

Abstract
Introduction
Many therapeutics are limited to parenteral administration. Oral administration is a desirable alternative because of the convenience and increased compliance by patients, especially for chronic diseases that require frequent administration. Polymeric nanoparticles are one technology being developed to enable clinically feasible oral delivery.
Areas covered
This review discusses the challenges associated with oral delivery. Strategies used to overcome gastrointestinal barriers using polymeric nanoparticles will be considered, including mucoadhesive biomaterials and targeting of nanoparticles to transcytosis pathways associated with M cells and enterocytes. Applications of oral delivery technologies will also be discussed, such as oral chemotherapies, oral insulin, treatment of inflammatory bowel disease, and mucosal vaccinations.
Expert opinion
There have been many approaches used to overcome the transport barriers presented by the gastrointestinal tract, but most have been limited by low bioavailability. Recent strategies targeting nanoparticles to transcytosis pathways present in the intestines have demonstrated that it is feasible to efficiently transport both therapeutics and nanoparticles across the intestines and into systemic circulation after oral administration. Further understanding of the physiology and pathophysiology of the intestines could lead to additional improvements in oral polymeric nanoparticle technologies and enable the translation of these technologies to clinical practice.
Keywords: chemotherapy, inflammatory bowel disease, insulin, M cells, mucoadhesives, mucosal vaccinations, oral delivery, polymeric nanoparticles, transcytosis
1. Introduction
1.1 Motivation for Oral Delivery
Over the past 50 years, there has been an explosion in biomedical research that has led to a remarkable understanding of the pathophysiology associated with many diseases. Armed with this knowledge, many new therapies are being developed that could have a significant impact on disease treatment and patient outcomes. Many of these new therapies fall into a pharmaceutical class known as biologics, which include peptide hormones, antibodies, growth factors, enzymes, vaccines, and nucleic acids. In fact, more than 100 biologics have been developed over the past 30 years to treat diseases ranging from cancer and diabetes to rare genetic disorders, generating $64 billion in the US market in 2012 [1]. Furthermore, combining biological understanding with engineering and materials science principles has led to the development of nanoparticle (NP)-based therapeutics, or nanomedicines. This emerging class of therapeutics is now entering clinical trials [2–4], and could have a significant impact of the treatment of many diseases [5,6].
Many diseases that would benefit from biologics or nanomedicines are chronic, requiring frequent treatments over prolonged periods of time. Unfortunately, these therapeutics are currently restricted to parenteral administration methods, which could limit their use. Injection-based therapies can suffer from poor patient compliance and reduced efficacy due to the pain and inconvenience associated with treatment regimens. Therefore, alternate routes of administration, such as transdermal, nasal, buccal, pulmonary, and oral, are under investigation as a means to improve these therapies. Of these alternate routes, oral is considered the most desirable, especially for chronic diseases, because of the convenience and improved compliance [7–11]. In clinical studies, oral administration of chemotherapy is not only more convenient for patients, it has also been shown in some cases to have less drug-related adverse effects due to favorable pharmacokinetics [12]. In addition, oral formulations have advantages for physicians such as flexible dosing schedules, less demands on staff, and reduced costs through less hospital or clinic visits [13–17].
1.2 Physiology of the Gastrointestinal Tract
Oral delivery technologies can be used to treat diseases of the gastrointestinal (GI) tract locally or to enable therapeutics to reach systemic circulation. For therapeutics to reach the bloodstream after oral administration, they must pass through the gastrointestinal (GI) tract and be absorbed. The small intestine plays the primary role in regulating the absorption of material (Figure 1). It has several features specific for this role, with macroscopic folds on the inner surface and microscopic finger-like projections called villi that significantly increase the absorption surface area [18]. The villi epithelium is a polarized cell monolayer that acts as a barrier to tightly regulate the transport of material from the external environment (intestinal lumen) to the lamina propria [19]. The epithelial cells rest on top of an extracellular matrix called the basal lamina that divides the epithelium from the lamina propria. The lamina propria within each villus contains a network of capillaries for absorption of molecules transported across the epithelial barrier and a blunt-ended lymphatic vessel called a lacteal for absorption of larger particles such as fats. The capillaries converge into venules and, eventually, the portal vein that transports material to the liver. The lymphatic vessels carry material through the lymph nodes before entering into the bloodstream through the thoracic duct.
Figure 1.
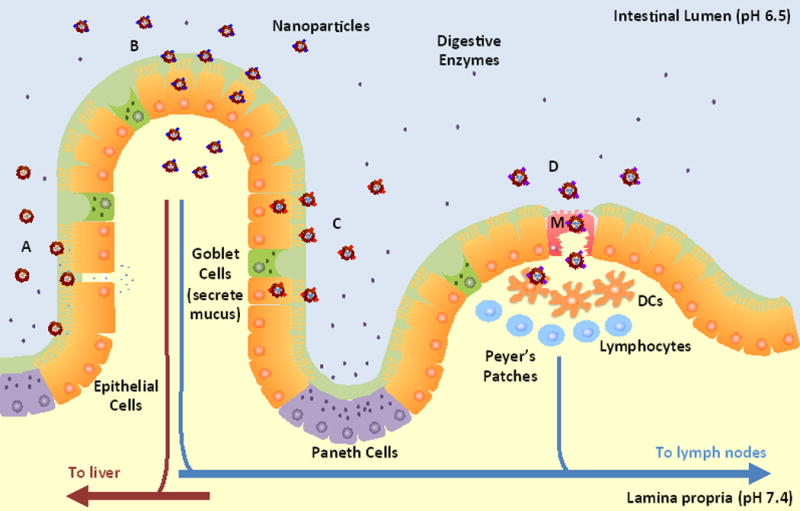
Schematic illustration of the intestinal epithelium and NP strategies for oral delivery. The epithelium includes absorptive epithelial cells, goblet cells that secrete mucus, and Paneth cells that secrete lysozyme and antimicrobial peptides. A Peyer’s Patch is shown with an M cell (M) and underlying immune cells such as dendritic cells (DCs) and lymphocytes. The barriers to oral delivery include acidic pH and enzymes present in the intestinal lumen, a mucus layer above the epithelial cells, the epithelial monolayer with restricted permeability due to tight junctions, and immune cells associated with the M cells. Strategies for oral delivery include: (A) NPs formed from mucoadhesive materials that adhere to the mucus layer and release drugs near the epithelial cells as well as reversibly open tight junctions for paracellular transport of therapeutics; (B) Receptor-mediated transcytosis of targeted NPs that enable NPs to cross the epithelium and reach the lamina propria; (C) Receptor-mediated uptake of targeted NPs for local drug delivery to epithelial cells instead of systemic delivery; (D) M cell transcytosis pathway that allows NPs to cross the epithelium, but with the potential for underlying DCs associated with Peyer’s Patches to endocytose NPs after crossing.
The intestinal epithelium consists of different cell types such as absorptive epithelial cells (or enterocytes), mucin-secreting goblet cells, endocrine cells, and Paneth cells that secrete lysozyme and antimicrobial peptides [18]. The enterocytes secrete digestive enzymes and have hair-like projections on the apical membrane called microvilli that further increase the absorptive surface area up to 300–400 m2 [20]. Another cell type, M cells, are only associated with Peyer’s Patches, which are organized components of the gut-associated lymphoid tissue (GALT). The role of M cells in the intestinal epithelium is to transport antigens through a non-degradative pathway to dendritic cells in the Peyer’s Patches [21].
1.3 Challenges of Oral Delivery
The challenge of oral delivery is a result of the obstacles presented by the GI tract. These obstacles include exposure to a wide range of pH environments, enzymatic degradation, and poor permeability across the intestinal epithelium. The pH in the GI tract can vary from 1 in the stomach to 8 in parts of the intestine [22]. Exposure to these pH values can result in pH-induced oxidation, deamidation, or hydrolysis of protein therapeutics, resulting in a loss of activity [23]. Enzymatic degradation is caused by proteases, nucleases, and lipases present in the GI tract for digestion of biological molecules prior to absorption [24]. If these obstacles are overcome, the therapeutic must then reach the epithelial barrier for absorption. The obstacles presented by the epithelium consist of extrinsic and intrinsic barriers [25].
The extrinsic barrier consists of the mucus layer covering the epithelial cells. The mucus layer is a complex hydrogel material composed of proteins, carbohydrates, lipids, salts, antibodies, bacteria, and cellular debris [26,27]. It consists of loosely and firmly adherent layers that vary in thickness along the GI tract and can fluctuate based on diet [28]. The mucus layer serves several roles. It aids in digestion by lubricating food particles to facilitate transport along the GI tract. It also protects epithelial surfaces by trapping pathogens and foreign particulates and rapidly clearing them [29]. Penetration of this mucus barrier is necessary in order to reach the absorptive epithelial cells.
The intrinsic barrier consists of the epithelial cell monolayer, which is the major transport barrier for material from the intestinal lumen to the lamina propria. Cells maintain this barrier by forming tight junctions, which are fusions between lateral membranes of adjacent cells [30,31]. Through specific combinations of different proteins, the permeability of the tight junctions can be modulated [32,33]. Intestinal cells also have metabolic systems and P-glycoprotein (P-gp) drug efflux pumps that can cause low bioavailability for many small molecule therapeutics such as chemotherapeutic agents [34].
For NPs, an additional obstacle is the immune system, which is intimately associated with the epithelium. Numerous types of immune cells patrol the lamina propria, including T cells, macrophages, and dendritic cells [21]. If NPs reach the bloodstream, they must also evade the mononuclear phagocytic system (MPS) while releasing their therapeutic cargo.
1.4 Intestinal Transport Pathways
There are several pathways across the epithelial barrier that could be used for oral delivery [19] (Figure 2). The transcellular pathway passes through the apical and basolateral cell membranes as well as the cytoplasm. This pathway is very restrictive to the passive flow of hydrophilic solutes because of the lipid bilayer membrane and is impermeable for large molecules. Mechanisms of transport for this pathway can be passive for hydrophobic molecules or active if a membrane pump is present for specific molecules such as ions. The paracellular pathway is the major passive permeation pathway and allows diffusion of small molecules in the space between epithelial cells. The tight junctions regulate the permeability of this pathway based on the size and charge of the molecules [31,35]. Transcytosis is an active transport pathway that relies on receptors specific for a molecule to guide the molecule through the cell without entering a degradation pathway. Because of their large hydrodynamic size, macromolecules, such as biologics, and NPs are restricted to this pathway for transport across the cellular barrier.
Figure 2.
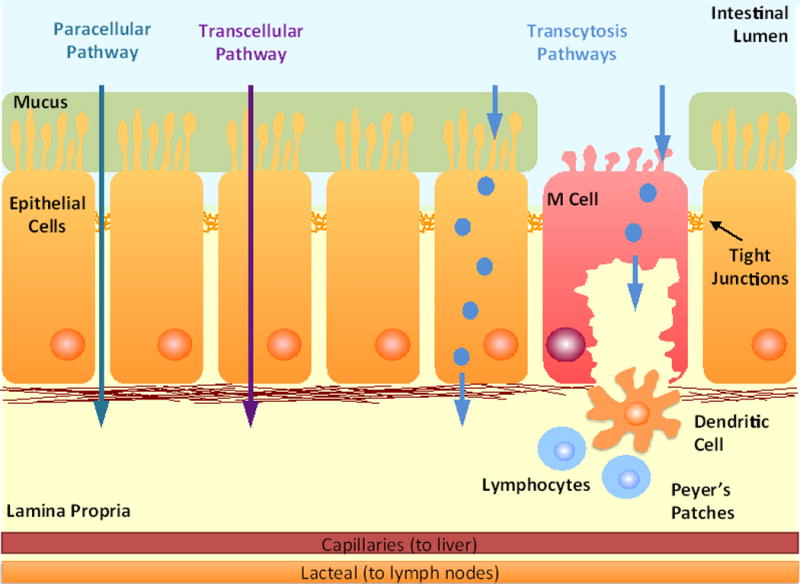
Pathways for intestinal transepithelial transport include paracellular, transcellular, and transcytosis pathways. The paracellular pathway allows diffusion of molecules in the space between epithelial cells and is regulated by tight junctions formed between the cells. The transcellular pathway passes through the apical and basolateral cell membranes as well as the cytoplasm. It is restricted to hydrophobic molecules or molecules that have membrane pumps on the cell surface. The transcytosis pathway is an active transport pathway that relies on receptors specific for a molecule to guide molecules through the cell without entering a degradation pathway. Transcytosis pathways are found in both epithelial and M cells.
1.5 Polymeric Nanoparticles
Polymeric NPs have been extensively studied for oral delivery and have several advantages over other technologies [36,37]. NPs are stable in the GI environment and can protect encapsulated therapeutics from the pH environment, enzyme degradation, and drug efflux pumps [7,38]. For example, when exposed to proteases, insulin and calcitonin stability was improved through encapsulation in polymeric NPs [39,40]. Using polymers to form the NPs provides considerable design flexibility by enabling modulation of physicochemical properties (size, surface charge, hydrophobilicity) and drug release properties (controlled or triggered by external stimuli) [41]. Furthermore, the surface properties can be modulated by using different polymer end groups or conjugating polymers to the NP surface [42]. Targeting ligands such as antibodies, peptides, or small molecules can be conjugated to the surface as well to allow specific interactions with tissue components or cellular receptors [43]. NPs are also able to encapsulate a broad range of therapeutics such as nucleic acids such as DNA or small interfering RNA (siRNA), small molecule drugs, or biologics. The small size of NPs increases the specific surface area, allowing increased contact area with the epithelial surface and a greater potential for non-specific uptake or receptor-mediated endocytosis. Finally, polymeric NPs can be composed of biodegradable materials, many of which are approved for human use by the US Food and Drug Administration [3,44].
2. Strategies for Oral Delivery
2.1 Mucoadhesives
There are two different approaches for oral drug delivery that involve the mucus overlaying the intestinal epithelial cells: mucus-penetrating materials and mucoadhesive materials (Figure 3). Mucus-penetrating materials enable NPs to diffuse through the mucus layers and interact directly with the epithelial cells. One method to overcome the mucus barrier is to modify the surface of the NPs with polyethylene glycol (PEG). Dense PEG coating of NP surfaces minimizes interactions between NPs and mucus, enabling penetration of mucus barriers [45].
Figure 3.

Schematic illustration of the two oral drug delivery approaches that involve mucus: mucus-penetrating particles (MPP) and conventional mucoadhesive particles (CP). MPP are able to diffuse through the mucus layers and reach the epithelium for direct interactions with the surface of epithelial cells. In contrast, CP adhere to the mucus and release therapeutics that can then diffuse to the epithelial surface. CP are cleared along with the mucus layers while MPP clearance depends on the interactions with the epithelial cells. Reproduced with permission from ref [26] and [47].
Mucoadhesives are biomaterials designed to interact with the intestinal mucus layer to increase the residence time and contact with the epithelium, resulting in an increase in the concentration of released therapeutics at the site of absorption [46] (Figure 1). Mucoadhesion can be achieved through several mechanisms, including hydrogen bonding, electrostatic interactions, polymer entanglements with mucus, or a combination of these mechanisms [47–49]. Some examples of polymers used as mucoadhesives include chitosan [50], polyacrylic acid (PAA) [49], and poly(fumaric-co-sebacic) anhydride [51].
Several polymer properties can affect mucoadhesion strength [52]. Studies with chitosan particle revealed that higher polymer molecular weight increases mucoadhesion [53]. In another study, PAA hydrogels had long flexible poly(ethylene) (PEG) chains conjugated to the surface. Mucoadhesion was enhanced due to PEG chain penetration and entanglement in the mucus network [54]. Because mucins are negatively charged, chitosan amine groups on the surface of NPs adhere to mucus through electrostatic interactions [26]. Since the protonation of the amines varies with pH, the mucoadhesive properties due to electrostatic interactions of chitosan NPs vary along the GI tract and may be mucoadhesive only in certain regions of the intestine with acidic pH values [55,56]. Finally, thiol functional groups can enhance mucoadhesion [57]. Studies of chitosan NPs with thiol groups on the surface showed enhanced mucoadhesion due to covalent bonds formed between thiols and cysteine residues on mucus glycoproteins [58].
Besides increasing the concentration of therapeutics near the epithelium, many mucoadhesive materials increase intestinal absorption of therapeutics by acting as permeation enhancers, reversibly opening tight junctions to enhance paracellular transport [59]. Because the tight junctions are less than 20 nm in diameter when opened, most NP drug delivery systems are still unable to pass through this pathway [60]. In a study where chitosan was conjugated to quantum dots (QDs) and orally administered to mice, transmission electron microscopy of intestinal sections revealed chitosan-QD aggregates in the microvilli, but few particles in the paracellular spaces and none on the basolateral side of the intestines [61]. However, opening of the tight junctions does allow many therapeutics to cross using the paracellular pathway. In a study of NPs composed of chitosan and poly(γ-glutamic acid) (PGA) encapsulating aspart-insulin and orally administered to rats, biodistribution results showed that the aspart-insulin was absorbed and entered systemic circulation while the NPs were retained in the intestine [62].
One disadvantage of this approach is that the permeation enhancer activity is non-specific, potentially allowing toxins and other pathogens present in the intestines to cross the intestinal barrier once the tight junctions are open [63]. In a study of the effect of insulin-loaded chitosan NPs on the absorption of lipopolysaccharide (LPS), one of the most prevalent toxins in the GI tract, it was found that chitosan did not significantly increase the absorption of LPS but did enable insulin to reach circulation [64]. However, this may have been due to anionic LPS forming micelles that were repulsed by the negative charge of the mucus layer. When mucolytic agents were added, LPS absorption was observed, suggesting that toxins that are not repulsed by mucus could enter through openings in the tight junctions. Therefore, the issue of toxicity associated with permeation enhancers warrants further study. One other limitation of the use of paracellular pathway for transport is that the surface area for absorption from the paracellular pathway is less than 0.1% of the total intestinal epithelium surface area, which could limit the capacity for absorption of therapeutics [65].
2.2 Targeting M cells
Targeting NPs to natural transcytosis pathways is another approach used for oral delivery because it offers a way to cross the intestinal barrier without affecting barrier permeability. The most extensively studied for oral delivery is the M cell transcytosis pathway, which is used to transport antigens across the epithelium for immune surveillance [21,66] (Figure 1). This pathway is attractive because M cells have reduced protease activity, a lack of mucus secretion, and a sparse glycocalyx [67].
There are many ways to engineer NPs to target M cells. Hydrophobicity is one parameter that influences uptake, with studies indicating that NPs formed using hydrophobic polymers (polystyrene, polymethylmethacrylate, and polyhydroxybutrate) had significantly more uptake by M cells than less hydrophobic lactide and glycolide polymer particles [68]. However, hydrophobicity could also enhance interactions with mucus, so further optimization may be needed [26]. Particle size studies have indicated that larger NPs are more selectively taken up by M cells [69,70]. Optimization of the surface charge has been more difficult, and it remains unclear exactly how surface charge affects non-specific uptake by M cells. In one study, neutral NPs ranging in size from 130 to 950 nm were identified as ideal for uptake by M cells [71]. Others have hypothesized that a positive charge would enhance interactions with the negatively-charged surface of M cells. However, the negative charge of mucus could also interact with the NPs, reducing the effectiveness of M cell targeting [72]. Using targeting ligands conjugated to the NP surface, such as bacterial adhesins [73], IgA antibodies [74], and toxins [75], also enhances M cell transport. In one study, IgA absorbed to polystyrene particles was taken up by M cells 20–30 times more than polystyrene particles coated with bovine serum albumin (BSA) [74].
One potential problem with this approach is that M cells are closely associated with immune cells in the lamina propria such as dendritic cells and macrophages that are part of the Peyer’s Patches [76]. A recent study demonstrated that the majority of fluorescently-labeled glucan and PLGA NPs administered orally to mice were endocytosed by dendritic cells in the Peyer’s Patches after M cell transcytosis, preventing the NPs from reaching the bloodstream [77]. Besides limiting the ability of the NPs to deliver the therapeutics to the bloodstream, uptake by dendritic cells increases the risk that the NPs or therapeutics could induce an immune response [78]. While this may limit the use of this approach for long-term oral administration, it opens the possibility that NPs targeting M cells could be used for oral mucosal vaccination applications [79].
Absorption of NPs or therapeutics by M cells may also be limited because M cells only make up a small percentage (5–10%) of the non-absorptive epithelium in humans [80,81]. The number of Peyer’s Patches and surface properties of M cells also varies among species, which could make it difficult to use rodent data to predict performance in humans [82,83].
2.3 Targeting Enterocytes
Enterocytes are an attractive target for oral drug delivery because these cells form the majority of the absorptive surface area of the intestine. In the proximal jejunum of rats, the ratio of M cells to enterocytes was estimated to be 1:12 [84]. One of the challenges of targeting enterocytes is penetrating the mucus layer covering the cells so that the NPs can interact directly with the cell surface. As mentioned earlier, coating NPs with mucus-penetrating materials such as a dense outer layer of PEG enables the NPs to diffuse through the mucus barrier and reach the epithelial cell surface [45].
Several NP physicochemical parameters have been used to enhance enterocyte particle uptake. Particle size studies have indicated that smaller NPs (<50–100 nm) are taken up by enterocytes [69,70]. Studies of PLGA NPs in vitro with Caco-2 cells (human adenocarcinoma cell line) and in vivo in rats revealed that uptake of 100 nm particles was more efficient than larger particles (500 nm, 1 μm, 10 μm) [85,86].
In addition to physicochemical properties, there are several different ligands that have been used to target NPs to enterocytes. One example is lectins, a class of proteins that specifically bind to carbohydrates and have been investigated for receptor-mediated oral delivery because of their resistance to acidic pH and proteases as well as having a large number of binding sites on the glycocalyx of epithelial cells [87,88]. Studies with tomato lectin-conjugated NPs have shown that the targeted NPs have increased endocytosis through enhanced interactions with enterocytes [89,90]. Another lectin is wheat germ agglutinin (WGA), which binds to N-acetyl glucosamine and sialic acid found throughout the GI tract [91]. Studies with WGA demonstrated that it not only binds to the surface of enterocytes, but is taken up into the cells by receptor-mediated endocytosis through the epidermal growth factor receptor that is highly expressed on enterocytes [92]. One disadvantage of lectins is that they may also interact with components of the mucus layer, which causes them to act as bioadhesives and prevents NPs from reaching the epithelial surface [93,94]. In addition, lectins have not been shown to traffic through a transcytosis pathway. Therefore, NPs taken up either remain on the surface or, if endocytosed, may be trafficked through a degradation pathway to the lysosomes.
The folate receptor is another target of NPs for oral delivery that has demonstrated enhanced uptake of drugs encapsulated in folate-targeted NPs [95]. However, there is very limited evidence that folate receptors enable transcytosis across enterocytes. Lectins and folic acid ligands may be better suited for targeting of NPs directly to enterocytes for local delivery of therapeutics rather than systemic delivery after oral administration (Figure 1).
More recent NP targeting strategies have focused on receptor-mediated transcytosis pathways that are not associated with the GALT, which may help NPs evade immune cells after crossing epithelium (Figure 1). One example of this strategy is the vitamin B12 receptor. When vitamin B12 is ingested, it is bound by a protein called intrinsic factor (IF) in the small intestines. The vitamin B12-IF complex interacts with a receptor that traffics vitamin B12 across the intestinal epithelium [96]. As part of the pathway across the cell, the vitamin B12-IF complex enters the lysosomal compartment, which could potentially lead to inactivation of released therapeutics. Interestingly, conjugating vitamin B12 to NPs causes a switch in the transcytosis pathway used to cross the enterocytes. Studies have shown that vitamin B12-NP conjugates are able to avoid trafficking through lysosomes, while either vitamin B12 or non-targeted NPs pass through lysosomes in Caco-2 in vitro models [97]. Targeting of NPs to this pathway by conjugating vitamin B12 to the NP surface has been successful in delivering biologics to the bloodstream. Studies with insulin-loaded NPs targeted to the vitamin B12-IF receptor have demonstrated enhanced absorption of insulin and hypoglycemic effects in rat models after oral administration [98,99]. One potential drawback of this approach is that vitamin B12 absorption does not occur until the distal section of the ileum, requiring NPs to maintain stability while traveling through most of the small intestine before absorption.
Another receptor recently targeted by NPs for oral delivery is the neonatal Fc receptor (FcRn). FcRn is responsible for IgG transport across the intestinal epithelium [100,101], and is expressed throughout the intestine [102] (Figure 3). FcRn binds to the Fc region of IgG antibodies in a pH-dependent manner, with high affinity in acidic environments (pH <6.5) and low affinity at physiological pH (pH ~ 7.4) [103]. NPs can be targeted to FcRn by conjugating the Fc fragment of IgG to the surface of NPs. In acidic sections of the intestine such as the duodenum and jejunum [104], targeted NPs can bind to FcRn on the surface of enterocytes. Alternatively, in later sections of the intestine, targeted NPs can still be taken up through fluid-phase pinocytosis by enterocytes and be bound by FcRn once inside acidic endosomes [105]. Once bound to FcRn, targeted NPs can be trafficked across enterocytes. On the basolateral side of the cell, where the pH is neutral, targeted NP are released from FcRn and are free to diffuse into the lamina propria. In a recent study, Fc-targeted NPs administered orally to mice were absorbed 11.5 times more efficiently than non-targeted NPs, and the NPs were able to enter systemic circulation [106]. The Fc-targeted NPs were detected in the liver, spleen, lungs, and kidneys after absorption, which are all tissues that express the FcRn. Therefore, using IgG Fc to target the FcRn not only enables NPs to cross the intestines, but it also targets the NPs to several of the organs expressing FcRn. The study also demonstrated that administering Fc-targeted NPs containing insulin resulted in a hypoglycemic response for 15 hr with a clinically-relevant dose of only 1.1 U/kg [107], lower than other oral insulin delivery systems that require 10–100 U/kg to generate a glucose response [19]. One drawback of this approach is the potential for Fc fragments on the NP surface to interact with other Fc receptors in the body once absorbed, which could affect NP biodistribution and possibly cause an immune reaction.
3. Applications
3.1 Cancer Therapies
There are some chemotherapeutic agents that can be administered orally. However, many suffer from low bioavailability due to physicochemical properties or because they are substrates for P-gp proteins that pump drugs taken up by enterocytes back into the intestinal lumen. Encapsulating chemotherapeutic agents in NPs can enable oral administration of agents currently limited to intravenous administration. Taxanes are one example of a chemotherapeutic agents limited by low bioavailabiity due to P-gp pumps [108]. In a recent study, encapsulating docetaxel in NPs resulted in increased absorption after oral administration relative to oral Taxotere in mini pigs [109]. Furthermore, the study showed that docetaxel NPs were trafficked through the lymphatic system, avoiding first-pass liver metabolism effects such as degradation by cytochrome P450 3A4 (CYP3A4). This resulted in higher drug levels in the blood than intravenously administered Taxotere, reduced tumor growth, and increased survival in a mouse lung cancer metastatic model. Rapamycin was encapsulated in NPs composed of N-isopropylacrylamide, methylmethacrylate, and acrylic acid and administered orally to mice [110]. The NP formulation resulted in higher levels of drug in the blood and in xenograft pancreatic cancer tumors compared with free rapamycin administered orally. In addition, there was no evidence of toxicity in the intestines or other visceral organs from the oral rapamycin NPs after 4 weeks.
3.2 Proteins and Peptides
One of the biggest goals for oral delivery is insulin because of the chronic nature of the therapy and the need for multiple daily injections. In addition to the patient convenience, oral delivery is particularly beneficial for insulin because it more closely reproduces insulin’s physiological pharmacokinetic profile. Subcutaneous insulin injection targets tissues such as muscle and kidney because it enters systemic circulation first before reaching the liver [111]. The oral route replicates the pharmacodynamics of endogenous insulin release by entering the liver after intestinal absorption, similar to insulin secreted from the pancreas [50]. The liver metabolizes 50–75% of insulin secreted from the pancreas, but only 25% of subcutaneous insulin [112]. The liver is more sensitive to insulin and acts faster in response to insulin to lower blood glucose levels; therefore, less insulin is required to control blood glucose levels, even in diabetic patients [111].
Oral insulin delivery has been achieved using many approaches such as mucoadhesives, M cell targeting, and enterocyte targeting. However, many of these technologies have been limited by inefficiency, requiring high doses of insulin to generate a hypoglycemic response in rodent models. Previous studies have suggested that long-term oral administration of high doses of insulin could induce mitogenic changes in GI epithelial cells because insulin is a growth factor [113]. However, recent technologies are becoming more efficient and are using more clinically relevant insulin doses to achieve hypoglycemic response in mice [106]. Studies with insulin encapsulated in NPs have also demonstrated that controlled release of insulin from NPs results in a more prolonged hypoglycemic effect with less of a decrease in blood glucose than subcutaneous insulin administration [62].
There are several other examples of peptides and proteins being delivered orally using NPs. Interferon-β (INF-β) is used for the treatment of multiple sclerosis, but is limited to daily injections [114]. By encapsulating INF-β in NPs, INF-β was able to be administered orally to rabbits and achieved higher plasma concentration over 24 hrs compared with subcutaneous injection [115]. Salmon calcitonin is a peptide that causes a decrease in blood calcium levels and is used for the treatment of bone diseases such as osteoporosis [116]. When encapsulated in Eudragit®-PLGA NPs, it was able to be administered orally to rats and achieve a greater decline in blood calcium levels for 24 hrs compared with subcutaneous injections of free salmon calcitonin [117]. Finally, cyclosporine is a potent immunosuppressive agent used for the prevention of graft rejection [118]. When encapsulated in PLGA NPs and administered orally to rats, the cyclosporine NPs resulted in a sustained blood concentration of cyclosporine over 5 days compared with only 3 days for the commercial formulation and had markedly less nephrotoxicity [119].
3.3 Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) is a target for oral delivery because the affected tissue is the intestines. As opposed to most oral delivery applications that require the therapeutic to reach the bloodstream, the goal for IBD treatment is local delivery of therapeutics to immune cells in the intestines. Oral delivery strategies for IBD have attempted to take advantage of the pathophysiological processes associate with the disease to deliver therapies only at inflamed intestinal regions. These processes include increased mucus production and enhanced permeability of the intestinal barrier at inflammation sites [120]. By delivering anti-inflammatory therapies locally instead of systemically, the therapies can be concentrated at inflammation sites for greater efficacy while minimizing systemic adverse side effects [121,122].
Physicochemical NP parameters can be used to target inflamed intestinal tissue. In a rat IBD model, smaller NP sizes were more effective at depositing in inflamed tissue [123]. Within the inflamed tissue, greater deposition was observed in regions of thicker mucus and ulcerations. NPs have been effective at delivering many different therapeutics, including rolipram [124], 5-aminosalicylic acid [125], FK506 (tacrolimus) [120], and dexamathesone [121], with each demonstrating efficacy in reducing inflammation in rodent IBD models.
Another approach to target IBD is to attach ligands to the surface of NPs. In one study, the Fab portion of the F4/80 antibody was conjugated to the surface of PLA-PEG NPs containing TNF-α siRNA [126]. When administered to mice with colitis, the targeted NPs demonstrated reduced inflammation and body weight loss compared to control groups. Lectins such as WGA and peanut agglutinin (PNA) have also been used to target IBD. When conjugated to PLGA NPs, both WGA and PNA caused increased adherence of NPs to inflamed regions of the intestine and improved clinical activity scores in murine colitis models [127].
Polymer engineering has also been used to target inflammation in IBD models. Thioketal polymers were synthesized that selectively degraded in the presence of reactive oxygen species (ROS) [128]. Because inflammatory sites have elevated levels of ROS, NPs composed of thioketal polymers would degrade at inflammation sites after oral administration and release the therapeutic cargo (Figure 5). TNF-α siRNA was encapsulated in thioketal NPs and administered to mice with induced colitis, and the results showed that the thioketal NPs reduced inflammation and body weight loss over 7 days.
Figure 5.
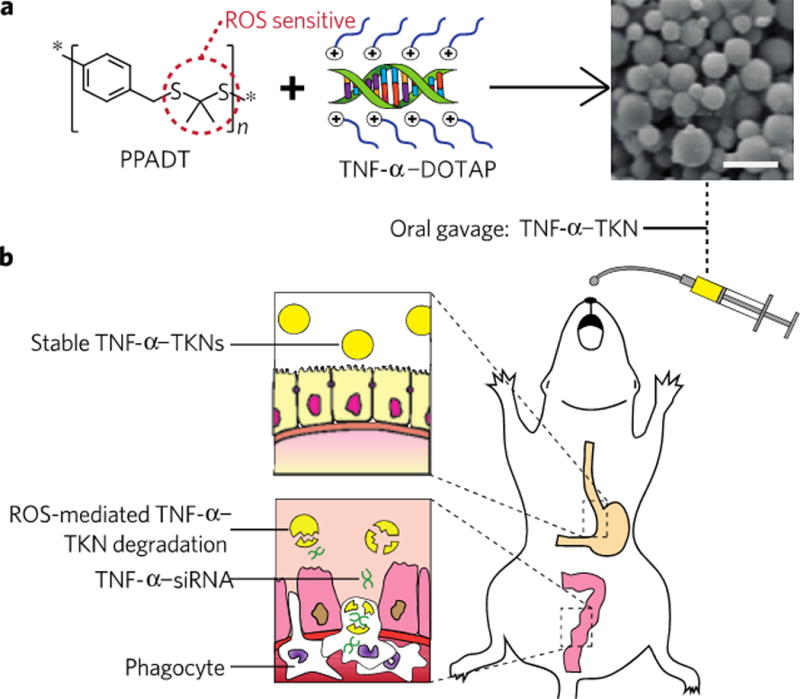
Targeting intestinal inflammation for IBD therapy using polymer engineering. (A) Thioketal linkages that are ROS-sensitive were incorporated into a polymer, causing the polymer to degrade in the presence of ROS. (B) Since inflammatory sites have elevated ROS levels, the NPs preferentially degrade and cause targeted release of the NP payload at sites of IBD. Reproduced with permission from [128].
3.4 Vaccines
NPs are also being developed for oral vaccination applications. Chitosan NPs containing DNA have been used to generate immune protection against several allergens. For example, mice administered orally with chitosan NPs containing a peanut allergen gene demonstrated reduced allergen-induced anaphylaxis compared with non-immunized mice [129]. Chitosan NPs containing a gene for allergens derived from house dust mites were also used for oral vaccination of mice [130]. Results showed that oral administration of the NPs led to expression of the gene in epithelial cells of the stomach and small intestines, preventing sensitization of a Th2 cell-regulated IgE response [131].
NPs have also been developed for vaccination against bacterial and viral infections. PLGA particles containing Staphylococcal enterotoxin B given orally to mice were taken up by M cells in the Peyer’s Patches and resulted in the production of circulating toxin-specific antibodies and secretory IgA-specific antibodies in saliva, gut wash fluids, and bronchial-alveolar wash fluids [68,132]. In contrast, soluble enterotoxoid administered orally was unable to generate an immune response. In another study, NPs encapsulated in pH-dependent microparticles were developed that could be targeted specifically to either the small or large intestine [133]. The system was used to vaccinate the colon against a viral infection challenge as an effective alternative to intracolorectal administration.
4. Conclusions
Many therapeutics are currently limited to parenteral administration. Alternate routes of administration, particularly oral, are considered favorable because they are more convenient and have improved patient compliance than injection-based administration. However, oral delivery is very challenging because of the barriers presented by the GI tract. Polymeric NPs can be used to overcome the pH and enzyme barriers, but the intestinal permeability barrier remains a significant hurdle.
There have been many approaches used to overcome the intestinal epithelial barrier to enable efficient oral delivery of biologics and nanomedicines. Mucoadhesives use the mucus layer of the intestines to prolong residence time in the intestines and increase the concentration of therapeutics near the surface of the epithelial cells. In addition, many mucoadhesives are permeation enhancers that open tight junctions between epithelial cells to enable drugs or biologics to cross the barrier. Other approaches have focused on targeting natural transcytosis pathways, such as M cells, the vitamin B12 pathway, and the FcRn pathway. Recent studies have demonstrated that targeting transcytosis pathways can efficiently deliver therapeutics and nanomedicines orally, but further studies are required before these technologies can be translated to the clinic.
Oral NP drug delivery systems are being developed for many different applications. These include oral administration of chemotherapeutics for cancer treatments, local delivery to the intestine for the treatment of IBD, and oral mucosal vaccination. Many proteins are being encapsulated in NPs for oral administration, in particular insulin for the treatment of diabetes. Successful development of NPs for oral administration could change the treatment paradigm for many of these diseases and have a significant impact on patient outcomes in the future.
5. Expert Opinion
Biologics and emerging nanomedicines have the potential to significantly alter the treatment of many diseases. However, both types of therapeutics are currently limited to parenteral administration because of intestinal barriers that limit absorption. Polymeric NPs are currently being investigated to enable efficient oral administration of these therapeutics. While polymeric NPs are able to overcome many of the challenges presented by the GI tract, the major hurdle of improving transport across the intestines remains.
There have been many different approaches developed to overcome the transport barrier presented by the GI tract. Mucoadhesives adhere to the mucus layer to increase therapeutic concentrations near epithelial cells and also cause reversible opening of tight junctions. While this approach utilizes the majority of intestinal surface area for adherence, there are safety issues associated with disrupting tight junction integrity. In addition, transport through tight junctions is limited by size to small molecules and biologics, and is not a valid approach for nanomedicines.
Another approach is to target NPs to the M cells in the Peyer’s Patches, which are part of the GALT. This strategy aims to take advantage of the transcytosis pathway used by M cells to transport antigens across the epithelium. Targeting transcytosis pathways could not only enable small molecules and biologics to cross the barrier, but it could also allow NPs to cross and reach systemic circulation. This would be a significant advance towards clinical development of nanomedicines for oral administration. However, the Peyer’s Patches only compose a small fraction of the surface area of the intestines and could limit absorption capacity. In addition, there is the potential for NPs that are transported across the epithelium to be taken up by these immune cells associated with M cells before entering systemic circulation. Alternatively, the association with immune cells makes this approach appealing for oral mucosal vaccination.
Recent approaches have tried to combine the advantages of these previous strategies by attempting to target transcytosis pathways present in intestinal epithelial cells. These cells are present throughout the majority of the intestines to maximize absorption capacity, but are not highly associated with the GALT so that interactions with immune cells after transcytosis may be reduced. Because this approach takes advantage of natural transcytosis pathways, it does not have to disrupt the integrity of the epithelial barrier. Examples include the vitamin B12 and the FcRn transcytosis pathways.
Despite all of the efforts focused on developing polymeric NPs for oral delivery, these technologies have historically been limited by low bioavailabilities. In the case of insulin delivery, this has resulted in the use of extremely high doses of insulin that would be impractical in clinical use. However, recent studies targeting transcytosis pathways such as the FcRn have achieved hypoglycemic effects in mouse models while significantly lowering insulin doses to levels approaching those used in the clinic for diabetic patients. In addition, this approach enabled NPs, not just insulin, to reach the bloodstream and target organs expressing the FcRn.
While these are positive developments for the oral delivery of biologics and NP-based therapies, there are still many challenges that remain for translation of these technologies to the clinic. Because none of the NP oral delivery technologies have been tested in humans, it will be important to study how predictive animal models are for performance in humans in order to better evaluate different technologies. For example, the number of M cells can vary between species [134]. These differences could significantly affect absorption rates in different species [106,135]. In addition, understanding how patient-to-patient variability, diet, fasting states, and disease states affect the performance of these technologies in humans will be important to determine the robustness of these technologies. It will also be necessary to evaluate the consequences of long-term oral administration of therapeutics since some agents can act as growth factors and cause changes in the epithelium, while others may cause intestinal toxicity. Finally, since many of these oral delivery strategies require attaching targeting ligands to the NP surface, it will be important to study how the presence of these ligands on the NPs affects pharmacological parameters such as the biodistribution and blood clearance after absorption.
Further improvements in polymeric NP oral delivery technologies will come from increased understanding of the physiology of the intestines, including transcytosis pathways. In addition, a better understanding of disease pathophysiology could provide insights that result in new approaches, such as the example of developing ROS-responsive polymers to target therapies directly to inflammation sites in IBD. NPs are currently entering clinical trials for the treatment of many diseases, and are demonstrating that nanomedicines are a valid therapeutic modality that can feasibly be implemented in the clinic. In the future, it is expected that more polymeric NP technologies will be developed for oral delivery to allow biologics and nanomedicines to realize their full potential in the treatment of many chronic diseases.
Figure 4.
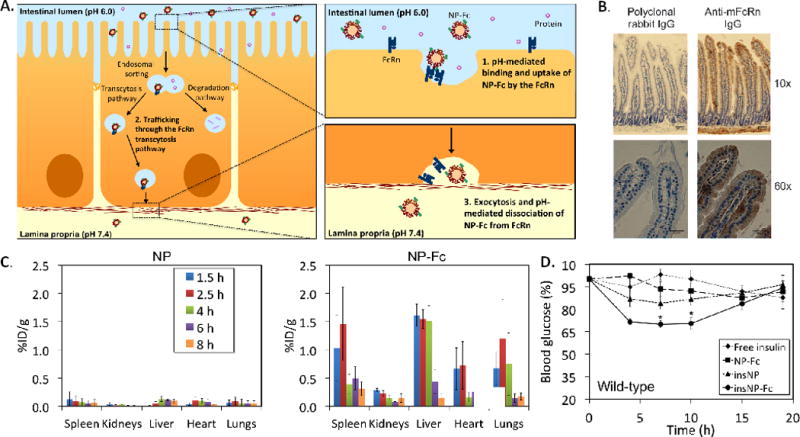
(A) Schematic of NP transport across the intestinal epithelium using the FcRn transcytosis pathway. (B) Immunohistochemistry on mouse duodenum tissue sections showing expression of the FcRn in the intestinal epithelium. Mouse FcRn appears brown in the images. The negative control for the staining was polyclonal IgG. (C) Biodistribution of 14C-labeled NPs after oral administration to mice demonstrating that NP-Fc were able to enter systemic circulation and reach the liver, lungs, and spleen significantly more than non-targeted NPs. (D) Blood glucose response of fasted mice to orally administered NPs. NP-Fc was able to generate a significantly greater hypoglycemic response than non-targeted NPs. Adapted with permission from ref. [106].
Article Highlights.
This article covers polymeric nanoparticle technologies being developed for the oral delivery of small molecule drugs, biologics, and nanomedicines.
Background will be provided on the challenges associated with oral delivery for different classes of therapeutics.
Different strategies used to overcome the barriers of the gastrointestinal tract by nanoparticles will be reviewed, including the use of mucoadhesive biomaterials and the targeting of nanoparticles to transcytosis pathways present in M cells and enterocytes.
Applications currently being evaluated for oral delivery using nanoparticles will be discussed such as oral chemotherapy, oral insulin administration, oral treatments for inflammatory bowel disease, and oral mucosal vaccinations.
Advantages and disadvantages of different approaches to oral delivery using nanoparticles will be considered.
Acknowledgments
Funding
Supported in part by a Koch-Prostate Cancer Foundation Award in Nanotherapeutics (R.L. and O.C.F.), the National Cancer Institute Center of Cancer Nanotechnology Excellence at MIT-Harvard (U54-CA151884), and a National Institutes of Health grant (R01 EB015419-01).
Financial disclosure
The authors were supported by Prostate Cancer Foundation and David Koch, National Cancer Institute, National Institute of Health. Eric Pridgen, Omid Farokhzad and Frank Alexis are inventors on the patent application related to neonatal Fc receptor technology, Omid Farokhzad discloses a financial interest in BIND therapeutics, Selecta Biosciences and Blend Therapeutics.
Footnotes
Declaration of Interest
The authors disclose the following: Omid Farokhzad has financial interests in BIND Therapeutics, Selecta Biosciences, and Blend Therapeutics, which are developing nanoparticle therapeutics; and Omid Farokhzad, Frank Alexis, and Eric Pridgen are inventors on patent applications related to the neonatal Fc receptor technology.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Aggarwal S. What’s fueling the biotech engine-2012 to 2013. Nat Biotechnol. 2014;32:32–9. doi: 10.1038/nbt.2794. [DOI] [PubMed] [Google Scholar]
- 2.Wang AZ, Langer R, Farokhzad OC. Nanoparticle Delivery of Cancer Drugs. Annu Rev Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 3.Hrkach J, Von Hoff D, Ali MM, et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci Transl Med. 2012;4:128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 4.Davis ME, Zuckerman JE, Choi CHJ, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lanza GM, Winter PM, Caruthers SD, et al. Nanomedicine opportunities for cardiovascular disease with perfluorocarbon nanoparticles. Nanomed. 2006;1:321–329. doi: 10.2217/17435889.1.3.321. [DOI] [PubMed] [Google Scholar]
- 6.Krol S, Ellis-Behnke R, Marchetti P. Nanomedicine for treatment of diabetes in an aging population: state-of-the-art and future developments. Nanomedicine Nanotechnol Biol Med. 2012;8:S69–S76. doi: 10.1016/j.nano.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Peppas NA, Kavimandan NJ. Nanoscale analysis of protein and peptide absorption: insulin absorption using complexation and pH-sensitive hydrogels as delivery vehicles. Eur J Pharm Sci. 2006;29:183–197. doi: 10.1016/j.ejps.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Liu G, Franssen E, Fitch MI, Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol. 1997;15:110–115. doi: 10.1200/JCO.1997.15.1.110. [DOI] [PubMed] [Google Scholar]
- 9.Borner MM, Schoffski P, de Wit R, et al. Patient preference and pharmacokinetics of oral modulated UFT versus intravenous fluorouracil and leucovorin: a randomised crossover trial in advanced colorectal cancer. Eur J Cancer. 2002;38:349–358. doi: 10.1016/s0959-8049(01)00371-9. [DOI] [PubMed] [Google Scholar]
- 10.Von Pawel J, Gatzemeier U, Pujol JL, et al. Phase ii comparator study of oral versus intravenous topotecan in patients with chemosensitive small-cell lung cancer. J Clin Oncol. 2001;19:1743–1749. doi: 10.1200/JCO.2001.19.6.1743. [DOI] [PubMed] [Google Scholar]
- 11.DiMeglio LA, Peacock M. Two-Year Clinical Trial of Oral Alendronate Versus Intravenous Pamidronate in Children With Osteogenesis Imperfecta. J Bone Miner Res. 2006;21:132–140. doi: 10.1359/JBMR.051006. [DOI] [PubMed] [Google Scholar]
- 12.Pfeiffer P, Mortensen JP, Bjerregaard B, et al. Patient preference for oral or intravenous chemotherapy: A randomised cross-over trial comparing capecitabine and Nordic fluorouracil/leucovorin in patients with colorectal cancer. Eur J Cancer. 2006;42:2738–2743. doi: 10.1016/j.ejca.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 13.Findlay M, Von Minckwitz G, Wardley A. Effective oral chemotherapy for breast cancer: pillars of strength. Ann Oncol. 2008;19:212–222. doi: 10.1093/annonc/mdm285. [DOI] [PubMed] [Google Scholar]
- 14.James R, Blanco C, Farina C. Savings in staff time as a result of switching from de Gramont to oral capecitabine for patients with advanced colorectal cancer. Eur J Cancer. 2003;1(Suppl 5):S83. (Abstr 271) [Google Scholar]
- 15.De Portu S, Mantovani LG, Ravaioli A, et al. Cost Analysis of Capecitabine vs 5-Fluorouracil-Based Treatment for Metastatic Colorectal Cancer Patients. J Chemother. 2010;22:125–128. doi: 10.1179/joc.2010.22.2.125. [DOI] [PubMed] [Google Scholar]
- 16.Jansman F, Postma M, van Hartskamp D, et al. Cost-benefit analysis of capecitabine versus 5-fluorouracil/leucovorin in the treatment of colorectal cancer in the Netherlands. Clin Ther. 2004;26:579–589. doi: 10.1016/s0149-2918(04)90060-4. [DOI] [PubMed] [Google Scholar]
- 17.Yabroff K, Warren J, Knopf K, et al. Estimating Patient Time Costs Associated with Colorectal Cancer Care. Med Care. 2005;43:640–648. doi: 10.1097/01.mlr.0000167177.45020.4a. [DOI] [PubMed] [Google Scholar]
- 18.Tortora G. Principles of Human Anatomy. New York: John Wiley & Sons, Inc.; 2005. [Google Scholar]
- 19.Chen M-C, Sonaje K, Chen K-J, Sung H-W. A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials. 2011;32:9826–9838. doi: 10.1016/j.biomaterials.2011.08.087. [DOI] [PubMed] [Google Scholar]
- 20.Schenk M, Mueller C. The mucosal immune system at the gastrointestinal barrier. Best Pr Res Cl Ga. 2008;22:391–409. doi: 10.1016/j.bpg.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Macdonald T, Monteleone G. Immunity, Inflammation, and Allergy in the Gut. Science. 2005;307:1920–1925. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 22.Kararli T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory-animals. Biopharm Drug Dispos. 1995;16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 23.Sood A, Panchagnula R. Peroral route: an opportunity for protein and peptide drug delivery. Chem Rev-Columb. 2001;101:3275–3304. doi: 10.1021/cr000700m. [DOI] [PubMed] [Google Scholar]
- 24.Ganapathy V, Gupta N, Martindale R. Protein digestion and absorption Physiology of the gastrointestinal tract. 4th. Burlington: Academic Press; 2006. [Google Scholar]
- 25.Johnson L, Christensen J, Jackson M, et al. Physiology of the Gastrointestinal Tract. New York: Raven Press; 1987. [Google Scholar]
- 26**.Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv Drug Deliv Rev. 2012;64:557–570. doi: 10.1016/j.addr.2011.12.009. Excellent review of the properties of the intestinal mucus barrier and the drug delivery technologies developed to overcome the barrier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai SK, Wang Y-Y, Wirtz D, Hanes J. Micro-and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61:86–100. doi: 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atuma C, Strugala V, Allen A, Holm L. The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol-Gastrointest Liver Physiol. 2001;280:G922–G929. doi: 10.1152/ajpgi.2001.280.5.G922. [DOI] [PubMed] [Google Scholar]
- 29.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61:75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Banan A. theta Isoform of Protein Kinase C Alters Barrier Function in Intestinal Epithelium through Modulation of Distinct Claudin Isotypes: A Novel Mechanism for Regulation of Permeability. J Pharmacol Exp Ther. 2005;313:962–982. doi: 10.1124/jpet.104.083428. [DOI] [PubMed] [Google Scholar]
- 31.Mitic LL, Van Itallie CM, Anderson JM. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol-Gastrointest Liver Physiol. 2000;279:G250–G254. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- 32.Stamatovic SM, Keep R, Kunkel S, Andjelkovic A. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 33.Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2:285–293. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- 34.DeMario MD, Ratain MJ. Oral chemotherapy: rationale and future directions. J Clin Oncol. 1998;16:2557–2567. doi: 10.1200/JCO.1998.16.7.2557. [DOI] [PubMed] [Google Scholar]
- 35.Salama N, Eddington N, Fasano A. Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev. 2006;58:15–28. doi: 10.1016/j.addr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 36.Galindo-Rodriguez S, Allemann E, Fessi H, Doelker E. Polymeric nanoparticles for oral delivery of drugs and vaccines: a critical evaluation of in vivo studies. Crit Rev Ther Drug Carr Syst. 2005;22:419–464. doi: 10.1615/critrevtherdrugcarriersyst.v22.i5.10. [DOI] [PubMed] [Google Scholar]
- 37.Moore T, Graham E, Mattix B, Alexis F. Biomaterials Science: An Integrated Clinical and Engineering Approach. 1st. Boca Raton, Fl: Taylor and Francis Group, LLC; 2012. [Google Scholar]
- 38.Carino G, Mathiowitz E. Oral insulin delivery. Adv Drug Deliv Rev. 1999;35:249–257. doi: 10.1016/s0169-409x(98)00075-1. [DOI] [PubMed] [Google Scholar]
- 39.Lowe PJ, Temple CS. Calcitonin and insulin in isobutylcyanoacrylate nanocapsules — protection against proteases and effect on intestinal-absorption in rats. J Pharm Pharmacol. 1994;46:547–552. doi: 10.1111/j.2042-7158.1994.tb03854.x. [DOI] [PubMed] [Google Scholar]
- 40.Damge C, Vranckx H, Balschmidt P, Couvreur P. Poly (alkyl cyanoacrylate) nanospheres for oral administration of insulin. J Pharm Sci. 1997;86:1403–1409. doi: 10.1021/js970124i. [DOI] [PubMed] [Google Scholar]
- 41.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol Pharm. 2008 Aug;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valencia PM, Pridgen EM, Rhee M, et al. Microfluidic Platform for Combinatorial Synthesis and Optimization of Targeted Nanoparticles for Cancer Therapy. ACS Nano. 2013;7:10671–10680. doi: 10.1021/nn403370e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu MK, Park J, Jon S. Targeting Strategies for Multifunctional Nanoparticles in Cancer Imaging and Therapy. Theranostics. 2012;2:3–44. doi: 10.7150/thno.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansour HM, Rhee Y-S, Wu X. Nanomedicine in pulmonary delivery. Int J Nanomed. 2009;4:299–319. doi: 10.2147/ijn.s4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang BC, Dawson M, Lai SK, et al. Biodegradable polymer nanoparticles that rapidly penetrate the human mucus barrier. Proc Natl Acad Sci U S A. 2009;106:19268–19273. doi: 10.1073/pnas.0905998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smart J. The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev. 2005;57:1556–1568. doi: 10.1016/j.addr.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 47.Lai SK, Wang Y-Y, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dünnhaupt S, Barthelmes J, Hombach J, et al. Distribution of thiolated mucoadhesive nanoparticles on intestinal mucosa. Int J Pharm. 2011;408:191–199. doi: 10.1016/j.ijpharm.2011.01.060. [DOI] [PubMed] [Google Scholar]
- 49.Makhlof A, Werle M, Tozuka Y, Takeuchi H. A mucoadhesive nanoparticulate system for the simultaneous delivery of macromolecules and permeation enhancers to the intestinal mucosa. J Controlled Release. 2011;149:81–88. doi: 10.1016/j.jconrel.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Chen M-C, Mi F-L, Liao Z-X, et al. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv Drug Deliv Rev. 2013;65:865–879. doi: 10.1016/j.addr.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 51.Furtado S, Abramson D, Burrill R, et al. Oral delivery of insulin loaded poly(fumaric-co-sebacic) anhydride microspheres. Int J Pharm. 2008;347:149–155. doi: 10.1016/j.ijpharm.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 52.Boddupalli BM, Mohammed ZNK, Nath RA, Banji D. Mucoadhesive drug delivery system: An overview. J Adv Pharm Technol Res. 2010;1:381–387. doi: 10.4103/0110-5558.76436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeuchi H, Thongborisute J, Matsui Y, et al. Novel mucoadhesion tests for polymers and polymer-coated particles to design optimal mucoadhesive drug delivery systems. Adv Drug Deliv Rev. 2005;57:1583–1594. doi: 10.1016/j.addr.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 54.Huang Y, Leobandung W, Foss A, Peppas NA. Molecular aspects of muco-and bioadhesion:: Tethered structures and site-specific surfaces. J Controlled Release. 2000;65:63–71. doi: 10.1016/s0168-3659(99)00233-3. [DOI] [PubMed] [Google Scholar]
- 55.Kotze AF, Luessen HL, De Boer AG, et al. Chitosan for enhanced intestinal permeability: prospects for derivatives soluble in neutral and basic environments. Eur J Pharm Sci. 1999;7:145–151. doi: 10.1016/s0928-0987(98)00016-5. [DOI] [PubMed] [Google Scholar]
- 56.Sandri G, Bonferoni MC, Rossi S, et al. Insulin-Loaded Nanoparticles Based on N-Trimethyl Chitosan: In Vitro (Caco-2 Model) and Ex Vivo (Excised Rat Jejunum, Duodenum, and Ileum) Evaluation of Penetration Enhancement Properties. AAPS PharmSciTech. 2010;11:362–371. doi: 10.1208/s12249-010-9390-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krauland AH, Guggi D, Bernkop-Schnürch A. Oral insulin delivery: the potential of thiolated chitosan-insulin tablets on non-diabetic rats. J Controlled Release. 2004;95:547–555. doi: 10.1016/j.jconrel.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 58.Bravo-Osuna I, Vauthier C, Farabollini A, et al. Mucoadhesion mechanism of chitosan and thiolated chitosan-poly(isobutyl cyanoacrylate) core-shell nanoparticles. Biomaterials. 2007;28:2233–2243. doi: 10.1016/j.biomaterials.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 59.Sonaje K, Lin Y-H, Juang J-H, et al. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials. 2009;30:2329–2339. doi: 10.1016/j.biomaterials.2008.12.066. [DOI] [PubMed] [Google Scholar]
- 60.Adson A, Burton PS, Raub TJ, et al. Passive diffusion of weak organic electrolytes across Caco-2 cell monolayers: uncoupling the contributions of hydrodynamic, transcellular, and paracellular barriers. J Pharm Sci. 1995;84:1197–204. doi: 10.1002/jps.2600841011. [DOI] [PubMed] [Google Scholar]
- 61*.Sonaje K, Chuang E-Y, Lin K-J, et al. Opening of Epithelial Tight Junctions and Enhancement of Paracellular Permeation by Chitosan: Microscopic, Ultrastructural, and Computed-Tomographic Observations. Mol Pharm. 2012;9:1271–1279. doi: 10.1021/mp200572t. Recent study demonstrating the reversible opening of tight junctions by mucoadhesive materials and how size limitations allow biologics but not nanoparitcles to cross the epithelium. [DOI] [PubMed] [Google Scholar]
- 62.Sonaje K, Lin K-J, Wey S-P, et al. Biodistribution, pharmacodynamics and pharmacokinetics of insulin analogues in a rat model: Oral delivery using pH-Responsive nanoparticles vs subcutaneous injection. Biomaterials. 2010;31:6849–6858. doi: 10.1016/j.biomaterials.2010.05.042. [DOI] [PubMed] [Google Scholar]
- 63.Yeh T-H, Hsu L-W, Tseng MT, et al. Mechanism and consequence of chitosan-mediated reversible epithelial tight junction opening. Biomaterials. 2011;32:6164–6173. doi: 10.1016/j.biomaterials.2011.03.056. [DOI] [PubMed] [Google Scholar]
- 64.Sonaje K, Lin K-J, Tseng MT, et al. Effects of chitosan-nanoparticle-mediated tight junction opening on the oral absorption of endotoxins. Biomaterials. 2011;32:8712–8721. doi: 10.1016/j.biomaterials.2011.07.086. [DOI] [PubMed] [Google Scholar]
- 65.Cano-Cebrian MJ, Zornoza T, Granero L, Polache A. Intestinal absorption enhancement via the paracellular route by fatty acids, chitosans and others: a target for drug delivery. Curr Drug Deliv. 2005;2:9–22. doi: 10.2174/1567201052772834. [DOI] [PubMed] [Google Scholar]
- 66**.Bakhru SH, Furtado S, Morello AP, Mathiowitz E. Oral delivery of proteins by biodegradable nanoparticles. Adv Drug Deliv Rev. 2013;65:811–821. doi: 10.1016/j.addr.2013.04.006. Excellent review of the development of mucoadhesive materials for oral drug delivery. [DOI] [PubMed] [Google Scholar]
- 67.Jang MH, Kweon M-N, Iwatani K, et al. Intestinal villous M cells: an antigen entry site in the mucosal epithelium. Proc Natl Acad Sci U S A. 2004;101:6110–6115. doi: 10.1073/pnas.0400969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eldridge JH, Hammond CJ, Meulbroek JA, et al. Controlled vaccine release in the gut-associated lymphoid tissues. I. Orally administered biodegradable microspheres target the Peyer’s patches. J Controlled Release. 1990;11:205–214. [Google Scholar]
- 69.Des Rieux A, Fievez V, Garinot M, et al. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Controlled Release. 2006;116:1–27. doi: 10.1016/j.jconrel.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 70.Des Rieux A, Fievez V, Théate I, et al. An improved in vitro model of human intestinal follicle-associated epithelium to study nanoparticle transport by M cells. Eur J Pharm Sci. 2007;30:380–391. doi: 10.1016/j.ejps.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 71.Roger E, Lagarce F, Garcion E, Benoit J-P. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomed. 2010;5:287–306. doi: 10.2217/nnm.09.110. [DOI] [PubMed] [Google Scholar]
- 72.Slütter B, Hagenaars N, Jiskoot W. Rational design of nasal vaccines. J Drug Target. 2008;16:1–17. doi: 10.1080/10611860701637966. [DOI] [PubMed] [Google Scholar]
- 73.Clark M, Jepson MA, Hirst BH. Exploiting M cells for drug and vaccine delivery. Adv Drug Deliv Rev. 2001;50:81–106. doi: 10.1016/s0169-409x(01)00149-1. [DOI] [PubMed] [Google Scholar]
- 74.Porta C, James PS, Phillips AD, et al. Confocal analysis of fluorescent bead uptake by mouse Peyer’s patch follicle-associated M cells. Exp Physiol. 1992;77:929–932. doi: 10.1113/expphysiol.1992.sp003662. [DOI] [PubMed] [Google Scholar]
- 75.Brayden DJ. Oral vaccination in man using antigens in particles: current status. Eur J Pharm Sci. 2001;14:183–189. doi: 10.1016/s0928-0987(01)00175-0. [DOI] [PubMed] [Google Scholar]
- 76.Kammona O, Kiparissides C. Recent advances in nanocarrier-based mucosal delivery of biomolecules. J Controlled Release. 2012;161:781–94. doi: 10.1016/j.jconrel.2012.05.040. [DOI] [PubMed] [Google Scholar]
- 77.De Jesus M, Ostroff GR, Levitz SM, et al. A Population of Langerin-Positive Dendritic Cells in Murine Peyer’s Patches Involved in Sampling β-Glucan Microparticles. PLoS ONE. 2014;9:e91002. doi: 10.1371/journal.pone.0091002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Khafagy E-S, Morishita M, Onuki Y, Takayama K. Current challenges in non-invasive insulin delivery systems: a comparative review. Adv Drug Deliv Rev. 2007;59:1521–1546. doi: 10.1016/j.addr.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 79.Fievez V, Plapied L, des Rieux A, et al. Targeting nanoparticles to M cells with non-peptidic ligands for oral vaccination. Eur J Pharm Biopharm. 2009;73:16–24. doi: 10.1016/j.ejpb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 80.Jepson MA, Clark MA. Studying M cells and their role in infection. Trends Microbiol. 1998;6:359–65. doi: 10.1016/s0966-842x(98)01337-7. [DOI] [PubMed] [Google Scholar]
- 81.Miller H, Zhang J, KuoLee R, et al. Intestinal M cells: the fallible sentinels? World J Gastroenterol WJG. 2007;13:1477. doi: 10.3748/wjg.v13.i10.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pappo J, Ermak TH, Steger HJ. Monoclonal antibody-directed targeting of fluorescent polystyrene microspheres to Peyer’s patch M cells. Immunology. 1991;73:277. [PMC free article] [PubMed] [Google Scholar]
- 83.Jepson MA, Clark MA, Foster N, et al. Targeting to intestinal M cells. J Anat. 1996;189:507. [PMC free article] [PubMed] [Google Scholar]
- 84.Sass W, Dreyer HP, Seifert J. Rapid insorption of small particles in the gut. Am J Gastroenterol. 1990;85:255–60. [PubMed] [Google Scholar]
- 85.Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13:1838–1845. doi: 10.1023/a:1016085108889. [DOI] [PubMed] [Google Scholar]
- 86.Desai MP, Lab V, Walter E, et al. The mechanism of uptake of biodegradable microparticles in Caco-2 cells is size dependent. Pharm Res. 1997;14:1568–73. doi: 10.1023/a:1012126301290. [DOI] [PubMed] [Google Scholar]
- 87.Gabor F, Schwarzbauer A, Wirth M. Lectin-mediated drug delivery: binding and uptake of BSA-WGA conjugates using the Caco-2 model. Int J Pharm. 2002;237:227–239. doi: 10.1016/s0378-5173(02)00049-2. [DOI] [PubMed] [Google Scholar]
- 88.Gabor F, Wirth M, Jurkovich B, et al. Lectin-mediated bioadhesion: Proteolytic stability and binding-characteristics of wheat germ agglutinin and Solanum tuberosum lectin on Caco-2, HT-29 and human colonocytes. J Controlled Release. 1997;49:27–37. [Google Scholar]
- 89.Kilpatrick DC, Pusztai A, Grant G, et al. Tomato lectin resists digestion in the mammalian alimentary canal and binds to intestinal villi without deleterious effects. FEBS Lett. 1985;185:299–305. doi: 10.1016/0014-5793(85)80927-3. [DOI] [PubMed] [Google Scholar]
- 90.Hussain N, Jani PU, Florence AT. Enhanced oral uptake of tomato lectin-conjugated nanoparticles in the rat. Pharm Res. 1997;14:613–618. doi: 10.1023/a:1012153011884. [DOI] [PubMed] [Google Scholar]
- 91.Weissenboeck A, Bogner E, Wirth M, Gabor F. Binding and uptake of wheat germ agglutinin-grafted PLGA-nanospheres by caco-2 monolayers. Pharm Res. 2004;21:1917–1923. doi: 10.1023/b:pham.0000045247.09724.26. [DOI] [PubMed] [Google Scholar]
- 92.Lochner N, Pittner F, Wirth M, Gabor F. Wheat germ agglutinin binds to the epidermal growth factor receptor of artificial Caco-2 membranes as detected by silver nanoparticle enhanced fluorescence. Pharm Res. 2003;20:833–839. doi: 10.1023/a:1023406224028. [DOI] [PubMed] [Google Scholar]
- 93.Clark M, Hirst BH, Jepson MA. Lectin-mediated mucosal delivery of drugs and microparticles. Adv Drug Deliv Rev. 2000;43:207–223. doi: 10.1016/s0169-409x(00)00070-3. [DOI] [PubMed] [Google Scholar]
- 94.Bies C, Lehr C-M, Woodley JF. Lectin-mediated drug targeting: history and applications. Adv Drug Deliv Rev. 2004 Mar;56:425–435. doi: 10.1016/j.addr.2003.10.030. [DOI] [PubMed] [Google Scholar]
- 95.Roger E, Kalscheuer S, Kirtane A, et al. Folic Acid Functionalized Nanoparticles for Enhanced Oral Drug Delivery. Mol Pharm. 2012;9:2103–2110. doi: 10.1021/mp2005388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Petrus AK, Fairchild TJ, Doyle RP. Traveling the Vitamin B12 Pathway: Oral Delivery of Protein and Peptide Drugs. Angew Chem Int Ed. 2009;48:1022–1028. doi: 10.1002/anie.200800865. [DOI] [PubMed] [Google Scholar]
- 97.Fowler R, Vllasaliu D, Trillo FF, et al. Nanoparticle Transport in Epithelial Cells: Pathway Switching Through Bioconjugation. Small. 2013;9:3282–94. doi: 10.1002/smll.201202623. [DOI] [PubMed] [Google Scholar]
- 98.Chalasani KB, Russell-Jones GJ, et al. Effective oral delivery of insulin in animal models using vitamin B12-coated dextran nanoparticles. J Controlled Release. 2007;122:141–150. doi: 10.1016/j.jconrel.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 99*.Chalasani KB, Russell-Jones GJ, Yandrapu SK, et al. A novel vitamin B12-nanosphere conjugate carrier system for peroral delivery of insulin. J Controlled Release. 2007;117:421–429. doi: 10.1016/j.jconrel.2006.12.003. First studies using nanoparticles targeted to the vitamin B12 pathway for oral insulin delivery. [DOI] [PubMed] [Google Scholar]
- 100.Yoshida M, Claypool SM, Wagner JS, et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004;20:769–783. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 101.Claypool SM, Dickinson BL, Wagner JS, et al. Bidirectional transepithelial IgG transport by a strongly polarized basolateral membrane Fcγ-receptor. Mol Biol Cell. 2004;15:1746–1759. doi: 10.1091/mbc.E03-11-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Israel EJ, Taylor S, Wu Z, et al. Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology. 1997;92:69–74. doi: 10.1046/j.1365-2567.1997.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raghavan M, Gastinel LN, Bjorkman PJ. The class I major histocompatibility complex related Fc receptor shows pH-dependent stability differences correlating with immunoglobulin binding and release. Biochemistry (Mosc) 1993;32:8654–8660. doi: 10.1021/bi00084a037. [DOI] [PubMed] [Google Scholar]
- 104.Lalezari D. Gastrointestinal pH profile in subjects with irritable bowel syndrome. Ann Gastroenterol. 2012;25:333. [PMC free article] [PubMed] [Google Scholar]
- 105.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 106*.Pridgen EM, Alexis F, Kuo TT, et al. Transepithelial Transport of Fc-Targeted Nanoparticles by the Neonatal Fc Receptor for Oral Delivery. Sci Transl Med. 2013;5:213ra167. doi: 10.1126/scitranslmed.3007049. First studies using nanoparticles targeted to the FcRn transcytosis pathway for oral delivery, enabling biologics and nanoparticles to cross the epithelium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cochran E, Musso C, Gorden P. The use of U-500 in patients with extreme insulin resistance. Diabetes Care. 2005;28:1240–1244. doi: 10.2337/diacare.28.5.1240. [DOI] [PubMed] [Google Scholar]
- 108.Oostendorp RL, Huitema A, Rosing H, et al. Coadministration of Ritonavir Strongly Enhances the Apparent Oral Bioavailability of Docetaxel in Patients with Solid Tumors. Clin Cancer Res. 2009;15:4228–4233. doi: 10.1158/1078-0432.CCR-08-2944. [DOI] [PubMed] [Google Scholar]
- 109.Attili-Qadri S, Karra N, Nemirovski A, et al. Oral delivery system prolongs blood circulation of docetaxel nanocapsules via lymphatic absorption. Proc Natl Acad Sci U S A. 2013 Oct 22;110:17498–17503. doi: 10.1073/pnas.1313839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bisht S, Feldmann G, Koorstra J-BM, et al. In vivo characterization of a polymeric nanoparticle platform with potential oral drug delivery capabilities. Mol Cancer Ther. 2008 Dec 3;7:3878–3888. doi: 10.1158/1535-7163.MCT-08-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arbit E. The physiological rationale for oral insulin administration. Diabetes Technol Ther. 2004;6:510–517. doi: 10.1089/1520915041705929. [DOI] [PubMed] [Google Scholar]
- 112.Quellhorst E. Insulin therapy during peritoneal dialysis: pros and cons of various forms of administration. J Am Soc Nephrol. 2002;13:S92–S96. [PubMed] [Google Scholar]
- 113.Brogden RN, Heel RC. Human insulin. A review of its biological activity, pharmacokinetics and therapeutic use. Drugs. 1987;34:350–371. doi: 10.2165/00003495-198734030-00003. [DOI] [PubMed] [Google Scholar]
- 114.Jongen PJ, Hengstman G, Hupperts R, et al. Drug adherence and multidisciplinary care in patients with multiple sclerosis: Protocol of a prospective, web-based, patient-centred, nation-wide, Dutch cohort study in glatiramer acetate treated patients (CAIR study) BMC Neurol. 2011;11:40. doi: 10.1186/1471-2377-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kondiah PPD, Tomar LK, Tyagi C, et al. A novel pH-sensitive interferon-β (INF-β) oral delivery system for application in multiple sclerosis. Int J Pharm. 2013;456:459–472. doi: 10.1016/j.ijpharm.2013.08.038. [DOI] [PubMed] [Google Scholar]
- 116.Guggi D, Kast CE, Bernkop-Schnürch A. In vivo evaluation of an oral salmon calcitonin-delivery system based on a thiolated chitosan carrier matrix. Pharm Res. 2003;20:1989–1994. doi: 10.1023/b:pham.0000008047.82334.7d. [DOI] [PubMed] [Google Scholar]
- 117.Cetin M, Aktas MS, Vural I, Ozturk M. Salmon calcitonin-loaded Eudragit® and Eudragit®-PLGA nanoparticles: in vitro and in vivo evaluation. J Microencapsul. 2012;29:156–166. doi: 10.3109/02652048.2011.635426. [DOI] [PubMed] [Google Scholar]
- 118.Italia JL, Bhardwaj V, Ravi Kumar MNV. Disease, destination, dose and delivery aspects of ciclosporin: the state of the art. Drug Discov Today. 2006;11:846–854. doi: 10.1016/j.drudis.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 119.Italia JL, Bhatt DK, Bhardwaj V, et al. PLGA nanoparticles for oral delivery of cyclosporine: Nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral®. J Controlled Release. 2007;119:197–206. doi: 10.1016/j.jconrel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 120.Lamprecht A. Nanoparticles Enhance Therapeutic Efficiency by Selectively Increased Local Drug Dose in Experimental Colitis in Rats. J Pharmacol Exp Ther. 2005;315:196–202. doi: 10.1124/jpet.105.088146. [DOI] [PubMed] [Google Scholar]
- 121.Nakase H, Okazaki K, Tabata Y, et al. An oral drug delivery system targeting immune-regulating cells ameliorates mucosal injury in trinitrobenzene sulfonic acid-induced colitis. J Pharmacol Exp Ther. 2001;297:1122–1128. [PubMed] [Google Scholar]
- 122.Wachsmann P, Lamprecht A. Polymeric Nanoparticles for the Selective Therapy of Inflammatory Bowel Disease. Methods Enzym. 2012;508:377–397. doi: 10.1016/B978-0-12-391860-4.00019-7. [DOI] [PubMed] [Google Scholar]
- 123.Lamprecht A, Schäfer U, Lehr C-M. Size-dependent bioadhesion of micro-and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18:788–793. doi: 10.1023/a:1011032328064. [DOI] [PubMed] [Google Scholar]
- 124.Lamprecht A, Ubrich N, Yamamoto H, et al. Biodegradable nanoparticles for targeted drug delivery in treatment of inflammatory bowel disease. J Pharmacol Exp Ther. 2001;299:775–781. [PubMed] [Google Scholar]
- 125.Pertuit D, Moulari B, Betz T, et al. 5-amino salicylic acid bound nanoparticles for the therapy of inflammatory bowel disease. J Controlled Release. 2007;123:211–218. doi: 10.1016/j.jconrel.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 126.Laroui H, Viennois E, Xiao B, et al. Fab’-bearing siRNA TNFα-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J Controlled Release. 2014;186:41–53. doi: 10.1016/j.jconrel.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Moulari B, Béduneau A, Pellequer Y, Lamprecht A. Lectin-decorated nanoparticles enhance binding to the inflamed tissue in experimental colitis. J Controlled Release. 2014;188:9–17. doi: 10.1016/j.jconrel.2014.05.046. [DOI] [PubMed] [Google Scholar]
- 128**.Wilson DS, Dalmasso G, Wang L, et al. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater. 2010;9:923–928. doi: 10.1038/nmat2859. Excellent example of how insight into the pathophysiology of a disease and polymer engineering can be combined to develop a smart nanoparticle for targeted drug release. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roy K, Mao H-Q, Huang S-K, Leong KW. Oral gene delivery with chitosan–DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;5:387–391. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 130.Chew JL, Wolfowicz CB, Mao H-Q, et al. Chitosan nanoparticles containing plasmid DNA encoding house dust mite allergen, Der p 1 for oral vaccination in mice. Vaccine. 2003;21:2720–2729. doi: 10.1016/s0264-410x(03)00228-7. [DOI] [PubMed] [Google Scholar]
- 131.Li GP, Liu ZG, Liao B, Zhong NS. Induction of Th1-type immune response by chitosan nanoparticles containing plasmid DNA encoding house dust mite allergen Der p 2 for oral vaccination in mice. Cell Mol Immunol. 2009;6:45–50. doi: 10.1038/cmi.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eldridge JH, Meulbroek JA, Staas JK, et al. Vaccine-containing biodegradable microspheres specifically enter the gut-associated lymphoid tissue following oral administration and induce a disseminated mucosal immune response. Adv Exp Med Biol. 1989;251:191–202. doi: 10.1007/978-1-4757-2046-4_18. [DOI] [PubMed] [Google Scholar]
- 133.Zhu Q, Talton J, Zhang G, et al. Large intestine–targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nat Med. 2012;18:1291–1296. doi: 10.1038/nm.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gebert A. The role of M cells in the protection of mucosal membranes. Histochem Cell Biol. 1997;108:455–470. doi: 10.1007/s004180050186. [DOI] [PubMed] [Google Scholar]
- 135.Martín MG, Wu SV, Walsh JH. Ontogenetic development and distribution of antibody transport and Fc receptor mRNA expression in rat intestine. Dig Dis Sci. 1997 May;42:1062–1069. doi: 10.1023/a:1018853506830. [DOI] [PubMed] [Google Scholar]