

ABSTRACT
Autophagy is activated early after human cytomegalovirus (HCMV) infection but, later on, the virus blocks autophagy. Here we characterized 2 HCMV proteins, TRS1 and IRS1, which inhibit autophagy during infection. Expression of either TRS1 or IRS1 was able to block autophagy in different cell lines, independently of the EIF2S1 kinase, EIF2AK2/PKR. Instead, TRS1 and IRS1 interacted with the autophagy protein BECN1/Beclin 1. We mapped the BECN1-binding domain (BBD) of IRS1 and TRS1 and found it to be essential for autophagy inhibition. Mutant viruses that express only IRS1 or TRS1 partially controlled autophagy, whereas a double mutant virus expressing neither protein stimulated autophagy. A mutant virus that did not express IRS1 and expressed a truncated form of TRS1 in which the BBD was deleted, failed to control autophagy. However, this mutant virus had similar replication kinetics as wild-type virus, suggesting that autophagy inhibition is not critical for viral replication. In fact, using pharmacological modulators of autophagy and inhibition of autophagy by shRNA knockdown, we discovered that stimulating autophagy enhanced viral replication. Conversely, inhibiting autophagy decreased HCMV infection. Thus, our results demonstrate a new proviral role of autophagy for a DNA virus.
KEYWORDS: autophagy, BECN1, cytomegalovirus, EIF2AK2/PKR, IRS1, TRS1
Introduction
Autophagy is an evolutionarily conserved degradation process, which has been described as a self-defense mechanism against intracellular microorganisms.1,2 Indeed, autophagy can participate in the control of viral infection by direct degradation of viral components, by regulating the intensity of the inflammatory response or by facilitating the processing of viral antigens for presentation by major histocompatibility complex (MHC).3 Autophagy induction can therefore be detrimental for viral infections, but also beneficial, when it is hijacked and regulated by viruses. Indeed, in addition to its recognized role as a scaffold platform for replication of positive-strand RNA viruses, autophagy contributes to viral infectivity through multiple mechanisms. This cellular process can provide energy, lipid membranes, or a vehicle for the virus to get out the cell and can also improve the survival of the infected cell and thereby increase viral production.4,5 Viruses can also specifically redirect cellular signaling mediators to autophagosomal degradation to dampen the inflammatory response.6 On the other hand, autophagy enhances the delivery of viral antigens to MHC class I and II complexes and improves antigen presentation. For example, presentation of 3 antigens of human cytomegalovirus (HCMV), classically processed in a proteasome-dependent manner, has been described to also occur via a vacuolar pathway involving autophagosomes, lysosomal proteases, and recycling HLA-molecules.7 Interestingly, we and others have previously demonstrated that HCMV modulates autophagy during its life cycle, stimulating autophagic flux shortly after its entry.8,9 Then, after 18 to 24 h of infection, autophagosome formation is blocked by expression of viral proteins.8
HCMV is a ubiquitous opportunistic pathogen virus, which, like other viruses in the Herpesviridae family, has the ability to persist in the host in an inactive state known as latency after the primary infection subsides. HCMV infections in immunocompromised patients cause substantial morbidity and mortality, especially among transplant recipients, while infection in immunocompetent individuals is generally mild or asymptomatic. Studies of HCMV multiplication in vitro showed that viral cycle occurs in a series of stages. After entry of the nucleocapsid into the cell, the viral genome is delivered to the nucleus to be transcribed and replicated. Transcription is a complex process with 3 classes of proteins that need to be made for production of mature virions. Synthesis of immediate early proteins, which are nonstructural proteins involved in transcriptional regulation, is followed by expression of early genes, encoding proteins mainly involved in viral DNA replication. Synthesis of late proteins, structural components of the virus, is initiated after replication of the viral genome. Viral nucleocapsids assemble within the nucleus and bud across both the inner and outer nuclear membranes to transit to the cytoplasm. Naked cytoplasmic nucleocapsids acquire their tegument and then their final envelope from the trans-Golgi network or from the endocytic pathway.10 Mature virions are transported inside vacuoles to the cell surface to be secreted.
HCMV has a linear 235-kbp double-stranded DNA genome with an estimated coding capacity between 160 and 200 open reading frames (ORFs) or even possibly as many as ∼750 ORFs, as predicted by recent ribosomal profiling studies.11 The genome consists of 2 regions of unique sequences, flanked by 2 sets of inverted repeats (TRL-IRL) and (IRS-TRS). Partially located in one set of inverted repeats, IRS1 and TRS1 ORFs encode 2 immediate early proteins of 847 and 795 amino acids, respectively, with identical N-terminal domains and divergent C-terminal regions.12 IRS1 and TRS1 have been reported to inhibit the phosphorylation of the translation initiation factor EIF2S1, preventing the shutoff of cellular protein synthesis that occurs upon infection.13,14 They are able to rescue the function of the vaccinia virus E3L protein, in VVΔE3L infected cells, to prevent activation of the kinase EIF2AK2/PKR (eukaryotic translation initiation factor 2-α kinase 2).13 When EIF2AK2 binds to double-stranded RNA (dsRNA), it dimerizes and autophosphorylates and then, it phosphorylates its substrate EIF2S1. Phosphorylated EIF2S1 inhibits guanine nucleotide exchange factor EIF2B, resulting in a shutdown of protein synthesis that in turn hinders viral production. Herpesviruses produce dsRNA during infection, likely as a result of hybridization of convergent overlapping mRNAs. IRS1 and TRS1 bind to both dsRNA and EIF2AK2 to block EIF2AK2 and that these interactions require their carboxy termini.15-17 Because TRS1 antagonized EIF2AK2 and the product of activated EIF2AK2, phosphorylated EIF2S1, can activate autophagy,18 we tested the impact of TRS1 and found that it inhibited autophagy.8
Here we explored the functions of TRS1 and IRS1 as regulators of autophagy by HCMV using recombinant viruses. We show that each protein is able to block autophagy in the context of HCMV replication and that an N-terminal domain that is identical in the 2 proteins is essential for this function. Coexpression of both TRS1 and IRS1 is necessary to block autophagic flux. However, blocking autophagy appears not to be essential for viral replication. In fact, analyses employing pharmacological modulators of autophagy and depletion of ATG16L1 suggest that autophagy plays a proviral role in the HCMV cell cycle.
Results
IRS1 and TRS1 both inhibit starvation-induced autophagy
We used several assays to investigate the impact of the 2 HCMV proteins TRS1 and IRS1 on autophagy. First, we transiently transfected HeLa cells that stably express GFP-LC3B with an IRS1 or TRS1 expression vector or with an empty vector. After inducing autophagy by starvation for 4 h prior to fixation, we quantified GFP-LC3 dots in cells expressing viral proteins. Figure 1A and B shows that the number of LC3 dots per cell was lower in HeLa cells expressing IRS1 and TRS1 than in control cells. To confirm these findings, we optimized an automated quantification of GFP-LC3 dots using a Cellomics ArrayScan microscope (Fig. 1C). We measured several parameters including the number of GFP-LC3 dots per cell, the total intensity per cell, and the total area in the cells that was covered by GFP-LC3 dots, and the average area of individual GFP-LC3 dots. Cells transfected with IRS1 or TRS1 displayed reduction of these 3 former parameters, confirming a global decrease in the number of autophagosomes. Only the size of the autophagosomes (GFP-LC3 spot average area) was not modified by IRS1 or TRS1 expression, indicating that the decrease in the average number of dots is not simply due to fusion of several autophagosomes.
Figure 1.

Inhibition of starvation-induced autophagy by ectopic expression of TRS1 and IRS1. (A) Representative images of GFP-LC3 HeLa cells transfected with an empty vector (vector), IRS1, or TRS1 plasmids and then fixed after starvation. (B) The number of GFP-LC3-dots in TRS1 or IRS1-expressing cells in the transfected HeLa cells was quantified. The results are the mean of 6 independent experiments; 50 to 100 cells were analyzed per assay. (C) (Microscopy panels). (Left) Cellomics ArrayScan images of GFP-LC3 HeLa cells transfected with empty vector, IRS1 or TRS1. (Right) Image analysis software detected DAPI-labeled nuclei (blue), cell outline (red line), GFP-LC3 dots (green dots) and nontransfected cell nuclei (yellow line). The number of GFP-LC3 dots per cell, total intensity, total area and average area were quantified using Cellomics spot detector software. (D) (Top panel) SQSTM1 staining in HeLa cells under starvation or after 3-methyladenine (3MA) treatment. (Lower panel) SQSTM1 staining in HeLa cells expressing IRS1 or TRS1 and after starvation. (E) Autophagy was quantified by counting the number of SQSTM1 dots per transfected cell. The results are the mean of 3 independent experiments; 20 cells were analyzed per assay. (F) Immunoblot analysis of SQSTM1 and LC3 proteins in HeLa cells transfected with IRS1, TRS1 or ICP34.5 plasmids. CM, complete medium. ACTB was used as a loading control. **, P<0.01; ***, P<0.001 (One-way ANOVA).
We also used immunofluorescence studies to measure the abundance of SQSTM1/p62 (sequestosome 1), an autophagic substrate located within autophagosomes (Fig. 1D and E). In our system, we observed that starvation increased the number of SQSTM1 dots compared to basal conditions, whereas 3-methyladenine, a classic autophagy inhibitor, diminished them (Fig. 1D). A reduction of SQSTM1 dots in HeLa cells expressing IRS1 or TRS1 confirmed that these proteins inhibited autophagy. Finally, we monitored SQSTM1 and LC3-II by immunoblot in normal conditions (complete medium) and after starvation (Fig. 1F). We observed an accumulation of SQSTM1 and a decrease of LC3-II, by immunoblot after IRS1 or TRS1 expression, compared to starved cells and even to cells cultured in normal conditions, which correspond to an inhibition of autophagy. We used the viral protein ICP34.5, an HSV-1 protein that has been previously reported to block autophagy, as a positive control in this experiment.19 Taken together, these results demonstrate that IRS1 and TRS1 are each able to block the formation of autophagosomes.
IRS1 and TRS1 inhibit autophagy independently of EIF2AK2
In order to investigate the mechanism of the autophagy inhibition by these 2 proteins, we investigated the role of the double-stranded RNA (dsRNA)-dependent kinase EIF2AK2. Indeed, we have recently identified a viral protein encoded by HSV-1 that blocks autophagy by interaction with EIF2AK2.20 Moreover, TRS1 and IRS1 bind to dsRNA and EIF2AK2 to prevent the shutoff of protein synthesis.14-16 In order to explore whether the interaction between EIF2AK2 and the viral proteins is involved in the inhibition of autophagy, we performed assays in murine embryonic fibroblasts (MEFs) that have or lack EIF2AK2. We transiently cotransfected Eif2ak2+/+ and eif2ak2−/− MEFs with GFP-LC3 and either IRS1 or TRS1 expression vectors then induced autophagy by starvation. Consistent with our previous analyses of TRS1, we observed that both IRS1 and TRS1 decreased the number of autophagosomes, independently of the expression of EIF2AK2 (Fig. 2A and B).8 We also monitored SQSTM1 accumulation by immunoblot assay of lysates of Eif2ak2+/+ and eif2ak2−/− MEFs in normal conditions and after starvation (Fig. 2C). We observed that expression of TRS1 or IRS1 induced accumulation of SQSTM1 in both cell lines. These results suggest that IRS1, like TRS1, blocks autophagy independently of EIF2AK2.
Figure 2.
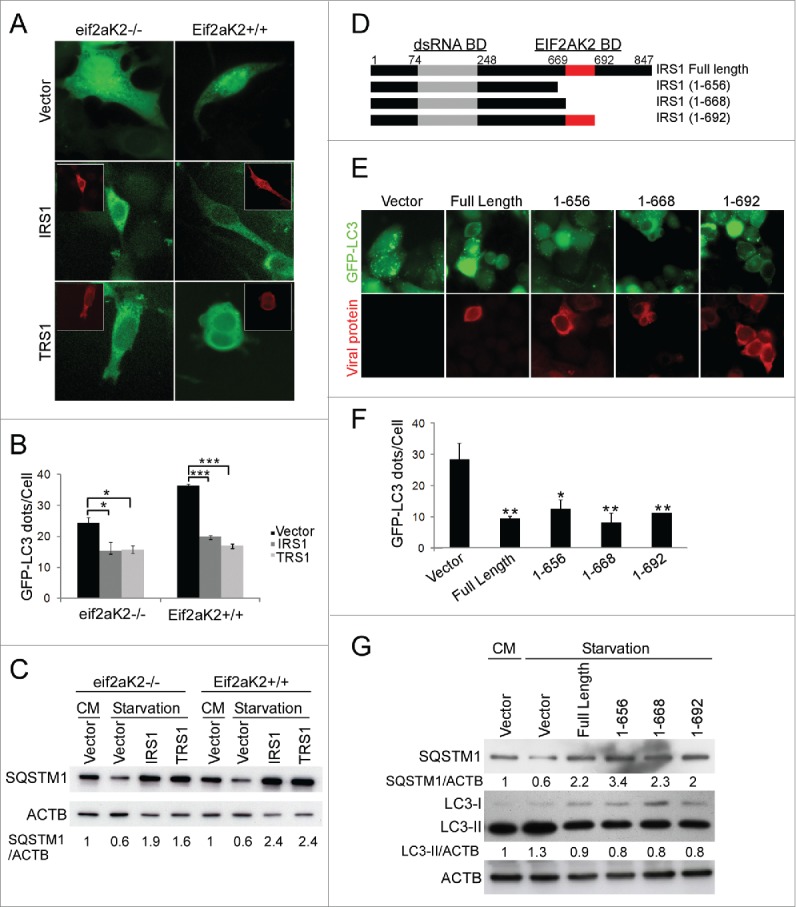
IRS1 and TRS1 inhibit autophagy independently of EIF2AK2. (A and B) eif2ak2−/− and Eif2ak2+/+ MEFs were cotransfected with GFP-LC3 and IRS1 or TRS1 and 48 h later were grown in starvation medium for 4 h before fixation. (A) Representative images of GFP-LC3 with insets showing TRS1 or IRS1 expression (red), and (B) quantification of GFP-LC3 dots per cell. (C) Immunoblot analysis of SQSTM1 protein in lysates of eif2ak2−/− and Eif2ak2+/+ MEFs transfected with IRS1 or TRS1 plasmids. (D) Schematic representation of full-length and truncated IRS1 expression plasmids, showing positions of the dsRNA-binding domain (amino acids 74 to 248) and the EIF2AK2 binding domain (amino acids 669 to 692). (E) Representative images of GFP-LC3 HeLa cells transfected with the indicated IRS1 constructs and then starved for 4 h and (F) quantification of GFP-LC3 dots. (G) Immunoblot analysis of SQSTM1 and LC3 proteins in HeLa cells transfected with the indicated IRS1 constructs. CM, complete medium. *, P<0.05; **, P<0.01; ***, P<0.001 (One-way ANOVA).
IRS1 contains a EIF2AK2 binding domain located in the C-terminal domain (an essential component of which lies between amino acids 669 and 692).16 We transiently transfected HeLa cells stably expressing GFP-LC3, with full length or C-terminal truncations of IRS1 (Fig. 2D). We observed that the truncated forms of IRS1 blocked LC3 dots accumulation (Fig. 2E and F). We also observed by immunoblot an accumulation of SQSTM1 and a decrease of LC3-II in HeLa cells transfected with the different IRS1 constructs (Fig. 2G). Taken together, our results showed that all C-terminally truncated IRS1 proteins inhibited starvation-induced autophagy (Fig. 2E to G). Notably, the region of IRS1 between codon 655 and 692, which is essential for EIF2AK2-binding, is not required for autophagy inhibition. We obtained similar results with TRS1.8
IRS1 and TRS1 inhibit autophagy by interaction of their N-terminal domain with BECN1
With the finding that the EIF2AK2 binding domain of IRS1 and TRS1 is dispensable to their inhibitory effects, we tested whether the autophagy machinery protein BECN1 is involved, since several viral proteins interact with it. BECN1, the mammalian ortholog of yeast Vps30/Atg6, along with PIK3C3 (mammalian ortholog of yeast Vps34), which is the catalytic subunit of the class III phosphatidylinositol 3-kinase (PtdIns3K), and with other core autophagy proteins, forms several complexes which are required for the initiation of autophagosome formation and endocytic trafficking.21 The activity of BECN1 is modulated by diverse cellular stimuli leading to phosphorylation and ubiquitination modifications or to relocalization of BECN1 or via protein-protein interactions.21 For example, the antiapoptotic BCL2 family members can interact with BECN1 and inhibit autophagy.22 We analyzed by confocal microscopy the distribution of the 2 viral proteins and BECN1 in MRC5 cells infected with HCMV for 24 h. The staining pattern of IRS1 and TRS1 was widespread in the cytoplasm and BECN1 was labeled in punctate structures (Fig. 3A). Importantly, TRS1 and IRS1 showed substantial overlap with BECN1.
Figure 3.

IRS1 interacts with BECN1 and inhibits autophagy via its N-terminal region. (A) Colocalization of IRS1 and TRS1 with BECN1 in MRC5 cells infected with HCMV 24 h pi by confocal microscopy. (B) Schematic representations of different IRS1 N-terminal-deleted fragments and BECN1-deleted fragments. (C) Immunoprecipitation assays of BECN1 in HeLa cells transfected with EGFP-His, different constructs of His-IRS1 or full length His-TRS1. (D) Immunoprecipitation assays of FLAG-BECN1 in HeLa cells transfected with full-length His-IRS1 and different constructs of BECN1. (E) GFP-LC3 dots were quantified in GFP-LC3 HeLa cells transfected for 48 h with the indicated IRS1 constructs. (F) Immunoblot analysis of SQSTM1 levels in HeLa cells transfected with the indicated constructs and then grown in starvation medium for 4 h before cell lysis. The results are the mean of 3 experiments; 100 cells were analyzed per assay. **, P<0.01 (One-way ANOVA).
To investigate whether IRS1 interacts with BECN1, we performed a coimmunoprecipitation experiments with 6×His-tagged IRS1 and BECN1 plasmids. We found that IRS1 bound to BECN1 (Fig. 3C). In order to delineate the domain of IRS1 which interacts with BECN1, we analyzed 2 truncations of IRS1 from the amino termini, IRS1(45 to 847) and IRS1(93 to 847) (Fig. 3B). Coimmunoprecipitation experiments revealed that the 2 amino-terminal mutants of IRS1 did not bind to BECN1 (Fig. 3C). EGFP-His was used as a negative control and full length IRS1 and TRS1 as positive controls. These results demonstrated that deletion of the N-terminal domain of IRS1 abolishes the interaction with BECN1. BECN1 contains 3 major protein binding domains, an N-terminal BH3 (BCL2 homology 3) domain, a central coiled-coil domain (CCD), and an evolutionarily conserved domain at the C terminus.21 Using different fragments of FLAG-BECN1 (Fig. 3B), we determined the BECN1 domain that interacts with IRS1. IP assays showed that the (141 to 450) and (141 to 265) BECN1 fragments immunoprecipitated IRS1, indicating that IRS1 interacts with the CCD domain of BECN1 (Fig. 3D). The (1 to 255) BECN1 fragment did not interact with IRS1, possibly because of effects of the N terminus on the folding of the truncated CCD.
We then assessed the ability of the amino-terminal truncations mutants of IRS1 to inhibit starvation-induced autophagy. HeLa cells stably expressing GFP-LC3 were transiently transfected with full-length IRS1, TRS1 or with 2 IRS1 N-terminal-deleted fragments and GFP-LC3 dots were analyzed by immunofluorescence. TRS1 was used as positive control. We observed that IRS1 mutants (45 to 847) and (93 to 847) failed to inhibit autophagy as efficiently as full-length IRS1 or TRS1 (Fig. 3E). We also observed that cellular levels of SQSTM1 only partially increased in cells transfected with the 2 IRS1 mutants compared to cells transfected with full-length IRS1 or TRS1 (Fig. 3F). Thus, the inhibition of autophagy by IRS1 requires its N-terminus. These results suggest that IRS1 and TRS1 inhibit autophagy as a result of their interaction with BECN1 by this N-terminal domain.
IRS1 and TRS1 are both involved in the control of autophagy during infection
In order to investigate the role of IRS1 and TRS1 in autophagy blocking in the context of viral infection, we analyzed autophagy in cells infected with HCMV mutants lacking either IRS1 or TRS1 genes or both (Fig. 4A and B). In HCMV-infected cells, we observed, as previously reported,8,23 that the number of autophagosomes was significantly decreased. In contrast, the number of autophagosomes was significantly increased after infection with mutant HCMV lacking both IRS1 and TRS1 (HCMV [ΔI-ΔT]), compared to mock-infected cells and HCMV-infected cells. Furthermore, the IRS1 and TRS1 single mutant viruses each inhibited autophagosome formation relative to HCMV [ΔI-ΔT], but not as strongly as the wild-type (WT) virus. Accumulation of SQSTM1 confirmed that whereas HCMV [ΔI-ΔT] drastically stimulated autophagy, WT HCMV blocked autophagy and the 2 single mutant viruses had an intermediate phenotype (Fig. 4C). Autophagic flux can be measured by inferring LC3-II turnover by western blot in the presence and absence of chloroquine (CQ). CQ neutralizes the lysosomal pH and by inhibiting endogenous protein degradation causes the accumulation of LC3-II in either autophagosomes or autolysosomes.24 The relevant parameter in this assay is the ratio in the amount of LC3-II in the presence and absence of CQ, which can be used to examine the transit of LC3-II through the autophagic pathway. When autophagic flux is occurring, the amount of LC3-II is higher in the presence of CQ. As is evident in Figure 4D, HCMV infection leads to a similar accumulation of LC3-II with and without CQ, whereas none of the mutant viruses was able to do so. We previously reported the accumulation of LC3-II in WT HCMV-infected cells, which is not correlated with the number of autophagosomes.8,23 This result suggests that the 2 viral proteins are necessary to inhibit autophagic flux. In support of this hypothesis, we found that cotransfection of IRS1 and TRS1 in HeLa cells clearly led to LC3-II accumulation and a complete block of autophagic flux (Fig. 4E). Taken together, these results demonstrate that IRS1 and TRS1 are both involved in a complementary manner in the regulation of autophagy by HCMV.
Figure 4.

IRS1 and TRS1 are involved in the inhibition of autophagy after HCMV infection. (A) MRC5 cells were mock-infected or infected with HCMV, ΔIRS1-ΔTRS1, ΔIRS1 and ΔTRS1 mutant viruses at an MOI of 1, transfected with GFP-LC3 and then fixed at 24 h pi. Cells were fixed and immunostained for pp65 or IEA viral proteins as indicated (scale bar: 10 µm). (B) GFP-LC3 dots were quantified in pp65- or IEA-positive cells. The results are the mean of 6 independent experiments. Twenty-five cells were counted per assay. **, P<0.01; ***, P<0.001 (One-way ANOVA). (C and D) Immunoblot analysis of SQSTM1 and LC3 proteins in mock-infected MRC5 cells or infected with WT HCMV or the indicated mutant viruses for 24 h. (E) Immunoblot analysis of LC3 protein in cells transfected with the indicated constructs for 24 h and grown in starvation medium for 4 h before cell lysis. (D and E) Chloroquine was added 4 h before lysis to monitor autophagic flux.
The BECN1-binding-deficient TRS1 mutant virus is unable to control autophagy
Next, we investigated the functional significance of TRS1 and IRS1 interaction with BECN1 in the context of HCMV infection. Based on a previously described BAC-derived HCMV strain AD169 lacking both TRS1 and IRS1,14 we constructed mutant viruses in which an HA-tagged version of the TRS1 gene was reintroduced either with a deletion of amino acids 1 to 44 (referred to as TRS1(45–795)-ΔIRS1 HCMV) or the complete gene (referred to as TRS1HA-ΔIRS1 HCMV) (Fig. 5A). TRS1(45–795)- ΔIRS1 HCMV and TRS1HA- ΔIRS1 HCMV express a protein of the predicted molecular weight (80 kDa and 85 kDa, respectively) that reacts with an anti-TRS1 and anti-IRS1 antibody (Fig. 5B). Whereas TRS1 coimmunoprecipitates with BECN1 in TRS1HA-ΔIRS1 HCMV infected cells, interaction between TRS1(45–795) and BECN1 is greatly reduced (Fig. 5C). We evaluated the ability of this BECN1-binding-deficient TRS1 mutant virus to control autophagy measuring accumulation of GFP-LC3 dots and SQSTM1 and LC3 immunoblots. We observed that infection with TRS1(45–795)-ΔIRS1 HCMV did not decrease the number of autophagosomes compared to mock-infected cells while the repaired virus, TRS1HA- ΔIRS1 HCMV, partially inhibited autophagy (Fig. 5D). Moreover, TRS1(45–795)-ΔIRS1-infected cells accumulated less SQSTM1 protein and more LC3-II compared to TRS1HA- ΔIRS1-infected cells (Fig. 5E). Adding CQ showed that only the parental BAC-derived AD169 blocked autophagic flux, in agreement with our previous results (Fig. 4). Taken together, these results show that the BECN1-binding-deficient-TRS1 mutant virus is not able to inhibit autophagy.
Figure 5.

The BECN1-binding-deficient TRS1 mutant virus does not inhibit autophagy. (A) Schematic representation of the HA-tagged TRS1 recombinant viruses. (B) Expression of TRS1 and IRS1, detected by immunoblotting with anti-TRS1 and IRS1 antibody of cells lysates after infection with the indicated viruses. (C) MRC5 cells were infected with TRS1(45–795)-ΔIRS1 and TRS1HA-ΔIRS1 recombinant viruses for 48 h. Cells were immunoprecipitated with a goat anti BECN1 antibody, followed by immunoblotting with an anti-HA antibody and anti-BECN1. (D) MRC5 cells were infected with the indicated viruses, transfected with GFP-LC3 and then fixed 24 h pi. Autophagy was quantified in IEA-positive cells by counting the number of GFP-LC3 dots per cell. The results are the mean of 3 independent experiments. Twenty cells were counted per assay. ***, P<0.001 (One-way ANOVA). (E) Immunoblot analysis of SQSTM1 and LC3 levels in MRC5 cells infected with the indicated viruses at MOI 1. ACTB was used as a loading control. CQ, chloroquine.
An HCMV recombinant virus containing a mutation in TRS1 that abrogates binding to BECN1 does not have a growth defect
We sought to determine whether the interaction of TRS1 with BECN1 and the ability to block autophagy is important for virus replication. We hypothesized that a virus that is unable to control autophagy by virtue of deletion of all of IRS1 and the BBD of TRS1 would replicate less efficiently. We therefore analyzed the growth kinetics of the different mutant viruses in human fibroblasts (Fig. 6). The multiple-step growth kinetics of wild-type (AD169-BAC) and deletion mutant viruses were examined in cells infected at a low multiplicity of infection and virus titers were determined. The TRS1(45–795)-ΔIRS1 recombinant virus achieved titers comparable to those of TRS1HA-ΔIRS1 and AD169-BAC (Fig. 6A). When MRC5 cells were infected with HCMV [ΔI-ΔT], no progeny virus was detected for the duration of the assay (20 days), consistent with the previous report.25 For single-step growth analysis, the production of extracellular virus and intracellular virus was studied at the indicated times after infection.26 The amount of infectious virus produced at various times post infection (pi) was determined by using an immunoenzymatic assay in which the number of immediate early antigen (IEA)-positive cells was determined at 2 d pi (see Materials and Methods). Compared to the wild type (AD169-BAC), the TRS1(45–795)-ΔIRS1 mutant virus produced similar amounts of infectious intracellular and extracellular virus at 4 and 6 d pi (Fig. 6B). This set of experiments indicates that lack of control of autophagy confers no growth disadvantage to the mutant virus.
Figure 6.

Characterization of a HCMV recombinant virus containing a mutation in TRS1 that abrogates binding to BECN1. (A) Multiple-step growth analysis. MRC5 cells were infected at MOI of 0.02 with the indicated viruses. Cultures were harvested at the indicated times pi, and viral titers were determined by TCID50 assay. Results represent the averages of 3 independent experiments. (B) Single step growth analysis was performed to examine the production of extracellular virus and intracellular virus. MRC5 cells were infected at MOI 0.5 with AD169-BAC (WT), TRS1(45–795)-ΔIRS1, or TRS1HA-ΔIRS1 recombinant viruses. Infected cell culture medium were harvested at the indicated times pi (cell-free), and intracellular virus was isolated by freezing and thawing cell pellets (cell associated). The amount of virus present in each sample was determined by infecting MRC5 cells with these samples and counting the number of IEA-positive cells. Results represent the averages of 2 independent experiments. (C) Lysates from HCMV-infected cells were prepared at 1, 2 and 5 d pi and immunoblotted using antibodies specific for IEA, αp999 antiserum specific for pIRS1 and pTRS1, and for pp28. (D) Viral DNA genome after infection of MRC5 cells with the indicated viruses, 2 to 4 d pi.
HCMV transcribes its genes in a temporal manner, which is classified into 3 stages: immediate-early, early, and late. We used immunoblot assays to monitor expression of the immediate early (IE1 and IE2) proteins, TRS1 and IRS1 proteins and a late (pp28) protein in cells infected for 1, 2 or 5 d with the various viruses (Fig. 6C). IE1 and IE2 proteins were expressed at identical levels in cells infected with any of the viruses, at any time pi. We also detected similar expression of the late protein pp28 5 d pi. We then examined viral genome accumulation relative to cellular gene copies at different times pi (Fig. 6D). Viral DNA of TRS1(45–795)-ΔIRS1 mutant virus accumulated to similar levels as WT virus. Together, these results suggest that the IRS1 and TRS1 autophagy modulating activity does not have a direct role in promoting viral replication in terms of viral protein expression, viral DNA replication, viral production or release in human fibroblasts.
Inhibition of autophagy decreases viral production
Finally, with the aim of clarifying the impact of autophagy modulation on viral replication, we sought to determine whether modulation of autophagy by pharmacological approaches could impact the viral multiplication. We used serum starvation, rapamycin and methyl β cyclodextrin (MβCD) treatment to induce the formation of autophagosomes.27 These treatments stimulated autophagy in MRC5 cells (Fig. 7A).8,23 Then, we infected cells with HCMV at low multiplicity of infection (MOI) and treated them from 18 h to 4 d pi. This protocol enabled us to avoid nonspecific effects of the drugs on viral entry and early stages of the viral cycle. We observed that extracellular and intracellular viral production was higher in starved cells and in cells treated with MβCD or rapamycin than in nontreated cells (Fig. 7B). Next, we evaluated the effect of Spautin 1, a potent and specific inhibitor of autophagy (Fig. 7A), on viral production.28 As shown in Figure 7C, we observed that Spautin 1-repressed autophagy inhibited viral replication. In cells infected at low MOI and treated with Spautin 1, both intra and extra cellular viral productions were strongly decreased compared to nontreated cells. These results suggest that autophagy is able to modulate viral production but does not affect viral exit from infected cells, since the effect is similar on extra and intracellular production. To investigate whether cellular autophagy regulates HCMV at the replication level, we examined by qPCR the effects of these drugs added 18 h pi on viral genome accumulation. Figure 7D shows that Spautin 1 treatment profoundly decreased viral DNA replication. No effect of starved-induced autophagy induction was observed whereas MβCD and rapamycin decreased DNA replication by 40%. Finally, quantification of infected cells 4 d pi after different modulating treatments confirmed that induction of autophagy increased the level of infection and conversely, inhibition strongly reduced it (Fig. 7E).
Figure 7.

Autophagy affects HCMV infection. (A) SQSTM1 immunoblots and LC3 staining in MRC5 cells under starvation or after the indicated treatments. (B) MRC5 cells were infected with HCMV (MOI 0.02), treated with MβCD, rapamycin or serum starved 18 h pi and analyzed for viral production after 4 d. (C) MRC5 cells were infected in the same conditions then treated with Spautin 1 and viral production was analyzed. (D) Viral DNA genome replication after the indicated treatments 4 d pi. (E) Images and quantification of IEA expression in MRC5 cells infected with HCMV for 4 d and treated with the indicated drugs. Representative assay of 3 independent experiments (F) Immunoblot analysis of ATG16L1, LC3, SQSTM1 and viral proteins expression in ATG16L1 shRNA- or scrambled RNA-expressing cells. (G) Viral DNA genome replication and IEA expression after 4 d. NT no treatment.
To complement these studies using pharmacological modulation of autophagy, we also conducted genetics experiments. ATG16L1 forms a complex with ATG5 and ATG12 that is essential for autophagosome biogenesis. We therefore established human foreskin fibroblasts stably expressing ATG16L1 shRNA or scrambled shRNA. We confirmed successful knockdown of the ATG16L1 protein and analyzed its effects on autophagy by ATG16L1, SQTSM1 and LC3 immunoblot assays (Fig. 7F). Knocking down ATG16L1 with either of 2 shRNAs decreased autophagy compared to scrambled shRNA control. After infection with WT HCMV at MOI 0.02, we observed that viral DNA replication was decreased in both ATG16L1 knockdown cells compared to the control cell line (Fig. 7G). We observed that expression of TRS1 and IRS1 was similar in the 3 cell lines. We examined the effect of shRNA-mediated downregulation on expression of immediate early proteins 4 d pi, which reflect level of infection. Blocking autophagosome formation significantly reduced HCMV infection (Fig. 7G). These results provide additional evidence that inhibition of autophagy reduces HCMV infection.
Discussion
In this report, we demonstrated that HCMV blocks both autophagosome formation and maturation, dependently of viral protein synthesis. We previously identified an antiautophagic function of the viral protein TRS1 and we report here that IRS1, which has homologous regions with TRS1, is also able to decrease the accumulation of autophagosomes.8,9 Based on several readouts to monitor autophagic activity, we demonstrated that autophagy is inhibited by both TRS1 and IRS1 proteins in starved and in infected cells.24 The use of a high throughput microscopy approach, previously developed by McKnight et al.,29 allowed us to precisely quantify the ability of TRS1 or IRS1 expression to decrease not only the number of GFP-LC3 dots but also the intensity of the dots and the total area covered by the GFP-LC3 positive dots. Only the size of autophagosomes remained unchanged. Moreover, we noticed that simultaneous expression of TRS1 and IRS1 leads to a block of the autophagic flux.
In order to examine the respective roles of TRS1 and IRS1 in the context of infection, we constructed mutant viruses that do not express TRS1, or IRS1 or either protein. Whereas the 2 viruses lacking either TRS1 or IRS1 each partially control autophagy, deletion of both proteins leads to a marked stimulation of autophagy. However, it is important to note that HCMV [ΔI-ΔT] does not replicate in wild-type fibroblasts, as a result of protein synthesis shutoff.14 Most viral proteins are therefore not expressed. In the same way, when all de novo viral protein expression is prevented by UV-inactivation, HCMV stimulates autophagy.8 We also demonstrated that HCMV infection leads to a profound inhibition of the autophagic flux, resulting of an accumulation of LC3-II. Both TRS1 and IRS1 are necessary to clearly establish this blockade, since none of the single deletion mutant viruses is able to act the same way as WT virus.
We characterized the mechanism by which the viral proteins modulate autophagy. TRS1 and IRS1 have been identified as viral tegument proteins that localize to the nucleus and cytoplasm in infected cells.30,31 They both have been reported to activate transcription in cooperation with other viral activators and TRS1 might be involved in the assembly of virus capsids.12,32-34 Both TRS1 and IRS1 interact with UL44, a processivity factor of the DNA polymerase, but not simultaneously.35 TRS1 has recently been reported to bind to mRNA caps and stimulate translation.36 However, the best-characterized function known for these 2 large proteins is the repression of EIF2AK2 activity.14 In addition to a dsRNA binding domain that is identical in TRS1 and IRS1, they both have homologous but not identical C-terminal regions that are required for binding to EIF2AK2. Interestingly, we have previously demonstrated that the herpes simplex virus type 1 (HSV-1) late protein Us11 is able to block autophagy, by direct interaction with EIF2AK2.20 However, we demonstrated here that the mechanism by which TRS1 and IRS1 inhibit autophagy is independent of their activity on EIF2AK2. Instead, TRS1 and IRS1 colocalize with and bind to the autophagy machinery protein BECN1. Moreover, their identical N-terminal regions are required both for this interaction and for their antiautophagic activity. Interestingly, this region of IRS1 or TRS1 is not required for interaction with UL44, EIF2AK2 or dsRNA.15,16,35
Several viral proteins that are able to modulate autophagy act by interaction with BECN1.21 Whereas the adenovirus E1B19K protein triggers autophagy by displacement of the antiautophagic protein, BCL2, from the BECN1 interactome,37 all the other viral BECN1-interacting proteins disrupt autophagy, either at the autophagosome formation or at the maturation step.21 The Nef protein of HIV-1,38 M2 protein of influenza virus39 and ICP34.5 of HSV-140 suppress autophagosome maturation into autolysosomes, likewise via their interaction with BECN1.22 Viral encoded BCL2 proteins block autophagosome formation through a direct interaction with the BH3 domain of BECN1, as does cellular BCL2,22 while HSV-1 ICP34.5 and HCMV IRS1 do not interact with the BH3 domain of BECN1.19 We found that the CCD domain in BECN1, a universal oligomerization domain, is the binding domain of IRS1. Notably, this CCD domain allows ATG14 and UVRAG (UV radiation resistance associated) to interact with BECN1 respectively in 2 distinct PtdIns3K-containing complexes that function differentially in autophagy formation and in maturation of the endosome and the autophagosome.41 Interestingly, our results suggest that individually IRS1 and TRS1 may block autophagosome biogenesis through the PtdIns3K-BECN1-ATG14 complex, and coexpression of them may block the maturation process through the PtdIns3K-BECN1-UVRAG complex.
Infection with HCMV [ΔI-ΔT] virus leads to a global shutdown of proteins synthesis because of the lack of control of EIF2AK2.25 In order to retain the inhibitory activity of TRS1 on EIF2AK2 (but not on BECN1), we constructed an HCMV ΔIRS1 recombinant virus containing a mutation in TRS1 that abrogates binding to BECN1 (TRS1(45–795)-ΔIRS1). We found that this virus is not able to inhibit autophagy. This finding supports that conclusion that HCMV uses both TRS1 and IRS1 to block autophagy.
Autophagy can contribute to antiviral defenses and it has been reported in others studies that autophagy can directly reduce viral multiplication. For example, autophagy has an inhibitory effect on chikungunya virus propagation.42 In the case of HCMV, our results show that the inability to control of autophagy by the mutant virus HCMV TRS1(45–795)-ΔIRS1 has no impact on viral multiplication. Viral protein expression, viral replication, intracellular viral production, and viral release were not different in HCMV TRS1(45–795)-ΔIRS1-infected cells compared to WT virus, suggesting that, at least in cultured human fibroblasts, autophagy is not detrimental to viral propagation. It is interesting to note that HSV-1 lacking the ICP34.5 BECN1-binding domain grows as well as WT HSV-1 in fibroblasts in vitro but is clearly neuroattenuated in mice.19 It is possible that inhibition of autophagy by HCMV may be immune-response related and therefore apparent in natural infections of humans but not in fibroblast culture. It will be interesting to explore whether HCMV has different consequences in other cells types. We noted that the TRS1(45–795)-ΔIRS1 recombinant virus, unlike the HCMV [ΔI-ΔT], does not stimulate autophagy after 24 h of infection although autophagy is induced by this virus quickly after infection, in the same manner as WT virus (data not shown). Among the possible explanations for these results is that another viral protein, not expressed in HCMV [ΔI-ΔT]-infected cells but expressed by this virus, participates to the control of autophagy. This might explain the lack of effect on viral replication. Further investigations will be needed to determine whether HCM encodes additional autophagy-modulating proteins.
Since the recombinant virus TRS1(45–795)-ΔIRS1 seems not able to fully block autophagy, we decided to use pharmacological approaches and ATG16L1 knockdown cells to study the impact of the modulation of autophagy on viral multiplication. Surprisingly, we observed that activation of autophagy enhanced HCMV infectivity, whereas its inhibition decreased viral production. We noticed similar effects of the drugs on extra and intracellular viral yields, suggesting that autophagy does not influence release of the virus. Viral DNA replication was severely repressed by autophagy inhibition but was not increased by autophagy induction. These findings suggest that stimulation of autophagy may contribute to HCMV infection after viral DNA replication. Conversely, inhibition of autophagy seems to have an impact earlier in the viral replication cycle. Taken together, these results show that autophagy has a proviral role in HCMV replication. These unexpected findings in HCMV exhibit similarities with recent report of a proviral role for autophagy during Epstein Barr virus reactivation, a gammaherpesvirus.43 The Epstein Barr virus inhibits autophagosome degradation and seems to utilize the molecular machinery of autophagy for its envelope. Another Herpesvirus, varicella zoster virus induces an autophagic response which has a proviral effect, that may be related to the fact that autophagy improves biosynthesis and processing of the viral glycoprotein gE.44 In fact, one has to wonder whether HCMV subverts autophagosome formation and maturation in order to avoid degradation in some cell types and to utilize the autophagic machinery to its own profit. Indeed, recent studies have demonstrated that several DNA viruses utilize autophagosomes or the autophagic process to benefit their viral life cycle. For example, autophagy positively affects infection of adenovirus, hepatitis B virus, and human BK polyomavirus.45–47 Interestingly, inhibition of autophagy is now considered to be possibly used as a therapeutic strategy in cancer and in neurodegenerative diseases,48,49 and it could be attractive to explore that possibility for HCMV.
Materials and methods
Cells and virus
Primary human embryonic lung fibroblasts MRC5 were purchased from Biomérieux, and used between passages 23 and 28 post isolation. These cells were maintained in minimum essential medium (Gibco®, 21090–022) supplemented with 10% fetal calf serum (FCS), penicillin G (100U/ML), streptomycin sulfate (100µg/ml), l-glutamine (1%), and nonessential amino acids (1%). HeLa cells were cultured in RPMI (Gibco®, 61870–10) 10% FCS. GFP-LC3 stably-transfected HeLa cells50 were provided by Aviva Tolkovsky, (Cambridge Centre for Brain Repair, Cambridge, UK) and were grown in RPMI 10% FCS with 500 µg/ml of G418. Eif2ak2+/+ and eif2ak2−/− MEFs kindly provided by B. R. G. Williams (Monash University, Victoria, Australia), and human foreskin fibroblasts, provided by Thomas Shenk (MolBio Department, Princeton University, Princeton USA) were maintained in DMEM (Gibco®, 41965–039) supplemented with 10% FCS. The pTRS1-expressing cell line, HF-2.7βTRS1 was described elsewhere.14 The WT virus used in this study was the AD169 strain. The AD169 strain of HCMV was obtained from ATCC and was propagated in MRC5 cells as previously described.51 All mutant viruses were constructed on the basis of the AD169 bacterial artificial chromosome (BAC), which contains the full-length genome of the HCMV AD169 strain.52 The AD169-BAC (WT) virus and the mutants AD169 ΔIRS1, AD169 ΔTRS1, and AD169 ΔIRS1-ΔTRS1 are previously described.14 The mutants TRS1HA-ΔIRS1 and TRS1(45–795)-ΔIRS1 were generated by reinserting either the full-length TRS1 coding sequence or a truncated TRS1 sequence lacking the first 44 codons into the AD169 ΔIRS1-ΔTRS1 BAC. Briefly, oligonucleotide primers containing 50-nucleotide homologies immediately up- and downstream of TRS1 were used to amplify HA-tagged TRS1 and a kanamycin resistance marker flanked by FLP recombination target (FRT) sites. Forward and reverse primers for full-length TRS1 were 5′-TGACGCGGGTTTGCTTCCTATATAGTGGACGTCGGAGGTGTCCGGCGCCCATGGCCCAGCGCAACGGCAT-3′ and 5′-GGATGTCTGGTACTTATCACTGGCGTCGTTATAACATTGTAAAACAAGTTTTCGAAACATAACGACAGCTGCAAAAGAAAACCAGT-3′. For the truncated TRS1, 5′-TGACGCGGGTTTGCTTCCTATATAGTGGACGTCGGAGGTGTCCGGCGCCCATGACTGGTGCGAGTGCTGC-3′ and the same reverse primer as for full-length TRS1 was used. Plasmid pBS-TRS1HA-fk served as PCR template. The PCR product was used to insert the full-length or truncated TRS1 gene into the AD169 ΔIRS1-ΔTRS1 BAC by homologous recombination in Escherichia coli strain EL250.53 The kanamycin resistance marker was subsequently removed by arabinose-induced FLP recombinase as described.53 All the recombinant viruses, except the AD169 ΔIRS1-ΔTRS1 virus, were propagated and their titers were determined in MRC5 cells. The AD169 ΔIRS1-ΔTRS1 virus was grown and its titer was determined in HF-2.7βTRS1 cells.
Pharmacological modulators of autophagy
The compounds 3MA (M9281), Spautin 1 (SML0440), rapamycin (R8781) and MβCD (C4555) were purchased from Sigma and the concentrations used were 10 mM, 10 µM, 5 nM and 5 mM, respectively. When indicated, drugs were applied to cultured infected cells 18 h pi. Chloroquine (Sigma, C6628) was used at 50 µM 4 h previous cell lysis to block autophagic degradation.
Plasmids
The GFP-LC3 expression vector was kindly provided by Tamotsu Yoshimori (Research Institute for Microbial Diseases, Osaka University, Osaka, Japan).54 The FLAG-ICP34.5 plasmid was a gift from Bin He (University of Illinois, Chicago, USA).55 The FLAG-tagged BECN1 plasmids were a kind gift of Beth Levine (UT Southwestern, Dallas, USA). The pEQ1180 construct expresses full-length HCMV TRS1 protein cDNA.56 The pEQ1100 construct expresses enhanced green fluorescent protein (EGFP) (5). The pEQ1007 construct, containing a full-length HCMV IRS1 protein cDNA and the pEQ1002 (1 to 656), pEQ1010 (1 to 668), pEQ1033 (1 to 692) constructs, containing the indicated lengths of IRS1, have been previously described.16 The plasmids pEQ1455 (45 to 847) and pEQ1456 (93 to 847) were constructed by PCR amplification of pEQ1007 using forward primers 5′ ACCATGGGTGCAAGTACTGCGGGTTCG-3′ or 5′-ACCATGGTGGA-GCGGCAGGCGCTG-3′, respectively, with the reverse primer 5′-ATGATGAACGTGGTGAGGG-GCGTGT-3′.16 The resulting PCR product was cloned into pcDNA3.1/V5-His-TOPO (Invitrogen). All of these constructs have a carboxyl–terminal 6-His tag. Transfections were performed using the FuGENE HD transfection reagent (Roche), as previously described.8 Starvation-induced autophagy was carried out by culturing the cells in Earle's Balanced Salt Solution (EBSS, GIBCO) for 4 h before fixation.
Antibodies
To detect HCMV-infected cells, we used a murine monoclonal antibody directed against the viral proteins IE1 and IE2 (clone E13; Biomérieux, 11–003) and a murine monoclonal antibody directed against the tegument protein pp65 (Biomérieux, 11–002). A mouse monoclonal antibody against the late protein pp28 (clone CH19, sc-69749) was purchased from Santa Cruz. To detect His- and FLAG-tagged constructions, we used a rabbit antibody directed against 6xHis (Cell Signaling Technology, 2365) or a mouse antibody against FLAG (Sigma, F3165). Additional primary antibodies used in this study included anti-SQSTM1 (Abnova clone 2C11, H00008878-M01), anti-BECN1 (BD bioscience, 612112), anti-ATG16L1 (Clinisciences, PM040), and anti-ACTB/β-actin (Merck Millipore MAB1501 clone C4). For immunoprecipitation of BECN1, we used a goat antibody provided by Santa Cruz Biotechnology (sc-16647). Secondary fluorescein isothiocyanate (FITC), tetramethyl rhodamine isothiocyanate (TRITC)-conjugated, horseradish peroxidase-labeled goat anti-mouse or anti-rabbit secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (115–035–003, 111–035–003). The IRS1-TRS1 rabbit polyclonal antiserum αp999 directed against amino acids 74 to 248 of TRS1 has been previously described.14
Coimmunoprecipitation assays
To study the interaction between BECN1 and IRS1 or TRS1, HeLa cells were transfected to coexpress BECN1, and the various molecular partners (EGFP-His, His-IRS1, His-TRS1, IRS1(45 to 847) and IRS1(93 to 847) proteins). To identify the interaction domain of BECN1 with IRS1, HeLa cells were cotransfected with different constructions of FLAG-BECN1 and His-IRS1. To study the interaction between TRS1 and endogenous BECN1 in the context of infection, MRC5 cells were infected with a mutant virus TRS1(45–795)-ΔIRS1 or the repair virus TRS1HA-ΔIRS1. Forty-eight h after transfection or infection, the cells were washed with cold, sterile phosphate-buffered saline (PBS; Gibco®, 14200–067) and then lysed at 4°C for 2 h in lysis buffer (50 mM Tris HCl, 50 mM NaCl, 0.5% Triton X-100 (Sigma, T-8787) 0.5% deoxycholic acid (Sigma, D-6750), 0.2% bovine serum albumin (Sigma, A1595), 25 mM NaPPi, 50 mM NAF, 1 mM Na3VO4) followed by ultracentrifugation at 274,000xg at 4°C for 30 min to remove cell debris. Immunoprecipitation was performed overnight at 4°C with a goat polyclonal anti-BECN1 antibody or a mouse anti-FLAG antibody. Protein G-Sepharose beads were added for 1 h at 4°C and were washed 3 times with lysis buffer and 2 times with wash buffer (20 mM Tris HCl, 50 mM NaCl, 0.2% bovine serum albumin). The immune complexes were finally boiled for 5 min in loading buffer (62.5 mM Tris, pH 6.8, 10% glycerol, 1.5% SDS, 0.025% bromophenol blue, 8% β-mercaptoethanol) before being analyzed by SDS-PAGE.
Immunoblot analysis
MRC5 and HeLa cells were lysed in 65 mM Tris, pH 6.8, 4% SDS, 1.5% β-mercaptoethanol, and held at 100°C for 5 min. After SDS-PAGE, the proteins were electrotransferred onto a polyvinylidene difluoride membrane. After incubation in blocking buffer, the blots were probed overnight with specific antibodies, then incubated with secondary antibodies, followed by chemiluminescent detection, according to the manufacturer's instructions (Immobilon, Millipore). Scanning for quantification was monitored using ImageJ software.
Immunofluorescence analysis
Cell monolayers were washed 3 times with PBS, and then fixed with 3.5% paraformaldehyde in PBS or acetone. The cells were permeabilized using 0.2% Triton X-100 in PBS and incubated for 1 h in blocking buffer, and then with appropriate primary antibodies for 1 h or overnight. The cells were washed 3 times, and then incubated with appropriate secondary antibodies. To detect IRS1 or TRS1 expression, cells were stained with mouse anti-His Mab or rabbit anti-IRS1-TRS1 serum. Coverslips were mounted in Glycergel (Dako, C0563) and examined using a Nikon Eclipse 80i epifluorescence microscope (Nikon instruments, Champigny sur Marne, France) or a LSM510 Zeiss confocal microscope (Carl Zeiss Microscopy, Iena, Germany). Digitized images were stored, and overlaid to evaluate 2-color experiments. Photographic images were resized, organized, and labeled using Adobe Photoshop software.
Optimized automated quantification of GFP-LC3 dots by high throughput microscopy
GFP-LC3 HeLa cells were transfected with empty vector, IRS1 or TRS1 and fixed after 4 h of starvation. GFP-LC3 dots were quantified using a Thermo Cellomics ArrayScan VTI HCS Reader (Thermo Fisher, Pittsburgh, PA, USA). The analysis program detected DAPI-labeled nuclei, cell outline, GFP-LC3 dots and nontransfected rejected cells. The number of GFP-LC3 dots per cell, the total intensity of GFP-LC3 dots per cell, the total area covered by GFP-LC3 dots per cell and the average area covered by GFP-LC3 dots per well were determined by the ID view software.
Virus production
For multistep growth analysis, supernatant fractions of infected cells were harvested at the indicated times after infection at a MOI of 0.02 and virus titers were determined in triplicate by 50% tissue culture infective dose (TCID50) assay. For single-step growth analysis, viruses were collected from both cell-free supernatant fractions and infected cells at various time points pi and quantified as previously described.26 Cell-associated virus was isolated through 3 rounds of freezing and thawing in a liquid nitrogen bath. Serial dilutions of virus samples were plated on MRC5 cells. Infected cells were fixed 48 h pi and permeabilized in acetone/water at −20°C for 20 min. IEA-positive cells were labeled using the primary mouse antibody against IEA (clone E13) and the secondary goat anti-mouse antibody conjugated with horseradish peroxidase, and positive cells were quantified at the appropriate dilution. To determine viral DNA levels in infected cells, cells were harvested and lysed in the presence of proteinase K and total DNA was purified using the DNeasy Blood and Tissue kit (Qiagen, 69506) according to the manufacturer's instructions. Real-time PCR was performed using Taqman probes and primers specific for the UL123 viral gene, and ACTB or CXCR4 cellular genes, as previously described.57
Lentiviral delivery of shRNA
Stable ATG16L1 and scrambled shRNA cells were generated using MISSION shRNA lentiviruses, which were kindly provided by Patrice Codogno (INEM, Paris, France). Stable cells were cultured in puromycin-containing media (5 µg/ml; InvivoGen anti-pr-1).
Statistics
Data are expressed as means ± standard error of the means (SEM) and were analyzed with Prism software (GraphPad version 6.0) by using one-way analysis of variance (ANOVA), followed by the Dunnett test comparisons. P values less than 0.05 were considered statistically significant. Experiments were performed a minimum of 3 times.
Abbreviations
- 3MA
3-methyladenine
- ATG
autophagy-related
- BAC
bacterial artificial chromosome
- BBD
BECN1- binding domain
- CCD
coiled-coil domain
- CQ
chloroquine; dsRNA, double-stranded RNA
- EIF2AK2/PKR
eukaryotic translation initiation factor 2-α kinase 2
- EIF2S1
eukaryotic translation initiation factor 2 subunit α
- FCS
fetal calf serum
- HCMV
human cytomegalovirus
- HSV-1
herpes simplex virus type 1
- ICP34.5
infected cell protein 34.5
- IE1 and IE2
immediate early proteins
- IRS1
internal repeat short protein 1
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MβCD
methyl β cyclodextrin
- MEF
mouse embryonic fibroblast
- MOI
multiplicity of infection
- ORF
open reading frame
- pi
post infection
- PtdIns3K
phosphatidylinositol 3-kinase
- SQSTM1/p62
sequestosome 1
- TRS1
terminal repeat short protein 1
- UVRAG
UV radiation resistance associated
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Michael Crawford for assistance in constructing IRS1-deletion plasmids. We are also very grateful to Beth Levine for providing us with the BECN1 constructs. We thank the FHCRC Genomics Core for technical assistance. We thank Valérie Nicolas from cellular imaging MIPSIT facility and Claudine Delomenie from the Transprot facility for technical assistance. We thank Patrice Codogno for insightful discussions and help for shRNA knockdown.
Funding
This work was supported by institutional funding from the Institut National de la Santé et de la Recherche Médicale (INSERM), from Univ. Paris-Sud, from Agence Nationale de la Recherche (to A.E.), from NIH AI027762 (to A.P.G.), from DFG BR1730/3–2 (to W.B.), and from Tichrine University-Syria (to L.M.). L.D. was supported by an EMBO fellowship.
References
- 1.Richetta C, Faure M. Autophagy in antiviral innate immunity. Cell Microbiol 2013; 15:368-76; PMID:23051682; http://dx.doi.org/ 10.1111/cmi.12043 [DOI] [PubMed] [Google Scholar]
- 2.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/ 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol 2012; 24:21-31; PMID:22118953; http://dx.doi.org/ 10.1016/j.coi.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richetta C, Gregoire IP, Verlhac P, Azocar O, Baguet J, Flacher M, Tangy F, Rabourdin-Combe C, Faure M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathog 2013; 9:e1003599; PMID:24086130; http://dx.doi.org/ 10.1371/journal.ppat.1003599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird SW, Maynard ND, Covert MW, Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci U S A 2014; 111:13081-6; PMID:25157142; http://dx.doi.org/ 10.1073/pnas.1401437111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, Hohenberg H, Ghazal P, Brune W. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog 2012; 8:e1002517; PMID:22319449; http://dx.doi.org/ 10.1371/journal.ppat.1002517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tey SK, Khanna R. Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood 2012; 120:994-1004; PMID:22723550; http://dx.doi.org/ 10.1182/blood-2012-01-402404 [DOI] [PubMed] [Google Scholar]
- 8.Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, et al.. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol 2012; 86:2571-84; PMID:22205736; http://dx.doi.org/ 10.1128/JVI.05746-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol 2011; 85:4212-21; PMID:21325419; http://dx.doi.org/ 10.1128/JVI.02435-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cepeda V, Esteban M, Fraile-Ramos A. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol 2010; 12:386-404; PMID:19888988; http://dx.doi.org/ 10.1111/j.1462-5822.2009.01405.x [DOI] [PubMed] [Google Scholar]
- 11.Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, et al.. Decoding human cytomegalovirus. Science 2012; 338:1088-93; PMID:23180859; http://dx.doi.org/ 10.1126/science.1227919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romanowski MJ, Shenk T. Characterization of the human cytomegalovirus irs1 and trs1 genes: a second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J Virol 1997; 71:1485-96; PMID:8995674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol 2004; 78:197-205; PMID:14671101; http://dx.doi.org/ 10.1128/JVI.78.1.197-205.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall EE, Bierle CJ, Brune W, Geballe AP. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J Virol 2009; 83:4112-20; PMID:19211736; http://dx.doi.org/ 10.1128/JVI.02489-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakki M, Geballe AP. Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol 2005; 79:7311-8; PMID:15919885; http://dx.doi.org/ 10.1128/JVI.79.12.7311-7318.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hakki M, Marshall EE, De Niro KL, Geballe AP. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 2006; 80:11817-26; PMID:16987971; http://dx.doi.org/ 10.1128/JVI.00957-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bierle CJ, Semmens KM, Geballe AP. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology 2013; 442:28-37; PMID:23601785; http://dx.doi.org/ 10.1016/j.virol.2013.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 2002; 99:190-5; PMID:11756670; http://dx.doi.org/ 10.1073/pnas.012485299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007; 1:23-35; PMID:18005679; http://dx.doi.org/ 10.1016/j.chom.2006.12.001 [DOI] [PubMed] [Google Scholar]
- 20.Lussignol M, Queval C, Bernet-Camard MF, Cotte-Laffitte J, Beau I, Codogno P, Esclatine A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J Virol 2013; 87:859-71; PMID:23115300; http://dx.doi.org/ 10.1128/JVI.01158-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine B, Liu R, Dong X, Zhong Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol 2015; 25(9):533-44; PMID:26071895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 23.Chaumorcel M, Souquere S, Pierron G, Codogno P, Esclatine A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 2008; 4:46-53; PMID:18340111; http://dx.doi.org/ 10.4161/auto.5184 [DOI] [PubMed] [Google Scholar]
- 24.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marshall EE, Geballe AP. Multifaceted evasion of the interferon response by cytomegalovirus. J Interferon Cytokine Res 2009; 29:609-19; http://dx.doi.org/ 10.1089/jir.2009.0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 2007; 81:3109-23; PMID:17202209; http://dx.doi.org/ 10.1128/JVI.02124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng J, Ohsaki Y, Tauchi-Sato K, Fujita A, Fujimoto T. Cholesterol depletion induces autophagy. Biochem Biophys Res Commu 2006; 351:246-52; PMID:17056010; http://dx.doi.org/ 10.1016/j.bbrc.2006.10.042 [DOI] [PubMed] [Google Scholar]
- 28.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al.. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011; 147:223-34; PMID:21962518; http://dx.doi.org/ 10.1016/j.cell.2011.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKnight NC, Jefferies HB, Alemu EA, Saunders RE, Howell M, Johansen T, Tooze SA. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J 2012; 31:1931-46; PMID:22354037; http://dx.doi.org/ 10.1038/emboj.2012.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith RM, Kosuri S, Kerry JA. Role of human cytomegalovirus tegument proteins in virion assembly. Viruses 2014; 6:582-605; PMID:24509811; http://dx.doi.org/ 10.3390/v6020582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romanowski MJ, Garrido-Guerrero E, Shenk T. pIRS1 and pTRS1 are present in human cytomegalovirus virions. J Virol 1997; 71:5703-5; PMID:9188653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blankenship CA, Shenk T. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J Virol 2002; 76:12290-9; PMID:12414969; http://dx.doi.org/ 10.1128/JVI.76.23.12290-12299.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamo JE, Schroer J, Shenk T. Human cytomegalovirus TRS1 protein is required for efficient assembly of DNA-containing capsids. J Virol 2004; 78:10221-9; PMID:15367587; http://dx.doi.org/ 10.1128/JVI.78.19.10221-10229.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stasiak PC, Mocarski ES. Transactivation of the cytomegalovirus ICP36 gene promoter requires the α gene product TRS1 in addition to IE1 and IE2. J Virol 1992; 66:1050-8; PMID:1370547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strang BL, Geballe AP, Coen DM. Association of human cytomegalovirus proteins IRS1 and TRS1 with the viral DNA polymerase accessory subunit UL44. J Gen Virol 2010; 91:2167-75; PMID:20444996; http://dx.doi.org/ 10.1099/vir.0.022640-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziehr B, Lenarcic E, Vincent HA, Cecil C, Garcia B, Shenk T, Moorman NJ. Human cytomegalovirus TRS1 protein associates with the 7-methylguanosine mRNA cap and facilitates translation. Proteomics 2015; 15:1983-94; PMID:25894605; http://dx.doi.org/ 10.1002/pmic.201400616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piya S, White EJ, Klein SR, Jiang H, McDonnell TJ, Gomez-Manzano C, Fueyo J. The E1B19K oncoprotein complexes with Beclin 1 to regulate autophagy in adenovirus-infected cells. PloS One 2011; 6:e29467; PMID:22242123; http://dx.doi.org/ 10.1371/journal.pone.0029467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al.. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 2009; 186:255-68; PMID:19635843; http://dx.doi.org/ 10.1083/jcb.200903070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, et al.. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6:367-80; PMID:19837376; http://dx.doi.org/ 10.1016/j.chom.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gobeil PA, Leib DA. Herpes simplex virus gamma34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. MBio 2012; 3:e00267-12; PMID:23073763; http://dx.doi.org/ 10.1128/mBio.00267-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al.. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/ 10.1038/ncb1846 [DOI] [PubMed] [Google Scholar]
- 42.Joubert PE, Werneke SW, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow DJ, Albert ML. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med 2012; 209:1029-47; PMID:22508836; http://dx.doi.org/ 10.1084/jem.20110996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowag H, Guhl B, Thriene K, Romao S, Ziegler U, Dengjel J, Münz C. Macroautophagy Proteins Assist Epstein Barr Virus Production and Get Incorporated Into the Virus Particles. EBioMedicine 2014; 1:116-25; PMID:26137519; http://dx.doi.org/ 10.1016/j.ebiom.2014.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buckingham EM, Carpenter JE, Jackson W, Grose C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J Virol 2014; 88:890-902; PMID:24198400; http://dx.doi.org/ 10.1128/JVI.02646-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouley SJ, Maginnis MS, Derdowski A, Gee GV, O'Hara BA, Nelson CD, Bara AM, Atwood WJ, Dugan AS. Host cell autophagy promotes BK virus infection. Virology 2014; 456-457:87-95; PMID:24889228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol 2011; 85:6319-33; PMID:21507968; http://dx.doi.org/ 10.1128/JVI.02627-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez-Rocha H, Gomez-Gutierrez JG, Garcia-Garcia A, Rao XM, Chen L, McMasters KM, Zhou HS. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011; 416:9-15; PMID:21575980; http://dx.doi.org/ 10.1016/j.virol.2011.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/ 10.1038/nrd3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sehgal AR, Konig H, Johnson DE, Tang D, Amaravadi RK, Boyiadzis M, Lotze MT. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia 2015; 562:40-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 2005; 1:23-36; PMID:16874023; http://dx.doi.org/ 10.4161/auto.1.1.1495 [DOI] [PubMed] [Google Scholar]
- 51.Esclatine A, Bellon A, Michelson S, Servin AL, Quero AM, Geniteau-Legendre M. Differentiation-dependent redistribution of heparan sulfate in epithelial intestinal Caco-2 cells leads to basolateral entry of cytomegalovirus. Virology 2001; 289:23-33; PMID:11601914; http://dx.doi.org/ 10.1006/viro.2001.1122 [DOI] [PubMed] [Google Scholar]
- 52.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski UH. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J Virol 2000; 74:7720-9; PMID:10933677; http://dx.doi.org/ 10.1128/JVI.74.17.7720-7729.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 2001; 73:56-65; PMID:11352566; http://dx.doi.org/ 10.1006/geno.2000.6451 [DOI] [PubMed] [Google Scholar]
- 54.Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1-12; PMID:19200872; http://dx.doi.org/ 10.1016/S0076-6879(08)03601-X [DOI] [PubMed] [Google Scholar]
- 55.Verpooten D, Ma Y, Hou S, Yan Z, He B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J Biol Chem 2009; 284:1097-105; PMID:19010780; http://dx.doi.org/ 10.1074/jbc.M805905200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Child SJ, Brennan G, Braggin JE, Geballe AP. Species specificity of protein kinase r antagonism by cytomegalovirus TRS1 genes. J Virol 2012; 86:3880-9; PMID:22278235; http://dx.doi.org/ 10.1128/JVI.06158-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leruez-Ville M, Ouachee M, Delarue R, Sauget AS, Blanche S, Buzyn A, Rouzioux C. Monitoring cytomegalovirus infection in adult and pediatric bone marrow transplant recipients by a real-time PCR assay performed with blood plasma. J Clin Microbiol 2003; 41:2040-6; PMID:12734246; http://dx.doi.org/ 10.1128/JCM.41.5.2040-2046.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]