

Abstract
This paper is motivated by a phase I–II clinical trial of a targeted agent for advanced solid tumors. We study a stylized version of this trial with the goal to determine optimal actions in each of two cycles of therapy. A design is presented that generalizes the decision-theoretic two-cycle design of Lee and others (2015. Bayesian dose-finding in two treatment cycles based on the joint utility of efficacy and toxicity. Journal of the American Statistical Association, to appear) to accommodate ordinal outcomes. Backward induction is used to jointly optimize the actions taken for each patient in each of the two cycles, with the second action accounting for the patient's cycle 1 dose and outcomes. A simulation study shows that simpler designs obtained by dichotomizing the ordinal outcomes either perform very similarly to the proposed design, or have much worse performance in some scenarios. We also compare the proposed design with the simpler approaches of optimizing the doses in each cycle separately, or ignoring the distinction between cycles 1 and 2.
Keywords: Adaptive design, Bayesian design, Decision theory, Dynamic treatment regime, Latent probit model, Ordinal outcomes, Phase I–II clinical trial
1. Introduction and motivation
This paper is motivated by the problem of designing a dose-finding trial of a new agent for cancer patients with advanced solid tumors. The agent aims to inhibit a kinase, which regulates cell metabolism and proliferation, in the cancer cells to reduce or eradicate the disease. The agent is given orally each day of a 28-day cycle at one of five doses, 2, 4, 6, 8, or 10 mg, combined with a fixed dose of standard chemotherapy. Because both efficacy and toxicity are used for dose-finding, it is a phase I–II trial (Thall and Cook, 2004; Yin and others, 2006; Zhang and others, 2006; Thall and Nguyen, 2012). Both outcomes are 3-level ordinal variables, with toxicity defined as None/Mild (grade 0,1), Moderate (grade 2), or Severe (grade 3,4) and efficacy defined in terms of disease status compared with baseline, with possible values progressive disease (PD), stable disease (SD), or partial or complete response (PR/CR).
We study a stylized version of this trial with the more ambitious goal to determine optimal doses or actions for each patient in each of two cycles of therapy. This is a major departure from conventional dose-finding designs, which focus on choosing a dose for only the first cycle. While virtually all clinical protocols for dose-finding trials include rules for making within-patient dose adjustments in cycles after the first, this aspect usually is ignored in the trial design. In practice, each patient's doses in cycle 2, or later cycles, are chosen subjectively by the attending physician. To choose a patient's cycle 2 dose using a formal rule, it is desirable to use the patient's dose-outcome data from cycle 1, as well as data from other patients treated previously in the trial. Thus, ideally, a decision rule that is adaptive both within and between patients is needed.
Recent papers on designs accounting for multiple treatment cycles include Cheung and others (2014) and Lee and others (2015). In this paper, we build on the latter, who use a decision-theoretic approach for dose-finding in two cycles based on joint utilities of binary outcomes in each cycle. We extend the model to accommodate ordinal outcomes, and use a decision criterion that accounts for the many possible (efficacy,toxicity) outcomes in each of the two treatment cycles, including the risk-benefit trade-offs between the levels of efficacy and toxicity. In the stylized version of the trial described above, since there are 3-level ordinal toxicity and efficacy outcomes in each cycle, accounting for two cycles there are 81 possible elementary outcomes for each patient. Consequently, dose-finding is a much more complex problem than in a conventional phase I–II trial with two binary outcomes that chooses a dose for cycle 1 only.
Aside from the issue of accounting for two cycles, an important question is whether the additional complexity required to account for ordinal outcomes provides practical benefits compared with the common approach of dichotomizing efficacy and toxicity, which would allow the two-cycle design of Lee and others (2015) to be applied. Simulations, described in Section 4.4 of the main text, Figure 2, and Section 3 of Supplementary Material (available at Biostatistics online), show that reducing ordinal outcomes to binary variables produces a design that either performs very similarly to the proposed design, or has much worse performance in certain scenarios. Moreover, the behavior of the simplified design depends heavily on how one chooses to reduce the two ordinal outcomes to two binary variables.
Fig. 2.

Plots of 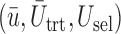 for a comparison of DTD-O2 vs. a design with binary outcomes. Here,
for a comparison of DTD-O2 vs. a design with binary outcomes. Here, 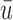 ,
, 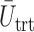 , and
, and 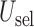 represent empirical mean utilities of patients treated in the trial, true mean utilities of treatments given to patients in the trial, and true expected utilities chosen for future patients, respectively. (a)
represent empirical mean utilities of patients treated in the trial, true mean utilities of treatments given to patients in the trial, and true expected utilities chosen for future patients, respectively. (a) 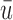 . (b)
. (b) 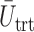 . (c)
. (c) 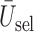 .
.
A naive design might aim to optimize the doses given in the two cycles separately. This may be not optimal. To see this, denote a patient's toxicity outcome by 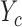 and efficacy outcome by
and efficacy outcome by 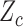 for
for 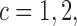 and denote the current data from
and denote the current data from  patients by
patients by 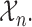 We include
We include 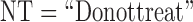 as a possible action in either cycle for cases where it has been determined that no dose is acceptable, so the action
as a possible action in either cycle for cases where it has been determined that no dose is acceptable, so the action 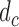 in each of cycles
in each of cycles 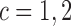 may be either to choose a dose or
may be either to choose a dose or  , that is,
, that is, 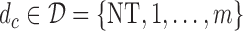 with 1 and
with 1 and 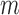 denoting the minimum dose and the maximum dose levels, respectively. Suppose that some optimality criterion has been defined. If one derives optimal adaptive actions
denoting the minimum dose and the maximum dose levels, respectively. Suppose that some optimality criterion has been defined. If one derives optimal adaptive actions 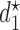 for cycle 1 and
for cycle 1 and 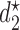 for cycle 2 separately, each based on the current data
for cycle 2 separately, each based on the current data 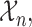 an inherent flaw is that in choosing one
an inherent flaw is that in choosing one 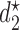 for all patients it ignores each patient's cycle 1 data. As in Lee and others (2015), we derive optimal decision rules
for all patients it ignores each patient's cycle 1 data. As in Lee and others (2015), we derive optimal decision rules 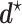 =
= 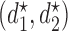 with the important property that
with the important property that 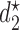 =
= 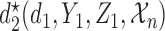 is a function of the first cycle decision
is a function of the first cycle decision 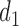 and response
and response 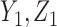 . This is implemented by applying backward induction (Bellman, 1957, etc.). The method accounts for the patient's cycle 1 dose and outcomes, as well as other patient's data, in making an optimal decision for cycle 2.
. This is implemented by applying backward induction (Bellman, 1957, etc.). The method accounts for the patient's cycle 1 dose and outcomes, as well as other patient's data, in making an optimal decision for cycle 2.
Iasonos and others (2011) and Van Meter and others (2012) studied the use of ordinal toxicity outcomes for a generalized continual reassessment method and reported that gains in performance of their ordinal toxicity designs are not substantial in comparison to binary toxicity designs. However, the comparison looks quite different for the model-based two-cycle design for bivariate ordinal (efficacy, toxicity) outcomes that we propose in this paper. In simulations described in Section 4.4, we compare the proposed design with designs that do not properly model association between cycles. In simulations reported in Section 4.5, we show that the use of ordinal rather than binary outcomes can substantially improve design performance in our setting.
Section 2 describes the proposed decision-theoretic method for ordinal outcomes in two cycles (DTD-O2). Sections 3 and 4 include decision criteria using utilities and a simulation study. The last section concludes with a final discussion.
2. A decision-theoretic design
2.1. Actions and optimal sequential decisions
For notational convenience, we denote the possible levels of toxicity by 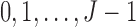 and efficacy by
and efficacy by 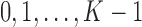 . For the motivating trial, these are
. For the motivating trial, these are 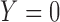 for None/Mild, 1 for Moderate, and 2 for Severe, and
for None/Mild, 1 for Moderate, and 2 for Severe, and 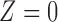 for PD, 1 for SD, and 2 for CR/PR, so
for PD, 1 for SD, and 2 for CR/PR, so 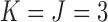 . If the adaptively chosen cycle 1 action
. If the adaptively chosen cycle 1 action 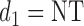 for any patient, then the trial is stopped and no more patients are enrolled. Otherwise, the patient receives a dose
for any patient, then the trial is stopped and no more patients are enrolled. Otherwise, the patient receives a dose 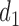 of the agent in cycle 1. A cycle 2 action is a function mapping the cycle 1 dose and outcomes,
of the agent in cycle 1. A cycle 2 action is a function mapping the cycle 1 dose and outcomes, 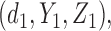 to an action in
to an action in 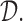 For example, if the cycle 1 action
For example, if the cycle 1 action 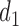 produced None/Mild toxicity (
produced None/Mild toxicity (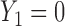 ), one possible cycle 2 action is
), one possible cycle 2 action is 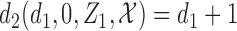 if
if 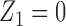 , and
, and 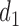 if
if 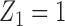 or 2. That is, if there was little or no toxicity but PD in cycle 1, then the action
or 2. That is, if there was little or no toxicity but PD in cycle 1, then the action 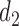 increases the dose in cycle 2, but if the patient had SD or better then it repeats the cycle 1 dose. The design thus involves an alternating sequence of decisions and observed outcomes,
increases the dose in cycle 2, but if the patient had SD or better then it repeats the cycle 1 dose. The design thus involves an alternating sequence of decisions and observed outcomes, 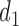 ,
, 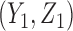 ,
, 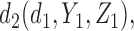 and
and 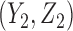 .
.
We apply a Bayesian decision-theoretic paradigm to determine an optimal decision rule. First, focus on cycle 1, and temporarily ignore cycle 2. The general setup of a Bayesian decision problem involves actions 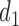 , observable data
, observable data 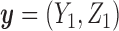 , parameters
, parameters 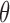 that index a sampling model
that index a sampling model 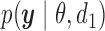 for the data, and a prior probability model
for the data, and a prior probability model 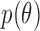 for the parameters. We discuss specification of
for the parameters. We discuss specification of 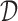 in more detail below. A utility function
in more detail below. A utility function 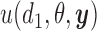 formalizes relative preferences for alternative actions under hypothetical outcomes
formalizes relative preferences for alternative actions under hypothetical outcomes 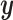 and assumed truth
and assumed truth 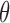 . Starting from first principles, one can then argue ((Robert, 2007, Chapter 2)) that a rational decision-maker chooses the action
. Starting from first principles, one can then argue ((Robert, 2007, Chapter 2)) that a rational decision-maker chooses the action 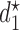 that maximizes utility in expectation, that is
that maximizes utility in expectation, that is
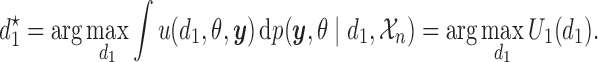 |
(2.1) |
The integral is the expected utility 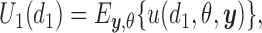 with the expectation taken with respect to
with the expectation taken with respect to 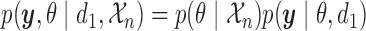 . To simplify notation, we will henceforth suppress conditioning on
. To simplify notation, we will henceforth suppress conditioning on 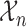 in the notation.
in the notation.
In the two-cycle dose-finding problem, the sequential nature of the within-patient decisions complicates the solution. In the second cycle, the utility 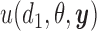 is replaced by the expected utility under optimal continuation. Denote
is replaced by the expected utility under optimal continuation. Denote 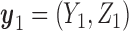 and
and 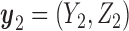 . We get an alternating sequence of optimization and expectation
. We get an alternating sequence of optimization and expectation
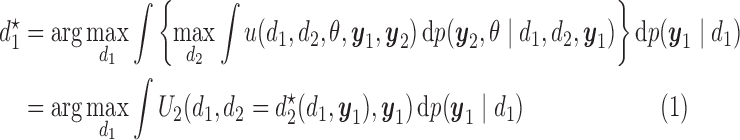 |
(2.2) |
with the second cycle expected total utility as a function of 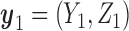 ,
, 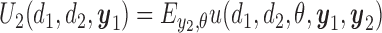 and the optimal second cycle decision
and the optimal second cycle decision 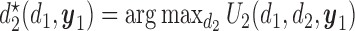 . When we substitute
. When we substitute 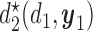 and take the expectation with respect to
and take the expectation with respect to 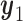 we obtain
we obtain
 |
(2.3) |
which is maximized to determine the optimal decision for cycle 1, 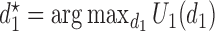 . This alternating sequence of maximization and expectation, called dynamic programming, is characteristic of sequential decision problems. While it often leads to intractable computational problems ((Parmigiani and Inoue, 2009, Chapter 12)), in the present setting with ordinal outcomes the problem is solvable. Dynamic programming recently has been applied in other clinical trial design settings (Murphy, 2003; Zhao and others, 2011; Lee and others, 2015; Cheung and others, 2014).
. This alternating sequence of maximization and expectation, called dynamic programming, is characteristic of sequential decision problems. While it often leads to intractable computational problems ((Parmigiani and Inoue, 2009, Chapter 12)), in the present setting with ordinal outcomes the problem is solvable. Dynamic programming recently has been applied in other clinical trial design settings (Murphy, 2003; Zhao and others, 2011; Lee and others, 2015; Cheung and others, 2014).
2.2. Utility function
We construct a utility function
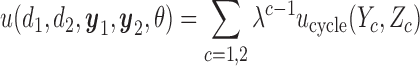 |
(2.4) |
as a sum over cycle-specific utilities 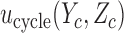 ,
, 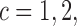 where
where 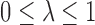 is a scale parameter. If
is a scale parameter. If 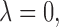 then the cycle 2 utility is ignored in selecting
then the cycle 2 utility is ignored in selecting 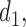 while
while 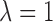 corresponds to treating utilities in the two cycles equally. Optimal decisions may change under different values of
corresponds to treating utilities in the two cycles equally. Optimal decisions may change under different values of 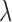 . Even with
. Even with 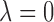 , however, the importance of jointly modeling the two cycles remains in that inference on
, however, the importance of jointly modeling the two cycles remains in that inference on 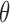 can be enhanced through borrowing information across cycles. For the simulations in Section 4, we used
can be enhanced through borrowing information across cycles. For the simulations in Section 4, we used 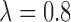 . A sensitivity analysis in
. A sensitivity analysis in  is reported in the Supplementary Materials (available at Biostatistics online). The utility function (2.4) focuses on the clinical outcomes and is a function of
is reported in the Supplementary Materials (available at Biostatistics online). The utility function (2.4) focuses on the clinical outcomes and is a function of 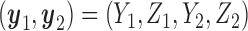 only. That is, the inference on
only. That is, the inference on 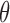 does not affect utility, and we do not initially consider preferences across doses
does not affect utility, and we do not initially consider preferences across doses 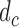 . We thus drop
. We thus drop 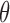 and
and 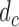 from the arguments of
from the arguments of 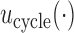 hereafter.
hereafter.
In practice, numerical utilities of the 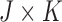 elementary must be elicited from the clinical collaborators, with specific numerical values reflecting physicians' relative preferences (cf. Thall and Nguyen, 2012). In our stylized illustrative trial, we fix the utilities of the best and worst possible outcomes to be
elementary must be elicited from the clinical collaborators, with specific numerical values reflecting physicians' relative preferences (cf. Thall and Nguyen, 2012). In our stylized illustrative trial, we fix the utilities of the best and worst possible outcomes to be 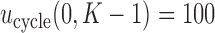 and
and 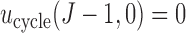 . In general, any convenient function with
. In general, any convenient function with 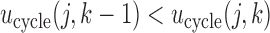 and
and 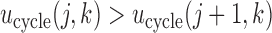 that gives higher utilities to more desirable outcomes may be used. For future reference, we note that
that gives higher utilities to more desirable outcomes may be used. For future reference, we note that 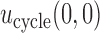 is the expected utility corresponding to
is the expected utility corresponding to 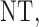 i.e. do not treat the patient. Table 1 shows the utilities that will be used for our simulation studies.
i.e. do not treat the patient. Table 1 shows the utilities that will be used for our simulation studies.
Table 1.
An example of elicited utilities, 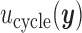
| Toxicity severity level |
|||
|---|---|---|---|
| Efficacy scores | Mild | Moderate | Severe |
| PD | 25 | 10 | 0 |
| SD | 70 | 50 | 25 |
| PR/CR | 100 | 80 | 50 |
To reduce notation, we denote the utility 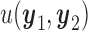 as a function of hypothetical outcomes
as a function of hypothetical outcomes 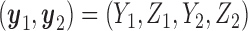 , and drop the arguments
, and drop the arguments 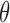 and
and 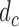 . Upper case
. Upper case 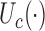 denotes expected utility, with data
denotes expected utility, with data 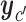 ,
,  removed by marginalization and decisions
removed by marginalization and decisions 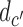 ,
, 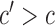 substituted by maximization, as in (2.2). In addition to the cycle index
substituted by maximization, as in (2.2). In addition to the cycle index 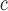 , the arguments of
, the arguments of 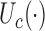 clarify the level of marginalization and maximization. Maximizing
clarify the level of marginalization and maximization. Maximizing 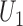 in (2.1) and
in (2.1) and 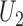 inside the integral in (2.2) yields the optimal action pair
inside the integral in (2.2) yields the optimal action pair 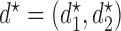 , where
, where 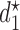 is either a dose or
is either a dose or  ,
, 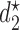 is applicable only when
is applicable only when 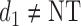 is a dose, and
is a dose, and 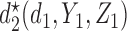 is a function of
is a function of 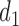 and the patient's cycle 1 outcomes,
and the patient's cycle 1 outcomes, 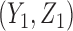 . Assuming that the utility function takes the additive form (2.4), we define cycle-specific expected utilities, with the expected utility for cycle 2 given by
. Assuming that the utility function takes the additive form (2.4), we define cycle-specific expected utilities, with the expected utility for cycle 2 given by
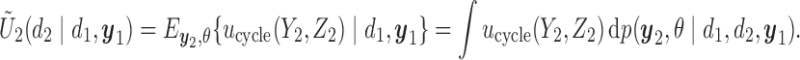 |
(2.5) |
Figure 1(a)–(c) illustrates 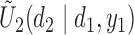 under the assumed simulation truth of Scenario 3 (discussed in Section 4.2), and shows how
under the assumed simulation truth of Scenario 3 (discussed in Section 4.2), and shows how 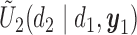 changes with
changes with 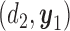 , given
, given 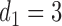 . Figure 1(d) illustrates the assumed true
. Figure 1(d) illustrates the assumed true 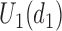 over
over 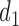 for the simulation scenarios discussed in Section 4.2.
for the simulation scenarios discussed in Section 4.2.
Fig. 1.

(a)–(c) The true expected cycle 2 utilities of taking 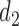 given
given 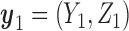 ,
, 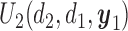 with
with 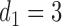 for scenario 3. Each panel corresponds to one of the three possible outcomes of
for scenario 3. Each panel corresponds to one of the three possible outcomes of 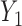 .
. 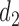 is acceptable only when its expected utility is greater than that of
is acceptable only when its expected utility is greater than that of  ,
, 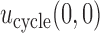 .
. 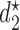 is marked with a bold circle for each
is marked with a bold circle for each 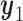 given
given 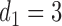 if the corresponding expected utility is greater than
if the corresponding expected utility is greater than 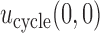 . If none of
. If none of 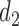 has an expected utility greater than
has an expected utility greater than 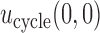 for
for 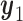 ,
, 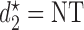 and none of
and none of 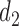 is marked with a bold circle. (d) Illustrates total expected utilities of
is marked with a bold circle. (d) Illustrates total expected utilities of 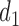 ,
, 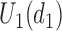 for the simulation scenarios assuming that the true
for the simulation scenarios assuming that the true 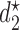 will be taken in cycle 2.
will be taken in cycle 2. 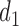 is acceptable only when its utility is greater than that of
is acceptable only when its utility is greater than that of 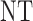 in the two cycles,
in the two cycles, 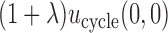 (red dashed horizontal line at 45). (a)
(red dashed horizontal line at 45). (a) 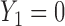 (mild toxicity). (b)
(mild toxicity). (b) 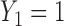 (moderate toxicity). (c)
(moderate toxicity). (c) 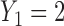 (severe toxicity). (d)
(severe toxicity). (d) 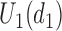 .
.
Some practical guidelines of using utility functions for a design with ordinal outcomes in the two-cycle setting are provided in Section 1 of the Supplementary Material (available at Biostatistics online).
2.3. Action set
Equation (2.2) includes two maximizations to determine 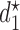 and
and 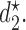 In the discussion thus far, we have not used the particular elements of
In the discussion thus far, we have not used the particular elements of 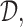 and they might have been any actions. In actual dose-finding, ethical and practical constraints are motivated by the knowledge that, in general, higher doses carry a higher risk of more severe toxicity. We thus require a more restrictive action set, with additional conditions for the acceptability of a dose assignment.
and they might have been any actions. In actual dose-finding, ethical and practical constraints are motivated by the knowledge that, in general, higher doses carry a higher risk of more severe toxicity. We thus require a more restrictive action set, with additional conditions for the acceptability of a dose assignment.
The first additional criterion is that we do not skip untried dose levels when escalating. This rule is imposed almost invariably in actual trials with adaptive dose-finding methods. Let 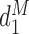 denote the highest dose level among the dose levels that have been tried in cycle 1 and
denote the highest dose level among the dose levels that have been tried in cycle 1 and 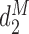 the highest dose level among those that have been tried in either cycle 1 or cycle 2. The search for the optimal actions is constrained such that
the highest dose level among those that have been tried in either cycle 1 or cycle 2. The search for the optimal actions is constrained such that 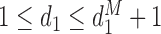 and
and 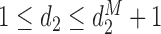 . In addition, we do not escalate a patient's dose level in cycle 2 if severe toxicity was observed in cycle 1 (
. In addition, we do not escalate a patient's dose level in cycle 2 if severe toxicity was observed in cycle 1 (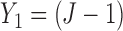 ). Both restrictions are due to safety concerns.
). Both restrictions are due to safety concerns.
A third safety restriction is defined implicitly in terms of the cycle-specific utility  A patient is not treated (
A patient is not treated (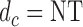 ) if there is no dose with expected utility
) if there is no dose with expected utility 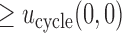 . For
. For 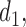 the expected utility
the expected utility 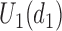 is compared with the expected utility of not receiving any treatment in both cycles,
is compared with the expected utility of not receiving any treatment in both cycles, 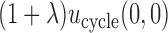 (horizontal dotted line in Figure 1(d)). Any
(horizontal dotted line in Figure 1(d)). Any 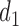 with
with 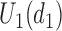 below the line is not considered acceptable treatment. For
below the line is not considered acceptable treatment. For  , the expected utility
, the expected utility 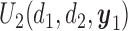 is similarly compared with the expected utility of
is similarly compared with the expected utility of 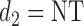 ,
, 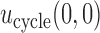 (horizontal dotted line in Figure 1(a)–(c)), and any
(horizontal dotted line in Figure 1(a)–(c)), and any 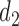 with
with 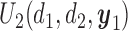 below the line is not acceptable.
below the line is not acceptable.
At any interim point in the trial, let 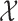 denote the current data, including dose assignments for previously enrolled patients. The three conditions together make the action sets for
denote the current data, including dose assignments for previously enrolled patients. The three conditions together make the action sets for 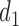 and
and 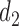 dependent on
dependent on  ,
, 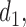 and
and 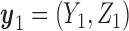 . We let
. We let 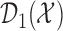 and
and 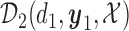 denote the action sets for
denote the action sets for 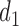 and
and 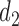 , respectively, that are implied by these three restrictions.
, respectively, that are implied by these three restrictions.
2.4. Inference model
Thus far, our discussion of optimal decisions has not included a particular probability model. We will assume a 4D ordinal probit model for 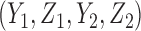 with a regression on doses
with a regression on doses 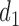 and
and 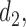 standardized to the domain [0, 1], with
standardized to the domain [0, 1], with 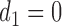 and
and 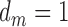 . Let
. Let 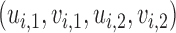 denote a vector of latent probit scores for the
denote a vector of latent probit scores for the 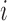 th patient and let
th patient and let 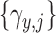 and
and 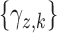 denote fixed cutoffs that define
denote fixed cutoffs that define 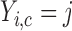 if
if 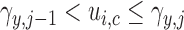 and
and 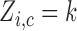 if
if 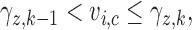
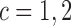 . While varying the mean of distributions of
. While varying the mean of distributions of 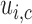 and
and 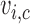 across cycles, the same cutoffs are used for all cycles. The
across cycles, the same cutoffs are used for all cycles. The 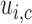 and
and 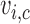 are multivariate normal probit scores,
are multivariate normal probit scores,
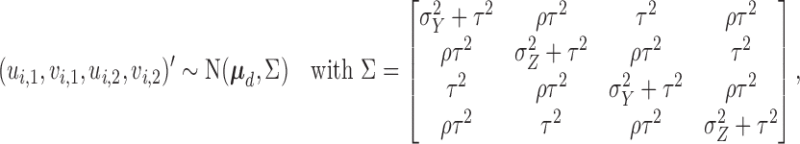 |
(2.6) |
and 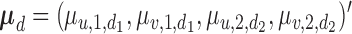 . The covariance matrix implies associations across cycles and across outcomes through
. The covariance matrix implies associations across cycles and across outcomes through 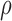 and
and 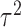 . Given that the ordinality of the outcomes is accounted for by the latent probit scores and fixed cutoff parameters
. Given that the ordinality of the outcomes is accounted for by the latent probit scores and fixed cutoff parameters 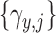 and
and 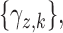 a simple yet flexible model for regression on dose is obtained by assuming
a simple yet flexible model for regression on dose is obtained by assuming 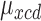 =
= 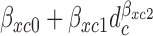 with
with 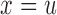 for toxicity and
for toxicity and 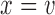 for efficacy. A discussion of nonlinear dose–response models is given by Bretz and others (2005). We assume that the toxicity and efficacy probabilities increase monotonic in dose by requiring
for efficacy. A discussion of nonlinear dose–response models is given by Bretz and others (2005). We assume that the toxicity and efficacy probabilities increase monotonic in dose by requiring 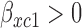 and
and 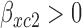 . Denote
. Denote 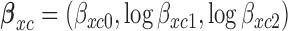 ,
, 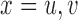 and
and 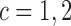 . We complete the model with a normal prior
. We complete the model with a normal prior 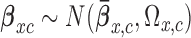 ,
, 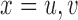 .
.
3. Trial design
3.1. Adaptive randomization
Denote 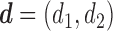 . Although, in terms of the utility-based objective function,
. Although, in terms of the utility-based objective function, 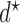 yields the best clinical outcomes for the next patient, the performance of the design, in terms of frequentist operating characteristics, can be improved by including adaptive randomization (AR) among actions giving values of the objective function near the maximum at
yields the best clinical outcomes for the next patient, the performance of the design, in terms of frequentist operating characteristics, can be improved by including adaptive randomization (AR) among actions giving values of the objective function near the maximum at 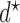 . Using AR decreases the probability of getting stuck at a suboptimal
. Using AR decreases the probability of getting stuck at a suboptimal 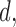 and also has the effect of treating more patients at doses having larger utilities, on average. The problem that a “greedy” search algorithm may get stuck at suboptimal actions, and the simple solution of introducing additional randomness into the search process, are well known in the optimization literature (cf. Tokic, 2010). This has been dealt with only very recently in dose-finding (Bartroff and Lai, 2010; Azriel and others, 2011; Braun and others, 2012; Thall and Nguyen, 2012).
and also has the effect of treating more patients at doses having larger utilities, on average. The problem that a “greedy” search algorithm may get stuck at suboptimal actions, and the simple solution of introducing additional randomness into the search process, are well known in the optimization literature (cf. Tokic, 2010). This has been dealt with only very recently in dose-finding (Bartroff and Lai, 2010; Azriel and others, 2011; Braun and others, 2012; Thall and Nguyen, 2012).
To implement AR, we first define 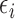 to be a function decreasing in patient index
to be a function decreasing in patient index 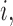 and denote
and denote 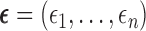 . We define the set of
. We define the set of 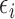 -optimal doses for cycle 1 to be
-optimal doses for cycle 1 to be
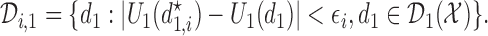 |
(3.1) |
The set, 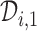 contains doses
contains doses 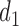 in
in 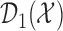 whose
whose 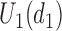 is within
is within 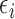 of the maximum posterior mean utility. Similarly, we define the set of
of the maximum posterior mean utility. Similarly, we define the set of 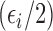 -optimal doses for cycle 2 given
-optimal doses for cycle 2 given 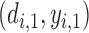 to be
to be
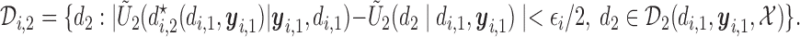 |
(3.2) |
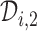 in (3.1) is based on (2.5). Our design randomizes patients uniformly among doses in
in (3.1) is based on (2.5). Our design randomizes patients uniformly among doses in 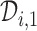 for
for 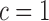 and
and 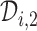 for
for 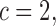 which we call AR(
which we call AR(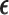 ). Numerical values of
). Numerical values of 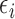 depend on the range of
depend on the range of 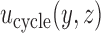 , and are determined by preliminary trial simulations in which
, and are determined by preliminary trial simulations in which 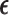 is varied.
is varied.
3.2. Illustrative trial
Our illustrative trial studied in the simulations is a stylized version of the phase I–II chemotherapy trial with five dose levels described in Section 1, but here accounting for two cycles of therapy. The maximum sample size is 60 patients with a cohort size of 2. Based on preliminary simulations, we set 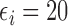 for the first 10 patients,
for the first 10 patients, 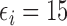 for the next 10 patients, and
for the next 10 patients, and 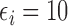 for the remaining 40 patients. An initial cohort of 2 patients is treated at the lowest dose level in cycle 1, their cycle 1 toxicity and efficacy outcomes are observed, the posterior of
for the remaining 40 patients. An initial cohort of 2 patients is treated at the lowest dose level in cycle 1, their cycle 1 toxicity and efficacy outcomes are observed, the posterior of 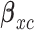 ,
, 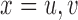 and
and 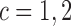 is computed, and actions are taken for cycle 2 of the initial cohort. If
is computed, and actions are taken for cycle 2 of the initial cohort. If 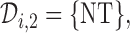 then patient
then patient 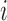 does not receive a second cycle of treatment. If
does not receive a second cycle of treatment. If 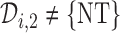 , then AR(
, then AR(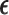 ) is used to choose an action for cycle 2 from
) is used to choose an action for cycle 2 from 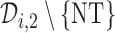 . When the toxicity and efficacy outcomes are observed from cycle 2, the posterior of
. When the toxicity and efficacy outcomes are observed from cycle 2, the posterior of 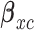 is updated. The second cohort is not enrolled until the first cohort has been evaluated for cycle 1. For all cohorts after the first, after the outcomes of all previous cohorts are observed, the posterior is updated, the posterior expected utility,
is updated. The second cohort is not enrolled until the first cohort has been evaluated for cycle 1. For all cohorts after the first, after the outcomes of all previous cohorts are observed, the posterior is updated, the posterior expected utility, 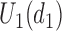 is computed using
is computed using 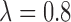 , and
, and 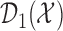 is determined. Using
is determined. Using 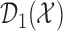 and
and 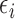 , we find
, we find 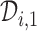 and search for
and search for 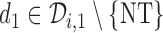 . If
. If 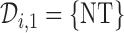 for any interim
for any interim 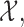 then
then 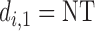 , and the trial is terminated. If
, and the trial is terminated. If 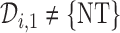 , we then choose a cycle 1 dose from
, we then choose a cycle 1 dose from 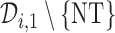 using AR(
using AR(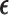 ). Once the outcomes in cycle 1 are observed, the posterior is updated. Using
). Once the outcomes in cycle 1 are observed, the posterior is updated. Using 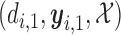 and
and 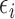 ,
, 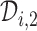 is searched. If
is searched. If 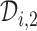 contains
contains  only, then
only, then 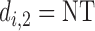 and no cycle 2 dose is given to patient
and no cycle 2 dose is given to patient 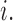 Otherwise,
Otherwise, 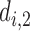 is selected from
is selected from 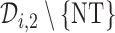 using AR(
using AR(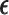 ). The toxicity and efficacy outcomes are observed from cycle 2 and the posterior of
). The toxicity and efficacy outcomes are observed from cycle 2 and the posterior of 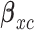 is updated. The above steps are repeated until either the trial has been stopped early or
is updated. The above steps are repeated until either the trial has been stopped early or 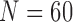 has been reached. At the end of the trial, we record
has been reached. At the end of the trial, we record 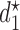 as recommended first cycle dose
as recommended first cycle dose 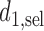 and
and 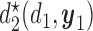 as optimal policy
as optimal policy 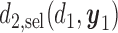 . If the trial is early terminated, let
. If the trial is early terminated, let 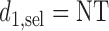 and
and 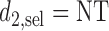 for all
for all 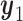 .
.
4. Simulation study
4.1. Designs for comparison
Let DTD-O2 denote the proposed decision-theoretic two-cycle design. We compare DTD-O2 with three other designs. The first is obtained by reducing each 3-level efficacy and toxicity outcome to a 2-category (binary) variable by combining categories, but using the same probability model to ensure a fair comparison. The next two comparators are single cycle designs. The first, called Single Cycle Comparator 1 (SCC1), assumes no association between cycles and optimizes 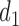 and
and 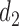 separately. The second, called Single Cycle Comparator 2 (SCC2), does not distinguish between cycles and treats the two cycles identically.
separately. The second, called Single Cycle Comparator 2 (SCC2), does not distinguish between cycles and treats the two cycles identically.
For SCC1, we assume patient-specific random probit scores, independent over cycles, 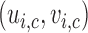
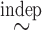
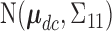 , where
, where 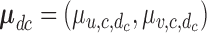 and
and 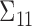 is the
is the 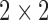 covariance matrix. We let
covariance matrix. We let 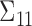 be the upper-left partition of
be the upper-left partition of 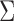 in (2.6). Owing to the independence of probit scores over cycles within a patient, SCC1 models the association between
in (2.6). Owing to the independence of probit scores over cycles within a patient, SCC1 models the association between 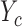 and
and 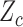 within the same cycle only and does not assume any association between outcomes in different cycles, for example,
within the same cycle only and does not assume any association between outcomes in different cycles, for example, 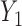 and
and 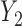 . The other model specification including the regression of
. The other model specification including the regression of 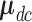 on the dose in Section 2.4 stays the same. For SCC2, in addition to having patient- and cycle-specific random probit scores as in SCC1, we assume that the mean dose effects are identical in the two cycles by dropping the cycle index from
on the dose in Section 2.4 stays the same. For SCC2, in addition to having patient- and cycle-specific random probit scores as in SCC1, we assume that the mean dose effects are identical in the two cycles by dropping the cycle index from 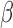 in Section 2.4, i.e. setting
in Section 2.4, i.e. setting 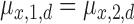 ,
, 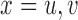 for all
for all 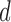 . For these two methods, we apply the acceptability rules in Section 2.3 and the AR rules in Section 3.1 for each cycle separately. For example, a trial is terminated if
. For these two methods, we apply the acceptability rules in Section 2.3 and the AR rules in Section 3.1 for each cycle separately. For example, a trial is terminated if 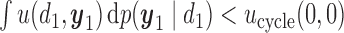 for all
for all 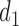 and
and 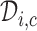 is defined with
is defined with 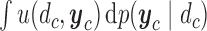 only. Also, the no-escalation rule after
only. Also, the no-escalation rule after  , no-skipping rule and AR similar to those implemented in the proposed method are implemented to SCC1 and SCC2.
, no-skipping rule and AR similar to those implemented in the proposed method are implemented to SCC1 and SCC2.
4.2. Simulation setup
We simulated trials under each of 8 scenarios using each of the designs. A total of 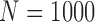 trials were simulated for each design under each scenario. The simulation scenarios were determined by fixing a set of marginal probabilities and regression coefficients on probit scores, given in Table 2 and Supplementary Material Table S1 (available at Biostatistics online). Each simulation scenario is specified by the marginal distributions of
trials were simulated for each design under each scenario. The simulation scenarios were determined by fixing a set of marginal probabilities and regression coefficients on probit scores, given in Table 2 and Supplementary Material Table S1 (available at Biostatistics online). Each simulation scenario is specified by the marginal distributions of  and
and 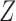 . Table 2 gives the true
. Table 2 gives the true 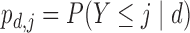 and
and 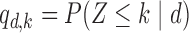 under each scenario. The corresponding probit scores are
under each scenario. The corresponding probit scores are 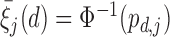 and
and 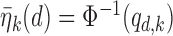 , where
, where 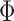 is the cumulative distribution function of the standard normal distribution. To ensure a fair comparison, we intentionally define a simulation truth that is different from the assumed model used by the design methodology. The simulation model is best described as a generative model, first for
is the cumulative distribution function of the standard normal distribution. To ensure a fair comparison, we intentionally define a simulation truth that is different from the assumed model used by the design methodology. The simulation model is best described as a generative model, first for 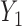 , then
, then 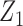 given
given 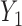 , and then
, and then 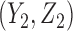 given
given 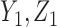 .
.
Table 2.
Assumed probabilities, 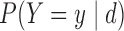 and
and 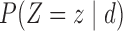 . These marginal probabilities are used to determine probit scores,
. These marginal probabilities are used to determine probit scores, 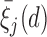 and
and 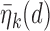
| Toxicity outcome |
Efficacy outcome |
||||||
|---|---|---|---|---|---|---|---|
| _______________________________ |
__________________________ |
||||||
| Scenarios | Dose | Mild | Moderate | Severe | PD | SD | PR/CR |
| 1, 2 | 1 | 0.23 | 0.52 | 0.25 | 0.44 | 0.44 | 0.12 |
| 2 | 0.225 | 0.515 | 0.26 | 0.35 | 0.42 | 0.23 | |
| 3, 4 | 3 | 0.20 | 0.530 | 0.27 | 0.18 | 0.40 | 0.42 |
| 4 | 0.18 | 0.40 | 0.42 | 0.10 | 0.445 | 0.455 | |
| 5 | 0.06 | 0.20 | 0.74 | 0.08 | 0.45 | 0.47 | |
| 5 | 1 | 0.53 | 0.39 | 0.08 | 0.35 | 0.515 | 0.135 |
| 2 | 0.38 | 0.47 | 0.15 | 0.325 | 0.52 | 0.155 | |
| 3 | 0.33 | 0.46 | 0.21 | 0.31 | 0.528 | 0.162 | |
| 4 | 0.315 | 0.455 | 0.23 | 0.225 | 0.505 | 0.27 | |
| 5 | 0.375 | 0.375 | 0.25 | 0.05 | 0.39 | 0.56 | |
| 6 | 1 | 0.55 | 0.30 | 0.15 | 0.51 | 0.31 | 0.18 |
| 2 | 0.475 | 0.31 | 0.215 | 0.45 | 0.275 | 0.275 | |
| 3 | 0.45 | 0.31 | 0.24 | 0.18 | 0.39 | 0.43 | |
| 4 | 0.44 | 0.31 | 0.25 | 0.15 | 0.40 | 0.45 | |
| 5 | 0.43 | 0.30 | 0.27 | 0.03 | 0.27 | 0.70 | |
| 7 | 1 | 0.65 | 0.20 | 0.15 | 0.18 | 0.33 | 0.49 |
| 2 | 0.52 | 0.20 | 0.28 | 0.175 | 0.325 | 0.50 | |
| 3 | 0.46 | 0.21 | 0.33 | 0.15 | 0.30 | 0.55 | |
| 4 | 0.37 | 0.27 | 0.36 | 0.125 | 0.25 | 0.625 | |
| 5 | 0.28 | 0.28 | 0.44 | 0.11 | 0.24 | 0.65 | |
| 8 | 1 | 0.19 | 0.43 | 0.38 | 0.85 | 0.12 | 0.03 |
| 2 | 0.13 | 0.22 | 0.65 | 0.78 | 0.14 | 0.08 | |
| 3 | 0.09 | 0.22 | 0.69 | 0.54 | 0.31 | 0.15 | |
| 4 | 0.03 | 0.23 | 0.74 | 0.43 | 0.39 | 0.18 | |
| 5 | 0.01 | 0.13 | 0.86 | 0.38 | 0.41 | 0.21 | |
Generating 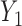 : We first generate
: We first generate 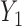 from the distribution specified by
from the distribution specified by 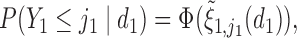 where
where 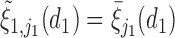 . For later reference, we define a rescaled variable
. For later reference, we define a rescaled variable 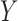 as
as 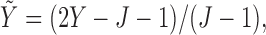 which is evenly spaced in
which is evenly spaced in 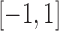 .
.
Generating 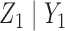 : Conditional on
: Conditional on 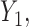 we specify a distribution of
we specify a distribution of 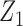 by letting
by letting
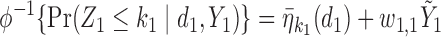 |
with coefficient 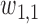 . Here,
. Here, 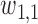 induces association between the cycle 1 outcomes,
induces association between the cycle 1 outcomes, 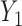 and
and 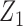 . A negative value of
. A negative value of 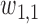 leads to a negative association between
leads to a negative association between 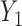 and
and 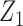 , that is,
, that is, 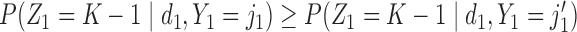 ,
, 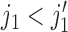 . For later use, we define
. For later use, we define 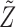 by rescaling
by rescaling 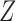 to be evenly spaced in
to be evenly spaced in 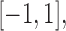 similarly to
similarly to 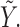
Generating
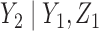 : We generate
: We generate 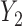 using
using
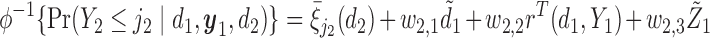 |
Here, 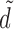 is a standardized dose in
is a standardized dose in 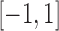 . We restrict
. We restrict 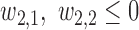 and
and 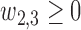 to induce a positive association of
to induce a positive association of 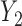 with
with 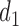 and
and 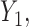 and negative association with
and negative association with 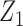 . Here,
. Here, 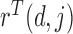 determines how
determines how 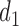 and
and 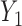 jointly affect
jointly affect 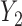 . A large negative value of
. A large negative value of 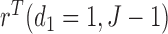 implies that given that
implies that given that 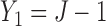 (severe toxicity) is observed at
(severe toxicity) is observed at 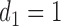 , the probability of observing
, the probability of observing 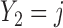 ,
, 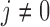 greatly increases for all
greatly increases for all 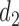 . Similarly, observing
. Similarly, observing 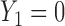 (mild toxicity) at
(mild toxicity) at 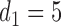 greatly increases the probability of observing
greatly increases the probability of observing 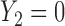 for all
for all 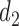 , implying a large positive value of
, implying a large positive value of 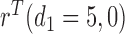 .
.
Generating 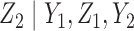 : We use
: We use
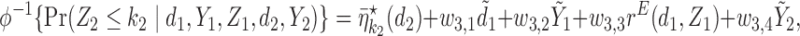 |
where 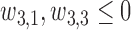 and
and 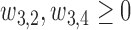 . Similar to
. Similar to 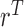 ,
, 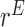 determines a joint effect of
determines a joint effect of 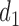 and
and 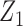 on
on 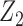 . The detailed specification of the coefficients,
. The detailed specification of the coefficients, 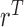 and
and 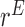 for each simulation scenario is described in the Supplementary Materials (available at Biostatistics online). Table 3 shows the optimal actions,
for each simulation scenario is described in the Supplementary Materials (available at Biostatistics online). Table 3 shows the optimal actions, 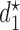 and
and 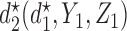 , over two cycles under each of the 8 simulation scenarios under the simulation truth. For example, in Scenario 3, the optimal cycle 1 action is to give dose level 3, and the optimal cycle 2 action is to treat patients with
, over two cycles under each of the 8 simulation scenarios under the simulation truth. For example, in Scenario 3, the optimal cycle 1 action is to give dose level 3, and the optimal cycle 2 action is to treat patients with 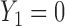 at
at 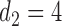 , and at
, and at 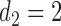 if
if 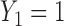 .
.
Table 3.
True optimal actions, 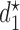 and
and 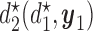
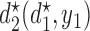 |
|||||
|---|---|---|---|---|---|
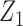 |
|||||
| Scenarios | 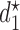 |
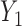 |
0 | 1 | 2 |
| 1 | 3 | 0 | 3 | 3 | 3 |
| 1 | 3 | 3 | 3 | ||
| 2 | 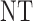 |
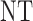 |
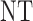 |
||
| 2 | 3 | 0 | 3 | 3 | 3 |
| 1 | 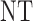 |
3 | 3 | ||
| 2 | 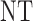 |
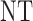 |
3 | ||
| 3 | 3 | 0 | 3 | 3 | 2 |
| 1 | 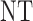 |
3 | 3 | ||
| 2 | 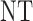 |
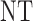 |
3 | ||
| 4 | 3 | 0 |  |
3 | 3 |
| 1 | 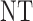 |
3 | 3 | ||
| 2 | 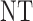 |
3 | 3 | ||
| 5 | 3 | 0 | 5 | 5 | 5 |
| 1 | 5 | 5 | 5 | ||
| 2 | 5 | 5 | 5 | ||
| 6 | 1 | 0 | 5 | 5 | 5 |
| 1 | 5 | 5 | 5 | ||
| 2 | 5 | 5 | 5 | ||
| 7 | 5 | 0 | 1 | 1 | 1 |
| 1 | 1 | 1 | 1 | ||
| 2 | 1 | 1 | 1 | ||
| 8 | 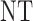 |
0 | 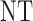 |
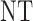 |
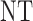 |
| 1 | 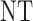 |
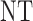 |
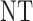 |
||
| 2 | 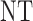 |
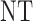 |
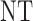 |
||
We calibrate the fixed hyperparameters, 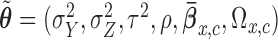 , for
, for 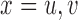 and
and 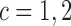 and the cutoff points,
and the cutoff points, 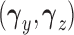 , using effective sample size (ESS), described in the Supplementary Materials (available at Biostatistics online). We set
, using effective sample size (ESS), described in the Supplementary Materials (available at Biostatistics online). We set 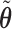 and the cutoffs,
and the cutoffs, 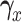 and
and 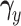 , and simulate 1000 pseudo-samples of
, and simulate 1000 pseudo-samples of 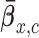 ,
, 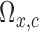 ,
, 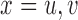 and
and 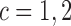 . We then compute probabilities of interest based on the pseudo-samples, such as
. We then compute probabilities of interest based on the pseudo-samples, such as 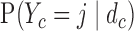 and
and 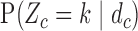 ,
,  . For all simulations, we determined
. For all simulations, we determined 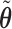 to give each prior ESS between 0.5 and 2, using the approximation obtained by matching moments with a Dirichlet distribution. We used the same
to give each prior ESS between 0.5 and 2, using the approximation obtained by matching moments with a Dirichlet distribution. We used the same 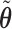 for SCC1 and SCC2.
for SCC1 and SCC2.
4.3. Evaluation criteria
We evaluate design performance for the patients treated in the trial using three different summary statistics, 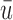 ,
, 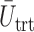 , and
, and 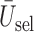 . Recall that in a trial we record the clinical outcomes of the
. Recall that in a trial we record the clinical outcomes of the 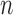 patients with their assigned doses and recommended doses for future patients,
patients with their assigned doses and recommended doses for future patients, 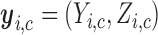 ,
, 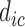 ,
, 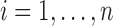 and
and 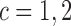 , and
, and 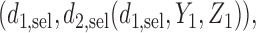 respectively. We index the
respectively. We index the 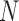 simulated replications of the trial by
simulated replications of the trial by 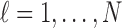 . We define average utility for the
. We define average utility for the 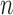 patients in the
patients in the 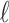 th simulated trial in two different ways;
th simulated trial in two different ways; 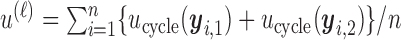 and
and 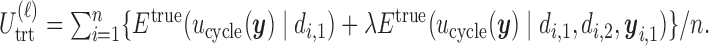 Note that
Note that 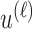 is a function only of occurred outcomes,
is a function only of occurred outcomes, 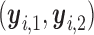 , whereas
, whereas 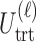 depends on the true utilities of assigned doses
depends on the true utilities of assigned doses 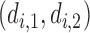 . For
. For 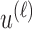 and
and 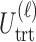 ,
, 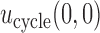 is used as the utility for patients with
is used as the utility for patients with 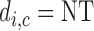 . The empirical mean total payoffs taken over all simulated trials are
. The empirical mean total payoffs taken over all simulated trials are
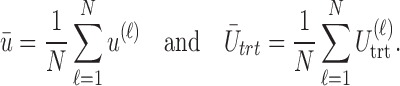 |
One may regard 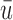 and
and 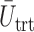 as indexes of the ethical desirability of the method, given
as indexes of the ethical desirability of the method, given 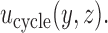
The proposed method gives an optimal action 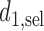 for cycle 1, and policy
for cycle 1, and policy 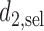 for cycle 2. We let
for cycle 2. We let 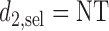 for all
for all 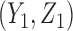 if
if  so the trial is terminated early. We use
so the trial is terminated early. We use 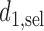 and
and 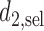 to evaluate performance in terms of future patient benefit. Under SCC1 and SCC2,
to evaluate performance in terms of future patient benefit. Under SCC1 and SCC2,  is not a function of
is not a function of 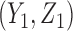 . For SCC2,
. For SCC2, 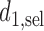 and
and 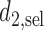 are identical. Assuming that the simulation truth is known, we define the expected payoff in cycle 1 of giving action
are identical. Assuming that the simulation truth is known, we define the expected payoff in cycle 1 of giving action 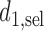 to a future patient as
to a future patient as 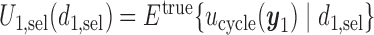 for
for 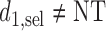 . That is the expected utility with respect to the assumed distribution of
. That is the expected utility with respect to the assumed distribution of 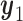 when
when 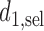 is given. For
is given. For 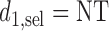 , let
, let 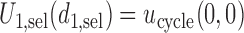 . This expectation is computed under the distribution of
. This expectation is computed under the distribution of 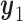 given
given 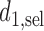 . If the rule
. If the rule 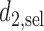 is used, the expected cycle 2 payoff is
is used, the expected cycle 2 payoff is
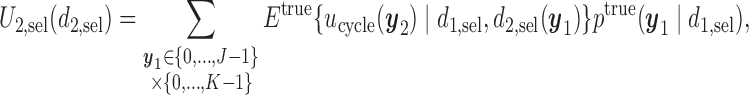 |
where 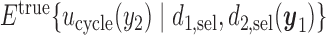 becomes
becomes 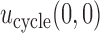 if
if 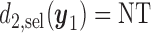 . The total expected payoff to a future patient treated using the optimal regime
. The total expected payoff to a future patient treated using the optimal regime 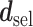 =
= 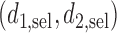 is defined to be
is defined to be 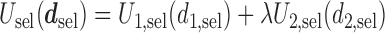 .
.
4.4. Comparison to designs with binary outcomes
We first compare DTD-O2 with designs obtained by collapsing each trinary toxicity and efficacy outcome to a binary variable. This mimics what often is done in practice in order to apply a phase I–II design based on binary efficacy and toxicity. We use an appropriately reduced version of our assumed underlying model to ensure a fair comparison. Since this reduction is not unique, we exhaustively define binary outcomes in four different ways, binary cases 1–4, given in Section 4 of the Supplementary Material (available at Biostatistics online). The utilities associated with the binary outcomes are defined accordingly based on the utilities in Table 1. The results, in terms of 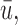
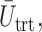 and
and 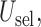 are summarized graphically in Figure 2. Scenario 8 is not included in Figure 2 because the optimal action is
are summarized graphically in Figure 2. Scenario 8 is not included in Figure 2 because the optimal action is 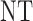 in both cycles, and in this case all designs stop the trial early with high probability, The figure shows that reducing to binary outcomes can produce designs with much worse performance than DTD-O2, while for some cases the performance may be comparable. The binary outcome design's performance also varies substantially with the particular dichotomization used. Since different physicians may combine ordinal categories in different ways, the practical implication is that the additional complexity of the ordinal outcome design is worthwhile, in terms of benefit to both the patients treated in the trial and future patients.
in both cycles, and in this case all designs stop the trial early with high probability, The figure shows that reducing to binary outcomes can produce designs with much worse performance than DTD-O2, while for some cases the performance may be comparable. The binary outcome design's performance also varies substantially with the particular dichotomization used. Since different physicians may combine ordinal categories in different ways, the practical implication is that the additional complexity of the ordinal outcome design is worthwhile, in terms of benefit to both the patients treated in the trial and future patients.
4.5. Comparison to single cycle designs
The simulation results for DTD-O2, SCC1, and SCC2 are summarized in Figure 3. Scenarios 1–4 have the same marginal toxicity and efficacy probabilities, but different values of coefficients (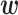 ), yielding different probit scores and different association structures of
), yielding different probit scores and different association structures of 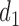 ,
, 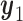 ,
, 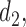 and
and 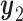 . Scenario 1 has large
. Scenario 1 has large 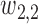 and
and 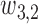 , so that the cycle 1 toxicity outcome greatly affects cycle 2 expected utilities in the simulation truth. As shown in Table 3, the optimal action in cycle 2 after observing severe toxicity in cycle 1 is
, so that the cycle 1 toxicity outcome greatly affects cycle 2 expected utilities in the simulation truth. As shown in Table 3, the optimal action in cycle 2 after observing severe toxicity in cycle 1 is 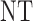 regardless of the cycle 1 efficacy outcome. Scenario 4 is similar to Scenario 1 but the cycle 1 efficacy outcome heavily affects the cycle 2 treatment in that all cycle 2 treatments are less desirable than
regardless of the cycle 1 efficacy outcome. Scenario 4 is similar to Scenario 1 but the cycle 1 efficacy outcome heavily affects the cycle 2 treatment in that all cycle 2 treatments are less desirable than 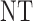 when PD is observed in cycle 1. In Scenarios 2 and 3, the two cycle 1 outcomes jointly determine the cycle 2 treatment as shown in the tables. Scenario 3 has larger association between
when PD is observed in cycle 1. In Scenarios 2 and 3, the two cycle 1 outcomes jointly determine the cycle 2 treatment as shown in the tables. Scenario 3 has larger association between 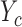 and
and 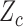 within each cycle. In Scenarios 1–4, modeling dependence across cycles improves the performance, as shown in Figure 3, where DTD-O2 is superior to SCC1 and SCC2 in terms of all the three criteria,
within each cycle. In Scenarios 1–4, modeling dependence across cycles improves the performance, as shown in Figure 3, where DTD-O2 is superior to SCC1 and SCC2 in terms of all the three criteria, 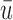 ,
, 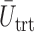 , and
, and 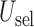 . Since the only difference between DTD-O2 and SCC1 is whether the two cycles are modeled jointly or separately, the results show that the joint modeling significantly improves the performance. Differences in the performance are smaller for Scenarios 1 and 4. This may be because the true structure that one cycle 1 outcome dominates cycle 2 decisions in the scenarios is not easily accommodated under the assumed covariance structure in (2.6) and each trial gets only a small number of patients. In such a case, separate estimation for the two cycles may not be a very poor approach. In addition, the three methods are compared using
. Since the only difference between DTD-O2 and SCC1 is whether the two cycles are modeled jointly or separately, the results show that the joint modeling significantly improves the performance. Differences in the performance are smaller for Scenarios 1 and 4. This may be because the true structure that one cycle 1 outcome dominates cycle 2 decisions in the scenarios is not easily accommodated under the assumed covariance structure in (2.6) and each trial gets only a small number of patients. In such a case, separate estimation for the two cycles may not be a very poor approach. In addition, the three methods are compared using 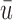 and
and 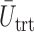 based on the last 20 patients in each trial for the three designs (not shown). This comparison shows that the improvement by DTD-O2 over the other two methods becomes greater, especially for Scenarios 1 and 4. It may imply that learning takes more patients for DTD-O2 when there is a discrepancy between the truth and the model assumption.
based on the last 20 patients in each trial for the three designs (not shown). This comparison shows that the improvement by DTD-O2 over the other two methods becomes greater, especially for Scenarios 1 and 4. It may imply that learning takes more patients for DTD-O2 when there is a discrepancy between the truth and the model assumption.
Fig. 3.

Plot of 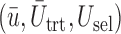 for a comparison with SCC1 and SCC2. Here,
for a comparison with SCC1 and SCC2. Here, 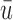 ,
, 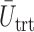 , and
, and 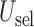 represent empirical mean utilities of patients treated in the trial, true mean utilities of treatments given to patients in the trial, and true expected utilities chosen for future patients, respectively. (a)
represent empirical mean utilities of patients treated in the trial, true mean utilities of treatments given to patients in the trial, and true expected utilities chosen for future patients, respectively. (a) 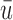 . (b)
. (b) 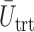 . (c)
. (c) 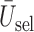 .
.
Scenarios 5–7 have different shapes for 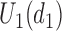 as a function of
as a function of 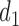 . The cycle 1 utilities are U-shaped in Scenario 5, monotone increasing in Scenario 6, and monotone decreasing in Scenario 7. Very mild associations between outcomes and between cycles are assumed for these scenarios. For Scenarios 5 and 6, DTD-O2 achieves notably better performance (see Figure 3), with
. The cycle 1 utilities are U-shaped in Scenario 5, monotone increasing in Scenario 6, and monotone decreasing in Scenario 7. Very mild associations between outcomes and between cycles are assumed for these scenarios. For Scenarios 5 and 6, DTD-O2 achieves notably better performance (see Figure 3), with 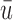 and
and 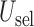 similar to each other for DTD-O2. This implies that DTD-O2 identifies desirable actions early in the trial, treats many of the patients with the desirable actions, and has a high probability of selecting truly optimal actions at the end of a trial. In Scenario 7, DTD-O2 shows slightly worse performance (see the rightmost of Figure 3). In the simulation truth of Scenario 7, the cycle 1 expected utility does not change much with
similar to each other for DTD-O2. This implies that DTD-O2 identifies desirable actions early in the trial, treats many of the patients with the desirable actions, and has a high probability of selecting truly optimal actions at the end of a trial. In Scenario 7, DTD-O2 shows slightly worse performance (see the rightmost of Figure 3). In the simulation truth of Scenario 7, the cycle 1 expected utility does not change much with 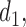 but the cycle 2 expected utility is very sensitive to
but the cycle 2 expected utility is very sensitive to 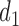 ,
, 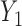 , and
, and 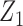 . This is a very challenging case for DTD-O2, and not modeling dependence between the cycles leads to better performance than incorrectly modeling in this particular scenario. Scenario 8 has no acceptable dose in either cycle. All the three methods terminate the trials with probability 1 in this case, with mean sample sizes 9.11, 8.33, and 8.29.
. This is a very challenging case for DTD-O2, and not modeling dependence between the cycles leads to better performance than incorrectly modeling in this particular scenario. Scenario 8 has no acceptable dose in either cycle. All the three methods terminate the trials with probability 1 in this case, with mean sample sizes 9.11, 8.33, and 8.29.
In all 8 scenarios, SCC2 yields better results than SCC1. This may be because 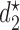 and
and 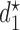 happen to be identical in many cases, so combining outcomes from the two cycles works well. However, the results for Scenarios 1–4 show that using each patient's cycle 1 dose and outcomes to select
happen to be identical in many cases, so combining outcomes from the two cycles works well. However, the results for Scenarios 1–4 show that using each patient's cycle 1 dose and outcomes to select 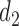 gives significantly superior performance in cases where there is significant dependence between the two cycles. More results are summarized using empirical toxicity and efficacy probabilities in Section 3 of Supplementary Material (available at Biostatistics online).
gives significantly superior performance in cases where there is significant dependence between the two cycles. More results are summarized using empirical toxicity and efficacy probabilities in Section 3 of Supplementary Material (available at Biostatistics online).
We carried out a sensitivity analysis in 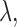 under Scenarios 2 and 5, including the four binary outcome designs, SCC1, SCC2, and DTD-O2, for
under Scenarios 2 and 5, including the four binary outcome designs, SCC1, SCC2, and DTD-O2, for 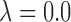 , 0.4, 0.8, and 1.0. The results, given in Section 5 of Supplementary Material (available at Biostatistics online), show that changes in design performance with
, 0.4, 0.8, and 1.0. The results, given in Section 5 of Supplementary Material (available at Biostatistics online), show that changes in design performance with 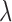 are very small, but
are very small, but 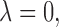 corresponding to no use of cycle 2 utility in making a decision at cycle 1, yields higher early termination probabilities for binary outcome cases 1 and 3.
corresponding to no use of cycle 2 utility in making a decision at cycle 1, yields higher early termination probabilities for binary outcome cases 1 and 3.
5. Discussion
We have extended the decision-theoretic two-cycle phase I–II dose-finding method in Lee and others (2015) to accommodate ordinal outcomes. Our simulations show that incorporating cycle 1 information into the cycle 2 treatment decision yields good performance for both patients treated in a trial and future patients. The simulations in Figure 2 show that this extension may greatly improve design performance, quantified by 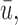
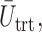 and
and 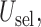 compared with using binary toxicity and efficacy indicators. The proposed model and method also compared quite favorably with either assuming the two cycles are independent or ignoring the distinction between cycles 1 and 2.
compared with using binary toxicity and efficacy indicators. The proposed model and method also compared quite favorably with either assuming the two cycles are independent or ignoring the distinction between cycles 1 and 2.
In theory, DTD-O2 could be extended to more than two cycles. For this to be tractable, additional modeling assumptions may required to control the number of parameters, since decisions must be made based on small sample sizes. Two possible approaches are to model dependence among cycles as a function of distance between cycles, or to make a Markovian assumption.
Supplementary material
Supplementary material is available at http://biostatistics.oxfordjournals.org.
Funding
Y.J. research is supported in part by NIH R01 CA132897. P.F.T. research was supported in part by NIH R01 CA 83932. P.M. research was supported in part by NIH 1-R01-CA157458-01A1. This research was supported in part by NIH through resources provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory, under grant S10 RR029030-01.
Supplementary Material
Acknowledgements
We specifically acknowledge the assistance of Lorenzo Pesce (University of Chicago). Conflict of Interest: None declared.
References
- Azriel D., Mandel M., Rinott Y. (2011). The treatment versus experimentation dilemma in dose finding studies. Journal of Statistical Planning and Inference 1418, 2759–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartroff J., Lai T. L. (2010). Approximate dynamic programming and its applications to the design of phase I cancer trials. Statistical Science 255, 245–257. [Google Scholar]
- Bellman R. (1957) Dynamic Programming, 1 edition Princeton, NJ, USA: Princeton University Press. [Google Scholar]
- Braun T. M., Kang S., Taylor J. M. G. (2012). A phase I/II trial design when response is unobserved in subjects with dose-limiting toxicity. Statistical Methods in Medical Research. 0962280212464541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretz F., Pinheiro J. C., Branson M. (2005). Combining multiple comparisons and modeling techniques in dose–response studies. Biometrics 613, 738–748. [DOI] [PubMed] [Google Scholar]
- Cheung Y. K., Chakraborty B., Davidson K. W. (2014). Sequential multiple assignment randomized trial (smart) with adaptive randomization for quality improvement in depression treatment program. Biometrics 712, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iasonos A., Zohar S., O'Quigley J. (2011). Incorporating lower grade toxicity information into dose finding designs. Clinical Trials 84, 370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Thall P. F., Ji Y., Müller P. (2015). Bayesian dose-finding in two treatment cycles based on the joint utility of efficacy and toxicity. Journal of the American Statistical Association 110510, 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. A. (2003). Optimal dynamic treatment regimes. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 652, 331–355. [Google Scholar]
- Parmigiani G., Inoue L. (2009) Decision Theory: Principles and Approaches. New York: Wiley. [Google Scholar]
- Robert C. P. (2007) The Bayesian Choice: From Decision-Theoretic Foundations to Computational Implementation, 2nd edition Berlin: Springer. [Google Scholar]
- Thall P. F., Cook J. D. (2004). Dose-finding based on efficacy–toxicity trade-offs. Biometrics 603, 684–693. [DOI] [PubMed] [Google Scholar]
- Thall P. F, Nguyen H. Q. (2012). Adaptive randomization to improve utility-based dose-finding with bivariate ordinal outcomes. Journal of Biopharmaceutical Statistics 224, 785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokic M. (2010). Adaptive
 -greedy exploration in reinforcement learning based on value differences. In: KI 2010: Advances in Artificial Intelligence. Berlin: Springer, pp. 203–210.
-greedy exploration in reinforcement learning based on value differences. In: KI 2010: Advances in Artificial Intelligence. Berlin: Springer, pp. 203–210. - Van Meter E. M., Garrett-Mayer E., Bandyopadhyay D. (2012). Dose-finding clinical trial design for ordinal toxicity grades using the continuation ratio model: an extension of the continual reassessment method. Clinical Trials 93, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G., Li Y., Ji Y. (2006). Bayesian dose-finding in phase i/ii clinical trials using toxicity and efficacy odds ratios. Biometrics 623, 777–787. [DOI] [PubMed] [Google Scholar]
- Zhang W., Sargent D. J., Mandrekar S. (2006). An adaptive dose-finding design incorporating both toxicity and efficacy. Statistics in Medicine 2514, 2365–2383. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Zeng D., Socinski M. A., Kosorok M. R. (2011). Reinforcement learning strategies for clinical trials in nonsmall cell lung cancer. Biometrics 674, 1422–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.