

Summary
Secondary lymphoid tissues share the important function of bringing together antigens and rare antigen-specific lymphocytes to foster induction of adaptive immune responses. Peyer’s patches (PPs) are unique compared to other secondary lymphoid tissues in their continual exposure to an enormous diversity of microbiome- and food-derived antigens and in the types of pathogens they encounter. Antigens are delivered to PPs by specialized microfold (M) epithelial cells and they may be captured and presented by resident dendritic cells (DCs). In accord with their state of chronic microbial antigen exposure, PPs exhibit continual germinal center (GC) activity. These GCs contribute to the generation of B cells and plasma cells producing somatically mutated gut antigen-specific IgA antibodies, but have also been suggested to support antigen-nonspecific diversification of the B cell repertoire. Here we review current understanding of how PPs foster B cell encounters with antigen, how they favor isotype switching to the secretory IgA isotype, and how their GC responses may uniquely contribute to mucosal immunity.
Keywords: B Cells, Dendritic Cells, Antibodies, Cell Trafficking, Repertoire Development, Mucosa
Introduction
Intestinal antibody has a crucial role in neutralization of orally invading pathogens, including viruses such as poliovirus and rotavirus and bacteria such as Cholera and Salmonella (1, 2). Recent advances in studying the intestinal microbiome have revealed critical influences of mucosal antibody on the host-commensal symbiosis (3). Given these broad ranging functions it is perhaps not surprising that IgA, the major mucosal immunoglobulin (Ig) isotype, is the most abundantly produced antibody in the body (4, 5). IgA is secreted in a dimeric form by plasma cells that are distributed throughout the small intestinal, and to a lesser extent large intestinal, lamina propria (LP) and it is transported into the intestinal lumen by the epithelial polymeric IgA receptor (polyIgR). Intestinal IgA producing cells can arise from a number of origins, including from B cells within mesenteric lymph nodes (LNs), spleen and intestinal isolated lymphoid follicles (ILFs), but Peyer’s patches (PPs) are the major source.
Peyer’s patches were named after Johann Conrad Peyer who described them in 1673 as ‘elevated areas composed of lymph nodules in the mucous membrane of the small intestine’, though they had been reported in earlier studies (reviewed in (6)). Distributed along the length of the small intestine, they number 100–200 in humans and 6–12 in mice (6, 7). PPs are organized into three major regions: a series of large B cell follicles; the overlying follicle associated epithelium (FAE) and associated sub-epithelial dome (SED) that lies between the follicles and the FAE, and; the small T cell zones that are situated adjacent to the B cell follicles (Fig. 1). A special property of the FAE is the presence of modified epithelial cells termed M cells that bind many luminal antigen types and transcytose them to the SED. As well as containing blood vessels, PPs have a rich content of lymphatic vessels that are used as lymphocyte and plasma cell exit portals.
Figure 1. Cross-sectional view of mouse Peyer’s patch showing main anatomical compartments.
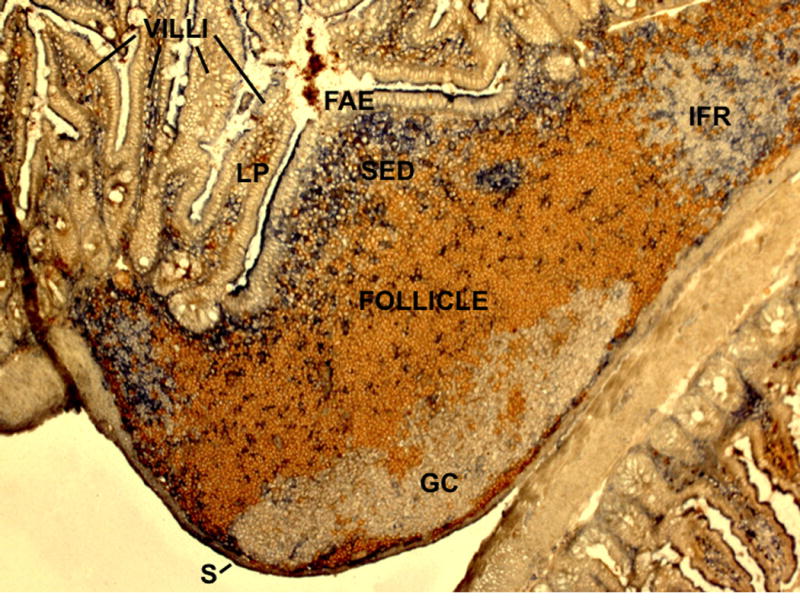
Stained to detect naïve B cells (IgD, brown) and dendritic cells (CD11c, blue). FAE, follicle associated epithelium; SED, subepithelial dome; GC, germinal center; IFR, interfollicular region (T cell zone); LP, lamina propria; S, serosa. During embedding of the tissue for preparation of the 7 μm frozen sections the PP became juxtaposed to another loop of small intestine (bottom right of the image).
PPs are continually exposed to mucosally-derived antigens and their follicles almost universally contain preformed germinal centers (GCs), sites of Ig gene somatic hypermutation (SHM) and B cell selection (8). The importance of the microbiome in promoting these responses is demonstrated by the much smaller tissue size and absence of GCs in PPs from germ-free mice (9–11). Many of the B cells present within PPs of conventionally housed animals have undergone isotype switching from IgM to IgA, and PPs give rise to IgA+ plasma cells, in most cases carrying somatic mutations in their Ig-genes, that home selectively to the intestinal LP.
While the importance of PPs in mucosal immunity is well appreciated, the specialized mechanisms by which these structures support the induction of B cell responses is less understood than for LNs and spleen. This is a significant dearth of knowledge given the important role of PP-derived IgA in host defense and in shaping properties of the microbiome. Here we review current understanding of PP development and organization, and we discuss how B cell antigen encounter may take place in these organs. We then consider what is currently known about the induction of IgA switching in PPs and discuss the properties and functions of PP GCs, including their possible role in antigen non-specific antibody diversification. Finally, we summarize properties of PP-derived IgA+ memory and plasma cell responses.
Peyer’s patch development and lymphocyte trafficking
Fetal and neonatal PP development
Peyer’s patch development in mice begins around embryonic day 12.5–13.5 with the appearance of hematopoietic cells in the gut (12, 13). Human PP development also begins quite early in gestation and the reader is referred to Heel et al., (6) for a detailed description of this ontogeny. The first evidence of cellular organization during mouse PP induction is the appearance at E15.5–16.5 of VCAM1+ spots distributed at intervals along the anti-mesenteric side of the intestine. Induction of these PP anlagen is promoted by two types of hematopoietic cells, cKit+CD11c+Ret+ lymphoid tissue initiator (LTin) cells and ID2+RORγt+IL7Rα+ lymphoid tissue inducer (LTi) cells, also known as type-3 innate lymphoid cells (ILC3). A current model posits that RET signaling initiates PP formation by promoting VCAM1-dependent adhesion of LTin cells and their signaling to lymphoid tissue organizer (LTo) stromal cells to induce increased adhesion molecule (VCAM1, ICAM1, MADCAM1) and chemokine (CXCL13, CCL19, CCL21) expression. This then causes accumulation of the IL7-dependent LTi cells that trigger via LTβR signaling further expression of adhesion molecules and chemokines (12, 14). Recent work has found that a fetal Id2-dependent Arg1+ ILC precursor that expressed a transitional developmental phenotype (ftILCP) is the first appearing ILC type in the intestine (15). These cells are able to give rise to ILC3s as well as some other ILC types, and they accumulate at the earliest sites of cell clustering during PP development in a LTα1β2- and RORγt-dependent manner (15). These ILC precursor cells are suggested to act as a local source of LTi cells during PP development. Retinoic acid controls LTi cell maturation, at least in part by promoting RORγt expression and retinoic acid receptor or vitamin A deficiency is associated with defects in PP development (16). In contrast to LTi cells, the fILCP cells do not express CXCR5 and CCR7, leaving it unclear how their clustering is promoted.
Lymphocytes begin arriving in the nascent mouse PPs in the first few days after birth (17) and B cells make an important contribution to the growth and maturation of PPs (10). LTi cells also have a role in promoting organization of postnatal PPs, most likely by LTα1β2 signaling to stromal cells, and when LTi cell function is disrupted in the first days after birth, PPs fail to develop organized follicles and interfollicular T zones (17). This organizing activity of LTi cells is associated with a switch from CXCR5 expression and localization in CXCL13+ nascent follicles, to CXCR4 expression and positioning in nascent interfollicular T zones (17). TGFβ signaling promotes induction of CXCR4 on PP LTi cells, but the signals downregulating CXCR5 remain unclear.
While PPs and LNs share the requirement of LTα1β2 for their development, a second TNF family cytokine that is essential for LN development, TNFSF11 (RANKL, TRANCE), is not necessary for the early steps of PP development (13). However, TNFSF11-deficient mice do show a reduced size of PP (18). In situ staining revealed TNFSF11 in low amounts at E17.5, but more strongly at birth, in an area beneath the FAE of the developing PP (19, 20). Induction of M cells critically depends on TNFSF11 and PP M cells are severely reduced in TNFSF11-deficient or blocked mice (10, 21). TNFSFR11 is expressed by epithelial cells and the TNFSF11+ stromal cells below the FAE may be involved in triggering M cell differentiation. How B cells contribute to M cell maturation is unclear, though this does not appear to require TNF or LT-expression by the B cells, factors that are important for follicular stromal cell development (10, 22). Microbial stimuli can also increase the number of M cells and consistent with this, M cell deficiency in TNFSF11-deficient mice is less pronounced toward the distal end of the small intestine where the bacterial load is heavier. In accord with the specialized antigen-transport function of M cells (discussed below), TNFSF11 deficient mice have fewer GCs in their PP follicles and lower levels of fecal IgA (21).
Lymphocyte entry, transit and exit
Lymphocytes enter PPs from the bloodstream via HEVs (23). B cells are more frequent in PPs than in LNs and this reflects at least in part the specialized properties of PP HEVs. Like in LNs, many PP HEVs are situated in T cell-rich interfollicular zones. However, PPs also have HEVs located within follicles. PP HEVs have high expression of MADCAM1, the ligand for the α4β7 integrin. This integrin is more abundant on naive B cells than on naïve T cells and is one factor that contributes to the more efficient entry of B cells than T cells to PPs (23). Chemokine mediated attachment to PP HEVs involves CCL21 and CXCL12 as in LNs, but additionally the HEV within PP follicles display CXCL13 and selectively support the attachment and entry of B cells (24).
The organization of PPs into follicles and T zone is mediated by CXCR5-CXCL13 and CCR7-CCL21, respectively (25). The oxysterol receptor, EBI2, allows B cells to distinguish between the outer and center follicle in lymphoid tissues, though the significance of this guidance information in PPs is not known beyond the importance of EBI2 downegulation in fostering GC B cell clustering at the follicle center (26–28). Sphingosine-1-phosphate receptor-2 (S1PR2) induction also contributes to GC B cell clustering (29). The factors promoting positioning of DCs in the SED region of the PPs are not well defined. Although CCR6 was implicated in this process and the CCR6 ligand, CCL20, is made abundantly by FAE (30, 31), subsequent studies (32) including our own recent work (in preparation) have not provided support for this requirement. M cell-derived CCL9 may have a role, though deficiency in the CCL9 receptor CCR1 did not affect PP DC distribution (32, 33), making it likely that additional organizing cues will be involved. The FAE also produces CXCL16 and this may have a role in recruiting CXCR6-expressing activated or effector T cells to the SED (34).
B and T cell egress from PPs shows a similar dependence on sphingosine-1-phosphate receptor-1 (S1PR1) as egress from LNs and ablation of sphingosine kinase-1 (Sphk1) from lymphatic endothelial cells in Sphk2−/− mice causes a block in cell appearance in PP lymphatic sinuses (35, 36). Using mice expressing photoconvertible fluorescent proteins and newly developed PP photoconversion procedures, the transit time of B cells through PPs (t1/2~10hrs) was found to be shorter than through LNs (t1/2~16hrs) (37–39). This might in part reflect a distinct action of CXCR4 in guiding cell movements in PPs versus LNs. The CXCR4 ligand, CXCL12, is abundantly produced by stromal cells in the vicinity of subserosal lymphatic vessels and guides B cells to this peri-lymphatic location (38). This presumably then facilitates their encounter with lymphatic vessels, allowing S1PR1 signaling to trigger egress. Expression of the CXCR5 ligand, CXCL13, by contrast, is restricted to follicular regions and consistent with follicles being separated from lymphatic sinuses, CXCR5 promotes PP retention of B cells (38). The significance of transiting PPs more rapidly than LNs is unclear but might relate both to the more rapid access of entering cells to follicles, and the more ubiquitous presence of antigen perhaps limiting the scan time that is needed.
As in LNs, the lymphocyte activation marker CD69, a negative regulator of S1PR1 (40), can inhibit lymphocyte egress from PPs (39). Thus CD69 upregulation on B and T lymphocyte might cause their retention in an inflamed PP, promoting local cell accumulation and increased chance of antigen-specific lymphocytes being present and encountering the stimulus antigen. Elegant recent work has shown that additional mechanisms control cell egress from PPs. Salmonella infection caused egress shutdown and hypertrophy of PPs independently of type I IFNR and CD69. This egress block was persistent over multiple days and did not involve decreased CXCR4 expression (39). Further work will be needed to define the mechanism, but one possibility might involve regulation of local lymphatic flow (41). The chronic nature of this egress blockade suggests it may have functions beyond promoting antigen encounter, perhaps ensuring appropriate local maturation of effector or memory cells, but possibly also helping restrict spread of an invading pathogen or its toxins.
B cell antigen encounter
What we know in lymph nodes
B cell antigen encounter in LNs has been a topic of investigation for many years and a number of pathways have been defined. Lymph-borne antigens enter LNs via afferent lymphatics that connect with the subcapsular sinus (SCS) at locations overlying follicles. Small soluble antigens (~70kd or a few nm in hydrodynamic radius) can gain free access to the follicle or travel in via stromal cell-ensheathed collagen fibers (conduits) for direct B cell encounter (42, 43). Larger antigens do not have free access to follicles due to the filtering function of the lymphatic cells lining the SCS. A range of particle types as well as opsonized antigens can become bound and displayed by specialized (CD169+) SCS macrophages that extend between the sinus and underlying follicle (43). Cognate B cells can capture antigens from the tail processes of these macrophages. Alternatively, noncognate B cells pickup opsonized complexes using complement receptors and deliver them to FDCs for longer-term display (44). In some cases, antigens may be transported into interfollicular regions by migratory DCs and then become displayed for B cell antigen encounter (45). Some lymph-borne antigens elude capture by SCS macrophages but instead become bound by sinus-exposed DCs and these cells may deliver antigens to B cells (46). There is also evidence that B cells migrate through the macrophage-rich LN medullary regions and they may become exposed to antigen in this location (47). Finally, many B cells are expected to encounter antigens on FDCs since these cells serve as long-term reservoirs of opsonized antigens (22, 48, 49). The existence of multiple modes of antigen capture and display in LNs suggests that multiple mechanisms may exist to foster B cell antigen encounter within PPs where this process has been less studied.
M cells and antigen uptake in PPs
Antigen entry to PPs occurs predominantly via M cells (Fig. 2). These specialized epithelial cells can associate with and transcytose a range of antigen types including various bacteria, viruses, fungi, cholera toxin, horseradish peroxidase, immune complexes, prions, and inert particles (latex beads) (50–52). Their ability to interact with particulate antigens is promoted by a low glycocalyx covering, by their having short and irregular microvilli and a flattened surface and by being located in pits. However, the receptor systems allowing the breadth of antigen uptake are not clear. M cells do not express the polyIgR but are somehow able to bind IgA, though not other Ig isotypes, and IgA coating enhances the efficiency with which various antigens are captured and transcytosed (52–54). M cell-expressed Dectin-1 has been implicated in transcytosis of glycosylated sIgA-antigen complexes (55), though it was not required for uptake of IgA coated bacteria (53). M cells express glycoprotein-2 (GP2), recognizing FimH (of type I pili) that is present on a subset of commensal and pathogenic enterobacteria (E.coli, S.typhimurium) and deficiency in FimH or GP2 leads to defects in bacterial transcytosis (56). They also have unique surface glycosylation, allowing their detection by specific lectins (51, 52) and these sugars may facilitate binding of some microbes. Their expression of glycolipid GM1 supports binding of cholera toxin (50).
Figure 2. Diagram highlighting cellular complexity of the PP subepithelial dome (SED) and showing several possible mechanisms of B cell antigen exposure.
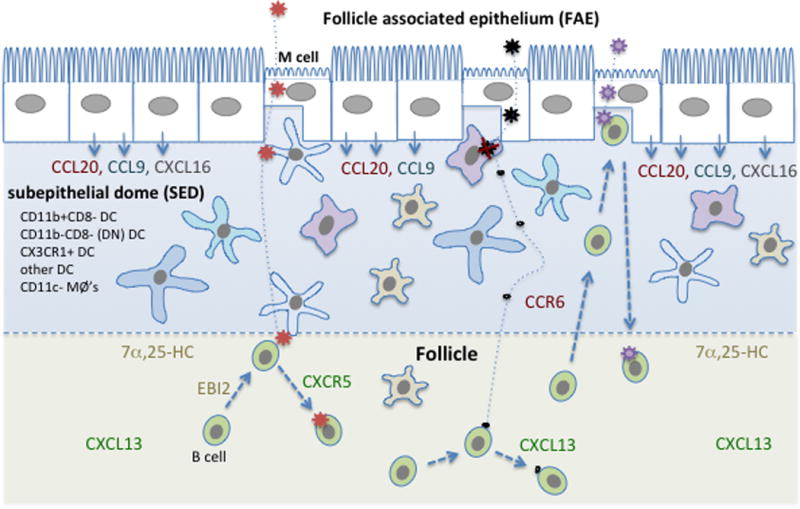
Major myeloid cell types present in the SED are listed and represented by different cell shapes. Distribution of major chemoattractant factors is indicated. Stromal cells, which include the cells that produce the CXCR5 ligand CXCL13 and the EBI2 ligand, 7α,25-HC, are not shown. Three example antigens are shown entering the SED after M cell-mediated transcytosis. The red antigen then travels via DC to reach the interface with the follicle. B cell movement to this interface is favored by EBI2 responding to 7α,25-HC. The black antigen is taken up by a macrophage and largely degraded, but some small fragments travel away passively and reach follicular B cells. The purple antigen is encountered directly by a B cell that is suggested to have accessed the SED and M cell pocket in a CCR6-CCL20 dependent manner, likely a recently activated or memory B cell.
On their basolateral side, M cells have large cell-containing pockets (51, 52). A range of cell types have been reported in M cell pockets, including IgD+ and IgD− B cells, T cells, DCs and macrophages (6, 57–60), though the small size of the pockets and irregular distribution of M cells in the FAE has made it difficult to obtain quantitative data on the associated cell types. Stimulation by peptidoglycan or LPS increases the uptake of antigens by M cells (61). These stimuli also increase DC association with the FAE, including the density of DCs that associate with M cells and project processes across the FAE, and this may augment or modify antigen uptake.
Complexities of the subepithelial dome (SED)
Once antigens have been delivered into the PP they seem to rapidly disperse through the SED (Fig. 2). The cells involved in this process are not clear but it might be anticipated based on the LN work that different cell types in the SED contribute to the display of different antigen types. SED contain a high density of DCs (Figs. 1 and 2), particularly CD11b+CD8− DCs and CD11b−CD8− DN DCs (62), lower densities of B cells and macrophages as well as a scattering of T cells and RORγt+ ILCs ((63) and unpubl. obs.). CX3CR1+ DCs have been reported associated with the FAE (60). It seems possible that DCs help deliver certain antigens from the SED into the adjacent follicles and it is notable that PP follicles have a higher density of CD11c+ cells than usually observed in LN and splenic follicles ((64) and unpubl. obs.). However, as pre-GC and memory B cells in PPs can express CD11c (63, 65) more work needs to be done to quantitate the density of DCs in PP follicles and to establish whether they act as antigen transport cells. A central role for SED DCs in capturing transcytosed antigens has been observed in a number of studies. More than 90% of translocated E.coli were captured by DCs (56). CD11b+ lysozyme+ DCs were involved in the uptake of Salmonella and dead epithelial cells including M cells (66). SIgA coated Shigella flexneri was bound and internalized by CD11b+ DCs (54). Reo virus-containing apoptotic epithelial cell fragments were detected within DN DCs (67) and apoptotic epithelial cells were detected within OX41− DCs in rat PPs (68). Candida albicans and β-glucan particles that reached the SED were selectively associated with langerin+ DCs (69). Translocated prion-containing particles were taken up by ferritin+ cells in the SED that were suggested to be macrophages (70). In some cases, DCs bearing transcytosed antigen were reported to move from the SED to T cell areas after exposure to maturation stimuli such as cholera toxin or live Salmonella (52). As well as presenting MHC-peptide complexes to T cells, these DCs might make antigen available for B cell encounter at the follicle-T zone interface.
Although IgD+ B cells have been observed in association with M cell pockets (51), whether sufficient numbers of naïve B cells travel to the SED and interact with M cells to provide a chance of encounter with specific antigen is not known, but given the rarity of antigen-specific B cells in the naïve compartment, this seems unlikely. Moreover, whether the pocket-associated IgD+ cells are all naïve or not is unclear. Our recent findings have shown that CCR6 function is critical for B cell access to the SED and likely to M cell pockets, and CCR6 function was upregulated on activated B cells but was minimal on naïve B cells (unpubl. obs.). However, there are cells with properties of memory B cells (such as being IgA+) in the SED (58). These cells are likely to have been selected for recognition of antigens of the type that enter into the SED and antigen pass-off from M cell to memory B cell seems possible. Direct encounter between transcytosed antigen (horseradish peroxidase) and pocket-lymphocytes has been observed in electron microscopy studies (71), but the type of lymphocyte and the antigen’s subsequent fate are unknown from these experiments.
Reaching the follicles
Molecules below ~70kD in size are able to access LNs via conduits and molecules the size of hen egg lysozyme (~10kD) may gain direct access to the LN follicle (72, 73). One study showed that larger complexes could become proteolyzed by the time of arrival in the LN subcapsular sinus, thereby allowing their direct access to the follicle (74). It seems likely that a range of small molecules or fragments of digested larger molecules will be delivered by M cells into the SED to then gain direct access to the underlying follicle by diffusion. Whether conduits travel between the SED and follicle has not been determined.
PP GCs contain well-developed FDC networks that are orientated toward the SED. FDCs retain and display antigens using complement and Fc receptors. As well as their role in GCs, FDCs in LN primary follicles display antigen for encounter by recirculating naïve B cells (48, 49); whether FDCs are an important site of antigen encounter by naïve B cells in PPs is not known but seems likely. Yet, how antigens reach these specialized antigen-presenting cells in PPs and the requirements for their retention and display are poorly defined. In LNs many complex antigens enter the SCS pre-coated in C3b due to activation of the complement cascade. This allows their complement receptor-mediated capture by non-cognate follicular B cells and delivery to FDCs (44). It is unlikely that antigens are coated in complement in the intestinal lumen and currently there is little evidence indicating that antigens become complement coated after they arrive in the SED, though this possibility merits further analysis. Complement components C1q, C4b and C3b have been detected on the surface of PP FDCs (75–77) though whether this reflects deposition of complement-coated mucosally-derived versus circulating antigens has not been established.
An antibody against an epithelial marker stained PP FDC in mice, leading to the suggestion that epithelial cell-derived exosomes may be deposited on FDCs as a mechanism of antigen display (70). In a possibly related observation, epithelial cell-derived carbonic anhydrase positive membrane particles were observed within follicles in sheep ileal PPs (78). Given that C1q can bind apoptotic membranes (79), it seems possible that complement is involved in FDC display of epithelial cell-derived vesicles. FDCs in PPs express the IgA- and IgM-binding Fcα/μR, though whether it contributes to immune complex retention is not known (80); contrary to this notion, absence of the receptor was associated with prolonged retention of certain antigens on splenic FDCs (81). PP FDCs abundantly express MADCAM1 (82). A recent study from our group on LN responses showed that integrins are not critical for GC responses to model protein antigens, but the work did reveal a reduced ability of integrin-deficient B cells to participate in chronic GC responses in MLNs (83) making it possible that integrin ligands on FDCs contribute to the efficiency of antigen acquisition by PP B cells.
We have recently observed that B cell access to the SED is strongly CCR6 dependent, and in vitro experiments suggest that BCR-activated but not naïve B cells respond to the CCR6 ligand, CCL20 (in preparation). Whether stimuli other than BCR engagement, for example ligands for pattern recognition receptors, can promote this migration has yet to be determined, but if it were the case, this might be a mechanism promoting B cell antigen encounter. EBI2 has a prominent ability to favor naïve B cell positioning in the outer follicle in a location abutting the SED (26, 28) making it important to assess whether EBI2 expression augments PP B cell antigen encounter (Fig. 2).
IgA isotype switching
Key requirements in PPs
Seminal work in rabbits established PPs as important follicular inductive sites for intestinal IgA production (84). The molecular factors that guide follicular B cells to differentiate to IgA+ cells are both intrinsic and extrinsic. Expression of activation-induced cytidine deaminase (AID) is required in B cells since this enzyme promotes class switch recombination (CSR) and affinity maturation through somatic hypermutation (SHM) (85). Environmental cues present in PPs, including cytokines, regulate the ability of B cell to undergo CSR, the most critical being transforming growth factor β (TGFβ) (Fig. 3). TGFβ induces germline Ig α-transcripts in B cells, initiating IgA CSR, and its deficiency, as well as the deficiency for TGFβ receptor II (TGFβRII) on B cells, virtually abrogates the production of IgA+ B cells (86–88). A key source of TGFβ is the B cells themselves (89), though T cells, FDCs and DCs may also produce this cytokine in the PP (4). TGFβ is made in a latent form and PP FDCs have been suggested to promote its activation through production of matrix metalloproteases, but this has not yet been demonstrated in vivo (90). In other studies, αvβ8 integrins on DCs are required for TGFβ activation during intestinal regulatory T cell responses (91). Further work is needed to establish the mechanisms responsible for TGFβ activation during IgA switching in PPs.
Figure 3. Factors promoting IgA switching in PPs.
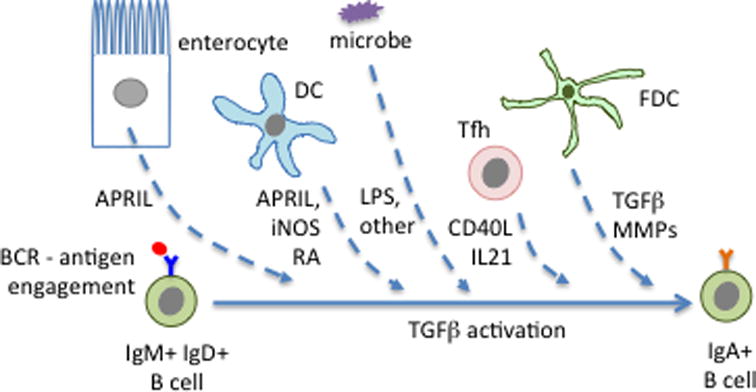
The highlighted factors are known to be produced by the indicated cell types but in several cases it has not yet been established which is the key cellular source of the factor and whether the action on the B cell is direct or indirect. Microbe-derived ligands for pattern recognition receptors, such as LPS, may also promote switching. See text for further details.
Consistent with the TGFβ requirement, intestinal DCs express inducible-nitric-oxide-synthase (iNOS) and this augments IgA CSR in PPs through a mechanism suggested to involve upregulation of TGFβRII on B cells (92). Although in vitro studies indicate NO can promote TGFβRII mRNA expression by naïve B cells in a PI3K and AKT-dependent manner, whether NO acts directly on naïve B cells in vivo or indirectly through changes in other cells remains undefined. PP DCs and epithelial cells are a source of a proliferation inducing ligand (APRIL) and mice deficient in APRIL and some humans with mutations in the receptor for APRIL (TACI), are characterized by reduced IgA production (93, 94). B cell-activating factor of the TNF family (BAFF) can be expressed by DCs, epithelial cells and FDCs and can augment IgA CSR in vitro (90, 95, 96), In vivo BAFF overexpression leads to an elevated commensal specific IgA response (97). However, BAFF and BAFF-R deficient mice show normal or mildly reduced IgA (98, 99), and patients with deletion of the BAFF-R gene have normal serum IgA (100). More work is needed to determine whether antigen-specific intestinal IgA levels are altered by BAFF deficiency. DCs also produce retinoic acid, which can promote IgA CSR and gut-homing receptor expression on B cells (101). Recent in vivo work indicates that intrinsic retinoic acid receptor signaling in B cells is essential to induce α4β7, but may be less critical for IgA CSR (102). Instead, retinoic acid may cause PP DCs to augment IgA induction by other mechanisms (95, 103)(unpubl. obs.).
The above observations converge on the view that DCs have a direct role in promoting B cell IgA CSR. This raises questions about the compartment where PP B cells are subjected to the influences of DCs, and a likely location is the DC-rich SED. As already noted, we have recently found that activated B cells upregulate CCR6 and efficiently migrate into the SED prior to entering the GC response (manuscript in preparation). Our data suggest that such movement is important for promoting interactions with SED DCs and inducing productive IgA CSR. Consistent with switching being initiated outside GCs, PP B cells with a GL7int activated phenotype had high amounts of Cα switch circles (104). However, whether switching at this stage is restricted to T-independent IgA production or can play a role in T-dependent IgA responses is still unknown.
The extent to which T cells directly promote IgA CSR in PPs remains an active area of investigation. T cell-deficient mice show about a 75% reduction in intestinal IgA establishing that the majority of IgA – at least in specific pathogen free mice – is T-dependent (TD), but also indicating there is a substantial amount of T-independent (TI) IgA (105–107). T cells likely promote IgA production through two mechanisms: by contributing signals that induce IgA CSR, and by promoting expansion of B cells that have already completed CSR. In vitro studies have established that CD40L promotes AID expression and switching to all Ig isotypes, and that it can cooperate with other signals to favor switching to IgA (108). Somewhat surprisingly, however, CD40−/− mice had almost normal intestinal IgA levels (108). It seems likely that the CD40L-CD40 contribution to IgA CSR will vary with the type of antigen driving the response since in vitro studies indicate that some innate signals, such as through TLR4, can substitute for CD40 to promote AID expression and CSR (109). Consistent with this view, CD40-deficient mice were severely compromised in their ability to mount an antigen-specific IgA response upon oral immunization with cholera toxin, a protein antigen (104, 108). However, it still remains unclear whether CD40 acts during the initial induction of isotype switching or during the expansion of cells that have already received inductive signals. The critical role of T cells and CD40 during the expansion phase is highlighted by the absence of PP GCs in TCR- and CD40-deficient mice (110–112). Evidence that PP GCs make a major contribution to the expansion of gut IgA+ cells comes from the findings that 85% or more of IgA encoding antibody sequences in intestinal plasma cells are somatically mutated, often heavily (105–107). Beyond expression of CD40L, PP T cells may influence IgA CSR through IL21 production. IL-21, produced by Tfh cells and CXCR5+ Th17 cells (113, 114) synergizes with TGFβ to drive mouse and human B cells toward IgA production (115, 116). In summary, while it seems almost certain that T cells will contribute to the inductive phase of IgA CSR, a lot more work is needed to understand the temporal order in which the signals that promote IgA switching are received during B cell activation, proliferation and differentiation in PPs.
T-independent switching
As noted above, the lack of T cells or CD40 leads to only a partial reduction in IgA, establishing that T cell independent (TI) IgA responses contribute to the total intestinal IgA level. Past and recent studies in T cell-deficient mice have shown TI-IgA responses to target preferentially commensal bacteria, with both B2 and B1b cells contributing to the response (111, 112). These data are consistent with the idea that microbe-derived signals can substitute for CD40 signals to promote IgA responses. Indeed, IgA was produced against largely the same constellation of commensals in wild-type and T cell-deficient mice, with the exception that a small number of epithelium-attaching commensals evaded the TI-response (112), However, given that the majority of IgA antibodies, including those against commensals (106), are somatically hypermutated, it seems likely that T cells are normally involved in the induction of antibody responses towards many commensals. In support of this view, mice harboring a mutant form of AID, AIDG23S, that supports CSR but not SHM had normal amounts of IgA but greatly reduced V-region mutation and this led to GC hyperplasia and bacterial expansion in the small intestine (117). In another informative study, mice that lacked the inhibitory co-receptor programmed cell death-1 (PD-1) gene generated an excess of abnormal Tfh cells and IgA with reduced affinity maturation and commensal binding capacity, and this was associated with alterations of gut microbial homeostasis (118). Taken together, the studies to date indicate that TI responses are capable of generating IgA that binds many commensals, but AID-dependent and Tfh-dependent responses make important contributions to the amount and quality of the commensal-specific IgA. A caveat with all the studies examining TI IgA responses is that they have been performed in mice unable to mount TD responses. It will be crucial in future work to determine the relative contribution of TD vs TI IgA responses to gut bacteria and other antigens in animals able to mount both types of response.
IgA switching outside the PP
Although PPs are the main inductive sites for IgA in the intestine, mice with reduced or absent PPs show a variable but not absent number of IgA plasma cells in their gut LP, indicating that other sites in the intestine besides PP can sustain IgA CSR. Isolated lymphoid follicles (ILFs) are single clusters of B cells, surrounded by many DCs and a few T cells, located in the terminal ileum and colon. ILF formation, similarly to PP formation, depends on RORγt+ LTi cells and on their interaction with stromal cells through the LTα1β2-LTβR axis. However, ILF formation occurs after birth, and their maturation can be promoted by recognition of peptidoglycan derived from commensal bacteria through NOD1 (11, 119). Mice deficient for the transcription factor RORγt lack ILFs as well as LNs and PPs. Upon irradiation and reconstitution with wild-type BM, RORγt-deficient mice show an increased frequency of IgA plasma cells in the LP. Since ILFs, but not PPs, can be generated in these chimeras, this result suggests that ILFs can play a role as an IgA inductive site. Similarly, mice lacking both αβ and γδ T cells, show an increase in AID induction and IgA CSR in ILFs compared to WT mice, accounting for the intestinal IgA level present in the animals (4). These observations suggest that while ILFs normally make only a small contribution to IgA production in mice housed in specific pathogen free animals facilities, physiological conditions likely exist where these structures make more substantial contributions to the IgA response.
In the absence of all gut-associated lymphoid tissue (GALT), IgA plasma cells in the LP can still be detected, suggesting IgA CSR may also be able to occur directly in the LP. Two sets of observations support this hypothesis: first, the expression of AID has been observed by in situ staining and the use of reporter mice (96, 120, 121); second, Ig Cα germline and Ig α circle transcripts has been detected in B cells isolated from LP (122). However, other studies failed to detect CSR in the LP (108, 123); this inconsistency might depend on the age, mouse strain, and commensal community.
Unique properties of PP GC responses
A signature feature of PP follicles is the presence of GC responses in the absence of immunization or infection by pathogens. These chronic responses are a consequence of the continual exposure to microbiome-derived molecules as they are largely absent in germ-free mice (9). Like GCs in other lymphoid tissues, PP GCs are organized into light and dark zones. The LZ contains a well-developed FDC network and the DZ contains a network of CXCL12+ reticular cells (CRCs) involved in attracting CXCR4hi GC B cells to this compartment. CRCs in PPs form a network that typically occupies the entire DZ, a distribution that is more extensive than found in LN and spleen GCs (124, 125). This may indicate that the chronic PP GC DZ requires more vigorous stromal support than is needed for the transient GCs forming in other lymphoid tissues.
Minimal antigen-recognition requirements for PP GCs
PP GC responses can be induced by a range of antigen types including commensals, pathogens and pathogen products. As discussed above, PP GC responses are strongly T cell- and CD40-dependent, like GC at other sites, and most studies indicate they have a conventional role in generating B cells with improved affinity for the inducing antigens while avoiding generating autoreactive B cells (126). The properties of conventional GC responses have been reviewed elsewhere (8). However, a small number of studies have pointed to the possibility that under some circumstances the antigen-recognition requirements for inducing and sustaining PP GCs responses may be less stringent than in other tissues.
A study of the requirements for GC B cell SHM made the surprising finding that RAG1-deficient mice carrying Ig and TCR transgenes contained GCs even without immunization with the cognate antigen (127). The GC B cells in these mice demonstrated SHM (127). Further support for the possibility that cognate antigen recognition may not be essential for PP GC formation emerged in a striking study in 2004 showing the presence of PP GCs in mice carrying the EBV LMP2A protein-encoding gene in place of their endogenous Ig heavy chain gene. LMP2A contains immunoreceptor tyrosine-based activation (ITAM) motifs and causes constitutive BCR pathway signaling when expressed in B cells (128). This signaling property might explain how the B cells become activated to enter GC responses, but leaves the puzzle of how the cells receive T cell help. What antigen(s) are being presented to what T cell type(s)? Could non-classical antigen recognition, such as by γδT cells, CD1d-restricted natural killer (NK) T cells or MR1-restricted mucosal-associated invariant T (MAIT) cells be involved? Consistent with an ability of cells other than CD4 T cells to have a role, MHC class II-deficient mice retain small PP GC responses (129). Or are LMP2A-driven GCs T cell independent? Similarly to the Bemark et al., study (127), these workers showed that mice carrying knocked in Ig heavy chains and transgenic light chains exhibited PP GC responses in the absence of immunization with cognate antigen, though this was not tested on a RAG-deficient background (128, 129). A more recent study found that mice carrying a knocked in hapten-specific heavy chain contained PP GC B cells that had undergone extensive SHM and this was again taken to favor the conclusion that a program of GC hypermutation can operate in PPs to promote Ig diversification (130). While the concept of PPs contributing to repertoire diversification is an intriguing idea, especially given the prominence of this process in a number of other species as discussed below, more work needs to be done to rigorously test this possibility in mice. At this point, putting the LMP2A transgenic mice aside as a special case, it seems most likely that BCR antigen binding is required for PP GC responses and that Tfh cells support most of these responses, but low affinity interactions with continually and abundantly available microbiome-derived antigens may be sufficient to drive them (131) (Fig. 4). This type of response might be especially prominent in contexts where the starting B cell clonal diversity is low or absent, as in the case of Ig transgenic mice, since the lack of commensal-reactive Ig in the gut may lead to a high microbial antigen burden in the PP.
Figure 4. Two models of GC function in PPs.
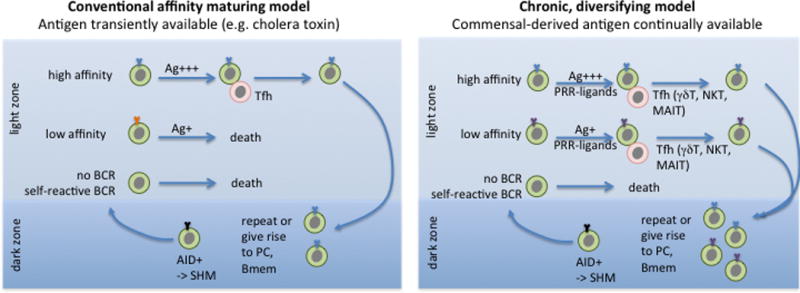
In the conventional model, GC B cells that acquire a high affinity BCR through AID-dependent SHM in the dark zone take up more of the transiently available antigen (Ag) than low affinity cells while in the light zone and win-out in receiving T cell help, leading to their selective survival and continuation on in the GC reaction or emergence as high affinity memory B cells (Bmem) or plasma cells (PC). In the diversifying model, there is continually available antigen, and sufficient T cell help (possibly of a range of types) to support the survival and differentiation of low and high affinity cells. Commensal-derived signals acting via pattern-recognition receptors (PRRs) likely cooperate with T cell-derived signals to support the chronic GC responses.
PPs as sites of antibody diversification in other species
A function for intestinal lymphoid tissue GC-like structures in the generation of B cell repertoire diversity has been established in rabbits, sheep, cattle and chicken (132–134). Newly generated B cells migrate to GALT that includes the appendix, PPs and sacculus rotundus (a type of ileal PP) in rabbits, a very large (100–200 cm long) ileal PP in sheep and cattle, and the Bursa of Fabricius in chicken. In rabbits and sheep, the Ig diversification involves SHM and intestinal microbiota are required (though in sheep the diversification begins in the sterile embryonic environment). This diversification mechanism may be particularly important in these species because they generate only limited B cell repertoire diversity during early B cell development; for example, 80–90% of the Ig heavy chain rearrangements in the rabbit use the 3′ most IGVH gene (133). The B cell proliferation underlying intestinal tissue-associated diversification is thought to be driven in an antigen non-specific manner (polyclonally) and based on the minimal presence of T cells in rabbit ileal PP’s and a study in embryonically thymectomized sheep, the diversification process may not require conventional T cell help (132, 133). TLRs have been suggested to be major drivers in some species, though involvement of a BCR superantigen has also been invoked (133). In rabbits, the diversification activity is developmentally timed to populate the periphery with diversified B cells at 3 weeks (weaning), when maternal Ab is waning (135); in sheep the ileal PP involutes starting from 3–4 months of life (whereas the jejunal PPs that have a typical secondary lymphoid tissue character, are maintained) (132). The findings suggesting mice may be able to mount PP GC responses in the absence of strong antigen recognition makes it attractive to think that related processes contribute to repertoire diversification in a broader range of species, possibly including mice and humans. Indeed, some antigen-independent SHM-based diversification of the human B cell repertoire has been suggested to occur in the first few years of life (136), though whether this is truly antigen-independent has been debated (137, 138) and the location where it takes place remains to be determined. The finding of RAG gene expression in neonatal mouse LP B cells and Ig locus rearrangement in this compartment (139) adds to the notion that mice share some features of gut-based B cell repertoire diversification with other species. More work is needed on the neonatal intestinal immune system to rigorously explore to what extent and in what form GALT-based antibody diversification contributes to B cell repertoire diversity in humans.
Exchange of cells across PP GCs
A recent IgA lineage tree analysis has suggested that B cell exchange between PP GCs plays a role in establishing synchronized oligoclonal antigen-specific responses along the length of the mouse small intestine (126, 140). Consistent with these data, next generation sequencing of IgA genes revealed very similar repertoire representation in different regions of the small intestine (131). The nature of the B cells exchanging between GCs via circulation is not fully defined, though an ability of transferred GL7+ B cells to selectively invade existing PP GCs was demonstrated (140). It seems unlikely that the exchanging cells are GC cells given their intrinsic propensity for apoptosis and inability to survive in vivo outside the GC niche (141). Instead, the circulatory cells maybe newly generated memory B cells that retain measurable GL7. In an experiment tracking the movements of endogenous PP B cells using photoconversion, significant memory B cell exchange took place between PPs over a 3 day period (131). Presumably the exchanging cells are programmed to favor homing back to mucosal sites, perhaps by having high α4β7 integrin expression. The unique presence of CXCL13+ HEVs within PP follicles (24) might also facilitate homing of α4β7hi CXCR5+ CCR7− B cells between PPs. Whatever the mechanism, this type of clonal exchange may ensure that the same clones are expanded in GCs along the length of the small intestine and give rise to a sufficiently large number of plasma cells to seed the entire LP, possibly enhancing the quality of the antigen-specific IgA response in a manner that augments host-commensal symbiosis or anti-pathogen responses. The observation that following haptenated-cholera toxin immunization up to 15% of plasma cells in the small intestine can be antigen-specific (140) highlights the large scale of the response.
At present it is hard to reconcile how conventional affinity-maturing GC responses and chronic, diversifying GC responses could co-occur in PPs (Fig. 4). In this regard it is interesting to consider recent findings suggesting that GC responses in LNs have an early phase that favors the generation of a diversity of low affinity cells and a late phase that generates affinity-matured cells ((142) and Shlomchik et al., in press). Perhaps the chronic commensal microbe-driven GC responses that are continually present in PP GCs share features with the early GC phase of LN responses, supporting diversification with limited affinity maturation. When a strong conventional-type response is provoked by a newly arriving T-dependent antigen it may ‘take over’ the existing GCs and lead to selection of high affinity plasma cells and memory B cells. Once this response has been successful and the antigen is diminished, the chronic commensal-driven GC response might re-occupy the structure and continue to generate a diversity of commensal-reactive B cells. Recent deep sequencing analysis of individual mice across a period of months revealed that expanded and hypermutated clones were a typical feature of the intestinal IgA repertoire, providing evidence that the diversification mechanism may principally act on memory B cells (131). The authors speculate that use of a limited set of B cell clones and diversifying them through SHM might help sustain stable symbiotic host-microbe interactions.
PP-derived plasma cells and memory B cells
IgA plasma cell homing
IgA plasma cells derived from the small and large intestines have different Ig repertoires, indicating that they likely arise from different sources (143). Plasma cells differentiating in PPs are marked by induction of α4β7 and CCR9, molecules that foster homing to the small intestinal LP since MADCAM1 is selective to mucosal post capillary venules and the CCR9 ligand CCL25 (TECK) is made broadly by small intestinal epithelium (144). An important factor promoting induction of these mucosal homing molecules is retinoic acid, derived from PP DCs and epithelial cells (144–146). In mesenteric LNs, lymphoid stromal cells as well as DCs are a source of retinoic acid (147, 148) and it seems possible that stromal cells will also produce retinoic acid in PPs. Retinoic acid may cooperate with TGFβ1 signaling in promoting CCR9 and α4β7 expression (149). Egress of plasma cells from spleen and LNs into blood involves S1PR1 (150) and a study that employed the S1PR1 modulating drug, FTY720, provided evidence that this is also the case for PPs (151).
The ligand for CCR10, CCL28 (MEC) is made by epithelium in both the small and large intestine and many LP B cells and plasma cells express CCR10 (152, 153). Although CCR10 deficiency does not affect IgA levels under homeostatic conditions, this was found to reflect increased IgA+ cell production by ILFs (152). These compensatory IgA+ cells carried reduced frequencies of somatic mutations in their Ig genes, consistent with a reduced contribution by cells derived from GC-bearing lymphoid tissues. The factors favoring CCR10 induction are not as well defined as for CCR9. IL21 and TGFβ may cooperate to promote CCR10 expression in human plasma cells (116). In mice, CCR10 induction occurred strongly on activated B cells in the caecal patch, and induction could be promoted by caecal patch-derived DCs (154). The mouse caecal patch has a similar organization to PPs, but cells from the caecal patch are able to travel to both the small intestine and large intestine whereas cells from the PP are less efficient at homing to the large intestine (154). Consistent with these findings, removal of the caecal patch by appendectomy reduced the ability to generate colonic but not small intestinal IgA plasma cells. Whether higher CCR10 expression could account for the difference in large intestine homing efficiency needs further investigation though it is notable that the impact of CCR10-deficiency on transferred cell homing appeared greater for large intestine than small intestine (152).
Plasma cell trophic support
Once they arrive in the LP, plasma cells are supported by multiple factors, the most studied being APRIL and IL-6. Depletion experiments have established a role for eosinophils in plasma cell maintenance (155). Eosinophils are a source of IL-1β and have multiple effects in the gut, including on the microbiome and on PP development, and may augment IgA responses in several ways (156, 157). The key cellular sources of APRIL in the intestine are not defined but production by epithelial cells, and production or presentation by DCs, seems likely to be important (92, 158, 159). Although some studies have suggested a role for IL-6, possibly from eosinophils, this contribution may be redundant with other factors since mice deficient for IL-6 show normal mucosal IgA levels in steady state and after cholera toxin immunization (157). Initial investigation into the kinetics of LP plasma cells in the small intestine of mice estimated their half-life to be 5 days, with a maximum persistence of 6–8 weeks (160). Those data suggested that LP plasma cells are short-lived, a finding that is in striking contrast with the half-life of BM plasma cells, which can survive over 1 year in mice (161). However, this discrepancy might be due to technical factors: for example, the radioactive labeling was performed only at an early time in the animal’s life. There is evidence that early LP plasma cells are mainly derived by TI-response and they get replaced later in life with plasma cells derived by TD-response (131, 162). Thus it is possible that the short life span observed was a function of their TI origin and replacement by plasma cells arising from GC responses. In addition, at least three recent studies suggest the existence of long-lived IgA plasma cells in the gut. First, when germ-free mice are colonized in a reversible way, IgA plasma cells specific for the given commensal persisted in the gut over 100 days, even after the animals were returned to a germ-free state (163). Second, adult mice maintained a stable clonal composition of their IgA repertoire over time even after antibiotic treatment (131). Third, oral immunization with CT generates IgA plasma cells in the gut which were maintained for at least 9 months (159). Gut plasma cells are also quite radioresistant (164), but can be eliminated by treatment with proteasome inhibitors (143). Such depletion is short lasting, since the plasma cell niche is completely repopulated 7 days after treatment. While these findings begin to provide a consensus in establishing intestinal plasma cells can be long-lived, more work is needed to define the intrinsic factors influencing plasma cell lifespan and the stromal cell types acting to provide trophic support for these cells in the LP.
IgA+ memory B cells
Mucosal memory B cells are generated by oral immunization and contribute to intestinal protection in humans (165, 166) and mice (167–169). Less is known about memory B cell regulation against commensal organisms. Pioneering investigation of the regulation of B cell memory was performed using a strain of E.coli, which allowed reversible intestinal colonization (163). When germ-free animal were reversibly colonized, an E.coli specific IgA response was mounted in the first 2 weeks and persisted for over 17 weeks. However, when animals with commensal flora were reversibly colonized, an E.coli specific IgA response was mounted in the first 2 weeks similarly to the germ-free animals, but it became attenuated over time. This result led to the speculation that intestinal IgA+ memory B cells were regulated differently from the memory B cells in peripheral tissue, since the dominant bacterial species currently present could shape the IgA response. Recent data have shown that the IgA+ memory B cell repertoire mirrors the intestinal IgA plasma cell repertoire and they both show similar frequencies of somatic mutations (131). The IgA repertoire of memory B cells in PPs and spleen showed clonal relationships, suggesting recirculation between these sites. Moreover, the IgA repertoire and mutation rate in lactating mammary glands largely mirrored that in the gut, indicating that the plasma cells in mammary glands likely arise from circulating memory B cells originating from gut immune responses (131).
IgA+ memory B cells are characterized by high expression of IL-17Rc and IL-22Rα2, suggesting a specific ability to respond to the cytokines IL-17 and IL-22, both abundant in intestinal tissues (170). TGFβ signaling molecules of the Smad family (Smad2, Smad3 and Smad4) and the TGFβ-associated adaptor Daxx are highly expressed in IgA+ memory B cells (170). RORα, a transcription factor acting downstream of TGFβ and IL-17 signaling, is also highly expressed and interfering with its expression affects survival and BCR expression in IgA+ memory B cells (170). IgA+ memory B cells express integrin α4β7 and likely also CCR9 and CCR10 (152), which contribute to their gut-tropism. Based on developmental requirements, signaling propensity, repertoire and tissue-homing potential, it can be concluded that IgA+ memory B cells generated in PPs are distinct from memory B cell generated in other peripheral lymphoid organs. Further investigations will be needed to elucidate the mechanisms that concur to generate and maintain IgA+ memory B cells.
Concluding Remarks
The recent advances in gut microbiome research have highlighted the deep interrelationship between host and commensals and have led to a realization that many factors work together to maintain a healthy symbiosis. That IgA is one crucial factor in this crosstalk has led to renewed interest in understanding how IgA production is induced, maintained and regulated. PPs are a nexus between host and commensal – and between host and gut pathogen – providing a compartment that ensures antigen encounter by rare antigen-specific B cells and helper T cells, and induction of antibody responses. While a lot of advances have been made in understanding the importance of PPs as sites of TI and TD B cell responses, many questions about PPs and IgA B cell biology remain to be explored, as highlighted throughout this review. The mechanisms by which antigens travel from M cells to B cells and the role of various SED DCs in this process are poorly understood. Recent work suggests that some members of the commensal population, possibly those that are more colitogenic, are more heavily coated with IgA (171). Further studies are needed to understand whether these commensals gain greater access to PPs or whether they drive stronger IgA responses by other mechanisms. Host genetic factors strongly influence the magnitude of the IgA response against commensal bacteria and this in turn impacts on the ability to resist pathogen infection (53). Much work needs to be done to decode the key genetic polymorphisms influencing the quality and magnitude of the IgA response. The central role of TGFβ in promoting IgA switching in PPs is well defined but the details of where the TGFβ signal is received and how the magnitude of this signal, and others that promote IgA switching, are regulated are only now coming to light. PPs are a site of continual GC activity and these structures function in a conventional way to support induction of high affinity antibody responses against some gut antigens, but they may also support sustained SHM in a manner that serves more to diversify the repertoire than to improve affinity. How these chronic GC responses are sustained and how such a diversification mechanism operates in the context of maintaining a stable commensal population remain outstanding questions. Finally, IgA deficiency is the most common form of human immunodeficiency (172), yet how this genetic condition impinges on the properties of the microbiome – and thus the health of the host – remains an open question. Reciprocally, elevated IgA levels in serum are associated with IgA nephropathy (97, 173) yet the requirements for production of serum versus mucosal IgA are incompletely defined. An increased understanding of the factors controlling IgA+ memory and plasma cell generation and maintenance will have wide ranging implications for therapies to improve human health.
Acknowledgments
We thank Jagan Muppidi and Lauren Rodda for comments on the manuscript and other members of the Cyster lab for helpful discussions. A.R. was a recipient of Irvington postdoctoral fellowship from the Cancer Research Institute. J.G.C. is an Investigator of the Howard Hughes Medical Institute. Work cited in this review from this laboratory was supported in part by a grant from the National Institutes of Health (AI45073).
Footnotes
The authors declare they have no financial conflict of interest
References
- 1.Brandtzaeg P. Role of secretory antibodies in the defence against infections. Int J Med Microbiol. 2003;293:3–15. doi: 10.1078/1438-4221-00241. [DOI] [PubMed] [Google Scholar]
- 2.Blutt SE, Conner ME. The gastrointestinal frontier: IgA and viruses. Front Immunol. 2013;4:402. doi: 10.3389/fimmu.2013.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macpherson AJ, Koller Y, McCoy KD. The bilateral responsiveness between intestinal microbes and IgA. Trends Immunol. 2015;36:460–470. doi: 10.1016/j.it.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 5.Macpherson AJ, Geuking MB, Slack E, Hapfelmeier S, McCoy KD. The habitat, double life, citizenship, and forgetfulness of IgA. Immulogical Reviews. 2011;245:132–146. doi: 10.1111/j.1600-065X.2011.01072.x. [DOI] [PubMed] [Google Scholar]
- 6.Heel KA, McCauley RD, Papadimitriou JM, Hall JC. Review: Peyer’s patches. J Gastroenterol Hepatol. 1997;12:122–136. doi: 10.1111/j.1440-1746.1997.tb00395.x. [DOI] [PubMed] [Google Scholar]
- 7.Cornes JS. Peyer’s patches in the human gut. Proc R Soc Med. 1965;58:716. doi: 10.1177/003591576505800930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 9.Weinstein PD, Cebra JJ. The preference for switching to IgA expression by Peyer’s patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J Immunol. 1991;147:4126–4135. [PubMed] [Google Scholar]
- 10.Pickard JM, Chervonsky AV. Sampling of the intestinal microbiota by epithelial M cells. Curr Gastroenterol Rep. 2010;12:331–339. doi: 10.1007/s11894-010-0128-x. [DOI] [PubMed] [Google Scholar]
- 11.Lecuyer E, et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity. 2014;40:608–620. doi: 10.1016/j.immuni.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Coles M, Kioussis D, Veiga-Fernandes H. Cellular and molecular requirements in lymph node and Peyer’s patch development. Prog Mol Biol Transl Sci. 2010;92:177–205. doi: 10.1016/S1877-1173(10)92008-5. [DOI] [PubMed] [Google Scholar]
- 13.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 14.Patel A, et al. Differential RET signaling pathways drive development of the enteric lymphoid and nervous systems. Sci Signal. 2012;5:ra55. doi: 10.1126/scisignal.2002734. [DOI] [PubMed] [Google Scholar]
- 15.Bando JK, Liang HE, Locksley RM. Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat Immunol. 2015;16:153–160. doi: 10.1038/ni.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van de Pavert SA, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508:123–127. doi: 10.1038/nature13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakagawa R, Togawa A, Nagasawa T, Nishikawa S. Peyer’s patch inducer cells play a leading role in the formation of B and T cell zone architecture. J Immunol. 2013;190:3309–3318. doi: 10.4049/jimmunol.1202766. [DOI] [PubMed] [Google Scholar]
- 18.Kong YY, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 19.Taylor RT, et al. Lymphotoxin-independent expression of TNF-related activation-induced cytokine by stromal cells in cryptopatches, isolated lymphoid follicles, and Peyer’s patches. J Immunol. 2007;178:5659–5667. doi: 10.4049/jimmunol.178.9.5659. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama M, et al. Expression pattern changes and function of RANKL during mouse lymph node microarchitecture development. Int Immunol. 2012;24:369–378. doi: 10.1093/intimm/dxs002. [DOI] [PubMed] [Google Scholar]
- 21.Knoop KA, et al. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J Immunol. 2009;183:5738–5747. doi: 10.4049/jimmunol.0901563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cyster JG, Von Andrian UH. Dynamics of B cell Migration to and within Secondary Lymphoid Organs. In: Honjo T, Alt FW, Neuberger MS, editors. Molecular Biology of B cells. London, U.K: Elsevier Academic Press; 2004. [Google Scholar]
- 24.Okada T, et al. Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J Exp Med. 2002;196:65–75. doi: 10.1084/jem.20020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B cell migration in the early phases of T-dependent antibody responses. Int Immunol. 2010;22:413–419. doi: 10.1093/intimm/dxq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature. 2009;460:1122–1126. doi: 10.1038/nature08226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity. 2009;31:259–269. doi: 10.1016/j.immuni.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 28.Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and nonlymphoid tissues. J Immunol. 2011;187:3026–3032. doi: 10.4049/jimmunol.1101262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green JA, Cyster JG. S1PR2 links germinal center confinement and growth regulation. Immunol Rev. 2012;247:36–51. doi: 10.1111/j.1600-065X.2012.01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cook DN, et al. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity. 2000;12:495–503. doi: 10.1016/s1074-7613(00)80201-0. [DOI] [PubMed] [Google Scholar]
- 31.Varona R, et al. CCR6-deficient mice have impaired leukocyte homeostasis and altered contact hypersensitivity and delayed-type hypersensitivity responses. J Clin Invest. 2001;107:R37–45. doi: 10.1172/JCI11297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X, et al. CCL9 is secreted by the follicle-associated epithelium and recruits dome region Peyer’s patch CD11b+ dendritic cells. J Immunol. 2003;171:2797–2803. doi: 10.4049/jimmunol.171.6.2797. [DOI] [PubMed] [Google Scholar]
- 33.Kanaya T, et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat Immunol. 2012;13:729–736. doi: 10.1038/ni.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hase K, et al. The membrane-bound chemokine CXCL16 expressed on follicle-associated epithelium and M cells mediates lympho-epithelial interaction in GALT. J Immunol. 2006;176:43–51. doi: 10.4049/jimmunol.176.1.43. [DOI] [PubMed] [Google Scholar]
- 35.Pham TH, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. 2010;207:17–27. doi: 10.1084/jem.20091619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 37.Tomura M, Itoh K, Kanagawa O. Naive CD4+ T lymphocytes circulate through lymphoid organs to interact with endogenous antigens and upregulate their function. J Immunol. 2010;184:4646–4653. doi: 10.4049/jimmunol.0903946. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt TH, Bannard O, Gray EE, Cyster JG. CXCR4 promotes B cell egress from Peyer’s patches. J Exp Med. 2013;210:1099–1107. doi: 10.1084/jem.20122574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulz O, et al. Hypertrophy of infected Peyer’s patches arises from global, interferon-receptor, and CD69-independent shutdown of lymphocyte egress. Mucosal Immunol. 2014;7:892–904. doi: 10.1038/mi.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiow LR, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–544. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 41.Yang CW, Unanue ER. Neutrophils control the magnitude and spread of the immune response in a thromboxane A2-mediated process. J Exp Med. 2013;210:375–387. doi: 10.1084/jem.20122183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gretz JE, Anderson AO, Shaw S. Cords, channels, corridors and conduits: critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol Rev. 1997;156:11–24. doi: 10.1111/j.1600-065x.1997.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 43.Phan TG, Gray EE, Cyster JG. The microanatomy of B cell activation. Curr Opin Immunol. 2009;21:258–265. doi: 10.1016/j.coi.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez SF, et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol. 2010;11:427–434. doi: 10.1038/ni.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity. 2015;42:172–185. doi: 10.1016/j.immuni.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 47.Park C, Hwang IY, Sinha RK, Kamenyeva O, Davis MD, Kehrl JH. Lymph node B lymphocyte trafficking is constrained by anatomy and highly dependent upon chemoattractant desensitization. Blood. 2012;119:978–989. doi: 10.1182/blood-2011-06-364273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki K, Grigorova I, Phan TG, Kelly L, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med. 2009;206:1485–1493. doi: 10.1084/jem.20090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. 2014;14:495–504. doi: 10.1038/nri3689. [DOI] [PubMed] [Google Scholar]
- 50.Neutra MR, Pringault E, Kraehenbuhl JP. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu Rev Immunol. 1996;14:275–300. doi: 10.1146/annurev.immunol.14.1.275. [DOI] [PubMed] [Google Scholar]
- 51.Owen RL. Uptake and transport of intestinal macromolecules and microorganisms by M cells in Peyer’s patches–a personal and historical perspective. Semin Immunol. 1999;11:157–163. doi: 10.1006/smim.1999.0171. [DOI] [PubMed] [Google Scholar]
- 52.Neutra MR, Mantis NJ, Kraehenbuhl JP. Collaboration of epithelial cells with organized mucosal lymphoid tissues. Nat Immunol. 2001;2:1004–1009. doi: 10.1038/ni1101-1004. [DOI] [PubMed] [Google Scholar]
- 53.Fransen F, et al. BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity. 2015;43:527–540. doi: 10.1016/j.immuni.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 54.Kadaoui KA, Corthesy B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer’s patches with restriction to mucosal compartment. J Immunol. 2007;179:7751–7757. doi: 10.4049/jimmunol.179.11.7751. [DOI] [PubMed] [Google Scholar]
- 55.Rochereau N, et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013;11:e1001658. doi: 10.1371/journal.pbio.1001658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hase K, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature. 2009;462:226–230. doi: 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- 57.Neutra MR, Phillips TL, Mayer EL, Fishkind DJ. Transport of membrane-bound macromolecules by M cells in follicle-associated epithelium of rabbit Peyer’s patch. Cell Tissue Res. 1987;247:537–546. doi: 10.1007/BF00215747. [DOI] [PubMed] [Google Scholar]
- 58.Farstad IN, Halstensen TS, Fausa O, Brandtzaeg P. Heterogeneity of M-cell-associated B and T cells in human Peyer’s patches. Immunology. 1994;83:457–464. [PMC free article] [PubMed] [Google Scholar]
- 59.Gebert A, Steinmetz I, Fassbender S, Wendlandt KH. Antigen transport into Peyer’s patches: increased uptake by constant numbers of M cells. Am J Pathol. 2004;164:65–72. doi: 10.1016/S0002-9440(10)63097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Gusti V, Saraswati A, Lo DD. Convergent and divergent development among M cell lineages in mouse mucosal epithelium. J Immunol. 2011;187:5277–5285. doi: 10.4049/jimmunol.1102077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chabot S, Wagner JS, Farrant S, Neutra MR. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol. 2006;176:4275–4283. doi: 10.4049/jimmunol.176.7.4275. [DOI] [PubMed] [Google Scholar]
- 62.Iwasaki A, Kelsall BL. Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer’s patch dendritic cells. J Immunol. 2001;166:4884–4890. doi: 10.4049/jimmunol.166.8.4884. [DOI] [PubMed] [Google Scholar]
- 63.Ebisawa M, et al. CCR6hiCD11c(int) B cells promote M-cell differentiation in Peyer’s patch. Int Immunol. 2011;23:261–269. doi: 10.1093/intimm/dxq478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelsall BL, Strober W. Distinct populations of dendritic cells are present in the subepithelial dome and T cell regions of the murine Peyer’s patch. J Exp Med. 1996;183:237–247. doi: 10.1084/jem.183.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwickert TA, Alabyev B, Manser T, Nussenzweig MC. Germinal center reutilization by newly activated B cells. J Exp Med. 2009;206:2907–2914. doi: 10.1084/jem.20091225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lelouard H, et al. Pathogenic bacteria and dead cells are internalized by a unique subset of Peyer’s patch dendritic cells that express lysozyme. Gastroenterology. 2010;138:173–184. e171–173. doi: 10.1053/j.gastro.2009.09.051. [DOI] [PubMed] [Google Scholar]
- 67.Fleeton MN, Contractor N, Leon F, Wetzel JD, Dermody TS, Kelsall BL. Peyer’s patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J Exp Med. 2004;200:235–245. doi: 10.1084/jem.20041132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang FP, et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. 2000;191:435–444. doi: 10.1084/jem.191.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Jesus M, Rodriguez AE, Yagita H, Ostroff GR, Mantis NJ. Sampling of Candida albicans and Candida tropicalis by Langerin-positive dendritic cells in mouse Peyer’s patches. Immunol Lett. 2015;168:64–72. doi: 10.1016/j.imlet.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 70.Kujala P, et al. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog. 2011;7:e1002449. doi: 10.1371/journal.ppat.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Owen RL. Sequential uptake of horseradish peroxidase by lymphoid follicle epithelium of Peyer’s patches in the normal unobstructed mouse intestine: an ultrastructural study. Gastroenterology. 1977;72:440–451. [PubMed] [Google Scholar]
- 72.Pape KA, Catron DM, Itano AA, Jenkins MK. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity. 2007;26:491–502. doi: 10.1016/j.immuni.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 73.Grigorova IL, Panteleev M, Cyster JG. Lymph node cortical sinus organization and relationship to lymphocyte egress dynamics and antigen exposure. Proc Natl Acad Sci U S A. 2010;107:20447–20452. doi: 10.1073/pnas.1009968107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Catron DM, Pape KA, Fife BT, van Rooijen N, Jenkins MK. A protease-dependent mechanism for initiating T-dependent B cell responses to large particulate antigens. J Immunol. 2010;184:3609–3617. doi: 10.4049/jimmunol.1000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McManus LM, Nakane PK. Mouse c1q: light and electron microscopic immunohistochemical localization. J Immunol. 1981;126:1421–1427. [PubMed] [Google Scholar]
- 76.Balogh P, Aydar Y, Tew JG, Szakal AK. Appearance and phenotype of murine follicular dendritic cells expressing VCAM-1. Anat Rec. 2002;268:160–168. doi: 10.1002/ar.10148. [DOI] [PubMed] [Google Scholar]
- 77.Yamakawa M, Imai Y. Complement activation in the follicular light zone of human lymphoid tissues. Immunology. 1992;76:378–384. [PMC free article] [PubMed] [Google Scholar]
- 78.Landsverk T, Trevella W, Nicander L. Transfer of carbonic anhydrase-positive particles from the follicle-associated epithelium to lymphocytes of Peyer’s patches in foetal sheep and lambs. Cell Tissue Res. 1990;261:239–247. doi: 10.1007/BF00318665. [DOI] [PubMed] [Google Scholar]
- 79.Kouser L, et al. Emerging and Novel Functions of Complement Protein C1q. Front Immunol. 2015;6:317. doi: 10.3389/fimmu.2015.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kurita N, Honda S, Shibuya A. Increased serum IgA in Fcalpha/muR-deficient mice on the (129 × C57BL/6) F1 genetic background. Mol Immunol. 2015;63:367–372. doi: 10.1016/j.molimm.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 81.Honda S, et al. Enhanced humoral immune responses against T-independent antigens in Fc alpha/muR-deficient mice. Proc Natl Acad Sci U S A. 2009;106:11230–11235. doi: 10.1073/pnas.0809917106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Szabo MC, Butcher EC, McEvoy LM. Specialization of mucosal follicular dendritic cells revealed by mucosal addressin-cell adhesion molecule-1 display. J Immunol. 1997;158:5584–5588. [PubMed] [Google Scholar]
- 83.Wang X, Rodda LB, Bannard O, Cyster JG. Integrin-mediated interactions between B cells and follicular dendritic cells influence germinal center B cell fitness. J Immunol. 2014;192:4601–4609. doi: 10.4049/jimmunol.1400090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Craig SW, Cebra JJ. Peyer’s patches: an enriched source of precursors for IgA-producing immunocytes in the rabbit. J Exp Med. 1971;134:188–200. doi: 10.1084/jem.134.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 86.van Ginkel FW, et al. Partial IgA-deficiency with increased Th2-type cytokines in TGF-beta 1 knockout mice. J Immunol. 1999;163:1951–1957. [PubMed] [Google Scholar]
- 87.Cazac BB, Roes J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13:443–451. doi: 10.1016/s1074-7613(00)00044-3. [DOI] [PubMed] [Google Scholar]
- 88.Borsutzky S, Cazac BB, Roes J, Guzman CA. TGF-beta receptor signaling is critical for mucosal IgA responses. J Immunol. 2004;173:3305–3309. doi: 10.4049/jimmunol.173.5.3305. [DOI] [PubMed] [Google Scholar]
- 89.Gros MJ, Naquet P, Guinamard RR. Cell intrinsic TGF-beta 1 regulation of B cells. J Immunol. 2008;180:8153–8158. doi: 10.4049/jimmunol.180.12.8153. [DOI] [PubMed] [Google Scholar]
- 90.Suzuki K, et al. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity. 2010;33:71–83. doi: 10.1016/j.immuni.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 91.Travis MA, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tezuka H, et al. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature. 2007;448:929–933. doi: 10.1038/nature06033. [DOI] [PubMed] [Google Scholar]
- 93.Castigli E, et al. Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U S A. 2004;101:3903–3908. doi: 10.1073/pnas.0307348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Castigli E, Geha RS. TACI, isotype switching, CVID and IgAD. Immunol Res. 2007;38:102–111. doi: 10.1007/s12026-007-8000-2. [DOI] [PubMed] [Google Scholar]
- 95.Massacand JC, et al. Intestinal bacteria condition dendritic cells to promote IgA production. PLoS One. 2008;3:e2588. doi: 10.1371/journal.pone.0002588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.He B, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 97.McCarthy DD, et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest. 2011;121:3991–4002. doi: 10.1172/JCI45563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schiemann B, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 99.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173:2245–2252. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 100.Warnatz K, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mora JR, et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314:1157–1160. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- 102.Pantazi E, Marks E, Stolarczyk E, Lycke N, Noelle RJ, Elgueta R. Cutting Edge: Retinoic Acid Signaling in B Cells Is Essential for Oral Immunization and Microflora Composition. J Immunol. 2015;195:1368–1371. doi: 10.4049/jimmunol.1500989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Villablanca EJ, et al. MyD88 and retinoic acid signaling pathways interact to modulate gastrointestinal activities of dendritic cells. Gastroenterology. 2011;141:176–185. doi: 10.1053/j.gastro.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bergqvist P, Stensson A, Lycke NY, Bemark M. T cell-independent IgA class switch recombination is restricted to the GALT and occurs prior to manifest germinal center formation. J Immunol. 2010;184:3545–3553. doi: 10.4049/jimmunol.0901895. [DOI] [PubMed] [Google Scholar]
- 105.Barone F, et al. IgA-producing plasma cells originate from germinal centers that are induced by B-cell receptor engagement in humans. Gastroenterology. 2011;140:947–956. doi: 10.1053/j.gastro.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Benckert J, et al. The majority of intestinal IgA+ and IgG+ plasmablasts in the human gut are antigen-specific. J Clin Invest. 2011;121:1946–1955. doi: 10.1172/JCI44447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bemark M, Boysen P, Lycke NY. Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann N Y Acad Sci. 2012;1247:97–116. doi: 10.1111/j.1749-6632.2011.06378.x. [DOI] [PubMed] [Google Scholar]
- 108.Bergqvist P, Gardby E, Stensson A, Bemark M, Lycke NY. Gut IgA class switch recombination in the absence of CD40 does not occur in the lamina propria and is independent of germinal centers. J Immunol. 2006;177:7772–7783. doi: 10.4049/jimmunol.177.11.7772. [DOI] [PubMed] [Google Scholar]
- 109.Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev. 2014;260:76–85. doi: 10.1111/imr.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dianda L, Gulbranson-Judge A, Pao W, Hayday AC, MacLennan IC, Owen MJ. Germinal center formation in mice lacking alpha beta T cells. Eur J Immunol. 1996;26:1603–1607. doi: 10.1002/eji.1830260729. [DOI] [PubMed] [Google Scholar]
- 111.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- 112.Bunker JJ, et al. Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity. 2015;43:541–553. doi: 10.1016/j.immuni.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Morita R, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34:108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cao AT, Yao S, Gong B, Nurieva RI, Elson CO, Cong Y. Interleukin (IL)-21 promotes intestinal IgA response to microbiota. Mucosal Immunol. 2015;8:1072–1082. doi: 10.1038/mi.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Seo GY, Youn J, Kim PH. IL-21 ensures TGF-beta 1-induced IgA isotype expression in mouse Peyer’s patches. J Leukoc Biol. 2009;85:744–750. doi: 10.1189/jlb.0708450. [DOI] [PubMed] [Google Scholar]
- 116.Dullaers M, et al. A T cell-dependent mechanism for the induction of human mucosal homing immunoglobulin A-secreting plasmablasts. Immunity. 2009;30:120–129. doi: 10.1016/j.immuni.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat Immunol. 2011;12:264–270. doi: 10.1038/ni.1991. [DOI] [PubMed] [Google Scholar]
- 118.Kawamoto S, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–489. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- 119.Bouskra D, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 120.Tsuji M, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity. 2008;29:261–271. doi: 10.1016/j.immuni.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 121.Crouch EE, et al. Regulation of AID expression in the immune response. J Exp Med. 2007;204:1145–1156. doi: 10.1084/jem.20061952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fagarasan S, Kinoshita K, Muramatsu M, Ikuta K, Honjo T. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature. 2001;413:639–643. doi: 10.1038/35098100. [DOI] [PubMed] [Google Scholar]
- 123.Shikina T, et al. IgA class switch occurs in the organized nasopharynx- and gut-associated lymphoid tissue, but not in the diffuse lamina propria of airways and gut. J Immunol. 2004;172:6259–6264. doi: 10.4049/jimmunol.172.10.6259. [DOI] [PubMed] [Google Scholar]
- 124.Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity. 2013;39:912–924. doi: 10.1016/j.immuni.2013.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rodda LB, Bannard O, Ludewig B, Nagasawa T, Cyster JG. Phenotypic and Morphological Properties of Germinal Center Dark Zone Cxcl12-Expressing Reticular Cells. J Immunol. 2015 doi: 10.4049/jimmunol.1501191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lycke NY, Bemark M. The role of Peyer’s patches in synchronizing gut IgA responses. Front Immunol. 2012;3:329. doi: 10.3389/fimmu.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bemark M, Sale JE, Kim HJ, Berek C, Cosgrove RA, Neuberger MS. Somatic hypermutation in the absence of DNA-dependent protein kinase catalytic subunit (DNA-PK(cs)) or recombination-activating gene (RAG)1 activity. J Exp Med. 2000;192:1509–1514. doi: 10.1084/jem.192.10.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Casola S, et al. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5:317–327. doi: 10.1038/ni1036. [DOI] [PubMed] [Google Scholar]
- 129.Casola S, Rajewsky K. B cell recruitment and selection in mouse GALT germinal centers. Curr Top Microbiol Immunol. 2006;308:155–171. doi: 10.1007/3-540-30657-9_7. [DOI] [PubMed] [Google Scholar]
- 130.Yeap L, et al. Sequence-Intrinsic Mechanisms that Target AID Mutational Outcomes on Antibody Genes. Cell. 2015 doi: 10.1016/j.cell.2015.10.042. In Press Correted Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lindner C, et al. Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat Immunol. 2015;16:880–888. doi: 10.1038/ni.3213. [DOI] [PubMed] [Google Scholar]
- 132.Weill JC, Weller S, Reynaud CA. A bird’s eye view on human B cells. Semin Immunol. 2004;16:277–281. doi: 10.1016/j.smim.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 133.Lanning DK, Knight KL. Intestinal bacteria and development of the antibody repertoire. Discov Med. 2005;5:393–398. [PubMed] [Google Scholar]
- 134.Yasuda M, Jenne CN, Kennedy LJ, Reynolds JD. The sheep and cattle Peyer’s patch as a site of B-cell development. Vet Res. 2006;37:401–415. doi: 10.1051/vetres:2006008. [DOI] [PubMed] [Google Scholar]
- 135.Zhai SK, Lanning DK. Diversification of the primary antibody repertoire begins during early follicle development in the rabbit appendix. Mol Immunol. 2013;54:140–147. doi: 10.1016/j.molimm.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 136.Weller S, et al. Somatic diversification in the absence of antigen-driven responses is the hallmark of the IgM+ IgD+ CD27+ B cell repertoire in infants. J Exp Med. 2008;205:1331–1342. doi: 10.1084/jem.20071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tarlinton D. Sheepish B cells: evidence for antigen-independent antibody diversification in humans and mice. J Exp Med. 2008;205:1251–1254. doi: 10.1084/jem.20081057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Budeus B, Schweigle de Reynoso S, Przekopowitz M, Hoffmann D, Seifert M, Kuppers R. Complexity of the human memory B-cell compartment is determined by the versatility of clonal diversification in germinal centers. Proc Natl Acad Sci U S A. 2015;112:E5281–5289. doi: 10.1073/pnas.1511270112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wesemann DR, et al. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature. 2013;501:112–115. doi: 10.1038/nature12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bergqvist P, et al. Re-utilization of germinal centers in multiple Peyer’s patches results in highly synchronized, oligoclonal, and affinity-matured gut IgA responses. Mucosal Immunol. 2013;6:122–135. doi: 10.1038/mi.2012.56. [DOI] [PubMed] [Google Scholar]
- 141.Muppidi JR, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516:254–258. doi: 10.1038/nature13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247:52–63. doi: 10.1111/j.1600-065X.2012.01124.x. [DOI] [PubMed] [Google Scholar]
- 143.Lindner C, et al. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J Exp Med. 2012;209:365–377. doi: 10.1084/jem.20111980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mora JR, von Andrian UH. Differentiation and homing of IgA-secreting cells. Mucosal Immunol. 2008;1:96–109. doi: 10.1038/mi.2007.14. [DOI] [PubMed] [Google Scholar]
- 145.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21:527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 146.Edele F, et al. Cutting edge: instructive role of peripheral tissue cells in the imprinting of T cell homing receptor patterns. J Immunol. 2008;181:3745–3749. doi: 10.4049/jimmunol.181.6.3745. [DOI] [PubMed] [Google Scholar]
- 147.Hammerschmidt SI, et al. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J Exp Med. 2008;205:2483–2490. doi: 10.1084/jem.20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Molenaar R, et al. Lymph node stromal cells support dendritic cell-induced gut-homing of T cells. J Immunol. 2009;183:6395–6402. doi: 10.4049/jimmunol.0900311. [DOI] [PubMed] [Google Scholar]
- 149.Seo GY, et al. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF-beta1 to enhance the overall IgA response. J Leukoc Biol. 2013;94:325–335. doi: 10.1189/jlb.0313128. [DOI] [PubMed] [Google Scholar]
- 150.Kabashima K, et al. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J Exp Med. 2006;203:2683–2690. doi: 10.1084/jem.20061289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gohda M, et al. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J Immunol. 2008;180:5335–5343. doi: 10.4049/jimmunol.180.8.5335. [DOI] [PubMed] [Google Scholar]
- 152.Hu S, Yang K, Yang J, Li M, Xiong N. Critical roles of chemokine receptor CCR10 in regulating memory IgA responses in intestines. Proc Natl Acad Sci U S A. 2011;108:E1035–1044. doi: 10.1073/pnas.1100156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kunkel EJ, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest. 2003;111:1001–1010. doi: 10.1172/JCI17244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Masahata K, et al. Generation of colonic IgA-secreting cells in the caecal patch. Nature communications. 2014;5:3704. doi: 10.1038/ncomms4704. [DOI] [PubMed] [Google Scholar]
- 155.Chu VT, et al. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity. 2014;40:582–593. doi: 10.1016/j.immuni.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 156.Jung Y, et al. IL-1beta in eosinophil-mediated small intestinal homeostasis and IgA production. Mucosal Immunol. 2015;8:930–942. doi: 10.1038/mi.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bromander AK, Ekman L, Kopf M, Nedrud JG, Lycke NY. IL-6-deficient mice exhibit normal mucosal IgA responses to local immunizations and Helicobacter felis infection. J Immunol. 1996;156:4290–4297. [PubMed] [Google Scholar]
- 158.Joo H, et al. Serum from patients with SLE instructs monocytes to promote IgG and IgA plasmablast differentiation. J Exp Med. 2012;209:1335–1348. doi: 10.1084/jem.20111644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, Hauser AE. Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol. 2015 doi: 10.1038/mi.2015.38. [DOI] [PubMed] [Google Scholar]
- 160.Mattioli CA, Tomasi TB., Jr The life span of IgA plasma cells from the mouse intestine. J Exp Med. 1973;138:452–460. doi: 10.1084/jem.138.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;23:367–386. doi: 10.1146/annurev.immunol.23.021704.115723. [DOI] [PubMed] [Google Scholar]
- 162.Rogosch T, et al. IgA response in preterm neonates shows little evidence of antigen-driven selection. J Immunol. 2012;189:5449–5456. doi: 10.4049/jimmunol.1103347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hapfelmeier S, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–1709. doi: 10.1126/science.1188454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nossal GJ. Plasma cell proliferation following whole body irradiation. Aust J Exp Biol Med Sci. 1959;37:499–504. doi: 10.1038/icb.1959.51. [DOI] [PubMed] [Google Scholar]
- 165.Herremans TM, Reimerink JH, Buisman AM, Kimman TG, Koopmans MP. Induction of mucosal immunity by inactivated poliovirus vaccine is dependent on previous mucosal contact with live virus. J Immunol. 1999;162:5011–5018. [PubMed] [Google Scholar]
- 166.Svennerholm AM, Lange S, Holmgren J. Intestinal immune response to cholera toxin: dependence on route and dosage of antigen for priming and boosting. Infect Immun. 1980;30:337–341. doi: 10.1128/iai.30.2.337-341.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lycke N, Holmgren J. Intestinal mucosal memory and presence of memory cells in lamina propria and Peyer’s patches in mice 2 years after oral immunization with cholera toxin. Scand J Immunol. 1986;23:611–616. doi: 10.1111/j.1365-3083.1986.tb01995.x. [DOI] [PubMed] [Google Scholar]
- 168.Lycke N, Holmgren J. Long-term cholera antitoxin memory in the gut can be triggered to antibody formation associated with protection within hours of an oral challenge immunization. Scand J Immunol. 1987;25:407–412. doi: 10.1111/j.1365-3083.1987.tb02207.x. [DOI] [PubMed] [Google Scholar]
- 169.Lycke N, Holmgren J. Adoptive transfer of gut mucosal antitoxin memory by isolated B cells 1 year after oral immunization with cholera toxin. Infect Immun. 1989;57:1137–1141. doi: 10.1128/iai.57.4.1137-1141.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, McHeyzer-Williams MG. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nat Immunol. 2012;13:604–611. doi: 10.1038/ni.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Palm NW, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hammarstrom L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clin Exp Immunol. 2000;120:225–231. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kiryluk K, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46:1187–1196. doi: 10.1038/ng.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]