, 2H), 7.20 (m, 3H), 2.96 (t, J = 7.5 Hz, 2H), 2.83 (s, 6H), 2.60 (t, J = 7.6 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.1, 139.8, 128.7, 128.5, 126.6, 38.2, 37.6, 30.6 ppm; IR (KBr) ν 3439, 2919 cm–1; HRMS (ES+) calculated for C11H16N2O3NaS [M + Na]+ 279.0779, found 279.0773.
N-(Benzylcarbamoyl)methanesulfonamide (14)
To a stirred solution of benzylamine (0.107 g, 1.00 mmol, 1.00 equiv) in CH2Cl2 (1.0 mL) was added 4 M HCl in dioxane (0.250 mL, 1.00 mmol, 1.00 equiv) and the solution was stirred at r.t. for 5 min under N2. The solvents were removed in a stream of air, then the residue was dried under high vacuum for 3 h. The residue was dissolved in anhydrous CH3CN (0.60 mL), then a solution of CDI (0.178 g, 1.1 mmol, 1.10 equiv) dissolved in DMF (0.30 mL) was added. The solution was stirred at r.t. for 2 h. The solvents were removed in a stream of air. Purification by silica gel column chromatography (5% MeOH in CH2Cl2) provided the benzylcarbamoylimidazole intermediate (0.050 g, 2.48 mmol, 25%): 1H NMR (500 MHz, CDCl3) δ 8.17 (d, J = 5.9 Hz, 1H), 7.98 (s, 1H), 7.39 (s, 1H), 7.32–7.12 (m, 5H), 6.76 (s, 1H), 4.45 (d, J = 5.5 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 149.3, 137.4, 136.0, 129.6, 128.9, 128.0, 116.8, 45.0 ppm.
To a stirred solution of methylsulfonamide (0.028 g, 0.298 mmol, 1.20 equiv) in DMF (1.0 mL) was added NaH (60 wt % in oil, 0.007 g, 0.298 mmol, 1.20 equiv). The solution was stirred for 15 min at r.t.. The benzylcarbamoylimidazole intermediate (0.050 g, 0.248 mmol) was added, and the resulting solution was stirred at r.t. for 16 h. The reaction was diluted with EtOAc, then extracted with H2O. The combined aqueous layers were acidified with 1 M HCl (until pH 2–3), then extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC provided the title compound (0.025 g, 0.110 mmol, 44%). X-ray quality crystals were obtained by slow evaporation from EtOAc (see Supporting Information): mp (EtOAc) 163–164 °C; 1H NMR (500 MHz, MeOH-d4) δ 7.47–7.19 (m, 5H), 4.41 (s, 2H), 3.29 (d, J = 1.3 Hz, 3H) ppm; 13C NMR (125 MHz, MeOH-d4) δ 153.3, 138.6, 128.4, 127.1, 127.1, 47.5, 47.3, 43.3, 40.6 ppm; IR (KBr) ν 3338, 2920, 1648, 1562 cm–1.
N-(Benzylcarbamoyl)acetamide (15)
To a suspension of N-benzylurea (0.415 g, 2.75 mmol, 1.00 equiv) in CH2Cl2 (20 mL), Ac2O (2.09 mL, 22.1 mmol, 8.00 equiv) was added dropwise, and the resulting suspension was stirred at 50 °C for 24 h, then at r.t. for 48 h. The solvents were removed in vacuo. Purification by reverse phase HPLC provided the title compound (0.396 g, 2.06 mmol, 75%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from CH2Cl2 (see Supporting Information): mp (CH2Cl2) 118–119 °C; 1H NMR (500 MHz, CDCl3) δ 10.54 (s, 1H), 8.94 (t, J = 5.3 Hz, 1H), 7.39–7.20 (m, 5H), 4.49 (d, J = 6.0 Hz, 2H), 2.09 (s, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.8, 155.3, 138.2, 128.7, 127.5, 127.5, 43.6, 23.9 ppm; IR (KBr) ν 3447, 3356, 2922, 1689, 1630 cm–1; HRMS (ES+) calculated for C10H12N2NaO2 [M + Na]+ 215.0796, found 215.0798.
5-Phenethyl-1H-tetrazole (16)
Method A. 3-phenylpropanenitrile 41 (0.100 g, 0.762 mmol, 1.00 equiv), 1 M aq. NaN3 (0.840 mL, 0.840 mmol, 1.10 equiv) and 1 M aq. ZnBr2 (0.760 mL, 0.760 mmol, 1.00 equiv) were added to a microwave vial. The vial was sealed, warmed to 150 °C and vigorously stirred for 24 h. The reaction was quenched with 3 M HCl (1.5 mL) and extracted with EtOAc (×3). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC provided the title compound (0.026 g, 0.149 mmol, 20%) as a colorless solid. Method B. A suspension of amide 42 (0.105 g, 0.519 mmol, 1.00 equiv) and PPh3 (0.340 g, 1.30 mmol, 2.50 equiv) in anhydrous CH3CN (5 mL) under N2, was cooled to 0 °C and stirred for 10 min. DIAD (0.250 mL, 1.30 mmol, 2.50 equiv) was added dropwise (discoloration after each drop) and the suspension was stirred at 0 °C for 5 min. TMSN3 (0.210 mL, 1.56 mmol, 3.00 mmol) was then added dropwise in 5 min, and the reaction was stirred at 0 °C for 30 min, then at r.t. for 2 h, and finally at 50 °C for 15 h. The reaction was cooled to 0 °C, quenched by addition of 3 M aq. NaNO2 (0.520 mL, 1.56 mmol, 1.00 equiv). After 20 min stirring at 0 °C, 0.5 M aq. CAN (1.45 mL, 0.726 mmol, 1.40 equiv) was added and the stirring was continued at 0 °C for 30 min. The reaction was then diluted with H2O and extracted with CH2Cl2 (×3). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (50–70% EtOAc in hexanes) afforded a mixture of 3-(5-phenethyl-1H-tetrazol-1-yl)propanenitrile, PPh3 and EtOAc (0.106 g, 90 wt % NMR purity), which was used directly in the next step: 1H NMR (500 MHz, CDCl3) δ 7.30–7.24 (m, 3H), 7.10–7.08 (m, 2H), 4.12 (t, J = 6.9 Hz, 2H), 3.22–3.14 (m, 4H), 2.68 (t, J = 6.9 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 154.9, 139.3, 129.1, 128.5, 127.1, 115.9, 42.1, 33.9, 25.5, 18.3 ppm.
To a stirred solution of 3-(5-phenethyl-1H-tetrazol-1-yl)propanenitrile (0.106 g, 0.418 mmol, 1.00 equiv) in CH2Cl2 (3.8 mL) under N2, freshly distilled DBU (0.440 mL, 2.93 mmol, 7.00 equiv) was added dropwise and the resulting clear yellow solution was stirred at r.t. for 6 h. The reaction was diluted with CH2Cl2 and washed with 1 M HCl (×2), brine, then dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (12–100% EtOAc in hexanes) provided the title compound (0.046 mg, 0.264 mmol, 63% over 2 steps) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from CH2Cl2 (see Supporting Information): mp (CH2Cl2) 97–99 °C; 1H NMR (500 MHz, CDCl3) δ 11.97 (s, 1H), 7.23 (t, J = 7.2 Hz, 2H), 7.20–7.13 (m, 3H), 3.40 (t, J = 7.8 Hz, 2H), 3.16 (t, J = 7.8 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.2, 139.2, 128.9, 128.5, 127.0, 33.8, 25.5 ppm; IR (KBr) ν 3354, 3130, 2919, 2722, 1565, 1495 cm–1; HRMS (ES+) calculated for C9H11N4 [M + H]+ 175.0984, found 175.0989.
5-Benzylthiazolidine-2,4-dione (17)
Thiazolidine-2,4-dione (0.586 g, 5.00 mmol, 1.00 equiv) was weighed in a flame-dried round-bottom flask. The flask was evacuated and backfilled with N2 (×5), then anhydrous THF (20 mL) was added. The resulting clear solution was cooled to −78 °C and stirred for 10 min. n-BuLi (2.34 M in hexanes, 4.30 mL, 10 mmol, 2.00 equiv) was added dropwise over 5 min under vigorous stirring (a precipitate formed during the addition). The resulting deep yellow solution was stirred at the same temperature for 15 min, then warmed to 0 °C and stirred for 20 min. The reaction was cooled to −78 °C, and benzyl bromide (0.600 mL, 5.00 mmol, 1.00 equiv) was added dropwise. The solution was stirred for 20 min at the same temperature, then warmed to r.t. and stirred for 1.5 h. The reaction was quenched with satd. aq. NH4Cl, then extracted with EtOAc (×3). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (5–40% EtOAc in hexanes) provided the title compound (0.549 g, 2.65 mmol, 53%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from an Et2O/hexanes solution (see Supporting Information): mp (Et2O/hexanes) 74–75 °C; 1H NMR (500 MHz, CDCl3) δ 8.90 (s, 1H), 7.35–7.23 (m, 5H), 4.54 (dd, J = 9.9, 3.8 Hz, 1H), 3.56 (dd, J = 14.1, 3.8 Hz, 1H), 3.13 (dd, J = 14.0, 10.0 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 174.7, 170.9, 135.9, 129.3, 129.0, 127.8, 53.6, 38.8 ppm; IR (KBr) ν 3320, 2943, 1748 cm–1; HRMS (ES–) calculated for C10H8NO2S [M–H]− 206.0281, found 206.0289.
5-Benzyloxazolidine-2,4-dione (18)
2-Hydroxyacetamide (0.300 g, 4.00 mmol, 1.00 equiv) was weighed in a flame-dried microwave vial, then dissolved in anhydrous MeOH (4.2 mL). Potassium tert-butoxide (0.450 g, 4.00 mmol, 1.00 equiv) was added, followed by diethyl carbonate (0.508 mL, 4.80 mmol, 1.20 equiv). The vial was sealed and stirred at 70 °C for 19 h, then at r.t. for 24 h. The solvents were removed in vacuo, the residue was taken up in H2O, then acidified to pH 2 with 3 M HCl and extracted with EtOAc (×3). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to afford oxazolidine-2,4-dione, which was directly used in the next step without further purification. To a solution of crude oxazolidine-2,4-dione (0.105 g, 1.00 mmol, 1.00 equiv) in anhydrous THF (3.6 mL) at −78 °C, n-BuLi (2.65 M in hexanes, 0.780 mL, 2.10 mmol, 2.10 equiv) was added dropwise and the mixture was stirred at −78 °C for 15 min, then at 0 °C for 30 min. The solution was then cooled to −78 °C and benzyl bromide (0.120 mL, 1.00 mmol, 1.00 equiv) was added dropwise. The resulting solution was warmed to r.t. and stirred for 2 h. The reaction was quenched with satd. aq. NH4Cl, then extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC provided the title compound (0.009 g, 0.05 mmol, 5%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from CHCl3 (see Supporting Information): mp (CHCl3) 85–87 °C; 1H NMR (500 MHz, CDCl3) δ 8.38 (s, 1H), 7.30 (ddd, J = 9.6, 6.2, 3.2 Hz, 3H), 7.26–7.20 (m, 2H), 5.08 (dd, J = 5.7, 4.3 Hz, 1H), 3.32 (dd, J = 14.8, 4.2 Hz, 1H), 3.16 (dd, J = 14.8, 5.7 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.7, 154.1, 133.0, 129.7, 128.9, 127.9, 81.4, 36.6 ppm; IR (KBr) ν 3355, 2916, 2848, 1823, 1743 cm–1; HRMS (ES–) calculated for C10H8NO3 [M–H]− 190.0504, found 190.0503.
3-Phenethyl-1,2,4-oxadiazol-5(4H)-one (19)
To a solution of amidoxime 43 (0.120 g, 0.700 mmol, 1.00 equiv) and pyridine (0.061 mL, 0.760 mmol, 1.10 equiv) in DMF (1.20 mL) at 0 °C, isobutyl chloroformate (0.091 mL, 0.700 mmol, 1.00 equiv) was added dropwise, and the solution was stirred for 16 h while slowly warming to r.t.. The reaction was diluted with H2O and extracted with EtOAc (×3). The combined organic extracts were washed with H2O, brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was suspended in toluene (1.50 mL) in a microwave vial and stirred at 120 °C for 2 h, then at 140 °C for 24 h. After cooling to r.t., the mixture was concentrated in vacuo. Purification by reverse phase HPLC provided the title compound (0.080 g, 0.448 mmol, 59%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from Et2O (see Supporting Information): mp (Et2O) 98–100 °C; 1H NMR (500 MHz, CDCl3) δ 10.53 (s, 1H), 7.32 (t, J = 7.4 Hz, 2H), 7.23 (dd, J = 7.6, 6.4 Hz, 3H), 3.02 (t, J = 7.8 Hz, 2H), 2.91 (t, J = 7.7 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 161.6, 158.9, 138.7, 128.9, 128.4, 127.1, 31.7, 27.0 ppm; IR (KBr) ν 3189, 2956, 1768, 1606, 1454 cm–1; HRMS (ES–) calculated for C10H9N2O2 [M–H]− 189.0664, found 189.0675.
3-Phenethyl-1,2,4-thiadiazol-5(4H)-one (20)
To a solution of amidoxime 43 (0.100 g, 0.610 mmol, 1.00 equiv) in anhydrous THF (1.00 mL) at r.t. under Ar, 1,1′-thiocarbonyldiimidazole (0.160 g, 0.910 mmol, 1.50 equiv) was added, and the solution was stirred for 40 min. The reaction was quenched with H2O, then extracted with CH2Cl2 (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was dissolved in anhydrous THF (1.0 mL), then BF3·OEt2 (48%, 0.230 mL, 1.80 mmol, 3.00 equiv) was added dropwise, and the resulting mixture was stirred at r.t. for 3 h. The reaction was diluted with H2O and extracted with CH2Cl2 (×3). The combined organic extracts were washed with 1 M HCl, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC afforded the title compound (0.035 g, 0.170 mmol, 28%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from CH2Cl2 (see Supporting Information): mp (CH2Cl2) 119–122 °C; 1H NMR (500 MHz, CDCl3) δ 11.43 (br s, 1H), 7.32–7.20 (m, 5H), 3.06 (t, J = 7.8 Hz, 2H), 2.92 (t, J = 7.7 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.9, 139.5, 128.9, 128.4, 126.8, 33.2, 32.4 ppm; IR (KBr) ν 3437, 2918, 1666, 1564, 1450 cm–1; HRMS (ES–) calculated for C10H9N2OS [M–H]− 205.0436, found 205.0427.
4-Phenethyl-3H-1,2,3,5-oxathiadiazole 2-oxide (21)
To a solution of amidoxime 43 (0.100 g, 0.610 mmol, 1.00 equiv) in THF (18 mL), pyridine (0.130 mL, 1.60 mmol, 2.60 equiv) in CH2Cl2 (3.5 mL) was added, and the solution was cooled to 0 °C. Thionyl chloride (0.058 mL, 0.800 mmol, 1.30 equiv) was then added dropwise, and the reaction was stirred at 0 °C for 1 h, during which a colorless precipitate was formed. The solvents were removed in vacuo, and the residue was diluted with H2O and extracted with CHCl3 (×3). The combined organic extracts were washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC afforded the title compound (0.012 g, 0.060 mmol, 10%) as a colorless solid: 1H NMR (500 MHz, CDCl3) δ 7.92 (br s, 1H), 7.35–7.31 (m, 2H), 7.28–7.25 (m, 1H), 7.23–7.21 (m, 2H), 3.00 (t, J = 7.60 Hz, 2H), 2.94–2.85 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 152.2, 139.2, 129.2, 128.6, 127.3, 32.7, 25.7 ppm; IR (KBr) ν 3223, 2925, 1606, 1404, 1176 cm–1; HRMS (ES–) calculated for C9H9N2O2S [M–H]− 209.0390, found 209.0385.
3-Phenethyl-1,2,4-oxadiazole-5(4H)-thione (22)
To a solution of amidoxime 43 (0.100 g, 0.610 mmol, 1.00 equiv) in CH3CN (5.5 mL), 1,1′-thiocarbonyldiimidazole (0.120 g, 0.670 mmol, 1.10 equiv) and DBU (0.360 mL, 2.40 mmol, 3.9 equiv) were added and the reaction was stirred at r.t. for 24 h. The solvents were removed in vacuo, and the residue was diluted with H2O. The pH was adjusted to pH 4 with 1 M HCl, and the mixture was extracted with EtOAc (×3). The combined organic extracts were concentrated in vacuo, and the residue was dissolved in 1 M NaOH and washed with Et2O. The pH was again adjusted to pH 4 with 1 M HCl, and extracted with EtOAc (×3). The combined organic extracts were washed with H2O, dried over Na2SO4, filtered and concentrated in vacuo. Purification by reverse phase HPLC afforded the title compound (0.056 g, 0.271 mmol, 43%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from CH2Cl2 (see Supporting Information): mp (CH2Cl2) 96–100 °C; 1H NMR (500 MHz, CDCl3) δ 10.37 (br s, 1H), 7.35 (t, J = 7.3 Hz, 2H), 7.29 (d, J = 7.3 Hz, 1H), 7.23–7.18 (m, 2H), 3.05 (dd, J = 8.3, 5.0 Hz, 2H), 3.03–2.96 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 187.2, 159.3, 138.3, 129.1, 128.4, 127.3, 32.2, 25.8 ppm; IR (KBr) ν 3420, 2920, 1603, 1472 cm–1; HRMS (ES–) calculated for C10H9N2OS [M–H]− 205.0436, found 205.0441.
5-Benzylisoxazol-3-ol (23)
To a stirred solution of 44 (0.571 g, 2.18 mmol, 1.00 equiv) in toluene (20 mL), N,O-di-Boc-hydroxylamine (0.508 g, 2.18 mmol, 1.00 equiv) was added. The solution was stirred at 65 °C for 16 h. After cooling to r.t., the solvents were removed in vacuo to provide the β-keto hydroxamic acid intermediate (0.962 g, 100%), which was used directly in the next step without further purification. Crude β-keto hydroxamic acid intermediate (0.268 g, 0.681 mmol, 1.00 equiv) was dissolved in MeOH (6.0 mL) and 4 M HCl (9.0 mL), and the resulting solution was stirred at r.t. for 16 h. The mixture was concentrated in vacuo, diluted with H2O, and extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (10% EtOAc in hexanes, buffered with 1% AcOH) provided the title compound (0.013 g, 0.074 mmol, 10%): 1H NMR (500 MHz, CDCl3) δ 7.39–7.22 (m, 5H), 5.65 (s, 1H), 3.98 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 173.0, 171.3, 135.4, 129.0, 128.9, 127.4, 94.4, 33.9 ppm; IR (KBr) ν 3399, 2919, 2848, 1620, 1524 cm–1; HRMS (ES+) calculated for C10H10NO [M + H]+ 176.0712, found 176.0714.
5-Benzylpyrrolidine-2,4-dione (24)
To a solution of N-Boc-(DL)-phenylalanine (2.00 g, 7.54 mmol, 1.00 equiv), DMAP (1.29 g, 10.6 mmol, 1.40 equiv) and freshly recrystallized Meldrum’s acid (1.20 g, 8.29 mmol, 1.10 equiv) in CH2Cl2 (50 mL) at 0 °C, EDCI·HCl (1.73 g, 9.05 mmol, 1.20 equiv) was added in one portion. The reaction mixture was stirred at 0 °C for 5 min, then at r.t. for 15 h. The solvents were removed in vacuo, then the residue was diluted in EtOAc, washed with 1 M KHSO4, 5 wt % aq. citric acid, brine (×3), then dried over MgSO4, filtered, and concentrated in vacuo. The resulting colorless solid was dissolved in EtOAc (100 mL) and stirred at reflux for 30 min. The solvents were removed in vacuo to yield a colorless foam. The residue was dissolved in CH2Cl2 under N2 at r.t., then TFA (10 mL) was added in a steady stream and the reaction was stirred at r.t. for 15 min. The solvents were removed in vacuo, using toluene (2 portions) to azeotrope the residual TFA. The crude solid was dissolved in a minimum amount of Et2O, then cooled to −78 °C and precipitated with hexanes. Filtration afforded the title compound (1.30 g, 6.88 mmol, 91%) as a colorless to pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.2 Hz, 2H), 7.28 (d, J = 7.2 Hz, 1H), 7.16 (d, J = 7.0 Hz, 2H), 6.54 (br s, 1H), 4.23 (dd, J = 7.7, 3.3 Hz, 1H), 3.16 (dd, J = 13.9, 3.7 Hz, 1H), 2.93 (d, J = 22.2 Hz, 1H), 2.84 (dd, J = 13.9, 8.3 Hz, 1H), 2.71 (d, J = 22.2 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 206.5, 170.8, 135.3, 129.5, 129.2, 127.6, 65.3, 40.9, 38.4 ppm; IR (KBr) ν 3174, 2944, 1770, 1695 cm–1; HRMS (ES+) calculated for C11H12NO2 [M + H]+ 190.0868, found 190.0865.
5-Benzyl-3-hydroxycyclopent-2-en-1-one (25)
To a solution of 3-isobutoxycyclopent-2-en-1-one (0.130 g, 0.844 mmol, 1.00 equiv) in anhydrous THF (2.00 mL) at −78 °C under N2, a freshly prepared solution of LDA (1 M in THF, 1 mL, 1 mmol, 1.18 equiv) was added dropwise. The resulting solution was stirred at −78 °C for 45 min, then a solution of benzyl bromide (0.100 mL, 0.844 mmol, 1.00 equiv) in THF (3 mL) was added dropwise. The solution was stirred for 1 h while slowly warming to r.t.. The reaction was quenched with satd. aq. NH4Cl and extracted with EtOAc (×2). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (3% MeOH in CH2Cl2) provided the 5-benzyl-3-isobutoxycyclopent-2-en-1-one intermediate (0.055 g, 0.225 mmol, 27%): 1H NMR (500 MHz, CDCl3) δ 7.38–7.25 (m, 2H), 7.21 (t, J = 6.8 Hz, 3H), 5.26 (s, 1H), 3.71 (d, J = 6.6 Hz, 2H), 3.27 (dd, J = 14.0, 4.1 Hz, 1H), 2.88–2.74 (m, 1H), 2.67–2.51 (m, 2H), 2.43–2.30 (m, 1H), 2.04 (hept, J = 6.6 Hz, 1H), 0.97 (d, J = 6.7 Hz, 6H) ppm; 13C NMR (125 MHz, CDCl3) δ 207.3, 189.4, 139.6, 129.0, 128.6, 126.4, 103.9, 78.1, 46.8, 37.3, 34.3, 28.0, 19.1, 19.1 ppm. To a mixture of 5-benzyl-3-isobutoxycyclopent-2-en-1-one (0.035 g, 0.143 mmol, 1.00 equiv) in acetone (2.0 mL) at r.t., 3 M HCl (1.0 mL) was added and the mixture was stirred for 16 h. The reaction was then concentrated in vacuo. Purification by reverse phase HPLC provided the title compound (0.018 g, 0.096 mmol, 67%) as a colorless solid: 1H NMR (500 MHz, MeOH-d4) δ 7.27 (t, J = 7.6 Hz, 2H), 7.23–7.15 (m, 3H) 3.15 (dd, J = 13.8, 4.2 Hz, 1H), 3.01–2.92 (m, 1H), 2.64 (dd, J = 13.8, 9.4 Hz, 1H), 2.51 (dd, J = 18.1, 6.9 Hz, 1H), 2.24 (dd, J = 18.1, 2.4 Hz, 1H) ppm; 13C NMR (125 MHz, MeOH-d4) δ 139.0, 128.9, 128.2, 126.2, 44.8, 36.9, 36.1 ppm; IR (KBr) ν 3026, 2920, 2680, 2565, 1644, 1553 cm–1; HRMS (ES+) calculated for C12H13O2 [M + H]+ 189.0916, found 189.0913.
5-Benzyl-3-hydroxy-2-methylcyclopent-2-en-1-one (26)
Prepared as 25 from 3-isobutoxy-2-methylcyclopent-2-en-1-one (0.130 g, 0.773 mmol, 1.00 equiv) and benzyl bromide (0.092 mL, 0.773 mmol, 1.00 equiv). Purification by silica gel column chromatography (3% MeOH in CH2Cl2) provided the 5-benzyl-3-isobutoxy-2-methylcyclopent-2-en-1-one intermediate (0.090 g, 0.348 mmol, 45%): 1H NMR (500 MHz, CDCl3) δ 7.28 (dd, J = 8.7, 6.7 Hz, 2H), 7.23–7.17 (m, 3H), 3.81 (d, J = 6.6 Hz, 2H), 2.82–2.73 (m, 1H), 3.31 (dd, J = 14.1, 4.1 Hz, 1H), 2.66–2.56 (m, 1H), 2.50 (dd, J = 14.0, 10.7 Hz, 1H), 2.28 (dt, J = 17.5, 2.0 Hz, 1H), 2.01–1.90 (m, 1H), 1.64 (d, J = 1.8 Hz, 3H), 0.95 (dd, J = 6.7, 1.6 Hz, 5H) ppm; 13C NMR (125 MHz, CDCl3) δ 206.6, 183.4, 139.8, 129.0, 128.6, 126.4, 115.3, 75.6, 46.2, 37.5, 31.2, 28.8, 19.0, 6.2 ppm. After hydrolysis of the 5-benzyl-3-isobutoxy-2-methylcyclopent-2-en-1-one intermediate (as for 25), purification by reverse phase HPLC provided the title compound (0.030 g, 0.148 mmol, 43%) as a colorless solid: 1H NMR (500 MHz, MeOH-d4) δ 7.27 (dd, J = 8.3, 7.1 Hz, 2H), 3.32 (dq, J = 3.0, 1.5 Hz, 1H), 4.94 (s, 2H), 3.17 (dd, J = 13.7, 4.1 Hz, 1H), 2.88 (dq, J = 10.5, 3.7, 3.2 Hz, 1H), 2.58 (dd, J = 13.7, 9.6 Hz, 1H), 7.23–7.16 (m, 3H), 2.47 (ddt, J = 17.8, 6.7, 1.1 Hz, 1H), 1.57 (s, 3H), 2.20 (ddt, J = 17.8, 2.3, 1.1 Hz, 1H) ppm; 13C NMR (125 MHz, MeOH-d4) δ 139.1, 128.8, 128.1, 126.1, 112.5, 48.3, 48.1, 47.9, 47.8, 47.6, 47.4, 47.2, 44.0, 37.1, 34.7, 4.3 ppm; IR (KBr) ν 3410, 3028, 2927, 1780, 1714 cm–1; HRMS (ES–) calculated for C13H13O2 [M–H]− 201.0916, found 201.0926.
2-Benzyl-3-hydroxycyclopent-2-en-1-one (27)
To a solution of cyclopentane-1,3-dione (0.300 g, 3.06 mmol, 1.03 equiv), diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (0.775 g, 2.98 mmol, 1.00 equiv) and (L)-proline (0.017 g, 0.148 mmol, 5 mol %) in CH2Cl2 (10 mL) was added benzaldehyde (0.915 mL, 9.00 mmol, 3.00 equiv), and the resulting mixture was allowed to stir at r.t. for 30 min. The reaction mixture was concentrated in vacuo. Purification by silica gel column chromatography (30–100% EtOAc in hexanes) provided the title compound (0.350 g, 1.86 mmol, 61%) as a colorless solid: 1H NMR (500 MHz, DMSO-d6) δ 11.75 (s, 1H), 7.21 (t, J = 7.5 Hz, 2H), 7.18–7.14 (m, 2H), 7.14–7.08 (m, 1H), 3.33 (s, 2H), 2.40 (s, 4H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 141.1, 128.7, 128.6, 126.1, 116.1, 27.1 ppm; IR (KBr) ν 3365, 2920, 2852, 1667, 1567 cm–1; HRMS (ES–) calculated for C12H11O2 [M–H]− 187.0759, found 187.0761.
3-Benzyl-2-hydroxycyclopent-2-en-1-one (28)
A mixture of 46 (0.155 g, 0.367 mmol, 1.00 equiv) in glacial AcOH (0.70 mL) and concentrated HCl (37 wt %, 0.70 mL) in a sealed tube was stirred at 130 °C for 3 h. After cooling to r.t., the mixture was carefully quenched with H2O, then diluted in aq. buffer (pH 3). The pH was adjusted to pH 3 with 20 wt % aq. NaOH, then the mixture was extracted with EtOAc (×5). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (5–20% EtOAc in hexanes) provided the title compound (0.049 g, 0.260 mmol, 71%) as a colorless solid: 1H NMR (500 MHz, CDCl3) δ 7.33–7.29 (m, 2H), 7.26–7.22 (m, 3H), 5.68 (s, 1H, OH), 3.74 (s, 2H), 2.39–2.38 (m, 2H), 2.36–2.34 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 203.7, 148.9, 146.2, 137.8, 129.1, 128.8, 126.7, 35.0, 32.1, 24.9 ppm; IR (KBr) ν 3327, 3024, 2923, 1695, 1652, 1387, 1107 cm–1.
2-Hydroxy-4-phenylcyclopent-2-en-1-one (29)
A solution of 47 (0.027 g, 0.160 mmol, 1.00 equiv) in a 10 wt % solution of Mg(OMe)2 in MeOH (15 mL) was stirred at reflux for 90 min. After cooling to r.t., the reaction mixture was concentrated in vacuo. The residue was taken up in Et2O and H2O, then the pH was adjusted to pH 7 with 1 M HCl. The organic layer was washed with H2O (×4), dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (50% EtOAc in hexanes) provided the title compound (0.015 g, 0.086 mmol, 53%) as a colorless solid. X-ray quality crystals were obtained by slow evaporation from CH2Cl2 (see Supporting Information): mp (CH2Cl2) 106–107 °C; 1H NMR (500 MHz, CDCl3) δ 7.35–7.32 (m, 2H), 7.27–7.24 (m, 1H), 7.20–7.18 (m, 2H), 6.61 (m, 1H), 6.25 (br s, 1H), 4.04–4.03 (m, 1H), 2.99 (dd, J = 19.4, 6.4 Hz, 1H), 2.36 (d, J = 19.5 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 153.1, 142.7, 129.0, 127.3, 127.1, 42.4, 40.1 ppm; IR (KBr) ν 3334, 3058, 3025, 1699, 1403 cm–1; HRMS (CI+) calculated for C11H10O2 [M]+ 174.0681, found 174.0688.
3-Benzyl-4-hydroxycyclobut-3-ene-1,2-dione (30)
To a solution of 48 (0.099 g, 0.460 mmol, 1.00 equiv) in acetone (5.0 mL) at r.t., 3 M HCl (5.0 mL) was added and the solution was stirred at r.t. for 3 h. The acetone was removed in vacuo, and the aqueous layer was extracted with Et2O (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC afforded the title compound (0.057 g, 0.303 mmol, 66%) as a colorless solid: 1H NMR (500 MHz, acetone-d6) δ 11.59 (br s, 1H), 7.36–7.30 (m, 4H), 7.25–7.23 (m, 1H), 3.98 (s, 2H) ppm; 13C NMR (125 MHz, acetone-d6) δ 197.6, 196.6, 181.3, 136.2, 129.3, 129.2, 127.4, 30.4 ppm; IR (KBr) ν 3214, 2974, 1812, 1644 cm–1; HRMS (ES–) calculated for C11H7O3 [M–H]− 187.0401, found 187.0405.
3-(Benzylamino)-4-hydroxycyclobut-3-ene-1,2-dione (31)
To a suspension of squaric acid (0.114 g, 1.00 mmol, 1.00 equiv) in H2O (3.0 mL) in a microwave vial under N2, benzylamine (0.220 mL, 2.00 mmol, 2.00 equiv) was added dropwise, the tube was sealed and heated to 200 °C under microwave irradiation (Biotage Initiator) for 20 min. Note: the internal pressure reached up to 17 bar. After cooling to r.t., the reaction was diluted with 3 M HCl (40 mL), and extracted with Et2O (×5). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo to afford the title compound (0.045 g, 0.221 mmol, 22%) as a colorless solid: 1H NMR (500 MHz, DMSO-d6) δ 8.87 (t, J = 6.0 Hz, 1H), 7.36 (t, J = 7.5 Hz, 2H), 7.37–7.27 (m, 6H), 7.30–7.28 (m, 3H), 4.59 (d, J = 6.3 Hz, 2H) ppm; 13C NMR (125 MHz, DMSO-d6) δ 184.8, 173.7, 138.7, 128.6, 127.4, 47.0 ppm (C=O carbons not observed due to rapid tautomeric exchange).
4-Benzyl-2,6-difluorophenol (32)
To a solution of 51 (0.310 g, 1.00 mmol, 1.00 equiv) in MeOH (10 mL) under N2, Pd/C (10 wt % (wet), 0.031 g) was added. The resulting suspension was purged with H2 for 10 min, then stirred for 1 h under an H2 atmosphere (balloon). The reaction mixture was then purged with N2 for 10 min, filtered through a pad of Celite and thoroughly washed with EtOAc. Removal of the volatiles in vacuo provided the title compound (0.220 g, 0.999 mmol, 99%) as a colorless solid. X-ray quality crystals were obtained by slow layer diffusion of hexanes into a solution of the product in Et2O at r.t. (see Supporting Information): mp (hexanes/Et2O) 60.5–61.5 °C; 1H NMR (500 MHz, CDCl3) δ 7.37 (t, J = 7.4 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.22 (d, J = 7.5 Hz, 2H), 6.80–6.74 (m, 2H), 5.16 (br s, 1H), 3.91 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 151.7 (dd, J = 242.5, 5.7 Hz), 140.0, 133.4 (t, J = 7.6 Hz), 130.9 (t, J = 16.2 Hz), 128.9, 128.8, 126.6, 112.1–111.9 (m), 41.0 ppm; IR (KBr) ν 3343, 2964, 1220 cm–1; HRMS (ES–) calculated for C13H9F2O [M–H]− 219.0627, found 219.0625.
3-Benzyl-2,6-difluorophenol (33)
To a solution of 53 (0.100 g, 0.322 mmol, 1.00 equiv) in EtOH (3.2 mL) under N2, Pd(OAc)2 (∼0.001 g, ∼ 1 mol %) and activated carbon (0.010 g) were added. The resulting suspension was purged with H2 for 10 min, then stirred for 45 min under an H2 atmosphere (balloon). The reaction mixture was then purged with N2 for 10 min, filtered through a pad of Celite and thoroughly washed with EtOAc. Removal of the volatiles in vacuo provided the title compound (0.069 g, 0.313 mmol, 97%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.30 (t, J = 7.4 Hz, 2H), 7.24–7.19 (m, 3H), 6.82 (td, J = 9.2, 1.7 Hz, 1H), 6.62 (td, J = 8.3, 5.9 Hz, 1H), 5.20 (s, 1H), 3.96 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 150.4 (dd, J = 240.8, 4.4 Hz), 150.1 (dd, J = 241.2, 4.7 Hz), 139.5, 132.9 (t, J = 16.3 Hz), 128.8, 128.7, 126.5, 124.7 (dd, J = 14.1, 3.5 Hz), 120.3 (dd, J = 8.2, 5.1 Hz), 111.1 (dd, J = 18.0, 3.7 Hz), 34.6 (d, J = 2.6 Hz) ppm; IR (KBr) ν 3390, 3085, 3063, 3029, 2929, 2853, 1604, 1501, 1471 cm–1; HRMS (ES–) calculated for C13H9OF2 [M–H]− 219.0621, found 219.0625.
2-(Benzylthio)phenol (34)
To a stirred suspension of KHCO3 (0.508 g, 5.08 mmol, 1.05 equiv) in anhydrous DMF (5.0 mL) at r.t. under N2, 2-hydroxythiophenol (0.500 mL, 4.83 mmol, 1.00 equiv) was added dropwise. The suspension was stirred for 5 min, then benzyl bromide (0.580 mL, 4.88 mmol, 1.01 equiv) was added dropwise and the mixture was stirred for 16 h. The reaction was quenched with satd. aq. NH4Cl, then extracted with Et2O (×5). The combined organic extracts were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (7% EtOAc in hexanes) provided the title compound (0.972 g, 4.49 mmol, 93%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.31–7.26 (m, 5H), 7.13–7.10 (m, 2H), 6.97 (d, J = 8.1 Hz, 1H), 6.83 (t, J = 7.5 Hz, 1H), 6.58 (s, 1H), 3.87 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 157.2, 137.7, 136.5, 131.5, 128.9, 128.6, 127.5, 120.7, 118.3, 114.9, 41.5 ppm; IR (KBr) ν 3407, 3062, 3029, 2924, 1573, 1470, 1455 cm–1; HRMS (ES–) calculated for C13H11OS [M–H]− 215.0531, found 215.0539.
2-(Benzylsulfinyl)phenol (35)
To a solution of 34 (0.100 g, 0.462 mmol, 1.00 equiv) in glacial AcOH (3.80 mL), UHP (0.049 g, 0.509 mmol, 1.10 equiv) was added in one portion at r.t., and the resulting solution was stirred at r.t. for 20 h. The reaction mixture was diluted with satd. aq. NaHCO3, and then extracted with CH2Cl2 (×4). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (30% EtOAc in hexanes) provided the title compound (0.105 g, 0.452 mmol, 98%) as a colorless crystalline solid. X-ray quality crystals were obtained by slow vapor diffusion of hexanes into a solution of the product in THF at r.t. (see Supporting Information): mp (THF/hexanes) 134–134.5 °C; 1H NMR (500 MHz, CDCl3) δ 10.07 (br s, 1H), 7.33–7.26 (m, 4H), 7.09 (d, J = 7.1 Hz, 2H), 6.94–6.88 (m, 2H), 6.83 (t, J = 7.5 Hz, 1H), 4.36 (d, J = 12.7 Hz, 1H), 4.27 (d, J = 12.7 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 158.2, 132.9, 130.6, 129.0, 128.6, 128.6, 125.9, 122.0, 119.6, 118.3, 60.5 ppm; IR (KBr) ν 3062, 3031, 2923, 2847, 2696, 2562, 1587, 1452 cm–1; HRMS (ES–) calculated for C13H11O2S [M–H]− 231.0480, found 231.0487.
2-(Benzylsulfonyl)phenol (36)
To a solution of 34 (0.100 g, 0.462 mmol, 1.00 equiv) in CH2Cl2, N-methylmorpholine N-oxide (0.135 g, 1.15 mmol, 2.50 equiv) and K2OsO4·2H2O (0.008 g, 0.023 mmol, 5 mol %) were added at r.t., and the resulting solution was stirred for 20 h. The solvents were removed in vacuo. Purification by silica gel column chromatography (30% EtOAc in hexanes) provided the title compound (0.109 g, 0.439 mmol, 95%) as a colorless crystalline solid. X-ray quality crystals were obtained by slow diffusion of hexanes into a solution of the product in EtOAc at r.t. (see Supporting Information): mp (EtOAc/hexanes) 105–105.5 °C; 1H NMR (500 MHz, CDCl3) δ 8.64 (s, 1H), 7.47 (td, J = 7.8, 1.4 Hz, 1H), 7.39 (dd, J = 7.9, 1.6 Hz, 1H), 7.35 (t, J = 7.4 Hz, 1H), 7.27 (t, J = 7.6 Hz, 3H), 7.09 (d, J = 7.4 Hz, 2H), 6.92 (td, J = 7.7, 1.1 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 4.36 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.8, 136.7, 130.9, 129.8, 129.2, 128.8, 127.2, 120.3, 119.9, 118.6, 63.4 ppm; IR (KBr) ν 3340, 2929, 2848, 1596, 1474, 1453 cm –1; HRMS (ES–) calculated for C13H11O3S [M–H]− 247.0429, found 247.0438.
tert-Butyl (benzyloxy)carbamate (37)
To a suspension of O-benzylhydroxylamine hydrochloride (0.798 g, 5.00 mmol, 1.00 equiv) in THF/H2O (1:1, 10 mL), NEt3 (0.700 mL, 5.00 mmol, 1.00 equiv) was added dropwise. A solution of di-tert-butyl dicarbonate (1.09 g, 5.00 mmol, 1.00 equiv) in THF (2.5 mL) was added dropwise at r.t. in 30 min, and the resulting solution was stirred at r.t. for 1.5 h. The volatiles were removed in vacuo, and the residue was taken up in EtOAc, then washed with 0.5 M citric acid (×2), H2O, dried over Na2SO4, filtered, and concentrated in vacuo to provide the title compound (0.500 g, 2.24 mmol, 45%), which was directly used in the next step without further purification: 1H NMR (500 MHz, CDCl3) δ 7.42–7.31 (m, 5H), 7.10 (s, 1H), 4.86 (s, 2H), 1.48 (s, 9H) ppm.
tert-Butyl (benzyloxy) (phenethyl)carbamate (38)
To a stirred solution of 37 (0.500 g, 2.24 mmol, 1.00 equiv) in anhydrous DMF (4.5 mL) under N2, NaH (60 wt %, 0.059 g, 2.46 mmol, 1.10 equiv) was added and the resulting solution was stirred at r.t. for 30 min. Phenethyl bromide (0.340 mL, 2.46 mmol, 1.10 equiv) was added dropwise and the reaction mixture was stirred at r.t. for 16 h. The reaction was poured into H2O and extracted with hexanes (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (10% EtOAc in hexanes) provided the title compound (0.388 g, 1.19 mmol, 53%): 1H NMR (500 MHz, CDCl3) δ 7.56–6.95 (m, 10H), 4.82 (s, 2H), 3.62 (t, J = 7.6 Hz, 2H), 2.89 (t, J = 7.9, 7.5 Hz, 2H), 1.46 (s, 9H) ppm.
N-(Benzyloxy)-N-phenethylacetamide (39)
A solution of 38 (0.264 g, 0.806 mmol, 1.00 equiv) in CH2Cl2/TFA (3:1 v/v, 2.00 mL) was stirred at r.t. for 18 h. The reaction mixture was made alkaline by addition of 1 M NaHCO3 (4.2 mL) and extracted with CH2Cl2 (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo to provide O-benzyl-N-phenethylhydroxylamine (0.162 g, 0.713 mmol, 88%), which was used directly in the next step without further purification: 1H NMR (500 MHz, CDCl3) δ 7.46 (m, 4H), 7.43–7.35 (m, 3H), 7.34–7.27 (m, 3H), 5.60 (s, 1H), 4.83 (s, 2H), 3.28 (t, J = 7.1 Hz, 2H), 2.95 (t, J = 7.1 Hz, 2H) ppm. To a solution of crude O-benzyl-N-phenethylhydroxylamine (0.060 g, 0.267 mmol, 1.00 equiv) in CH2Cl2 (7.0 mL), DMAP (0.016 g, 0.134 mmol, 0.50 equiv), pyridine (0.043 mL, 0.534 mmol, 2.00 equiv) and Ac2O (0.050 mL, 0.534 mmol, 2.00 equiv) were added successively. The solution was stirred at r.t. for 2 h. The reaction was diluted with CH2Cl2 and washed with satd. aq. NaHCO3. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (30% EtOAc in hexanes) provided the title compound (0.056 g, 0.208 mmol, 79%): 1H NMR (500 MHz, CDCl3) δ 7.45–7.33 (m, 5H), 7.33–7.27 (m, 2H), 7.25–7.18 (m, 3H), 4.78 (s, 2H), 3.86 (bs, 2H), 2.94 (t, J = 7.7 Hz, 2H), 2.08 (d, J = 6.2 Hz, 3H) ppm.
2-Phenylethane-1-sulfonyl chloride (40)
To a suspension of NCS (2.00 g, 14.9 mmol, 4.00 equiv) in CH2Cl2 (15 mL) at r.t., 2-phenylethanethiol (0.500 mL, 3.73 mmol, 1.00 equiv) was added dropwise, followed by H2O (7.5 mL). A rapid color change (from colorless to yellow) and a vigorous bubbling were observed for 5 min. The resulting colorless biphasic mixture was stirred at r.t. for 3.5 h. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (×2). The combined organic extracts were washed with satd. aq. NaHCO3 (×3), H2O, brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (5% EtOAc in hexanes) afforded the title compound (0.683 g, 3.34 mmol, 90%) as a low-melting colorless solid: mp (CH2Cl2) 30–30.5 °C; Lit.56 = 32–33 °C; 1H NMR (500 MHz, CDCl3) δ 7.39–7.36 (m, 2H), 7.33–7.30 (m, 1H), 7.26–7.25 (m, 2H), 3.93–3.90 (m, 2H), 3.37–3.33 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 135.7, 129.2, 128.6, 127.7, 66.3, 30.5 ppm; IR (KBr) ν 3030, 2924, 1497, 1456 cm–1.
3-Phenylpropanenitrile (41)
To a suspension of hydroxylamine hydrochloride (0.570 g, 8.20 mmol, 1.10 equiv) in DMSO (3.0 mL) at r.t., 3-phenylpropanal (0.999 g, 7.44 mmol, 1.00 equiv) was added. The resulting mixture was stirred at 90 °C for 3.5 h. After cooling to r.t., the reaction was diluted with H2O (5 mL) and extracted with EtOAc (×2). The combined organic extracts were washed with H2O, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (10–20% EtOAc in hexanes) provided the title compound (0.600 g, 4.57 mmol, 61%): 1H NMR (500 MHz, CDCl3) δ 7.37–7.34 (m, 2H), 7.31–7.28 (m, 1H), 7.27–7.24 (m, 2H), 2.97 (t, J = 7.4 Hz, 2H), 2.63 (t, J = 7.4 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 138.2, 129.1, 128.4, 127.4, 119.3, 31.7, 19.5 ppm; IR (KBr) ν 3063, 3030, 2933, 2869, 2247, 1603, 1496, 1453 cm–1; HRMS (ES+) calculated for C9H10N [M + H]+ 132.0813, found 132.0819.
N-(2-Cyanoethyl)-3-phenylpropanamide (42)
To a solution of 1 (0.082 g, 0.547 mmol, 1.00 equiv), EDCI·HCl (0.262 g, 1.37 mmol, 2.5 equiv), and HOBt·H2O (0.209 g, 1.55 mmol, 2.80 equiv) in DMF (2.8 mL) under N2 at r.t., DIEA (0.380 mL, 2.19 mmol, 4.00 equiv) was added dropwise, followed 5 min later by 3-aminopropionitrile (0.100 mL, 1.37 mmol, 2.5 equiv). The resulting solution was stirred at r.t. for 15 h. The reaction was diluted with EtOAc, washed with 1 M HCl, H2O, satd. aq. NaHCO3, brine, The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to provide the title compound (0.105 g, 0.519 mmol, 95%) as a pale yellow solid, which was used directly in the next step without further purification: 1H NMR (500 MHz, CDCl3) δ 7.34–7.22 (m, 5H), 6.52 (br s, 1H), 3.46 (q, J = 6.2 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H), 2.58–2.53 (m, 4H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.9, 140.6, 128.6, 128.3, 126.4, 118.3, 37.9, 35.6, 31.5, 18.3 ppm.
N′-Hydroxy-3-phenylpropanimidamide (43)
To a solution of 41 (0.466 g, 3.60 mmol, 1.00 equiv) in EtOH (3.5 mL) in a microwave vial, hydroxylamine (50 wt % in H2O, 0.440 mL, 7.10 mmol, 1.97 equiv) was added, the vial was sealed, and the mixture was stirred at 75 °C for 5.5 h. After cooling to r.t., the solvents were evaporated in vacuo to provide the title compound (0.548 g, 3.30 mmol, 92%), which was directly used in the next step without further purification: 1H NMR (500 MHz, CDCl3) δ 8.42 (br s, 1H), 7.31–7.21 (m, 5H), 4.55 (br s, 2H), 2.89 (t, J = 8.1 Hz, 2H), 2.46 (t, J = 8.1 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 153.7, 140.9, 128.7, 128.4, 126.4, 33.1 ppm.
5-(1-Hydroxy-2-phenylethylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (44)
To a solution of freshly recrystallized Meldrum’s acid (1.00 g, 6.94 mmol, 1.00 equiv) in CH2Cl2 (2.7 mL) at 0 °C under N2, anhydrous pyridine (1.36 mL, 16.8 mmol, 2.40 equiv) was added dropwise in 10 min. Then, a solution of freshly distilled phenylacetyl chloride (0.920 mL, 6.94 mmol, 1.00 equiv) in anhydrous CH2Cl2 (2.2 mL) was added over 2 h, and the resulting solution was stirred at 0 °C for 2 h. The reaction was diluted with CH2Cl2, acidified with 2 M HCl, and extracted with CH2Cl2 (×3). The combined organic extracts were washed with 2 M HCl (×2), dried over Na2SO4, filtered, and concentrated in vacuo to provide the title compound (1.60 g, 6.10 mmol, 91%): 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 7.3 Hz, 2H), 7.36–7.27 (m, 3H), 4.43 (bs, 2H), 1.72 (s, 6H) ppm.
Diethyl 1-benzyl-4-(benzyloxy)-5-oxocyclopent-3-ene-1,3-dicarboxylate (46)
To a solution of 45(51) (0.250 g, 0.785 mmol, 1.00 equiv) in anhydrous DMF (4.6 mL) under N2, benzyl bromide (0.280 mL, 2.36 mmol, 3.00 equiv) was added in one portion at r.t., and the solution was stirred at 120 °C for 2 h. The reaction was diluted with EtOAc, then carefully quenched with 30% aq. AcOH. The aqueous layer was extracted with EtOAc (×3). The combined organic layers were washed with brine/1 M HCl (2:1), brine (×3), dried over MgSO4, filtered, and concentrated in vacuo using toluene to azeotrope the residual DMF and AcOH. Purification by silica gel column chromatography (1–20% EtOAc in hexanes) provided the title compound (0.155 g, 0.367 mmol, 47%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.37–7.28 (m, 7H), 7.23–7.19 (m, 3H), 7.09–7.07 (m, 2H), 5.46 (d, J = 12.1 Hz, 1H), 5.40 (d, J = 12.1 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.24 (s, 2H), 3.07 (d, J = 17.9 Hz, 1H), 2.70 (d, J = 17.9 Hz, 1H), 1.27 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 200.8, 169.7, 163.8, 155.1, 136.6, 135.3, 130.0, 128.6, 128.4, 128.4, 128.2, 128.2, 128.1, 127.7, 127.1, 72.3, 66.3, 62.0, 61.1, 57.4, 39.4, 33.3, 32.0, 14.1, 14.0 ppm.
(E)-5-Phenylpent-4-ene-2,3-dione (47)
To a solution of 3,3-dimethoxybutan-2-one (2.00 mL, 14.9 mmol, 1.00 equiv) and freshly distilled benzaldehyde (1.51 mL, 14.9 mmol, 1.00 equiv) in MeOH (35 mL), a solution of NaOH (0.776 g, 19.4 mmol, 1.30 equiv) in H2O (11.5 mL) was added dropwise at r.t., and the resulting yellow solution was stirred for 24 h. The solvents were removed in vacuo, and the residue was extracted with EtOAc (×4). The combined organic extracts were washed with satd. aq. NaHSO3 (×2), brine, dried over MgSO4, filtered, and concentrated in vacuo. The yellow residue was dissolved in acetone (75 mL), then TsOH·H2O (0.567 g, 2.98 mmol, 20 mol %) was added and the resulting mixture was stirred at r.t. for 38 h. The solvents were removed in vacuo, the residue was taken up in toluene, and washed with H2O (until pH 7), brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (5–10% EtOAc in hexanes) afforded the title compound (1.89 g, 10.9 mmol, 73% over 2 steps) as a yellow crystalline solid: mp (CH2Cl2) 47–48 °C; 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 16.2 Hz, 1H), 7.63–7.60 (m, 4H), 7.44–7.37 (m, 4H), 2.43 (s, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 198.9, 186.8, 147.8, 134.5, 131.5, 129.1, 129.0, 118.0, 24.5 ppm; IR (KBr) ν 3446, 3058, 3026, 1714, 1683, 1605, 1576, 1449 cm–1; HRMS (CI+) calculated for C11H10O2 [M]+ 174.0681, found 174.0672.
3-Benzyl-4-ethoxycyclobut-3-ene-1,2-dione (48)
To a solution of diethyl squarate (0.441 g, 2.59 mmol, 1.00 equiv) in anhydrous THF (10 mL) at 0 °C, benzyl magnesium bromide (0.6 M in THF, 5.49 mL, 3.29 mmol, 1.27 equiv) was added dropwise, and the solution was stirred at 0 °C for 10 min, then at r.t. for 30 min. The reaction was quenched with 3 M HCl and extracted with Et2O (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (30% EtOAc in hexanes) afforded the title compound (0.460 g, 2.13 mmol, 82%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.42–7.15 (m, 5H), 4.75 (q, J = 7.1 Hz, 2H), 3.91 (s, 2H), 1.45 (t, J = 7.1 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 198.2, 194.6, 194.1, 181.2, 134.6, 128.9, 128.8, 127.3, 70.9, 30.8, 15.6 ppm.
4-Bromo-2,6-difluorophenol (49)
To a solution of 2,6-difluorophenol (1.30 g, 10.0 mmol, 1.00 equiv) in anhydrous DMF at 0 °C under N2, recrystallized NBS (1.87 g, 10.5 mmol, 1.05 equiv) was added in one portion, and the clear solution was stirred in the dark for 40 h while warming to r.t.. The reaction was quenched with H2O, then extracted with Et2O (×4). The combined organic extracts were washed with 1 M Na2SO3, brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (10% EtOAc in hexanes) provided the title compound (1.80 g, 8.63 mmol, 86%) as a yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.12–7.06 (m, 2H), 5.17 (s, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 151.8 (dd, J = 246.8, 6.3 Hz), 132.6 (t, J = 15.8 Hz), 115.8–115.5 (m), 110.4 (t, J = 10.9 Hz) ppm; IR (KBr) ν 3214, 2974, 1812, 1644 cm–1; HRMS (ES–) calculated for C6H2BrF2O [M–H]− 206.9257, found 206.9250.
2-(Benzyloxy)-5-bromo-1,3-difluorobenzene (50)
A mixture of 49 (0.879 g, 4.20 mmol, 1.00 equiv) and K2CO3 (0.697 g, 5.04 mmol, 1.2 equiv) in acetone (21 mL) in a sealed tube was stirred at 70 °C for 1 h. After cooling to approximately 40 °C, benzyl bromide (0.600 mL, 5.04 mmol, 1.20 equiv) was added in a steady stream. The tube was sealed, warmed to 70 °C and stirred for 3 h, then cooled to r.t. and stirred for 14 h. The reaction was diluted with acetone, filtered through a short pad of Celite, then concentrated in vacuo. Purification by silica gel column chromatography (0–5% EtOAc in hexanes) provided the title compound (1.24 g, 4.15 mmol, 99%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.55–7.49 (m, 2H), 7.48–7.38 (m, 3H), 7.16–7.06 (m, 2H), 5.24 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.2 (dd, J = 252.5, 6.5 Hz), 134.7 (t, J = 14.2 Hz), 116.1 (dd, J = 19.7, 6.9 Hz), 114.3 (t, J = 11.4 Hz), 76.1 (t, J = 3.2 Hz) ppm; IR (KBr) ν 3214, 2974, 1812, 1644 cm–1; HRMS (ES+) calculated for C13H10BrF2O [M + H]+ 298.9878, found 298.9876.
5-Benzyl-2-(benzyloxy)-1,3-difluorobenzene (51)
Anhydrous ZnBr2 (1.08 g, 4.80 mmol, 2.40 equiv) was weighed in a round-bottom flask and flame-dried under high vacuum, cooled to r.t., then dissolved in THF (9.4 mL, previously sparged with Ar for 15 min) under Ar. A solution of benzylmagnesium chloride (0.33 M in THF, 12.1 mL, 3.99 mmol, 2.00 equiv) was added dropwise (formation of a colorless precipitate) and the resulting suspension was vigorously stirred for 15 min at r.t.. A solution of 50 (0.623 g, 2.00 mmol, 1.00 equiv) and PEPPSI-IPr (0.068 g, 0.100 mmol, 5 mol %) in THF (6.0 mL, previously sparged with Ar for 15 min) was then cannulated dropwise, and the resulting orange mixture was stirred at r.t. for 16 h. The reaction was quenched carefully with 1 M HCl, then diluted with Et2O and filtered through a short pad of Celite. The layers were separated, and the aqueous layer was extracted with Et2O (×3). The combined organic extracts were washed with satd. aq. NaHCO3 (×2), brine, dried over MgSO4, decolorized with activated carbon, filtered through a short pad of Celite, and concentrated in vacuo. Purification by silica gel column chromatography (2–40% CH2Cl2 in hexanes) provided the title compound (0.459 g, 1.48 mmol, 74%) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 7.2 Hz, 2H), 7.40–7.32 (m, 5H), 7.26 (t, J = 7.3 Hz, 2H), 7.17 (d, J = 7.4 Hz, 2H), 6.74–6.69 (m, 2H), 5.14 (s, 2H), 3.89 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.1 (dd, J = 248.6, 6.1 Hz), 139.7, 137.2 (t, J = 8.1 Hz), 136.7, 133.3 (d, J = 14.3 Hz), 129.0, 128.8, 128.6, 128.5, 128.4, 126.7, 126.7, 112.5 (dd, J = 17.3, 5.4 Hz), 76.2 (t, J = 3.0 Hz), 41.3 ppm; IR (KBr) ν 3424, 1514 cm–1; HRMS (ES+) calculated for C13H10BrF2O [M + H]+ 298.9878, found 298.9876.
2-(Benzyloxy)-1,3-difluorobenzene (52)
Prepared as 50, starting from 2,6-difluorophenol (0.911 g, 7.01 mmol, 1.00 equiv), K2CO3 (1.16 g, 8.41 mmol, 1.2 equiv) and benzyl bromide (1.00 mL, 8.41 mmol, 1.20 equiv). Purification by silica gel column chromatography (0–10% CH2Cl2 in hexanes) provided the title compound (1.51 g, 6.87 mmol, 98%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.51–7.49 (m, 2H), 7.42–7.35 (m, 3H), 7.00–6.87 (m, 3H), 5.21 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.5 (dd, J = 248.2, 5.6 Hz), 136.6, 135.3 (t, J = 14.1 Hz), 128.6, 128.5, 128.4, 123.1 (t, J = 9.2 Hz), 112.2 (dd, J = 17.3, 5.4 Hz), 76.1 (t, J = 3.1 Hz) ppm; IR (KBr) ν 3067, 3034, 2946, 2890, 1593, 1496, 1475 cm–1.
1-Benzyl-3-(benzyloxy)-2,4-difluorobenzene (53)
To a solution of n-BuLi (2.34 M in hexanes, 1.28 mL, 3.00 mmol, 1.50 equiv) in THF (6.0 mL, previously sparged with Ar for 15 min) under N2 at −50 °C (dry ice/acetone bath) in a flame-dried round-bottom flask, a solution of 52 (0.440 g, 2.00 mmol, 1.00 equiv) in THF (3.0 mL, previously sparged with Ar for 15 min) was cannulated dropwise over 10 min at −50 °C. The resulting bright yellow solution was stirred at the same temperature for 35 min, during which the color changed from bright yellow to bright green, then pale yellow. Triisopropyl borate (0.740 mL, 3.20 mmol, 1.60 equiv) was added dropwise at −50 °C, then the reaction was warmed to r.t. and stirred for 1 h. To this crude mixture were added successively [(PPh3)2Pd(Br)(Succ)·0.5 CH2Cl2]54 (0.085 g, 0.100 mmol, 5 mol %), benzyl bromide (0.480 mL, 4.00 mmol, 2.00 equiv) and aq. NaHCO3 (3.3 M in H2O, previously sparged with Ar for 15 min, 1.50 mL, 5.0 mmol, 2.50 equiv). The resulting mixture was vigorously stirred at 60 °C for 14 h. After cooling to r.t., the reaction mixture was filtered through a pad of Celite, thoroughly washed with Et2O, then concentrated in vacuo. Purification by silica gel column chromatography (3–30% CH2Cl2 in hexanes) provided the title compound (0.259 g, 0.835 mmol, 42%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 7.1 Hz, 2H), 7.39–7.33 (m, 3H), 7.30 (t, J = 7.6 Hz, 2H), 7.23 (t, J = 7.3 Hz, 1H), 7.16 (d, J = 7.6 Hz, 2H), 6.82–6.74 (m, 2H), 5.18 (s, 2H), 3.95 (s, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 154.9 (dd, J = 246.7, 4.9 Hz), 154.5 (dd, J = 247.7, 5.4 Hz), 139.6, 136.7, 135.2 (t, J = 14.7 Hz), 128.8, 128.7, 128.6, 128.5, 128.4, 126.5, 125.0 (dd, J = 14.9, 3.6 Hz), 124.1 (dd, J = 8.8, 5.6 Hz), 111.6 (dd, J = 19.2, 3.8 Hz), 76.1 (t, J = 3.2 Hz), 34.6 (d, J = 3.0 Hz) ppm; IR (KBr) ν 3063, 3030, 2919, 1498, 1453 cm–1; HRMS (CI+) calculated for C20H16F2O [M]+ 310.1169, found 310.1171.
Determination of plasma unbound fraction
Plasma unbound fraction was determined using rapid equilibrium dialysis units (Thermo Scientific Pierce, Rockford, IL). Compounds at 50 mM in DMSO were diluted with pooled normal human plasma (Innovative Research, Novi, MI) to a final concentration of 5 μM. In duplicate, 100 μL aliquots of plasma were placed in sample chambers and dialyzed against 300 μL of phosphate buffered saline at 37 °C with gentle shaking. A dialysis unit was included for each compound at 5 μM in buffer to confirm stability and that equilibrium was reached over the length of the experiment. After 6 h, 20 μL aliquots were removed from each side of the dialysis membrane and mixed with an equal volume of plasma or buffer so that the matrix compositions were identical. Samples were extracted with 120 μL MeOH, vortexed and centrifuged at 6000g for 20 min at 4 °C. Supernatants were analyzed by LC-MS using an Acquity UPLC-TQ MS (Waters Corporation, Milford, MA). A number of H2O/CH3CN mobile phases were used to promote electrospray ionization. Methods were optimized for each compound using 0.1% formic acid, 10 mM ammonium formate, or 10 mM ammonium hydroxide. Sample injections, 2 μL, were separated using a gradient from 5% to 95% acetonitrile over 2 min on an Acquity BEH C18 column (1.7 μm, 2.1 × 50 mm) at 6 μL/min and 35 °C. Compounds were detected using selected ion recording (SIR). In 4 cases were matrix effects interfered with compound detection, multiple reaction monitoring (MRM) of a specific collision induced ion was used. Fraction unbound (fu) was calculated as the buffer chamber to plasma chamber peak area ratio.
Acknowledgments
Financial support for this work has been provided in part by the intramural program of the National Center for Advancing Translational Sciences (CT), the NIH/NIA (AG044332; AG034140 for PL, BG, MJ, KB, and CB), the NIH/NIGMS (GM87605 for MCK and VT), the Marian S. Ware Alzheimer Program, and the NSF (Grant CHE-0840438, X-ray facility). Finally, the authors would like to thank Mary Liang (University of Pittsburgh) for careful reading of the manuscript.
Glossary
Abbreviations Used
- CDI
1,1′-carbonyldiimidazole
- DIAD
diisopropyl azodicarboxylate
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- HOBt
hydroxybenzotriazole
- (PPh3)2Pd(Br)(Succ)
trans-bromo(N-succinimidyl)bis(triphenylphosphine)-palladium(II)
- UGTs
uridine 5′-diphospho-glucuronosyl-transferase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01963.
SMILES string structures along the full data set in tabular form (CSV)
NMR spectra of test compounds; X-ray crystal structures of compounds 1 (CCDC 1428182), 2 (CCDC 1427573), 3 (CCDC 1427548), 5 (CCDC 1427579), 6 (CCDC 1427789), 8 (CCDC 1427589), 10 (CCDC 1427790), 11 (CCDC 1428190), 12 (CCDC 1428042), 13 (CCDC 1427584), 14 (CCDC 1427791), 15 (CCDC 1428191), 16 (CCDC 1428195), 17 (CCDC 1427583), 18 (CCDC 1427834), 19 (CCDC 1428038), 20 (CCDC 1428058), 22 (CCDC 1428049), 26 (CCDC 1427564), 28 (CCDC 1436469), 32 (CCDC 1427567), 35 (CCDC 1427585), and 36 (CCDC 1427787); atomic coordinates and experimental data; experimental details for the permeability assay (PAMPA), logD7.4, and pKa determinations, as well as UV–vis titrations; (PDF)
The authors declare no competing financial interest.
Dedication
C.B. dedicates this article to the late Chris McGuigan, Professor of Medicinal Chemistry at Cardiff University, an outstanding mentor.
Supplementary Material
References
- Meanwell N. A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Patani G. A.; LaVoie E. J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96, 3147–3176. 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- Thornber C. W. Isosterism and Molecular Modification in Drug Design. Chem. Soc. Rev. 1979, 8, 563–580. 10.1039/cs9790800563. [DOI] [Google Scholar]
- Ballatore C.; Huryn D. M.; Smith A. B. 3rd. Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Daniels J. S.. Carboxylic Acids and their Bioisosteres. In Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET; RSC Drug Discovery series; Smith D. A., Ed.; Royal Society of Chemistry: Cambridge, 2010; pp 99–167. [Google Scholar]
- Ballatore C.; Soper J. H.; Piscitelli F.; James M.; Huang L.; Atasoylu O.; Huryn D. M.; Trojanowski J. Q.; Lee V. M.; Brunden K. R.; Smith A. B. 3rd Cyclopentane-1,3-dione: a Novel Isostere for the Carboxylic Acid Functional Group. Application to the Design of Potent Thromboxane (A2) Receptor Antagonists. J. Med. Chem. 2011, 54, 6969–6983. 10.1021/jm200980u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C.; Gay B.; Huang L.; Robinson K. H.; James M. J.; Trojanowski J. Q.; Lee V. M.; Brunden K. R.; Smith A. B. 3rd. Evaluation of the Cyclopentane-1,2-dione as a Potential Bio-isostere of the Carboxylic Acid Functional Group. Bioorg. Med. Chem. Lett. 2014, 24, 4171–4175. 10.1016/j.bmcl.2014.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soper J. H.; Sugiyama S.; Herbst-Robinson K.; James M. J.; Wang X.; Trojanowski J. Q.; Smith A. B. 3rd; Lee V. M.; Ballatore C.; Brunden K. R. Brain-penetrant Tetrahydronaphthalene Thromboxane A2-prostanoid (TP) Receptor Antagonists as Prototype Therapeutics for Alzheimer’s Disease. ACS Chem. Neurosci. 2012, 3, 928–940. 10.1021/cn3000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Liu L.; Huang L.; Herbst-Robinson K.; Cornec A. S.; James M. J.; Sugiyama S.; Bassetto M.; Brancale A.; Trojanowski J. Q.; Lee V. M.; Smith A. B. 3rd; Brunden K. R.; Ballatore C. Potent, Long-acting Cyclopentane-1,3-Dione Thromboxane (A2)-Receptor Antagonists. ACS Med. Chem. Lett. 2014, 5, 1015–1020. 10.1021/ml5002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March 10, 2016. http://www.chemaxon.com/product/pka.html.
- Huynh P. N. H.; Walvoord R. R.; Kozlowski M. C. Rapid Quantification of the Activating Effects of Hydrogen-Bonding Catalysts with a Colorimetric Sensor. J. Am. Chem. Soc. 2012, 134, 15621–15623. 10.1021/ja3050663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk P. J.; Bures M.; Praestgaard J.; Fesik S. W. Privileged Molecules for Protein Binding Identified from NMR-based Screening. J. Med. Chem. 2000, 43, 3443–3447. 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]
- Rowland A.; Miners J. O.; Mackenzie P. I. The UDP-glucuronosyltransferases: Their role in Drug Metabolism and Detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. 10.1016/j.biocel.2013.02.019. [DOI] [PubMed] [Google Scholar]
- Fung M.; Thornton A.; Mybeck K. Evaluation of the Characteristic of Safety Withdrawal of Prescription Drugs from Worldwide Pharmaceutical Markets - 1960 to 1999. Drug. Inf. J. 2001, 35, 293–317. 10.1177/009286150103500134. [DOI] [Google Scholar]
- Valko K.; Nunhuck S.; Bevan C.; Abraham M. H.; Reynolds D. P. Fast Gradient HPLC Method to Determine Compounds Binding to Human Serum Albumin. Relationships with Octanol/Water and Immobilized Artificial Membrane Lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. 10.1002/jps.10494. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P. Plasma Protein Binding Affinity and Its Relationship to Molecular Structure: An In-silico Analysis. J. Med. Chem. 2007, 50, 101–112. 10.1021/jm060981b. [DOI] [PubMed] [Google Scholar]
- Galinis-Luciani D.; Nguyen L.; Yazdanian M. Is PAMPA a Useful Tool for Discovery?. J. Pharm. Sci. 2007, 96, 2886–2892. 10.1002/jps.21071. [DOI] [PubMed] [Google Scholar]
- Allen F. H.; Groom C. R.; Liebeschuetz J. W.; Bardwell D. A.; Olsson T. S. G.; Wood P. A. The Hydrogen Bond Environments of 1H-Tetrazole and Tetrazolate Rings: The Structural Basis for Tetrazole–Carboxylic Acid Bioisosterism. J. Chem. Inf. Model. 2012, 52, 857–866. 10.1021/ci200521k. [DOI] [PubMed] [Google Scholar]
- Matta C. F.; Arabi A. A.; Weaver D. F. The Bioisosteric Similarity of the Tetrazole and Carboxylate Anions: Clues from the Topologies of the Electrostatic Potential and of the Electron Density. Eur. J. Med. Chem. 2010, 45, 1868–1872. 10.1016/j.ejmech.2010.01.025. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo L.. Exploring QSAR. Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, 1995. [Google Scholar]
- Herr R. J. 5-Substituted-1H-tetrazoles as Carboxylic Acid Isosteres: Medicinal Chemistry and Synthetic Methods. Bioorg. Med. Chem. 2002, 10, 3379–3393. 10.1016/S0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- Fichert T.; Yazdanian M.; Proudfoot J. R. A Structure–Permeability Study of Small Drug-Like Molecules. Bioorg. Med. Chem. Lett. 2003, 13, 719–722. 10.1016/S0960-894X(02)01035-1. [DOI] [PubMed] [Google Scholar]
- Young A. M.; Audus K. L.; Proudfoot J.; Yazdanian M. Tetrazole compounds: The Effect of Structure and pH on Caco-2 Cell Permeability. J. Pharm. Sci. 2006, 95, 717–725. 10.1002/jps.20526. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Duce P. P.; Prior D. V.; Barratt D. G.; Morris J. J.; Taylor P. J. Hydrogen bonding. Part 9. Solute Proton Donor and Proton Acceptor Scales for Use in Drug Design. J. Chem. Soc., Perkin Trans. 2 1989, 1355–1375. 10.1039/p29890001355. [DOI] [Google Scholar]
- Diamond J. M.; Wright E. M. Molecular Forces Governing Non-Electrolyte Permeation through Cell Membranes. Proc. R. Soc. London, Ser. B 1969, 172, 273–316. 10.1098/rspb.1969.0022. [DOI] [PubMed] [Google Scholar]
- Karls M.; Rush B.; Wilkinson K.; Vidmar T.; Burton P.; Ruwart M. Desolvation Energy: A Major Determinant of Absorption, But Not Clearance, of Peptides in Rats. Pharm. Res. 1991, 8, 1477–1481. 10.1023/A:1015882030289. [DOI] [PubMed] [Google Scholar]
- Gurka D.; Taft R. W. Studies of Hydrogen-Bonded Complex Formation with p-Fluorophenol. IV. Fluorine Nuclear Magnetic Resonance Method. J. Am. Chem. Soc. 1969, 91, 4794–4801. 10.1021/ja01045a037. [DOI] [Google Scholar]
- Birch A. M.; Kenny P. W.; Simpson I.; Whittamore P. R. O. Matched Molecular Pair Analysis of Activity and Properties of Glycogen Phosphorylase Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 850–853. 10.1016/j.bmcl.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Lázníček M.; Lázníčková A. The Effect of Lipophilicity on the Protein Binding and Blood Cell Uptake of Some Acidic Drugs. J. Pharm. Biomed. Anal. 1995, 13, 823–828. 10.1016/0731-7085(95)01504-E. [DOI] [PubMed] [Google Scholar]
- Thaler F.; Colombo A.; Mai A.; Amici R.; Bigogno C.; Boggio R.; Cappa A.; Carrara S.; Cataudella T.; Fusar F.; Gianti E.; di Ventimiglia S. J.; Moroni M.; Munari D.; Pain G.; Regalia N.; Sartori L.; Vultaggio S.; Dondio G.; Gagliardi S.; Minucci S.; Mercurio C.; Varasi M. Synthesis and Biological Evaluation of N-hydroxyphenylacrylamides and N-hydroxypyridin-2-ylacrylamides as Novel Histone Deacetylase Inhibitors. J. Med. Chem. 2010, 53, 822–839. 10.1021/jm901502p. [DOI] [PubMed] [Google Scholar]
- Elfarra A. A.; Yeh H. M.; Hanna P. E. Synthesis and Evaluation of N-(phenylalkyl)acetohydroxamic Acids as Potential Substrates for N-arylhydroxamic acid N,O-acyltransferase. J. Med. Chem. 1982, 25, 1189–1192. 10.1021/jm00352a018. [DOI] [PubMed] [Google Scholar]
- Grigg R.; Rankovic Z.; Thoroughgood M. Acyclic Oxyiminium Ions. Mannich Reactions and Addition of Grignard Reagents. Tetrahedron 2000, 56, 8025–8032. 10.1016/S0040-4020(00)00721-3. [DOI] [Google Scholar]
- Yoganathan S.; Miller S. J. N-Methylimidazole-catalyzed Synthesis of Carbamates from Hydroxamic Acids via the Lossen Rearrangement. Org. Lett. 2013, 15, 602–605. 10.1021/ol303424b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover S. A.; Goosen A.; McCleland C. W.; Schoonraad J. L. N-alkoxy-N-acylnitrenium Ions as Possible Intermediates in Intramolecular Aromatic Substitution: Novel Formation of N-acyl-3,4-dihydro-1H-2,1-benzoxazines and N-acyl-4,5-dihydro-1H,3H-2.1-benzoxazepine. J. Chem. Soc., Perkin Trans. 1 1984, 2255–2260. 10.1039/p19840002255. [DOI] [Google Scholar]
- Wrigglesworth J. W.; Cox B.; Lloyd-Jones G. C.; Booker-Milburn K. I. New Heteroannulation Reactions of N-Alkoxybenzamides by Pd(II) Catalyzed C–H Activation. Org. Lett. 2011, 13, 5326–5329. 10.1021/ol202187h. [DOI] [PubMed] [Google Scholar]
- Jackson P. F.; Maclin K. M.; Slusher B. S.; Tays K. L.. Certain Phosphinyl Derivatives Useful as Naaladase Inhibitors. 1997, WO1997048399 A1.
- Zhong Z.; Bibbs J. A.; Yuan W.; Wong C. H. Active-Site-Directed Modification of Subtilisin. J. Am. Chem. Soc. 1991, 113, 2259–2263. 10.1021/ja00006a052. [DOI] [Google Scholar]
- Ankner T.; Hilmersson G. Instantaneous Deprotection of Tosylamides and Esters with SmI2/Amine/Water. Org. Lett. 2009, 11, 503–506. 10.1021/ol802243d. [DOI] [PubMed] [Google Scholar]
- Librowski T.; Kubacka M.; Meusel M.; Scolari S.; Müller C. E.; Gütschow M. Evaluation of Anticonvulsant and Analgesic Effects of Benzyl- and Benzhydryl Ureides. Eur. J. Pharmacol. 2007, 559, 138–149. 10.1016/j.ejphar.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Duspara P. A.; Islam M. S.; Lough A. J.; Batey R. A. Synthesis and Reactivity of N-Alkyl Carbamoylimidazoles: Development of N-Methyl Carbamoylimidazole as a Methyl Isocyanate Equivalent. J. Org. Chem. 2012, 77, 10362–10368. 10.1021/jo302084a. [DOI] [PubMed] [Google Scholar]
- Ortar G.; Cascio M. G.; Moriello A. S.; Camalli M.; Morera E.; Nalli M.; Di Marzo V. Carbamoyl Tetrazoles as Inhibitors of Endocannabinoid Inactivation: A critical Revisitation. Eur. J. Med. Chem. 2008, 43, 62–72. 10.1016/j.ejmech.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Demko Z. P.; Sharpless K. B. Preparation of 5-Substituted 1H-tetrazoles from Nitriles in Water. J. Org. Chem. 2001, 66, 7945–7950. 10.1021/jo010635w. [DOI] [PubMed] [Google Scholar]
- Deng X.; Mani N.; Mapes C. M.. Alpha, Beta-unsaturated Esters and Acids by Stereoselective Dehydration. 2007, EP1781617 A2.
- Mendgen T.; Steuer C.; Klein C. D. Privileged Scaffolds or Promiscuous Binders: A Comparative Study on Rhodanines and Related Heterocycles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 743–753. 10.1021/jm201243p. [DOI] [PubMed] [Google Scholar]
- Kohara Y.; Kubo K.; Imamiya E.; Wada T.; Inada Y.; Naka T. Synthesis and Angiotensin II Receptor Antagonistic Activities of Benzimidazole Derivatives Bearing Acidic Heterocycles as Novel Tetrazole Bioisosteres. J. Med. Chem. 1996, 39, 5228–5235. 10.1021/jm960547h. [DOI] [PubMed] [Google Scholar]
- Sørensen U. S.; Falch E.; Krogsgaard-Larsen P. A Novel Route to 5-Substituted 3-Isoxazolols. Cyclization of N,O-DiBoc β-Keto Hydroxamic Acids Synthesized via Acyl Meldrum’s Acids. J. Org. Chem. 2000, 65, 1003–1007. 10.1021/jo991409d. [DOI] [PubMed] [Google Scholar]
- Sengoku T.; Nagae Y.; Ujihara Y.; Takahashi M.; Yoda H. A Synthetic Approach to Diverse 3-Acyltetramic Acids via O- to C-Acyl Rearrangement and Application to the Total Synthesis of Penicillenol Series. J. Org. Chem. 2012, 77, 4391–4401. 10.1021/jo300527f. [DOI] [PubMed] [Google Scholar]
- Koreeda M.; Liang Y.; Akagi H. Easy Generation of the Dianions of 3-Isobutoxycyclopent-2-en-1-ones and their Reactions. J. Chem. Soc., Chem. Commun. 1979, 449–450. 10.1039/c39790000449. [DOI] [Google Scholar]
- Ramachary D. B.; Kishor M. Direct Amino Acid-catalyzed Cascade Biomimetic Reductive Alkylations: Application to the Asymmetric Synthesis of Hajos-Parrish Ketone Analogues. Org. Biomol. Chem. 2008, 6, 4176–4187. 10.1039/b807999d. [DOI] [PubMed] [Google Scholar]
- Jõgi A.; Ilves M.; Paju A.; Pehk T.; Kailas T.; Müürisepp A.-M.; Lopp M. Asymmetric Synthesis of 4′-C-benzyl-2′,3′-dideoxynucleoside Analogues from 3-Benzyl-2-hydroxy-2-cyclopenten-1-one. Tetrahedron: Asymmetry 2008, 19, 628–634. 10.1016/j.tetasy.2008.01.028. [DOI] [Google Scholar]
- Aqad E.; Leriche P.; Mabon G.; Gorgues A.; Allain M.; Riou A.; Ellern A.; Khodorkovsky V. Base-catalyzed condensation of cyclopentadiene derivatives. Synthesis of fulvalene analogues: strong proaromatic electron acceptors. Tetrahedron 2003, 59, 5773–5782. 10.1016/S0040-4020(03)00877-9. [DOI] [Google Scholar]
- Muxfeldt H.; Weigele M.; Rheenen V. V. Magnesium Methoxide Cyclization of Biacetyl Derivatives1. J. Org. Chem. 1965, 30, 3573–3574. 10.1021/jo01021a511. [DOI] [Google Scholar]
- Lopez C.; Vega M.; Sanna E.; Rotger C.; Costa A. Efficient Microwave-Assisted Preparation of Squaric Acid Monoamides in Water. RSC Adv. 2013, 3, 7249–7253. 10.1039/c3ra41369a. [DOI] [Google Scholar]
- Fairlamb I. J. S.; Sehnal P.; Taylor R. J. K. Suzuki-Miyaura Cross-Couplings Mediated by trans-PdBr(N-Succ) (PPh3)2: A Convenient Synthetic Method for Diarylmethanes and Aryl(heteroaryl)methanes. Synthesis 2009, 2009, 508–510. 10.1055/s-0028-1083293. [DOI] [Google Scholar]
- Felpin F. X.; Fouquet E. A Useful, Reliable and Safer Protocol for Hydrogenation and the Hydrogenolysis of O-Benzyl Groups: The In Situ Preparation of an Active Pd0/C Catalyst with Well-Defined Properties. Chem.—Eur. J. 2010, 16, 12440–12445. 10.1002/chem.201001377. [DOI] [PubMed] [Google Scholar]
- Johnson T. B.; Sprague J. M. A New Method for the Preparation of Alkyl Sulfonyl Chlorides. J. Am. Chem. Soc. 1936, 58, 1348–1352. 10.1021/ja01299a011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.